Introduction
It has previously been suggested that reactive
oxygen species (ROS) are involved in the pathogenesis and
progression of atherosclerotic diseases (1). Antioxidants protect against
atherosclerosis by preventing ROS-induced injury to endothelial
cells, which appears to be mediated by improving antioxidant
defenses and preserving mitochondrial function (2). Atherosclerosis is a chronic
inflammatory disease entailing an initial activation of
pro-inflammatory cytokines, which facilitates leukocyte
transmigration (3). Vascular
endothelial growth factor is a key mediator in the development of
T-cell priming, and vascular endothelial cells modulate the
endothelial induction of many genes involved in immune cell
transmigration (4,5). Various oxidative stress stimuli such
as local hypoxia and vascular injury may cause endothelial cell
dysfunction and stimulate increased permeability, and encourage
leukocyte transmigration into areas of inflammation by enhancing
the expression of cell adhesion molecules (6,7).
Accordingly, the inhibition of endothelial adhesion molecule
expression by drugs/agents with antioxidant effects may serve as a
potential therapeutic strategy for clinical atherosclerosis
(8).
Superoxide dismutase (SOD) is an enzyme that
catalyzes the dismutation of superoxide anion radical
(O2−), one of the ROS in cells, into oxygen
and H2O2, and this is an important
antioxidant defense in nearly all cells exposed to oxygen. SOD
plays a critical role in inhibiting the oxidative inactivation of
nitric oxide, thereby preventing peroxynitrite formation and
endothelial and mitochondrial dysfunction (9). In addition, SOD exerts powerful
anti-inflammatory effects. Treatment with SOD reduces peroxidation
reactions in the inflamed colon and ameliorates colonic
inflammatory changes in experimental colitis, which is related to a
reduction in adhesion molecule expression and leukocyte recruitment
into the inflamed intestine (10). Therefore, SOD may be an important
new therapy for the treatment of inflammatory bowel disease. Since
oxidative stress is critical to endothelial adhesiveness in
atherogenesis (11), SOD therapy
may prevent atherogenesis, which entails endothelial activation by
cytokines and agonists. However, these atheroprotective effects
require the targeted delivery of SOD into the cytoplasm of
endothelial cells. A recent study has shown that SOD conjugated
with antibodies alleviated endotoxin-induced leukocyte adhesion in
the cerebral vasculature and protected the brain from
ischemia-reperfusion injury (12).
The protein transduction domains or cell-penetrating
peptides have been shown to be involved in the successful delivery
of exogenous full-length fusion proteins into living cells in
vitro and in vivo (13,14). In the present study, the delivery
of SOD protein inside endothelial cells and monocytes in
vitro was conducted using the cell-permeable transactivator of
transcription (Tat) peptide in order to deliver exogenous proteins
into cells, as has also been previously undertaken (15,16). We also examined the
atheroprotective effect of transduced Tat-SOD in inflammatory
cytokine-induced leukocyte-endothelial interaction and
transmigration involving oxidative stress. We investigated whether
leukocyte recruitment to inflamed human umbilical vein endothelial
cells (HUVECs) was blocked by the antioxidant Tat-SOD. Monocyte
extravasation was examined by studying the induction of
intercellular junction proteins and matrix metalloproteinase (MMP)
proteins in tumor necrosis factor-α (TNF-α)-activated and
SOD-treated HUVECs. Furthermore, the blockade of nuclear factor-κB
(NF-κB) signaling by transduced Tat-SOD was elucidated in relation
to TNF-α-triggered monocyte transmigration.
Materials and methods
Materials
M199 and RPMI-1640 mediums, human epidermal growth
factor (hEGF) and hydrocortisone were obtained from Sigma Chemical
Co. (St. Louis, MO, USA), as were all other reagents, unless
specifically stated. Fetal bovine serum (FBS),
penicillin-streptomycin and trypsin-EDTA were purchased from Lonza
(Walkersville, MD, USA). Restriction endonuclease and T4 DNA ligase
were purchased from Promega Corporation (Madison, WI, USA).
Oligonucleotide primers were synthesized from Gibco-BRL (Grand
Island, NY, USA). Ni2+-nitrilotriacetic acid sepharose
was purchased from Qiagen (Valencia, CA, USA). Human SOD cDNA was
isolated using the polymerase chain reaction (PCR) technique.
Isopropyl-β-D-thiogalactoside (IPTG) was obtained from Duchefa
Biochemie (Haarlem, The Netherlands). Plasmid pET-15b and
Escherichia coli strain BL21 (DE3) were obtained from
Novagen (Darmstadt, Germany). Anti-vascular cell adhesion
molecule-1 (VCAM-1; Cat. no. sc-8304) and anti-polyhistidine (Cat.
no. sc-803) were obtained from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Anti-integrin β1 (Cat. no. MAB1778) was
purchased from R&D Systems (Minneapolis, MN, USA). Anti-β-actin
was purchased from Sigma Chemical Co. The NF-κB inhibitor SN50
(Cat. no. BML-P600-0005) was obtained from Enzo Life Sciences
(Farmingdale, NY, USA).
Expression and purification of Tat-SOD
protein
A Tat expression vector was prepared in our
laboratory as described previously (17). A Tat-SOD expression vector was
constructed to express the Tat peptides (RKKRRQRRR) as a fusion
protein with human SOD. The cDNA sequence for human SOD was PCR
amplified using the following sense and antisense primers (Fig. 1A). The SOD sense primer,
5′-CTCGAGGCGA CGAAGGCCGTGTGCGTG-3′ contains an XhoI site,
and the SOD antisense primer, 5′-GGATCCTTATTGGGCGATCCCA ATTAC-3′,
contains a BamHI restriction site. The PCR product was
excised with XhoI and BamHI, eluted, ligated into a
TA-cloning vector and a pTat vector using T4 DNA ligase, and cloned
into Escherichia coli DH5α cells. The human SOD gene was
fused with a 21 amino acid-Tat peptide in a bacterial expression
vector in order to produce a genetic in-frame Tat-SOD fusion
protein. Similarly, the PCR product excised with XhoI and
BamHI was subcloned into the XhoI and BamHI
sites of pET-15b in order to construct control SOD that expressed
the SOD fusion protein without the Tat peptides.
To produce the SOD fusion proteins (Tat-SOD and
control SOD), the plasmid was transformed into E. coli BL21
cells, as previously described (15). Transformed bacterial cells grown
in 100 ml LB media at 37°C to a D600 value of 0.5–1.0 were induced
with 0.5 mM IPTG at 37°C for 4 h. Harvested cells were lysed by
sonication at 4°C in a binding buffer (5 mM imidazole, 500 mM NaCl,
20 mM Tris-HCl, pH 7.9), and the formed recombinant Tat-SOD was
purified using an Ni2+-nitrilotriacetic acid sepharose
affinity column (Qiagen) under native conditions. After the column
was washed with 10 volumes of a binding buffer and six volumes of a
wash buffer (60 mM imidazole, 500 mM NaCl, and 20 mM Tris-HCl, pH
7.9), the fusion proteins were eluted in a buffer (250 mM
imidazole, 500 mM NaCl, 20 mM Tris-HCl, pH 7.9). The fusion
proteins containing Tat-SOD fractions were combined and the salts
removed using PD-10 column chromatography (GE Healthcare
Biosciences, Pittsburgh, PA, USA). Protein concentration was
measured by the Bradford procedure using bovine serum albumin as a
standard.
Transduction of Tat-SOD protein into
HUVECs
HUVECs were isolated from the umbilical cords using
collagenase (Worthington Biochemicals, Lakewood, NJ, USA) and were
grown in M199 supplemented with 10% FBS, hEGF and hydrocortisone at
37°C in a humidified atmosphere of 5% CO2. Anonymous
umbilical cord tissues were obtained from the Hallym University
Hospital (Department of Obstetrics and Gynecology, Chuncheon Sacred
Heart Hospital, Hallym University Medical Center). This study was
approved by Hallym University Institutional Review Board
(HIRB-2011-007-4). For the transduction of Tat-SOD, HUVECs grown to
confluence on a 6-well plate were treated with 0.1–3 µM
Tat-SOD or SOD for various durations, of 10–120 min. In addition,
HUVECs were treated with 1 mM of antioxidant, N-acetyl-cysteine
(NAC), or 10 µM of NF-κB inhibitor, SN50, in the absence or
presence of 10 ng/ml TNF-α. The cells were harvested in order to
prepare cell extracts to perform western blot analysis.
To measure HUVEC viability, cells were exposed to 10
ng/ml TNF-α and 0.1–1 µM Tat-SOD for 6 h. A
3-(4,5-Dimet-ylthiazol-yl)-diphenyl tetrazolium bromide (MTT;
Duchefa Biochemie) assay was carried out to quantify cellular
viability.
