Introduction
Congenital heart disease (CHD) represents the most
common form of birth defect, accounting for approximately one-third
of all major congenital abnormalities, and each year, approximately
1.35 million infants are born with CHD worldwide (1). The estimated prevalence of CHD is 1%
in live births, and up to 10% in stillbirths (2–4).
In terms of specific anatomic or hemodynamic lesions, various CHDs
are clinically classified into at least 21 distinct categories,
including ventricular septal defect, atrial septal defect,
endocardial cushion defect, tetralogy of Fallot (TOF), Ebstein's
anomaly, double outlet of right ventricle, transposition of the
great arteries, patent ductus arteriosus, persistent truncus
arteriosus, coarctation of the aorta, aortic stenosis, pulmonary
atresia, tricuspid atresia, interrupted aortic arch, total
anomalous pulmonary venous connection and hypo-plastic left heart
syndrome, of which TOF is the most common type of cyanotic CHD,
accounting for approximately 10% of all CHD cases (4). Severe CHD may give rise to a
diminished quality of life, decreased exercise performance,
retarded fetal brain development, depression, infective
endocarditis, thromboembolism, pulmonary arterial hypertension,
Eisenmenger's syndrome, heart failure, arrhythmias and even death
(4–11). Hence, CHD is responsible for
substantial morbidity and mortality, which lays a heavy economic
burden on patients and health care systems (4). Despite important clinical
significance, the etiologies of CHD remain largely unknown.
Cardiogenesis from the early embryo to the formation
of a fully functional four-chambered heart is a complex and dynamic
process that necessitates a harmonious concerto of transcription
factors, adhesion molecules, ion channels, signaling molecules and
structural proteins, and both environmental and genetic risk
factors may disrupt this biological process of heart development,
resulting in a wide variety of CHDs (12). Although environmental exposures
are also relevant, a growing number of studies have demonstrated
that genetic defects are the leading cause of CHD, and thus far,
mutations in >60 genes have been causally linked to CHD
(13–25). Among these CHD-causative genes,
those encoding cardiac transcription factors, including
homeodomain-containing protein, NK2 homeobox 5 (NKX2.5),
GATA-binding protein 4 (GATA4) and T-box transcription factor 5
(TBX5), are the most commonly involved genes in the pathogenesis of
CHD, underscoring the pivotal roles of cardiac transcription
factors in cardiovascular development and disease (26).
The basic helix loop helix family of transcription
factors, including heart and neural crest derivatives expressed
(HAND)1 and HAND2, the only two members identified up to now, has
been substantiated to be essential for normal cardiovascular
development, with either Hand1- or Hand2-deficient
mice not surviving due to cardiovascular developmental
abnormalities (27). In humans,
gain- or loss-of-function mutations in HAND1 have been
associated with various CHDs, encompassing hypoplastic left heart
syndrome, ventricular septal defect, atrial septal defect and
atrioventricular septal defect (28–30). Considering that the expression
profiles and functional roles of HAND2 overlap at least in
proportion to those of HAND1 (27,31–35), we hypothesized that genetically
compromised HAND2 may contribute to the development of CHD
in a subset of patients.
Materials and methods
Study subjects
A total of 145 unrelated patients with CHD were
enrolled in this study. The available family members of the index
patient who carried an identified HAND mutation were also
included. A total of 200 unrelated individuals without CHD, who
were matched to the CHD patients in ethnicity and gender, were
recruited as the controls. All the study subjects were from the Han
Chinese population. They underwent a comprehensive clinical
evaluation, including medical history, physical examination,
transthoracic echocardiography, standard 12-lead electrocardiogram
and chest X-ray radiography. The clinical types of CHD were defined
with two-dimensional continuous wave Doppler and color Doppler
techniques on transthoracic echocardiography. When indicated,
transesophageal echocardiography, cardiac catheterization and
angiography were performed to further clarify the cardiovascular
anatomic malformations. Cardiac surgery was carried out in some of
the patients with CHD. The patients who suffered from chromosomal
abnormalities or syndromic cardiovascular anomalies, such as
Axenfeld-Rieger syndrome, DiGeorge syndrome, Alagille syndrome and
Holt-Oram syndrome, were excluded from the current study. This
study is in conformity with the principles of the Declaration of
Helsinki. The study protocol was reviewed and approved by the
Ethics Committee of Tongji Hospital, Tongji University, Shanghai,
China (ethical approval number for cases and controls: LL(H)-09-07;
date of approval: July 27, 2009). Written informed consent was
obtained from the participants or their guardians prior to the
commencement of the study.
Genetic analysis of HAND2
Whole blood samples from the patients with CHD and
the control individuals were collected. Genomic DNA was isolated
from blood leukocytes using the Wizard Genomic DNA purification kit
(Promega, Madison, WI, USA), according to the manufacture's
instructions. With the aid of online Primer 3 (http://primer3.ut.ee), the primers used for the
amplification of the coding exons and flanking introns of
HAND2 by polymerase chain reaction (PCR) were designed as
shown in Table I. The referential
genomic DNA sequence of HAND2 was from GenBank (accession
no. NC_000004). PCR was conducted using a standard procedure on a
Veriti Thermal Cycler (Applied Biosystems, Foster City, CA, USA).