Western blot analysis
HUVECs and Tat-SOD-transfected HUVECs were lysed
with a lysis buffer containing 1% β-mercaptoethanol, 1 M
β-glycerophosphate, 0.1 M Na3VO4, 0.5 M NaF
and protease inhibitor cocktail. Equal amounts of proteins from
cell extracts were then electrophoresed on 8–12% SDS-PAGE and
transferred onto a nitrocellulose membrane. After blocking
non-specific binding, the membrane was subsequently incubated
overnight at 4°C with anti-polyhistidine, anti-VCAM-1,
anti-integrin β1, anti-membrane type-1 (MT1)-MMP (Cat. no.
sc-12367; Santa Cruz Biotechnology, Inc.), anti-MMP-2 (Cat. no.
MAB902; R&D systems), anti-MMP-9 (Cat. no. MAB936; R&D
systems), anti-occludin-1 (Cat. no. sc-5526; Santa Cruz
Biotechnology, Inc.), anti-PECAM-1 (Cat. no. sc-1505; Santa Cruz
Biotechnology, Inc.), anti-vascular endothelial (VE)-cadherin (Cat.
no. ab-33168; Abcam, Cambridge, UK) and anti-NF-κB (Cat. no.
sc-7151; Santa Cruz Biotechnology, Inc.). After washing with
Tris-buffered saline-Tween-20, the membrane was incubated with an
anti-rabbit IgG conjugated to horseradish peroxidase. The protein
levels were subsequently determined using immobilon
chemiluminescent horseradish peroxidase substrate (Millipore Corp.,
Billerica, MA, USA) and Agfa X-ray film (Agfa-Gevaert, Mortsel,
Belgium). In addition, the bound antibodies were visualized by
enhanced ECL chemilluminescence (Amersham, Pittsburgh, PA, USA).
Incubation with monoclonal mouse β-actin antibody was also
performed for comparative controls.
Cell staining
For the direct detection of fluorescein-labeled
protein, purified Tat-SOD and control SOD were labeled using an
EZ-labeled fluorescein isothiocyanate (FITC) protein labeling kit
(Pierce, Rockford, IL, USA) according to the manufacturer's
instructions. HUVECs grown on glass coverslips were treated with 3
µM Tat-SOD and control SOD fusion proteins. Following
incubation for 2 h at 37°C, the cells were washed twice with
phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde
for 10 min. The distribution of fluorescence was analyzed using a
fluorescence microscope (Nikon, Tokyo, Japan).
Cell adhesion assay
The human monocytic leukemic cell line THP-1
(purchased from The American Type Culture Collection, Rockville,
MD, USA) were labeled with 5 µM calcein AM (Molecular
Probes, Eugene, OR, USA). HUVECs were treated with 0.1–0.5
µM Tat-SOD and 10 ng/ml TNF-α in a glass chamber (Nunc
Lab-Tek II; Thermo Fisher Scientific Inc., Waltham, MA, USA) for 6
h. Thereafter, labeled THP-1 cells were added to the 6 h-stimulated
HUVECs with 10 ng/ml TNF-α in the glass chamber, and these cells
were co-cultured in RPMI-1640 medium containing 10% FBS for 1 h.
After a thorough wash with PBS, the cultures were photographed with
a fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
Measurement of superoxide anion
production
Superoxide anions generated were detected using a
commercial O2− assay kit (Sigma Chemical
Co.), according to the manufacturer's instructions. This
measurement is based on the oxidation of luminol by O2−,
resulting in the formation of chemiluminescence light. HUVECs
seeded on 96-well plates were treated with 10 ng/ml TNF-α in the
absence and presence of Tat-SOD or control SOD. Simultaneously, the
luminol solution and enhancer solution included in the kit were
added, and the luminescence intensity was read every 10 min during
a 4 h-period.
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
Total RNA was isolated from HUVECs using a
commercially available TRIzol reagent kit (Invitrogen, Carlsbad,
CA, USA). RNA (5 µg) was then reverse transcribed with 200
units of reverse transcriptase and 0.5 mg/ml oligo(dT)15
primer (Bioneer, Daejeon, Korea). The expression levels of the mRNA
transcripts of the following primers were subsequently evaluated by
RT-PCR: MMP-2 forward, 5′-TGGCAAGTACGGCTGTC-3′ and reverse,
5′-TTCTTGTCGCGGTCGTAGTC-3′, 180 bp; MMP-9 forward,
5′-ACGTGGACATCTTCGACGC-3′ and reverse, 5′-CGAACCTCCAGAAGCTCTGC-3′,
709 bp; and β-actin forward, 5′-GACTACCTCATGAAGATC-3′ and reverse,
5′-GATCCACATCTGCTGGAA-3′, 513 bp. PCR was performed in 25 µl
of 10 mM Tris-HCl (pH 9.0), 25 mM MgCl2, 10 mM dNTP, 5
units of TaqDNA polymerase, and 10 µM of each primer and
terminated by heating at 94°C for 3 min. After 35 cycles of
thermocycling at 94°C (30 sec), 55°C (45 sec), 72°C (90 sec), and
72°C (10 min), and electrophoresis of the PCR products (25
µl) on 1% agarose gel, the bands were visualized using a
TFX-20 M model-UV transilluminator (Vilber Lourmat,
Marne-la-Vallée, France) and gel photographs were obtained. The
absence of contaminants was routinely checked by the RT-PCR assay
of negative control samples without the addition of a primer.
Gelatin zymography
HUVECs were plated at 90–95% confluence for all
experiments. To measure MMP-2 and MMP-9 activity, gelatin
zymography was performed. Culture media were electrophoresed on 8%
SDS-PAGE in Tris-HCl buffer [0.3 M Tris-HCl (pH 6.8), 4% SDS, 20%
glycerol, and 0.03% bromophenol blue] co-polymerized with 0.1%
gelatin as the substrate. The gel was incubated in a 2.5% Triton
X-100 solution for 1 h. After three washes in 50 mM Tris-HCl (pH
7.5) for 30 min, the gel was incubated for 20 h in 50 mM Tris-HCl
containing 10 mM CaCl2, 0.05% Brij-35, 200 mM NaCl at
37°C for 24 h. The gel was stained in a solution with 0.1%
Coomassie brilliant blue G-250, 2% acetic acid and 45% methanol for
1 h, and destained in a solution with 30% methanol and 10% acetic
acid, and the bands were visualized using a TFX-20 M model-UV
transilluminator.
Measurement of THP-1 monocyte
transmigration
The experimental models for monocyte transmigration
employed 24-Transwell inserts with pore sizes of 8 µm
(Corning Life Sciences, Corning, NY, USA). The lower surface of the
insert filter was coated with 15 µl of 1 mg/ml gelatin
solution and dried for 1 h. THP-1 cells (2×105) cultured
in serum-free RPMI-1640 were loaded on gelatin-coated Transwell
inserts. These cells were treated with 0.1–0.5 µM Tat-SOD
and 1 µM SOD, and exposed to 10 ng/ml TNF-α for 6 h.
Transmigrated THP-1 cells were collected for the 6 h-stimulation
with TNF-α, plated in a glass chamber, and treated with 50 ng/ml
phorbol 12-myristate 13-acetate for 24 h to attach suspended THP-1
cells. THP-1 cells attached on the slide glass were fixed with 4%
paraformaldehyde for 20 min and dyed with toluidine blue for 10
min. Images were obtained using light microscopy.
Preparation of nuclear protein
extracts
The cytosolic protein fraction and nuclear protein
extract were prepared using a detergent lysis procedure to assay
the translocation of NF-κB. HUVECs were lysed in a buffer of 20 mM
HEPES (pH 7.9), 1 mM EDTA, 10 mM NaCl, 1 mM dithiothreitol, 1 mg/ml
Nonidet P-40, 0.4 mM phenylmethylsulfonyl fluoride, 0.01 ng/ml
leupeptin, and 200 units aprotinin and incubated on ice for 10 min.
Proteins were extracted from nuclear pellets via incubation with a
high-salt buffer containing 420 mM NaCl, 1 mM EDTA, 20 mM HEPES (pH
7.9), 25% glycerol, 1 mM dithiothreitol, 0.4 mM
phenylmethylsulfonyl fluoride, 0.01 ng/ml leupeptin, and 200 units
of aprotinin with vigorous shaking. The nuclear debris was pelleted
by a brief centrifugation at 2,000 × g for 30 min to collect
supernatants. For the measurement of protein levels of NF-κB,
western blot analysis was conducted with nuclear protein extracts
using a human NF-κB primary antibody.
Immunocytochemical analysis
Following cell culture protocols, HUVECs were fixed
with 4% paraformaldehyde for 20 min. To make the cell membrane
permeable, 0.1% Triton X-100 and 0.1% sodium citrate were used to
treat the cells on ice for 1 min. After blocking for non-specific
binding with 20% FBS in PBS, primary anti-NF-κB antibody was added
and incubated overnight at 4°C. After washing with PBS-Tween-20,
HUVECs were incubated with anti-rabbit IgG conjugated with
fluorescent dye Cy3 and 4′,6-diamidino-2-phenylindole (DAPI) for
nuclear staining of HUVECs. Immunocytochemical images of cells
mounted on the slide were then observed by fluorescence microscopy
at λ=550 nm excitation and λ=570 nm emission (Carl Zeiss).