Basically, a PCR mixture consisted of 1X PCR buffer, 1X Q solution,
5 pmol of each primer pairs, 0.2 mM dNTPs, 50 ng of genomic DNA and
1 unit of HotStar TaqDNA polymerase (Qiagen, Hilden, Germany), to a
volume of 25 µl with double distilled water. A typical PCR
program was an initial activation of the polymerase (Qiagen) at
95°C for 15 min, followed by 35 cycles of denaturation at 94°C for
30 sec, annealing at 62°C for 1 min, and elongation at 72°C for 1
min, with a final extension at 72°C for 6 min. The PCR-amplified
fragments were purified and sequenced with HAND2-specific
primers using the BigDye® Terminator v3.1 Cycle
Sequencing kit on an ABI PRISM 3130 XL DNA Analyzer (both from
Applied Biosystems). For an identified mutation in the coding
region of HAND2, the numbering of it started with the
nucleotide A of the initial translation codon ATG (accession no.
NM_021973.2). To confirm the novelty of an identified sequence
variation, the single nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) database, the
human genome mutation database (HGMD; http://www.hgmd.org/), the 1000 genomes project
database (1000 Genomes; http://www.1000genomes.org) and the exome variant
server (EVS; http://evs.gs.washington.edu/EVS) database were
queried.
 | Table IPrimers to amplify the coding exons
and flanking introns of the HAND2 gene. |
Table I
Primers to amplify the coding exons
and flanking introns of the HAND2 gene.
| Coding exon | Forward primer | Reverse primer | Amplicon size
(bp) |
|---|
| 1-a |
5′-cgagaggattctgcctccgc-3′ |
5′-acagggccatgctgtagtcg-3′ | 550 |
| 1-b |
5′-ggtaggtggttttccccacca-3′ |
5′-gcccaattggaaagaggccg-3′ | 624 |
| 2 |
5′-ggttcactgtctcctccggc-3′ |
5′-cgggatcccttaccacacgg-3′ | 483 |
Alignment of multiple amino acids of
HAND2 proteins across species
The amino acids of HAND2 proteins from various
species were aligned with the online MUSCLE program (http://www.ncbi.nlm.nih.gov/homologene?cmd=Retrieve&dopt=MultipleAlignment&list_uids=32092).
In silico analysis of HAND2 mutation
The functional consequence of an identified sequence
variation on HAND2 protein was predicted by MutationTaster
(http://www.mutationtaster.org/),
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT
(http://sift.jcvi.org).
Expression plasmids and site-directed
mutagenesis
The human cardiac full-length cDNA was prepared as
previously described (22,23,36–38).
Human HAND2 harboring the whole coding region was generated
by PCR with the human heart cDNA as a template, cut with the
restriction enzymes, EcoRI and NotI, and then
subcloned at the EcoRI-NotI sites of the pcDNA3.1
vector (Invitrogen, Carlsbad, CA, USA). The identified mutation was
introduced into the wild-type HAND2-pcDNA3.1 construct by
site-directed mutagenesis using a complementary pair of primers and
the QuickChange II XL Site-Directed Mutagenesis kit (Stratagene, La
Jolla, CA, USA), and verified by direct sequencing. The recombinant
expression plasmids GATA4-pSSRa and NKX2.5-pEFSA, and the
ANF-luciferase (ANF-luc) reporter plasmid, which contains 2,600
base pairs upstream of the transcriptional start site of the
ANF gene and expresses Firefly luciferase, were generously
provided by Dr Ichiro Shiojima from Chiba University School of
Medicine, Chiba, Japan.
Cell culture and luciferase reporter
assays
HeLa cells (from a cell bank of our cardiovascular
research laboratory) were cultured in Dulbecco's modified Eagle's
medium supplemented with 10% fetal bovine serum, 2 mM glutamine,
100 µg/ml of penicillin and 100 µg/ml of streptomycin
in an atmosphere of 5% CO2 at 37°C. Cell transfection
was carried out in 6-well plates using Lipofectamine®
2000 reagent (Invitrogen) 24 h after plating. The internal control
plasmid, pGL4.75 (hRluc/CMV; Promega), which expresses
Renilla luciferase, was used in the transfection assays to
normalize the transfection efficiency. In the transient
transfection of HeLa cells, the same amount (0.6 µg) of
plasmid DNA (wild-type HAND2-pcDNA3.1, mutant HAND2-pcDNA3.1,
GATA4-pSSRa or NKX2.5-pEFSA) was used alone or in combination, in
the presence of 1.0 µg of ANF-luc and 0.04 µg of
pGL4.75. The cells were lysed 48 h after transfection, and the
Firefly and Renilla luciferase activities were measured
using the Dual-Glo luciferase assay system (Promega) according to
the manufacturer's instructions. The activity of the ANF
promoter was expressed as the fold activation of Firefly luciferase
relative to Renilla luciferase. Three independent
experiments were performed in triplicate for each cell
transfection, and each value presented was the average of
triplicate samples.
Statistical analysis
Continuous data are expressed as the means ±
standard deviation (SD). Differences in continuous variables
between 2 groups were compared using the Student's unpaired t-test.
Differences in categorical variables between 2 groups were compared
using the χ2 test or Fisher's exact test, as indicated.
The significance level was set at a two-tailed P-value of <0.05.
All statistical analyses were performed with SPSS version 18.0
(SPSS IBM, New York, NY, USA).
Results
Clinical characteristics of the study
participants
In this study, 145 unrelated patients with CHD were
clinically investigated in contrast to 200 unrelated control
individuals without CHD. All the patients with CHD had congenital
cardiac defects confirmed by an echocardiogram or further by
cardiac surgery. Based on the medical histories and
echocardiographic records, the control individuals had neither CHD
nor a positive family history of CHD. There were no differences in
ethnicity, gender and age between the patient and control groups.
The baseline clinical characteristics of the study patients with
CHD are presented in Table
II.
| Table IIBaseline clinical characteristics of
the study patients with CHD (n=145). |
Table II
Baseline clinical characteristics of
the study patients with CHD (n=145).
| Variables | Statistics |
|---|
| Age (years) | 3.68±1.52 |
| Male (%) | 78 (54) |
| Positive family
history of CHD (%) | 6 (4) |
| Distribution of
different types of CHD | |
| Isolated CHD
(%) | 93 (64) |
| VSD (%) | 32 (22) |
| ASD (%) | 26 (18) |
| PDA (%) | 12 (8) |
| DORV (%) | 5 (3) |
| ECD (%) | 4 (3) |
| PTA (%) | 4 (3) |
| Other isolated CHD
(%) | 10 (7) |
| Complex CHD
(%) | 52 (36) |
| TOF (%) | 25 (17) |
| ASD + VSD (%) | 8 (6) |
| VSD + DORV
(%) | 5 (3) |
| VSD + AS (%) | 4 (3) |
| VSD + TGA (%) | 3 (2) |
| Other complex CHD
(%) | 7 (5) |
| Arrhythmias | |
| Cardiac conduction
block (%) | 6 (4) |
| Atrial
fibrillation (%) | 3 (2) |
| Treatment | |
| Cardiac surgery
(%) | 67 (46) |
| Catheter-based
repair (%) | 46 (32) |
| Follow-up (%) | 32 (22) |
Identification of a novel mutation in
HAND2
By direct PCR-sequencing of the HAND2 gene in
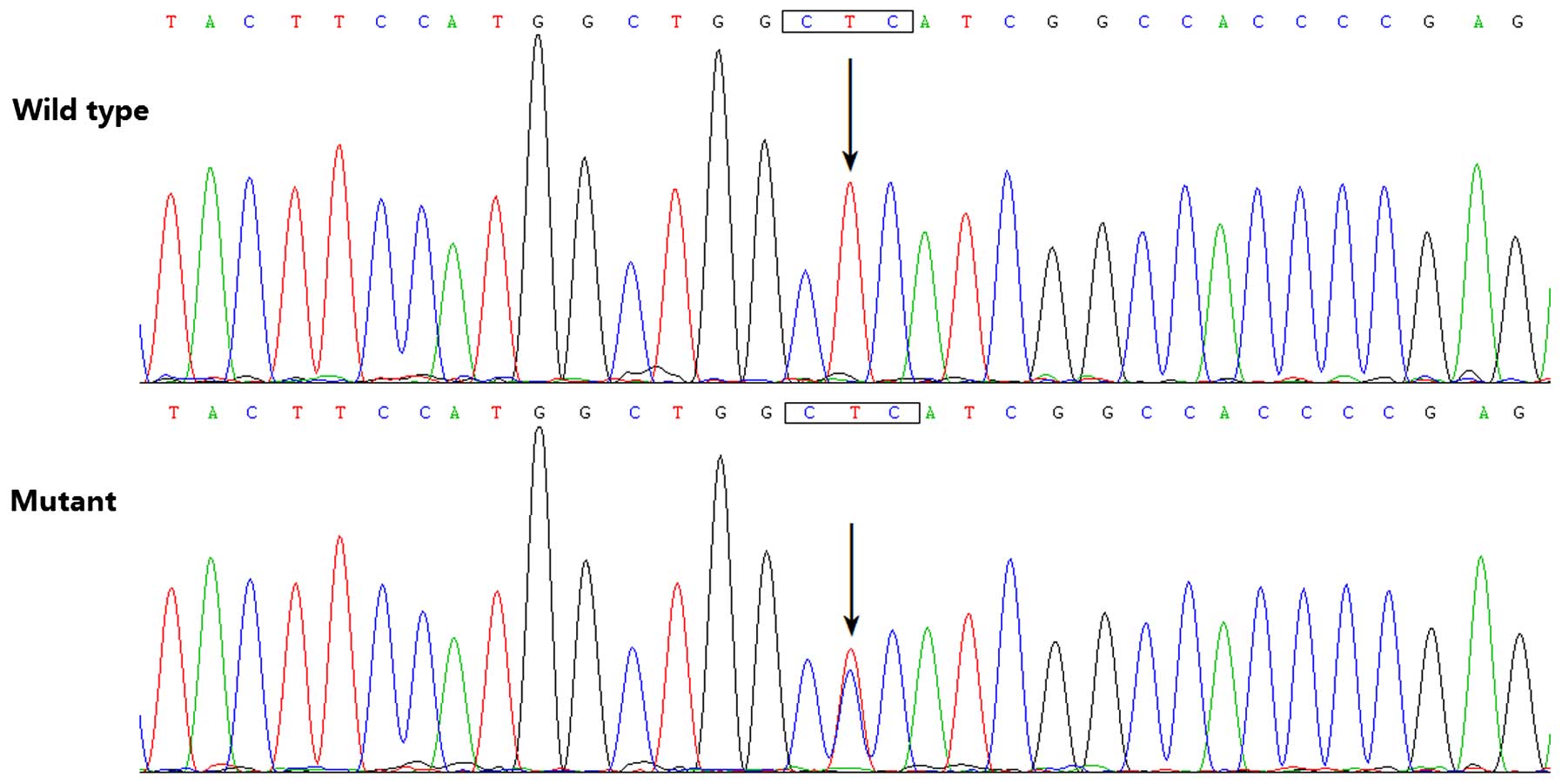
the 145 unrelated patients with CHD, a transition of thymine to
cytosine in the second nucleotide of codon 47 (c.140T>C),
predicting the substitution of proline at amino acid position 47
for leucine (p. L47P), was identified in a male patient with TOF,
who was half a year old without a positive family history of CHD.