Data analysis
The results are presented as the means ± SEM.
Statistical analysis was conducted using the Statistical Analysis
Statistical software package, version 6.12 (SAS Institute, Cary,
NC, USA). One-way ANOVA was used to determine the inhibitory
effects of Tat-SOD on endothelial trafficking and the migration of
monocytes. Differences between the treatment groups were analyzed
with Duncan's multiple range test, and a P-value <0.05 was
considered to indicate a statistically significant difference.
Results
Tat-SOD purification and
construction
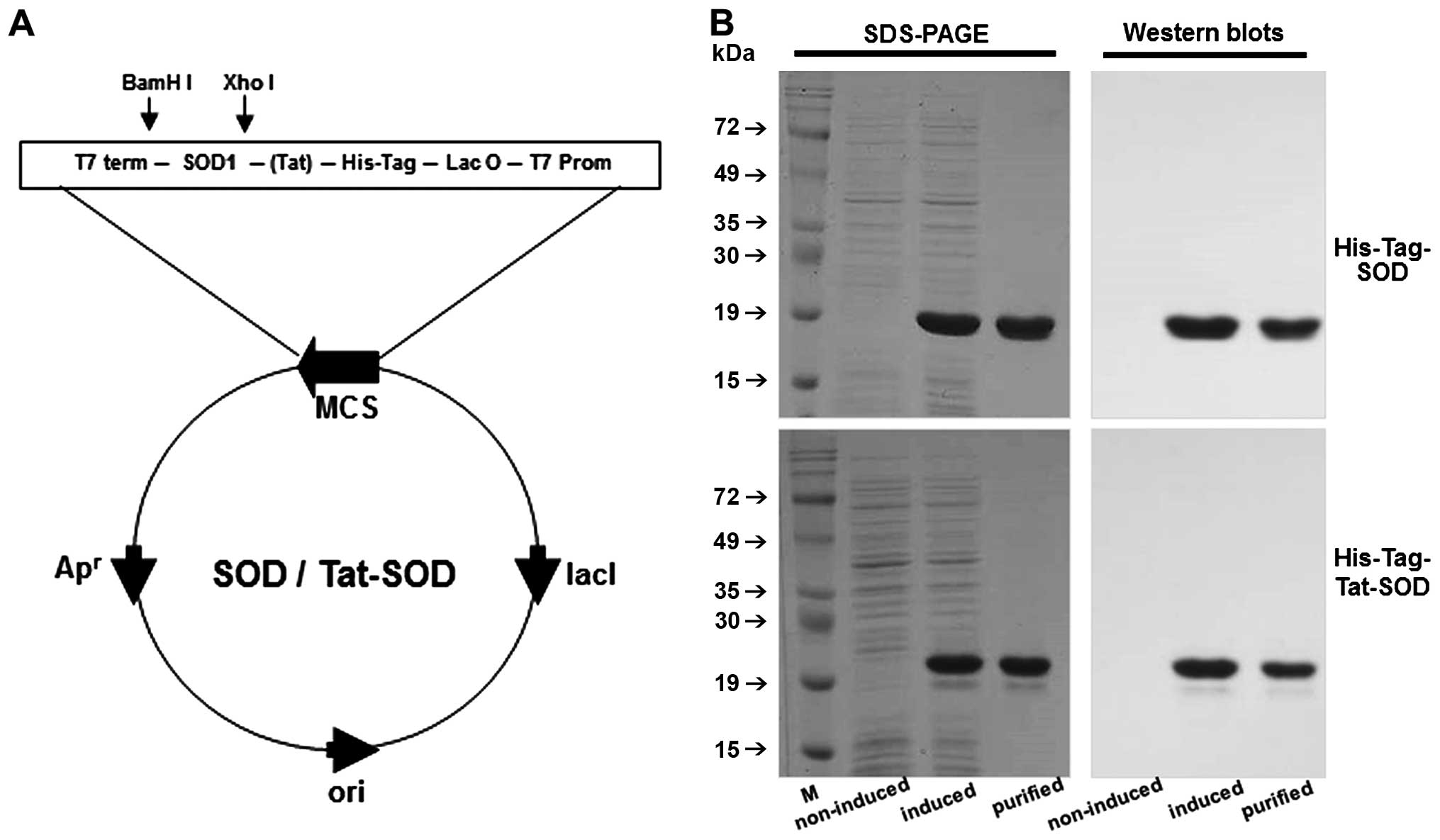
For the generation of the Tat-SOD vector, human SOD
cDNA was subcloned into a pET-15b plasmid reconstructed to contain
the Tat peptide. The Tat-SOD expression vector contained
consecutive cDNA sequences encoding human SOD, Tat peptide and six
histidine residues at the amino terminus. A SOD expression vector
was constructed to produce Tat peptide-free control SOD (Fig. 1A). Tat-SOD protein was purified
using an Ni2+-nitrilotriacetic acid sepharose affinity
column and PD-10 column chromatography, and was subsequently
confirmed by SDS-PAGE and western blot analysis using an
anti-rabbit polyhistidine antibody (Fig. 1B).
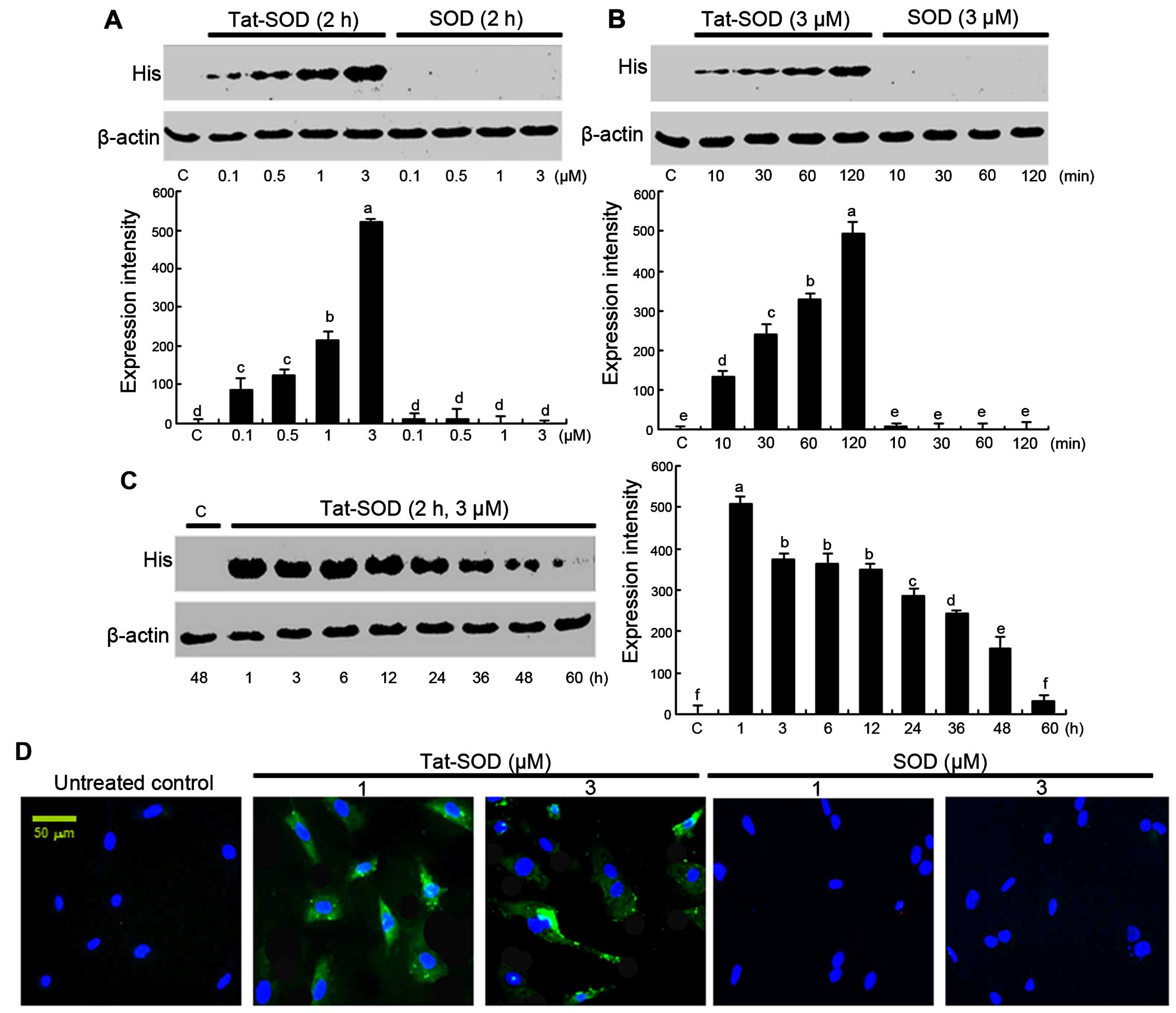
Transduction of Tat-SOD into HUVECs
The transduction of Tat-SOD into HUVECs was
performed by adding 0.1–3 µM Tat-SOD and control SOD to
HUVEC culture medium for 10–120 min. The dose dependency of Tat-SOD
transduction into HUVECs was observed at doses of 0.1–3 µM
(Fig. 2A), as evidenced by
western blot analysis with anti-histidine. The intracellular levels
of Tat-SOD transduced at 3 µM in the cells were rapidly and
markedly upregulated up to 120 min (Fig. 2B). By contrast, the control SOD
was not transduced into the cells (Fig. 2A and B).