Additionally, sequence analysis of HAND2 in the parents of
the mutation carriers revealed no mutation, indicating that the
identified mutation was a de novo mutation. The DNA
sequencing electropherograms showing the identified heterozygous
HAND2 mutation of c.140T>C in comparison with its control
sequence are shown in Fig. 1. A
schematic diagram of HAND2 depicting the functionally important
structural domains and the location of the mutation detected in
this study is presented in Fig.
2. The missense mutation was neither observed in the 200
control individuals nor found in the SNP, HGMD, 1000 Genomes and
EVS databases.
Alignment of multiple amino acids of
HAND2 proteins from various species
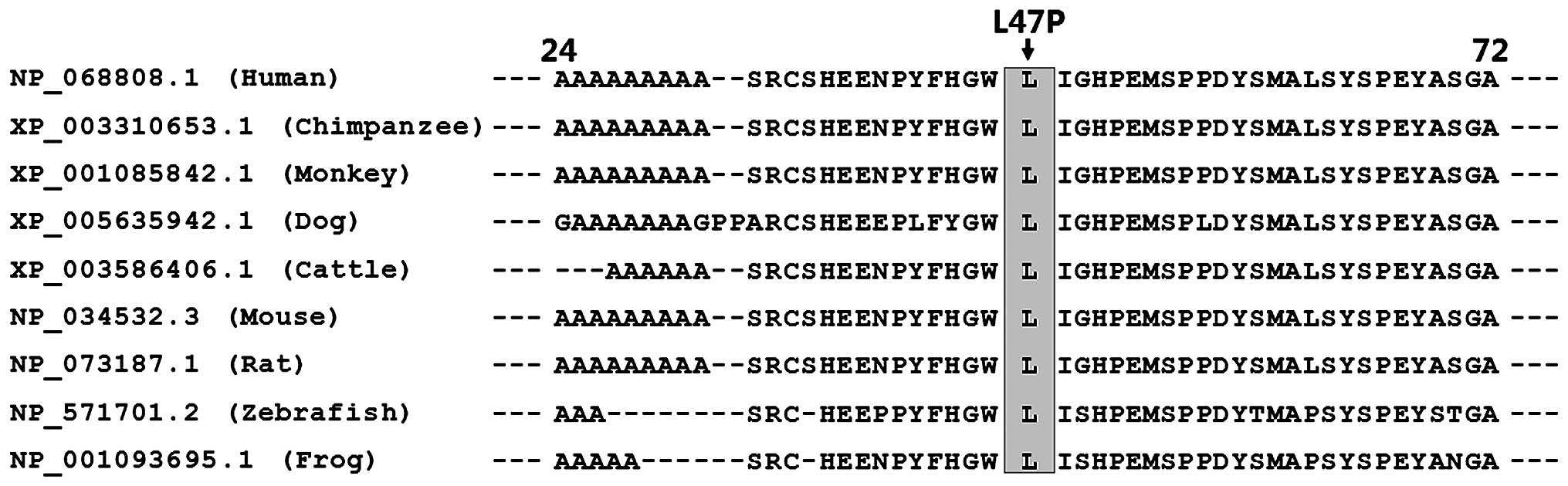
Alignment of the amino acids of the human HAND2
protein with those of chimpanzee, monkey, dog, cattle, mouse, rat,
zebrafish and frog exhibited that the altered leucine at amino acid
residue 47 of human HAND2 was completely conserved evolutionarily
(Fig. 3).
Causative potential of the identified
HAND2 sequence variation
The HAND2 sequence variation of c.140T>C
was predicted to be disease-causing by MutationTaster, with a
P-value of 1.0000, probably damaging by PolyPhen-2, with a score of
0.999 (sensitivity 0.14; specificity 0.99) and intolerated by SIFT,
with a score of 0.02.
Functional impairment of the HAND2
protein caused by the mutation
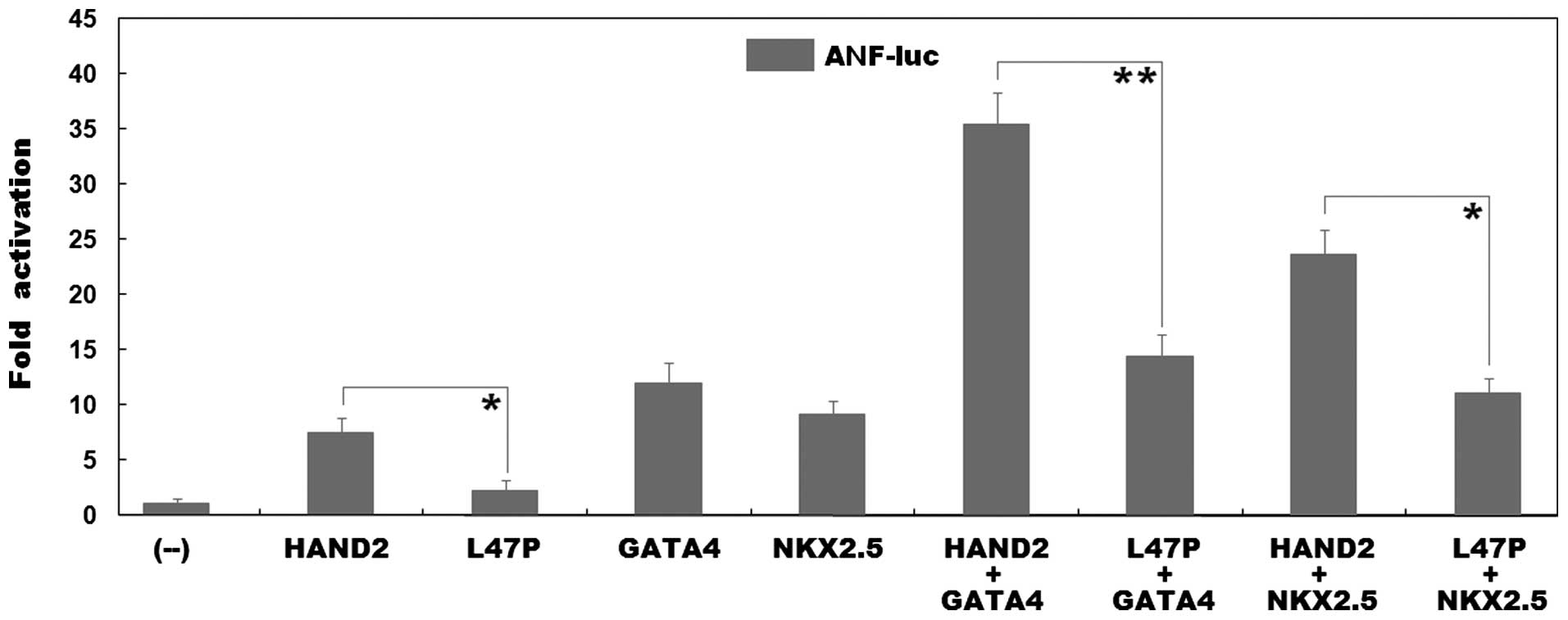
As shown in Fig.
4, the same amount (0.6 µg) of wild-type and L47P-mutant
HAND2 transcriptionally activated the ANF promoter by ~8- and
2-fold, respectively (wild-type vs. mutant, t=5.6462, P=0.0048). In
the presence of 0.6 µg of wild-type GATA4, the same
amount (0.6 µg) of wild-type and L47P-mutant HAND2 activated
the ANF promoter by ~35- and 14-fold, respectively
(wild-type vs. mutant, t=10.3947, P=0.0005); while in the presence
of 0.6 µg of wild-type NKX2.5, the same amount (0.6
µg) of wild-type and L47P-mutant HAND2 activated the
ANF promoter by ~24- and 11-fold, respectively (wild-type
vs. mutant, t=8.5137, P=0.0010). These results reveal that the
L47P-mutant HAND2 has a significantly reduced transcriptional
activity, and furthermore, the mutation markedly diminishes the
synergistic activation between HAND2 and GATA4 or between HAND2 and
NKX2.5.
Discussion
TOF, characterized by four distinct anatomic
features, including pulmonary outflow tract obstruction, overriding
aortic root, ventricular septal defect and right ventricular
hypertrophy, constitutes approximately 7–10% of all CHD cases,
corresponding to 3 of every 10,000 live births, with males being
affected slightly more often than females (39). If not treated surgically, 25% of
cases with severe obstruction succumb to the disease within the
first year, 40% succumb by the age of 3 years, 70% by the age of 10
years, and 95% by the age of 40 years (39). Therefore, it is of pronounced
clinical significance to ascertain the molecular basis of TOF. In
the present study, a novel heterozygous mutation, p.L47P, in HAND2
was identified in a child with TOF. The missense mutation was
absent in the 400 control chromosomes from a control population
matched for ethnicity and gender. The mutation, which altered the
amino acid conserved evolutionally across species, was predicted to
be pathogenic by MutationTaster, PolyPhen-2 and SIFT. Reporter gene
assays unveiled that the L47P-mutant HAND2 possessed a
significantly reduced transcriptional activity. Furthermore, the
L47P mutation markedly decreased the synergistic activation between
HAND2 and GATA4 or HAND2 and NKX2.5. Therefore, it is possible that
functionally compromised HAND2 predisposes to CHD in a subset of
patients.
In humans, HAND2 is located on chromosome
4q33, with a transcript of 2.3 kb in length encoding a protein of
217 amino acids, and is strongly expressed in the human heart
(40). The HAND2 protein harbors
two functionally important structural domains, a transcriptional
activation domain and a basic helix-loop-helix domain. The former
is required for the transcriptional activation of downstream genes,
and the latter is responsible for the binding to target DNAs and
the interactions with transcriptionally cooperative partners
(41). Previous studies have
demonstrated that HAND2 transcriptionally activates multiple target
genes highly expressed in the heart during embryogenesis, including
ANF, alone or in synergy with such cooperative partners as
GATA4, NKX2.5 and myocyte enhancer factor 2C (MEF2C) (42–45). In the present study, the mutation
identified in a patient with CHD was located in the transcriptional
activation domain of the HAND2 protein, and biological assays
revealed that the mutation significantly diminished the
transcriptional activation of the ANF promoter driven by
HAND2, and furthermore, the mutation markedly decreased the
synergistic activation between HAND2 and GATA4 or HAND2 and NKX2.5,
two other cardiac core transcription factors that are most commonly
linked to CHD in humans (13).