To confirm the pharmacological effect of transduced
Tat-SOD, the expression of transduced Tat-SOD in cells was
determined (Fig. 2C). The
intracellular level of transduced Tat-SOD protein in cells was
initially detected after 1 h, and Tat-SOD protein gradually
decreased up to 60 h. Furthermore, to identify the cellular
translocalization of Tat-SOD, cells transduced with FITC-stained
Tat-SOD were counter-stained with DAPI (Fig. 2D). Tat-SOD protein was detected in
the cytoplasm of transduced cells.
Inhibition of cell adhesion molecules of
activated HUVECs by Tat-SOD
To examine the cytotoxicity to HUVECs of Tat-SOD,
MTT analysis was performed. When Tat-SOD, at concentrations between
0.1 and 1 µM, was added to HUVECs, viability was not
significantly influenced at ≤0.5 µM Tat-SOD (Fig. 3A). However, Tat-SOD at 1 µM
caused cellular toxicity. Accordingly, non-toxic Tat-SOD at ≤0.5
µM was employed for the following experiments.
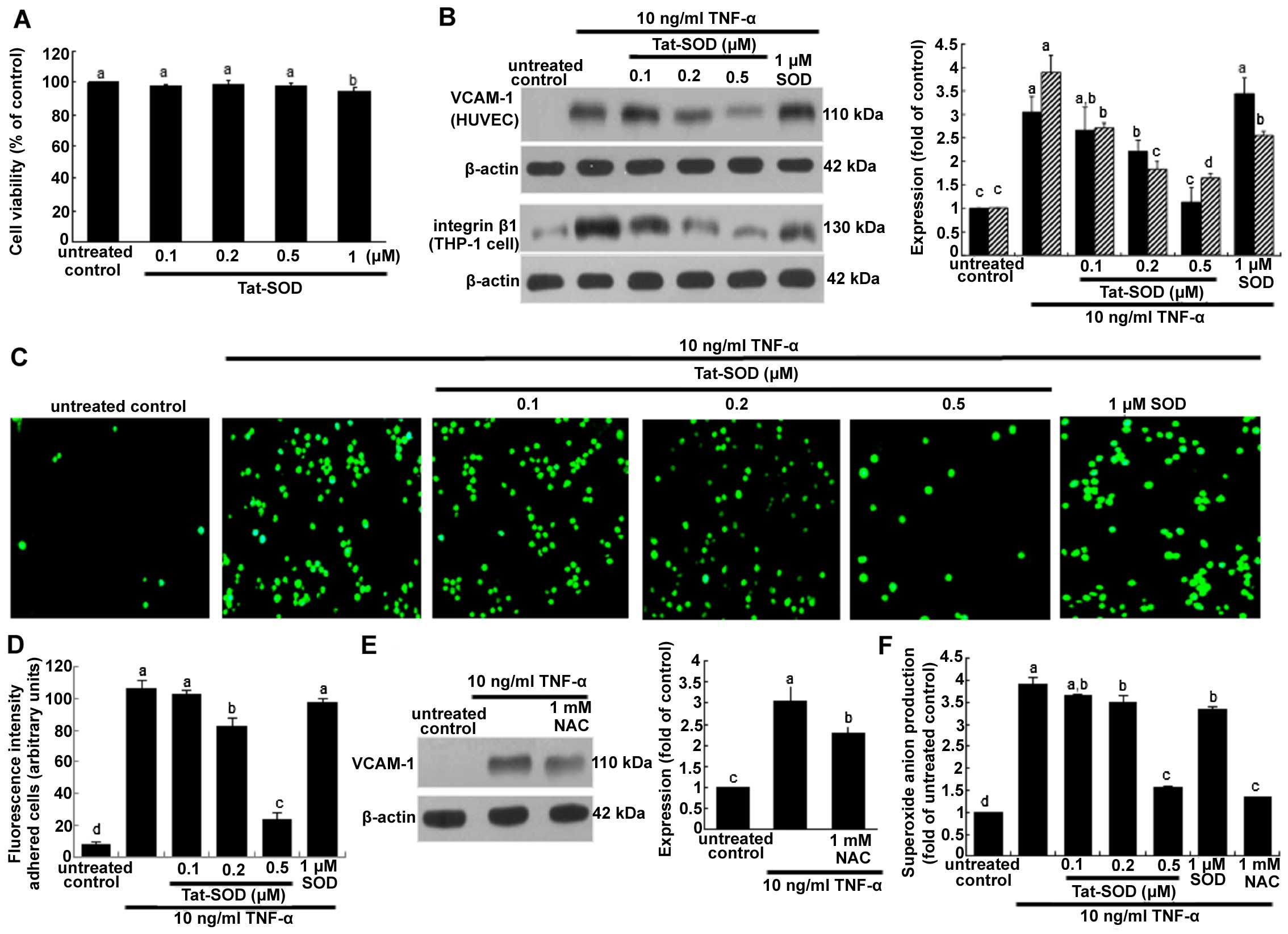 | Figure 3Cell viability (A), vascular cell
adhesion molecule-1 (VCAM-1) expression and monocyte adhesion in
human umbilical vein endothelial cells (HUVECs) treated with
transactivator of transcription (Tat)-superoxide dismutase (SOD) or
control SOD (B-D), and N-acetyl-cysteine (NAC) (E), and the release
of superoxide anion (F). HUVECs were treated with Tat-SOD, and cell
viability was measured by MTT assay (A). Cell viability (3 separate
experiments) was expressed as cell survival relative to untreated
controls in percentage (viability, 100%). To measure the cellular
protein expression of VCAM-1 and integrin β1 (n=5 experiments),
total cell lysates were subjected to western blot analysis with a
primary antibody against VCAM-1 or integrin β1 (B and E). To test
whether oxidative stress was responsible for VCAM-1 expression, the
antioxidant NAC was added to HUVECs activated by tumor necrosis
factor-α (TNF-α) (E). β-actin was used as an internal control. The
bar graph on the right represents quantitative results of blots
obtained from densitometric analysis. To examine THP-1 cell
adhesion to TNF-α-activated endothelial cells (C and D), HUVECs
were treated with 10 ng/ml TNF-α in the presence or absence of
Tat-SOD for 6 h and co-cultured with calcein-AM-labeled THP-1
monocytes for 1 h. Microscopic observation was conducted with a
fluorescence microscope and fluorescence intensity was quantified
(n=3 experiments). Magnification, x200. Superoxide anion radical
production was measured with a commercial superoxide anion assay
kit based on the oxidation of luminol (F). The columns represent
the means ± SEM, and variables without a common letter differ;
P<0.05. |
The present study investigated Tat-SOD-inhibited
monocyte trafficking onto activated HUVECs. As shown in Fig. 3B, VCAM-1 expression induced by 10
ng/ml TNF-α for 6 h was dose-dependently downregulated in the
presence of 0.1–0.5 µM Tat-SOD. By contrast, we noted no
marked inhibition of VCAM-1 expression triggered by 10 ng/ml TNF-α
for 6 h in HUVECs treated with 1 µM control SOD. It should
be noted that 0.5 µM Tat-SOD on its own did not stimulate
VCAM-1 induction. We noted that Tat-SOD suppressed the cellular
induction of integrin β1 in THP-1 monocytes exposed to 10 ng/ml
TNF-α (Fig. 3B). Some inhibition
of integrin β1 was observed with 1 µM control SOD. The
monocyte adhesion assay was further conducted in the co-culture
system of HUVECs and THP-1 monocytes. Monocyte adhesion onto
endothelial cells was augmented by TNF-α stimulation, which was
diminished by Tat-SOD in a dose-dependent manner (Fig. 3C and D). The inhibition of such
adhesion was not observed in control SOD-treated HUVECs which were
exposed to TNF-α. Thus, we suggest that Tat-SOD blocked the
inflammatory interaction of HUVECs and monocytes, a process which
may entail the induction of the adhesion molecules of endothelial
VCAM-1 and monocyte integrin β1.
Involvement of oxidative stress in
inducing endothelial adhesion molecules
The present study attempted to clarify that
oxidative stress was responsible for inflammatory VCAM-1
expression. When the antioxidant NAC at 1 mM was added to HUVECs
activated by TNF-α, VCAM-1 expression was significantly attenuated
(Fig. 3E). This study
demonstrated that 0.5 µM Tat-SOD abolished VCAM-1 induction
(Fig. 3B). Treatment with ≥0.2
µM Tat-SOD decreased TNF-α-induced superoxide anion
production, indicating that Tat-SOD moved to HUVEC cytoplasm and
affected antioxidant activity (Fig.
3F). We suggest that external stimulation of HUVECs with SOD
protein eliminates some of the extracellular superoxide anions
secreted from cells (Fig. 3F).
The inhibitory activity of Tat-SOD in relation to the adhesion of
monocytes to activated endothelium may result from the antioxidant
activity of intracellular Tat-SOD.
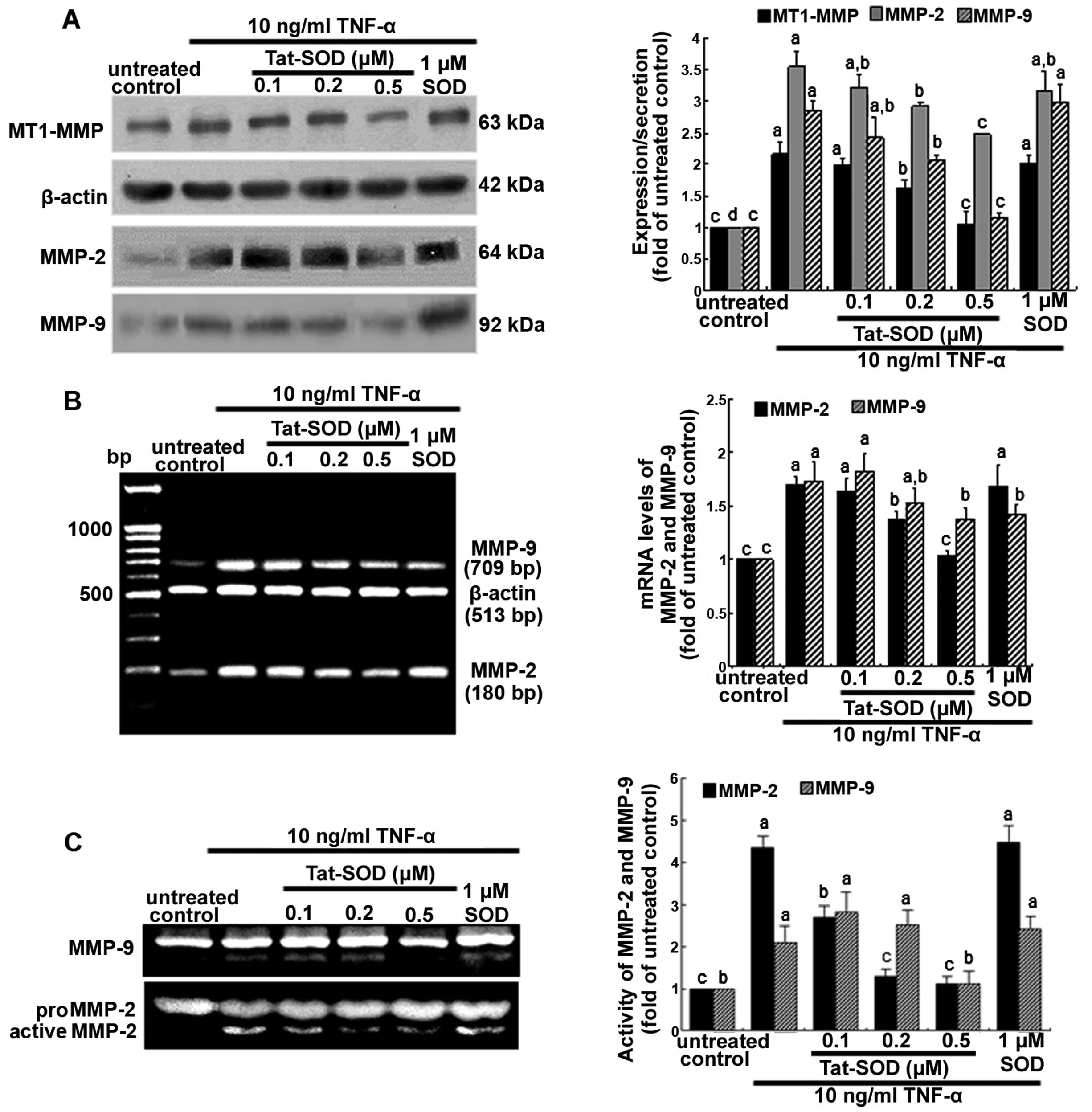
Suppression of TNF-α-induced MMP proteins
by Tat-SOD
It has previously been mentioned that MMP proteins
facilitate the passage of leukocytes across the matrix barriers of
the vascular basement membrane (18,19). In the present study, we examined
MMP induction in TNF-α-stimulated HUVECs using 0.1–0.5 µM
Tat-SOD. Tat-SOD downregulated MT1-MMP expression in HUVECs which
had been stimulated with 10 ng/ml TNF-α for 24 h (Fig. 4A). In addition, the secretory
protein level of MMP-2 and MMP-9 were enhanced by 24 h-treatment of
TNF-α, compared to the untreated control, and was dose-dependently
attenuated by Tat-SOD (Fig. 4A).
The diminution of protein production of MMP-2 and MMP-9 by Tat-SOD
required the inhibition of these MMP proteins at the
transcriptional levels (Fig. 4B).
By contrast, external SOD had no such effects on the production and
mRNA transcription of MMP-2 and MMP-9. Furthermore, we noted that
the gelatinolytic activity of MMP-2 and MMP-9 was markedly enhanced
by TNF-α, and this effect was attenuated by 0.5 µM Tat-SOD
(Fig. 4C).
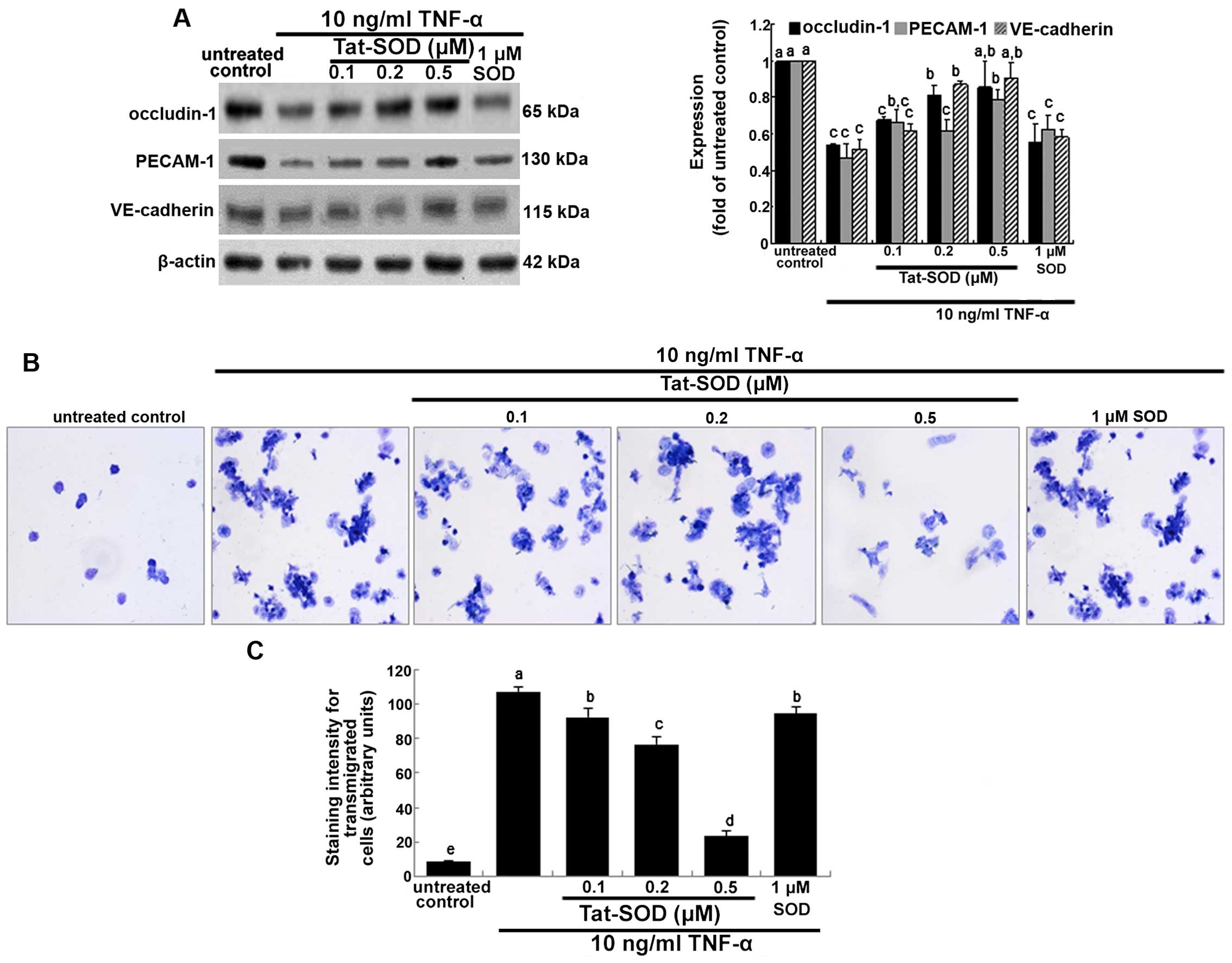
Effects of Tat-SOD on the cytokine
induction of intercellular junction proteins
Occludins are integral plasma-membrane proteins
located at the tight junctions and are distributed at low levels
and in a discontinuous fashion at cell-cell contacts (20). In the present study, we
investigated whether Tat-SOD-influenced occludin-1 expression was
downregulated by pro-inflammatory TNF-α. Occludin-1 was
downregulated at 24 h after treatment with TNF-α (Fig. 5A). By contrast, when HUVECs were
treated with 0.1–0.5 µM Tat-SOD, occludin-1 expression rose,
which is indicative of preserving the firm tight junction (Fig. 5A). However, 1 µM control
SOD did not have such an effect. Platelet/endothelial cell adhesion
molecule 1 (PECAM-1) and vascular endothelial (VE)-cadherin are
responsible for the endothelial adhesive junction and are crucial
to the process of leukocyte transmigration through intercel-lular
junctions of vascular endothelial cells (22). Western blot analysis indicated
that TNF-α suppressed the expression of PECAM-1 and VE-cadherin in
HUVECs, and this suppression was reversed by treatment with 0.5
µM Tat-SOD but not 1 µM control SOD (Fig. 5A). This indicates that transduced
Tat-SOD diminished the intercellular permeability of endothelial
cells, leading to concrete cell-cell contacts.