These findings suggest that haploinsufficiency caused by HAND2
mutation is likely an alternative mechanism underlying CHD.
The association of genetically defective
Hand2 with increased vulnerability to CHD has been
substantiated in animal models. In zebrafish, Hand2-mutant
embryos have been shown to have defects in myocardial development
from an early stage, with a reduced number of myocardial precursors
and an improperly patterned myocardial tissue, which were preceded
by the aberrant morphogenesis of the cardiogenic regions of the
lateral plate mesoderm (46).
Additionally, gene expression profiles in Hand2-mutant
embryos revealed an essential role of Hand2 in the
establishment of a favorable environment for cardiac fusion through
the negative regulation of fibronectin (47). In chicks, treatment of stage 8
chick embryos with Hand2 and Hand1 antisense
oligonucleotides demonstrated that either oligonucleotide alone did
not disrupt embryogenesis, whereas in combination, they inhibited
cardiac development at the looping heart tube stage (33). In mice, the targeted disruption of
Hand2 has been shown to give rise to embryonic lethality on
embryonic day 10.5, mainly due to right ventricular hypoplasia and
vascular deformities (34). In
rescued mouse embryos by activating adrenergic receptors, the
deletion of Hand2 has been shown to lead to the misalignment
of the outflow tract and aortic arch arteries, and ventricular
septal defect, double outlet right ventricle, interrupted aortic
artery, pulmonary stenosis, as well as retroesophageal right
subclavian artery (48).
Moreover, the conditional ablation of Hand2 alleles in specific
cardiac cell populations at defined developmental points
recapitulated the complete Hand2-null phenotype.
Specifically, the loss of Hand2 at later stages of
development and in restricted areas of the second heart field has
been shown to cause various cardiovascular abnormalities, including
hypo-plastic right ventricle, tricuspid atresia, truncus arteriosus
and ventricular septal defect (49). Besides, the endocardial
nullification of Hand2 contributes to the abnormal
development of tricuspid valve, intraventricular septum and
ventricles (50). By contrast,
mice with an increased copy number of Hand2, which were
generated by transgene with a bacterial artificial chromosome
containing Hand2, also presented with congenital heart
defects (51). Taken
collectively, these results suggest that Hand2 plays pivotal
roles in cardiovascular morphogenesis, and the imbalanced dosage of
HAND2 confers an increased predisposition to CHD.
In humans, previous studies have demonstrated that
patients with chromosomal deletion or duplication that involved
chromosome 4q33, the locus of HAND2, are liable to CHD,
including pulmonary atresia, ventricular septal defect, coarctation
of the aorta and TOF (40). In
addition, HAND2 sequence variations were also discovered in
patients with CHD, encompassing TOF, patent ductus arteriosus,
pulmonary atresia, atrial septal defect, atrioventricular septal
defect, pulmonary stenosis and double outlet right ventricle.
However, the functional roles of these CHD-related mutations remain
to be characterized (52).
In conclusion, to the best of our knowledge, this is
the first study on the association of HAND2 loss-of-function
mutation with an enhanced susceptibility to TOF in humans,
providing novel insight into the molecular mechanisms responsible
for the development of CHD, and implying potential implications for
the xgenetic counseling of families with CHD.
Acknowledgments
We are really thankful to the study participants for
their devotion to the study. This study was supported in part by
grants from the key program for Basic Research of Shanghai, China
(no. 14JC1405500), and the National Natural Science Fund of China
(nos. 81270161 and 81470372).
References
|
1
|
Fahed AC, Gelb BD, Seidman JG and Seidman
CE: Genetics of congenital heart disease: The glass half empty.
Circ Res. 112:707–720. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zheng JY, Tian HT, Zhu ZM, Li B, Han L,
Jiang SL, Chen Y, Li DT, He JC, Zhao Z, et al: Prevalence of
symptomatic congenital heart disease in Tibetan school children. Am
J Cardiol. 112:1468–1470. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marelli AJ, Ionescu-Ittu R, Mackie AS, Guo
L, Dendukuri N and Kaouache M: Lifetime prevalence of congenital
heart disease in the general population from 2000 to 2010.
Circulation. 130:749–756. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mozaffarian D, Benjamin EJ, Go AS, Arnett
DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ,
Howard VJ, et al American Heart Association Statistics Committee
and Stroke Statistics Subcommittee: Heart disease and stroke
statistics–2015 update: A report from the American Heart
Association. Circulation. 131:e29–e322. 2015. View Article : Google Scholar
|
|
5
|
Feltez G, Coronel CC, Pellanda LC and
Lukrafka JL: Exercise capacity in children and adolescents with
corrected congenital heart disease. Pediatr Cardiol. 36:1075–1082.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Williams IA, Fifer WP and Andrews H: Fetal
growth and neuro-developmental outcome in congenital heart disease.
Pediatr Cardiol. 36:1135–1144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barst RJ, Ivy DD, Foreman AJ, McGoon MD
and Rosenzweig EB: Four- and seven-year outcomes of patients with
congenital heart disease-associated pulmonary arterial hypertension
(from the REVEAL Registry). Am J Cardiol. 113:147–155. 2014.