It has previously been noted that MMPs facilitate
the passage of leukocytes across the matrix barriers of the
vascular basement membrane (21).
When THP-1 cells in the upper compartment of Transwell plates were
treated with TNF-α, a large number of toluidine blue-stained THP-1
cells appeared in their bottom compartments (Fig. 5B and C). This indicates that TNF-α
increased monocyte transmigration. However, the TNF-α-elevated
transmigration was markedly decreased in THP-1 cells treated with
0.5 µM Tat-SOD, almost to the level of the control.
Accordingly, we suggest that Tat-SOD, unlike control SOD, inhibited
cytokine-stimulated monocyte extrava-sation by suppressing MMP
activity and intercellular junction protein induction (Figs. 4A and 5A).
Downregulation of NF-κB translocation by
Tat-SOD
The NF-κB pathway was studied, and we noted that
Tat-SOD protein regulated the cellular expression of intercellular
junction proteins and MMP proteins, which are critical for
regulating the endothelial migration of monocytes. NF-κB p65 was
trans-located into the nucleus of HUVECs activated by 10 ng/ml
TNF-α for 6 h, as evidenced by western blot analysis using the
primary antibody for NF-κB p65 (Fig.
6A). When 0.1–0.5 µM Tat SOD was added to TNF-α-exposed
HUVECs, cyotosolic NF-κB level rose, but nuclear NF-κB level
declined in a dose-dependent manner. Thus, transduced Tat-SOD
inhibited the NF-κB translocation and activation caused by
inflammatory TNF-α. However, SOD used as a control against Tat-SOD
had no such effect on NF-κB p65 activation (Fig. 6A).
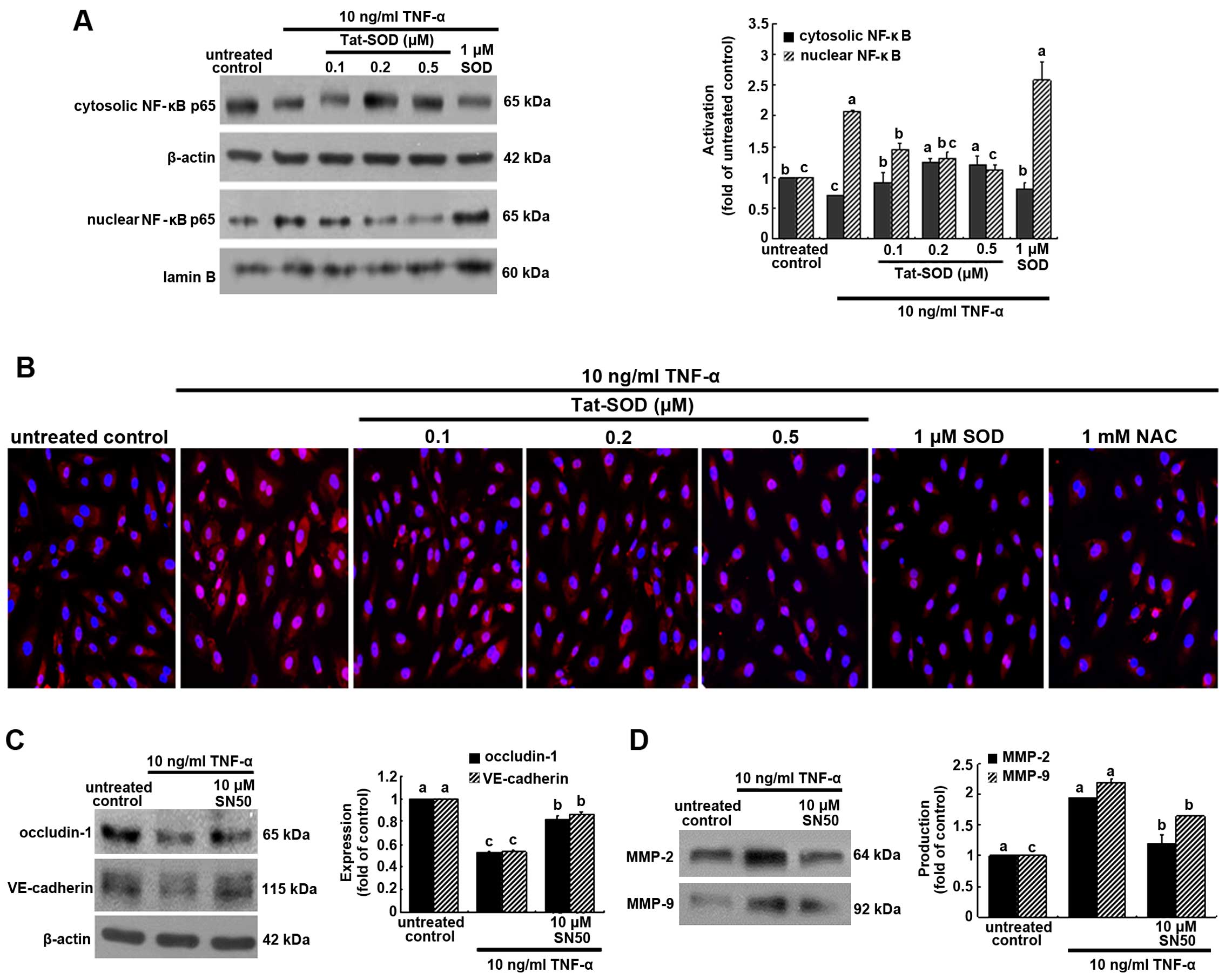 | Figure 6Inhibitory effects of transactivator
of transcription (Tat)-superoxide dismutase (SOD) on the
transactivation of nuclear factor-κB (NF-κB) (A and B), and the
restoration of occludin-1, vascular endothelial (VE)-cadherin and
matrix metalloproteinase (MMP) proteins by NF-κB inhibition (C and
D). Human umbilical vein endothelial cells (HUVECs) were treated
with 10 ng/ml tumor necrosis factor-α (TNF-α) for 6 h in the
presence or absence of Tat-SOD or control SOD. Cytosolic NF-κB and
nuclear NF-κB were examined by western blot analysis (A). Cytosolic
and nuclear fractions were subjected to western blot analysis with
a primary antibody against NF-κB. β-actin and lamin B were used as
internal controls. For the immunocytochemical staining of NF-κB
(B), HUVECs were immunostained and visualized using anti-NF-κB and
Cy3-conjugated IgG. Fluorescence microphotographs were obtained
with a fluorescence microscope. Each photograph is representative
of 3 separate experiments. Magnification, x200. The expression
levels of occludin-1, VE-cadherin, MMP-2 and MMP-9 were measured in
the presence of 10 µM NF-κB inhibitor SN50 (C and D).
Western blot analysis with a primary antibody against occludin-1,
VE-cadherin, MMP-2 and MMP-9 was performed. The bar graphs (means ±
SEM, n=3 experiments) for blots in the right panels represent
quantitative results obtained by densitometric analysis. The
columns represent the means ± SEM, and variables without a common
letter differ, P<0.05; n=3 experiments. |
This study further investigated NF-κB
transactivation in HUVECs treated with 10 ng/ml TNF-α for 6 h, as
measured by immunocytochemical staining. Heavy nuclear pinkish-red
staining was observed in TNF-α-exposed HUVECs (Fig. 6B). When 0.1–0.5 µM Tat SOD
or 1 mM NAC was applied to HUVECs treated with TNF-α, the cytosolic
staining became stronger in a dose-dependent manner (blue nuclear
staining). These data indicate that Tat-SOD retarded NF-κB
transacti-vation of HUVECs. We also noted that 0.5 µM SOD
did not block TNF-α-induced NF-κB activation (Fig. 6B).