View Article : Google Scholar
|
|
8
|
Wright LK, Ehrlich A, Stauffer N, Samai C,
Kogon B and Oster ME: Relation of prenatal diagnosis with one-year
survival rate for infants with congenital heart disease. Am J
Cardiol. 113:1041–1044. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Priromprintr B, Rhodes J, Silka MJ and
Batra AS: Prevalence of arrhythmias during exercise stress testing
in patients with congenital heart disease and severe right
ventricular conduit dysfunction. Am J Cardiol. 114:468–472. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ghosh RM, Gates GJ, Walsh CA, Schiller MS,
Pass RH and Ceresnak SR: The prevalence of arrhythmias, predictors
for arrhythmias, and safety of exercise stress testing in children.
Pediatr Cardiol. 36:584–590. 2015. View Article : Google Scholar
|
|
11
|
Walsh EP: Sudden death in adult congenital
heart disease: Risk stratification in 2014. Heart Rhythm.
11:1735–1742. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Srivastava D and Olson EN: A genetic
blueprint for cardiac development. Nature. 407:221–226. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Andersen TA, Troelsen KL and Larsen LA: Of
mice and men: Molecular genetics of congenital heart disease. Cell
Mol Life Sci. 71:1327–1352. 2014. View Article : Google Scholar :
|
|
14
|
Wang X, Li P, Chen S, Xi L, Guo Y, Guo A
and Sun K: Influence of genes and the environment in familial
congenital heart defects. Mol Med Rep. 9:695–700. 2014.
|
|
15
|
Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM,
Liu XY, Zhang XL, Li RG, Xu YJ, Hou XM, et al: A novel NKX2.5
loss-of-function mutation associated with congenital bicuspid
aortic valve. Am J Cardiol. 114:1891–1895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang X, Ji W, Wang J, Zhao P, Guo Y, Xu R,
Chen S and Sun K: Identification of two novel GATA6 mutations in
patients with nonsyndromic conotruncal heart defects. Mol Med Rep.
10:743–748. 2014.PubMed/NCBI
|
|
17
|
Al Turki S, Manickaraj AK, Mercer CL,
Gerety SS, Hitz MP, Lindsay S, D'Alessandro LC, Swaminathan GJ,
Bentham J, Arndt AK, et al: UK10K Consortium, Wilson DI, Mital S
and Hurles ME: Rare variants in NR2F2 cause congenital heart
defects in humans. Am J Hum Genet. 94:5745–5785. 2014.
|
|
18
|
Zhao L, Ni SH, Liu XY, Wei D, Yuan F, Xu
L, Xin-Li, Li RG, Qu XK, Xu YJ, et al: Prevalence and spectrum of
Nkx2.6 mutations in patients with congenital heart disease. Eur J
Med Genet. 57:579–586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Werner P, Paluru P, Simpson AM, Latney B,
Iyer R, Brodeur GM and Goldmuntz E: Mutations in NTRK3 suggest a
novel signaling pathway in human congenital heart disease. Hum
Mutat. 35:1459–1468. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei D, Gong XH, Qiu G, Wang J and Yang YQ:
Novel PITX2c loss-of-function mutations associated with complex
congenital heart disease. Int J Mol Med. 33:1201–1208.
2014.PubMed/NCBI
|
|
21
|
Cowan J, Tariq M and Ware SM: Genetic and
functional analyses of ZIC3 variants in congenital heart disease.
Hum Mutat. 35:66–75. 2014. View Article : Google Scholar :
|
|
22
|
Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu
L, Liu H, Li RG, Xu YJ, Wang Q, et al: GATA5 loss-of-function
mutations associated with congenital bicuspid aortic valve. Int J
Mol Med. 33:1219–1226. 2014.PubMed/NCBI
|
|
23
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J
Mol Med. 33:1227–1235. 2014.PubMed/NCBI
|
|
24
|
Racedo SE, McDonald-McGinn DM, Chung JH,
Goldmuntz E, Zackai E, Emanuel BS, Zhou B, Funke B and Morrow BE:
Mouse and human CRKL is dosage sensitive for cardiac outflow tract
formation. Am J Hum Genet. 96:235–244. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pan Y, Geng R, Zhou N, Zheng GF, Zhao H,
Wang J, Zhao CM, Qiu XB, Yang YQ and Liu XY: TBX20 loss-of-function
mutation contributes to double outlet right ventricle. Int J Mol
Med. 35:1058–1066. 2015.PubMed/NCBI
|
|
26
|
McCulley DJ and Black BL: Transcription
factor pathways and congenital heart disease. Curr Top Dev Biol.
100:253–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vincentz JW, Barnes RM and Firulli AB:
Hand factors as regulators of cardiac morphogenesis and
implications for congenital heart defects. Birth Defects Res A Clin
Mol Teratol. 91:485–494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Reamon-Buettner SM, Ciribilli Y, Inga A
and Borlak J: A loss-of-function mutation in the binding domain of
HAND1 predicts hypoplasia of the human hearts. Hum Mol Genet.
17:1397–1405. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Reamon-Buettner SM, Ciribilli Y, Traverso
I, Kuhls B, Inga A and Borlak J: A functional genetic study
identifies HAND1 mutations in septation defects of the human heart.