In order to confirm that intercellular junction
markers were mediated via NF-κB activation, we detected the
induction of occludin-1 and VE-cadherin using the NF-κB inhibitor
SN50. The inhibition of occludin-1 and VE-cadherin expression by
TNF-α was restored in the presence of 10 µM SN50 (Fig. 6C). In addition, the enhanced
production of MMP-2 and MMP-9 by TNF-α was attenuated by this NF-κB
inhibitor (Fig. 6D).
Discussion
Seven major findings were observed in the present
study. i) Tat-SOD protein was highly expressed as a major component
of the total soluble proteins in cells, as determined by SDS-PAGE.
ii) Purified Tat-SOD protein was efficiently transduced into HUVECs
in a time- and dose-dependent manner. iii) Transduced Tat-SOD,
which was non-toxic at ≤0.5 µM, inhibited the TNF-α-induced
induction of endothelial VCAM-1 and monocyte integrin β1,
indicating that intracellular SOD in the cytoplasm inhibited the
tight adhesion mediated by the leukocyte integrins and endothelial
adhesion molecules. iv) Endothelial VCAM-1 induction by TNF-α was
attributed to oxidative stress that was blocked by the antioxidant
Tat-SOD. v) Transduced Tat-SOD inhibited the matrix-degrading MMP
activity of MT1-MMP, MMP-2 and MMP-9 in HUVECs. vi) The induction
of intercellular occludin-1, PECAM-1 and VE-cadherin involved in
transendothelial monocyte migration was enhanced by treatment with
Tat-SOD. vii) Tat-SOD diminished the nuclear NF-κB transactivation
responsible for paracellular junction proteins and matrix-degrading
proteins in TNF-α-treated HUVECs. These results demonstrate that
transduced Tat-SOD prevented cytokine-induced endothelial
trafficking and the transmigration of monocytes. It should be noted
that external stimulation with SOD did not inhibit endothelial
adhesion or the transmigration of monocytes.
The SOD isoform enzymes are the major antioxidant
defense systems against superoxide anion radicals in each
subcellular location in mammals. SOD inhibits the oxidative
inactivation of nitric oxide, thereby blocking peroxynitrite
formation and endothelial dysfunction (9). Given the essential role which SOD
plays in cardiovascular diseases such as atherosclerosis,
hypertension and angiogenesis, the strengthening of endogenous
antioxidant defenses with SOD-dependent antioxidant therapies,
which will more effectively protect against oxidative stress, is of
considerable interest (22,23). In addition, studying the role of
manganese SOD, beyond its essential role for survival, may lead to
the development of a novel strategy for an antioxidant approach to
cancer intervention (24).
Catalytic scavengers of peroxides and their potential uses as
therapeutic agents for pulmonary, cardiovascular, neurodegenerative
and inflammatory disorders have been previously noted (25). SOD liposomes and mimetics have
been shown to be effective in animal models of cancer prevention
(26). Therapeutic strategies
which target oxidative stress with SOD mimetics such as tempol
boost the endogenous levels of antioxidants and scavenging
superoxide anions, as well as hydroxyl radical generation (27). However, clinical evidence remains
controversial.
Although catalytic SOD enzymes are considered to be
potential therapeutic agents for the treatement of atherosclerosis,
hypertension and cancer, the inability of SOD to transduce into
cells has limited their use in antioxidant therapy. Protein
transduction domains or cell-penetrating peptides are employed for
the successful delivery of exogenous full-length fusion proteins
into living cells in vitro and in vivo (13,14). Our previous study showed that
efficiently transduced Tat-glyoxalase protein protected against
sodium nitroprusside-induced RINm5F cell death and
streptozotocin-induced pancreatic β-cell destruction in diabetic
mice (15). In the present study,
Tat protein was used for SOD to become cell permeable, and Tat-SOD
protein was highly and almost homogeneously expressed as a major
component of the total soluble proteins in cells. Also, purified
Tat-SOD protein was efficiently transduced into HUVEC in a time-
and dose-dependent manner. Thus, we suggest that targeted SOD
delivery into the HUVEC cytoplasm provides atheroprotective
effects, with antioxidant activity against cytokine-induced
endothelial activation and dysfunction. In a recent study, the
targeted delivery of SOD to endothelium, but not non-targeted SOD,
alleviated acute vascular inflammation (12).
Previous studies have demonstrated that endothelial
activation, dysfunction and vascular inflammation occur when the
endothelium is exposed to oxidative stress, leading to the
pathogenesis of a range of disease states (28,29). Inflammatory atherosclerosis
requires the initial activation of pro-inflammatory cytokines to
facilitate leukocyte transmigration (3). Since oxidative stress is critical to
the endothelial adhesiveness of immune cells, we suggest that SOD
therapy hinders endothelial activation caused by cytokines and
agonists. Accordingly, it has also been suggested that the
inhibition of vascular cell-cell interaction by drugs/agents with
antioxidant effects serves as a potential therapeutic strategy for
clinical atherosclerosis (8).
Although the detailed mechanism remains to be further elucidated,
one may speculate that transduced Tat-SOD blocked monocyte
trafficking onto endothelium through moderating the intracellular
ROS production-associated induction of VCAM-1. As noted in the
present study, external stimulation of HUVEC and THP-1 cells with
non-targeted SOD did not result in such atheroprotection. In a
similar study (10), SOD
administration ameliorated inflammatory bowel disease by reducing
VCAM-1 expression and leukocyte recruitment into the inflamed
intestine in cases of experimental colitis. In addition, in mice,
the injection of SOD conjugated with PECAM-directed antibody
alleviated endotoxin-induced leukocyte adhesion in the cerebral
vasculature, thus protecting the brain from ischemia-reperfusion
injury (12). It has been
previously demonstrated that SOD conjugated with PECAM antibody
markedly alleviates abnormal endothelial permeability and
endothelial barrier function caused by exogenous ROS and vascular
endothelial growth factor (30).
In the present study, we showed that Tat-SOD decreased the cytokine
induction of occludin-1, PECAM-1, VE-cadherin and MMP in HUVECs,
compared to the untreated control. These results suggest that
intracellular SOD potentiated endothelial cell interaction and
basement membrane matrix function. A previous investigation has
shown that low-dose MnTBAP, a SOD mimetic and superoxide anion and
peroxynitrite scavenger, effectively decreases the MMP-9 and
MMP-9/TIMP-1 ratio, suggesting that MnTBAP exerts an
anti-inflammatory effect in cases of angiogenesis and
alveolarization in intermittent hypoxia of neonatal rats (31). However, how the cell adhesion
molecule genes are selectively modulated in response to the
antioxidant SOD and which signaling pathways are involved in the
selective regulation of these genes remain unknown.
Several signaling pathways, particularly
NF-κB-mediated signaling pathways, play crucial roles in a variety
of pathophysiological processes. There is evidence that the
SOD-quenched superoxide anion produced by endothelial cells in
response to pro-inflammatory agents mediates NF-κB activation
(12). Similarly, the present
study found that transduced Tat-SOD inhibited TNF-α-triggered
monocyte transmigration via the blockade of NF-κB signaling.
Treatment with Tat-SOD decreased the cytokine induction of
intercellular junction proteins and matrix-degrading proteins by
deterring NF-κB activation. In fact, the concept of reciprocity of
inflammation and oxidative stress is well-recognized. As ROS
production and oxidative stress resulting from activation of immune
cells lead to chronic inflammation through the activation of
transcription factors including NF-κB, activator protein 1(AP-1)
and peroxisome proliferator-activated receptor γ (PPAR-γ),
inflammation itself has a reciprocal relationship with oxidative
stress (32). Thus, the findings
of the present study, bridging inflammation and oxidation,
emphasized the novel mechanisms of Tat-SOD targeting inflammation-
and oxidative stress-driven atherosclerosis. It has previously been
noted that AMP-activated protein kinase activation is involved in
the SOD reduction of high glucose-induced brain microvascular
endothelial cell tight-junction proteins by suppressing the
induction of NAD(P)H oxidase-derived superoxide anions (33). Overexpression of SOD1 but not SOD2
prevented the increase in hypoxia-induced transepithelial
electrical conductance and reduction of the occluding junctions at
the plasma membrane in alveolar epithelial cells though blocking
protein kinase C, ζ (PKC-ζ) and protein phosphatase 2A (34).
In conclusion, this study demonstrated that
transduced Tat-SOD blocked the inflammatory process, and this
involved the expression of inducible adhesion proteins, the
production of matrix-degrading proteins and the induction of
intercellular junction proteins. Consequently, we suggest that
Tat-SOD, but not external control SOD, is effective in hampering
the trafficking and extravasation of circulating monocytes.