Hum Mol Genet. 18:3567–3578. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng Z, Lib L, Li Z, Liu M, Yan J, Wang B
and Ma X: Two novel HAND1 mutations in Chinese patients with
ventricular septal defect. Clin Chim Acta. 413:675–677. 2012.
View Article : Google Scholar
|
|
31
|
Thomas T, Yamagishi H, Overbeek PA, Olson
EN and Srivastava D: The bHLH factors, dHAND and eHAND, specify
pulmonary and systemic cardiac ventricles independent of left-right
sidedness. Dev Biol. 196:228–236. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thattaliyath BD, Livi CB, Steinhelper ME,
Toney GM and Firulli AB: HAND1 and HAND2 are expressed in the
adult-rodent heart and are modulated during cardiac hypertrophy.
Biochem Biophys Res Commun. 297:870–875. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Srivastava D, Cserjesi P and Olson EN: A
subclass of bHLH proteins required for cardiac morphogenesis.
Science. 270:1995–1999. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Srivastava D, Thomas T, Lin Q, Kirby ML,
Brown D and Olson EN: Regulation of cardiac mesodermal and neural
crest development by the bHLH transcription factor, dHAND. Nat
Genet. 16:154–160. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
McFadden DG, Barbosa AC, Richardson JA,
Schneider MD, Srivastava D and Olson EN: The Hand1 and Hand2
transcription factors regulate expansion of the embryonic cardiac
ventricles in a gene dosage-dependent manner. Development.
132:189–201. 2005. View Article : Google Scholar
|
|
36
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.
|
|
37
|
Wei D, Bao H, Zhou N, Zheng GF, Liu XY and
Yang YQ: GATA5 loss-of-function mutation responsible for the
congenital ventriculoseptal defect. Pediatr Cardiol. 34:504–511.
2013. View Article : Google Scholar
|
|
38
|
Zhang XL, Dai N, Tang K, Chen YQ, Chen W,
Wang J, Zhao CM, Yuan F, Qiu XB, Qu XK, et al: GATA5
loss-of-function mutation in familial dilated cardiomyopathy. Int J
Mol Med. 35:763–770. 2015.
|
|
39
|
Starr JP: Tetralogy of fallot: Yesterday
and today. World J Surg. 34:658–668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Russell MW, Kemp P, Wang L, Brody LC and
Izumo S: Molecular cloning of the human HAND2 gene. Biochim Biophys
Acta. 1443:393–399. 1998. View Article : Google Scholar
|
|
41
|
Dai YS and Cserjesi P: The basic
helix-loop-helix factor, HAND2, functions as a transcriptional
activator by binding to E-boxes as a heterodimer. J Biol Chem.
277:12604–12612. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dai YS, Cserjesi P, Markham BE and
Molkentin JD: The transcription factors GATA4 and dHAND physically
interact to synergistically activate cardiac gene expression
through a p300-dependent mechanism. J Biol Chem. 277:24390–24398.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thattaliyath BD, Firulli BA and Firulli
AB: The basic-helix-loop-helix transcription factor HAND2 directly
regulates transcription of the atrial naturetic peptide gene. J Mol
Cell Cardiol. 34:1335–1344. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zang MX, Li Y, Wang H, Wang JB and Jia HT:
Cooperative interaction between the basic helix-loop-helix
transcription factor dHAND and myocyte enhancer factor 2C regulates
myocardial gene expression. J Biol Chem. 279:54258–54263. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zang MX, Li Y, Xue LX, Jia HT and Jing H:
Cooperative activation of atrial naturetic peptide promoter by
dHAND and MEF2C. J Cell Biochem. 93:1255–1266. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yelon D, Ticho B, Halpern ME, Ruvinsky I,
Ho RK, Silver LM and Stainier DY: The bHLH transcription factor
hand2 plays parallel roles in zebrafish heart and pectoral fin
development. Development. 127:2573–2582. 2000.PubMed/NCBI
|
|
47
|
Garavito-Aguilar ZV, Riley HE and Yelon D:
Hand2 ensures an appropriate environment for cardiac fusion by
limiting Fibronectin function. Development. 137:3215–3220. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morikawa Y and Cserjesi P: Cardiac neural
crest expression of Hand2 regulates outflow and second heart field
development. Circ Res. 103:1422–1429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tsuchihashi T, Maeda J, Shin CH, Ivey KN,
Black BL, Olson EN, Yamagishi H and Srivastava D: Hand2 function in
second heart field progenitors is essential for cardiogenesis. Dev
Biol. 351:62–69. 2011. View Article : Google Scholar :
|
|
50
|
VanDusen NJ, Casanovas J, Vincentz JW,
Firulli BA, Osterwalder M, Lopez-Rios J, Zeller R, Zhou B,
Grego-Bessa J, De La Pompa JL, et al: Hand2 is an essential
regulator for two Notch-dependent functions within the embryonic
endocardium. Cell Rep. 9:2071–2083. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tamura M, Hosoya M, Fujita M, Iida T,
Amano T, Maeno A, Kataoka T, Otsuka T, Tanaka S, Tomizawa S, et al:
Overdosage of Hand2 causes limb and heart defects in the human
chromosomal disorder partial trisomy distal 4q. Hum Mol Genet.
22:2471–2481. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shen L, Li XF, Shen AD, Wang Q, Liu CX,
Guo YJ, Song ZJ and Li ZZ: Transcription factor HAND2 mutations in
sporadic Chinese patients with congenital heart disease. Chin Med J
(Engl). 123:1623–1627. 2010.
|