Oxidative stress blocked by transduced Tat-SOD appeared to
instigate NF-κB activation, which was responsible for monocyte
extrav-asation-associated vascular inflammation and
atherosclerosis. However, the specific mechanisms underlying the
inhibition of inflammatory responses by the antioxidant Tat-SOD are
not yet fully understood. Furthermore, the use of THP-1 cells
versus monocytes or other leukocytes isolated from blood requires
further study. Sufficient differences between these cell types
exist to recommend confirmation of any critical results obtained
with THP-1 cells and primary monocytes. Nevertheless, we suggest
that transduced Tat-SOD has implications for strategies which
attenuate monocyte/macrophage dysfunction-related inflammatory
atherosclerosis.
Acknowledgments
This study was supported by the National Research
Foundation of Korea (no. 2015R1A2A2A01006666) and by a Priority
Research Centers Program grant (no. NRF-2009-0093812) through the
National Research Foundation of Korea.
References
|
1
|
Magenta A, Greco S, Gaetano C and Martelli
F: Oxidative stress and microRNAs in vascular diseases. Int J Mol
Sci. 14:17319–17346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wen YD, Wang H, Kho SH, Rinkiko S, Sheng
X, Shen HM and Zhu YZ: Hydrogen sulfide protects HUVECs against
hydrogen peroxide induced mitochondrial dysfunction and oxidative
stress. PLoS One. 8:e531472013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Badimon L, Storey RF and Vilahur G: Update
on lipids, inflammation and atherothrombosis. Thromb Haemost.
105(Suppl 1): S34–S42. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rocha VZ and Libby P: Obesity,
inflammation, and atherosclerosis. Nat Rev Cardiol. 6:399–409.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sprague AH and Khalil RA: Inflammatory
cytokines in vascular dysfunction and vascular disease. Biochem
Pharmacol. 78:539–552. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Immenschuh S and Schröder H: Heme
oxygenase-1 and cardiovascular disease. Histol Histopathol.
21:679–685. 2006.PubMed/NCBI
|
|
7
|
Armstrong AW, Voyles SV, Armstrong EJ,
Fuller EN and Rutledge JC: Angiogenesis and oxidative stress:
common mechanisms linking psoriasis with atherosclerosis. J
Dermatol Sci. 63:1–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen YH, Lin SJ, Chen YL, Liu PL and Chen
JW: Anti-inflammatory effects of different drugs/agents with
antioxidant property on endothelial expression of adhesion
molecules. Cardiovasc Hematol Disord Drug Targets. 6:279–304. 2006.
View Article : Google Scholar
|
|
9
|
Fukai T and Ushio-Fukai M: Superoxide
dismutases: role in redox signaling, vascular function, and
diseases. Antioxid Redox Signal. 15:1583–1606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seguí J, Gironella M, Sans M, Granell S,
Gil F, Gimeno M, Coronel P, Piqué JM and Panés J: Superoxide
dismutase ameliorates TNBS-induced colitis by reducing oxidative
stress, adhesion molecule expression, and leukocyte recruitment
into the inflamed intestine. J Leukoc Biol. 76:537–544. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lubrano V and Balzan S: LOX-1 and ROS,
inseparable factors in the process of endothelial damage. Free
Radic Res. 48:841–848. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shuvaev VV, Han J, Tliba S, Arguiri E,
Christofidou-Solomidou M, Ramirez SH, Dykstra H, Persidsky Y,
Atochin DN, Huang PL and Muzykantov VR: Anti-inflammatory effect of
targeted delivery of SOD to endothelium: mechanism, synergism with
NO donors and protective effects in vitro and in vivo. PLoS One.
8:e770022013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zahid M and Robbins PD: Protein
transduction domains: applications for molecular medicine. Curr
Gene Ther. 12:374–380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bechara C and Sagan S: Cell-penetrating
peptides: 20 years later, where do we stand? FEBS Lett.
587:1693–1702. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim MJ, Kim DW, Lee BR, Shin MJ, Kim YN,
Eom SA, Park BJ, Cho YS, Han KH, Park J, et al: Transduced
Tat-glyoxalase protein attenuates streptozotocin-induced diabetes
in a mouse model. Biochem Biophys Res Commun. 430:294–300. 2013.
View Article : Google Scholar
|
|
16
|
Song HY, Ju SM, Goh AR, Kwon DJ, Choi SY
and Park J: Suppression of TNF-alpha-induced MMP-9 expression by a
cell-permeable superoxide dismutase in keratinocytes. BMB Rep.
44:462–467. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kwon HY, Eum WS, Jang HW, Kang JH, Ryu J,
Ryong Lee B, Jin LH, Park J and Choi SY: Transduction of
Cu,Zn-superoxide dismutase mediated by an HIV-1 Tat protein basic
domain into mammalian cells. FEBS Lett. 485:163–167. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kwon HM, Choi YJ, Choi JS, Kang SW, Bae
JY, Kang IJ, Jun JG, Lee SS, Lim SS and Kang YH: Blockade of
cytokine-induced endothelial cell adhesion molecule expression by
licorice isoliquiritigenin through NF-kappaB signal disruption. Exp
Biol Med (Maywood). 232:235–245. 2007.
|
|
19
|
Kim MS, Kim DS, Kim HS, Kang SW and Kang
YH: Inhibitory effects of luteolin on transendothelial migration of
monocytes and formation of lipid-laden macrophages. Nutrition.
28:1044–1054. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Steed E, Balda MS and Matter K: Dynamics
and functions of tight junctions. Trends Cell Biol. 20:142–149.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hordijk PL: Endothelial signalling events
during leukocyte transmigration. FEBS J. 273:4408–4415. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carillon J, Rouanet JM, Cristol JP and
Brion R: Superoxide dismutase administration, a potential therapy
against oxidative stress related diseases: several routes of
supplementation and proposal of an original mechanism of action.
Pharm Res. 30:2718–2728. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wong GH: Protective roles of cytokines
against radiation: induction of mitochondrial MnSOD. Biochim
Biophys Acta. 1271:205–209. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Holley AK, Dhar SK, Xu Y and St Clair DK:
Manganese superoxide dismutase: beyond life and death. Amino Acids.
42:139–158. 2012. View Article : Google Scholar
|
|
25
|
Day BJ: Catalase and glutathione
peroxidase mimics. Biochem Pharmacol. 77:285–296. 2009. View Article : Google Scholar :
|
|
26
|
Miriyala S, Spasojevic I, Tovmasyan A,
Salvemini D, Vujaskovic Z, St Clair D and Batinic-Haberle I:
Manganese superoxide dismutase, MnSOD and its mimics. Biochim
Biophys Acta. 1822:794–814. 2012. View Article : Google Scholar :
|
|
27
|
Samai M, Sharpe MA, Gard PR and Chatterjee
PK: Comparison of the effects of the superoxide dismutase mimetics
EUK-134 and tempol on paraquat-induced nephrotoxicity. Free Radic
Biol Med. 43:528–534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun X, Belkin N and Feinberg MW:
Endothelial microRNAs and atherosclerosis. Curr Atheroscler Rep.
15:3722013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Varga ZV, Giricz Z, Liaudet L, Haskó G,
Ferdinandy P and Pacher P: Interplay of oxidative,
nitrosative/nitrative stress, inflammation, cell death and
autophagy in diabetic cardiomyopathy. Biochim Biophys Acta.
1852:232–242. 2015. View Article : Google Scholar
|
|
30
|
Han J, Shuvaev VV and Muzykantov VR:
Catalase and superoxide dismutase conjugated with
platelet-endothelial cell adhesion molecule antibody distinctly
alleviate abnormal endothelial permeability caused by exogenous
reactive oxygen species and vascular endothelial growth factor. J
Pharmacol Exp Ther. 338:82–91. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chang M, Bany-Mohammed F, Kenney MC and
Beharry KD: Effects of a superoxide dismutase mimetic on biomarkers
of lung angiogenesis and alveolarization during hyperoxia with
intermittent hypoxia. Am J Transl Res. 5:594–607. 2013.PubMed/NCBI
|
|
32
|
Kim YW, West XZ and Byzova TV:
Inflammation and oxidative stress in angiogenesis and vascular
disease. J Mol Med Berl. 91:323–328. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu C, Wu J and Zou MH: Activation of
AMP-activated protein kinase alleviates high-glucose-induced
dysfunction of brain microvascular endothelial cell tight-junction
dynamics. Free Radic Biol Med. 53:1213–1221. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Caraballo JC, Yshii C, Butti ML, Westphal
W, Borcherding JA, Allamargot C and Comellas AP: Hypoxia increases
transepithelial electrical conductance and reduces occludin at the
plasma membrane in alveolar epithelial cells via PKC-ζ and PP2A
pathway. Am J Physiol Lung Cell Mol Physiol. 300:L569–L578. 2011.
View Article : Google Scholar : PubMed/NCBI
|