Introduction
Renal cell carcinoma (RCC) is the most common renal
cancer and most aggressive urologic tumor, and its incidence is
increasing (1). Approximately
15-20% of RCC patients already have metastases at diagnosis, while
up to 30% of patients develop metastases during therapy. Once
metastasized, the prognosis for patients is poor. Increased
knowledge of the molecular modes of action of RCC has contributed
to the development of targeted therapies during the last decade,
thus improving the outlook for patients in advanced stages of the
disease. However, despite therapeutic advances, prognosis for
patients with RCC remains poor, with 5-year survival remaining
between 5 and 12% (2,3).
Most patients with advanced RCC wish to actively
participate in their battle against cancer and/or to avoid adverse
side effects, which often occur during conventional therapy. Many
patients, therefore, turn towards complementary and alternative
medicine (CAM). More than 50% of cancer patients in Europe
(4) and up to 80% of cancer
patients in the United States (5)
use CAM together with or in place of conventional therapy.
Amygdalin (D-mandelonitrile-β-gentiobioside) is a
natural compound which is often used by cancer patients. It is
found in the fruit kernels of apricots, peaches, apples and in
bitter almonds (6–8). The first studies on amygdalin use by
cancer patients came from Russia in the 1840s (9). In the 1920s amygdalin was also
administered to cancer patients in the United States (10). In the 1950s a semi-synthetic,
chemically different derivate of amygdalin, laetrile, was
introduced. After the introduction of laetrile, the terms amygdalin
and laetrile were often used synonymously, making it difficult to
draw conclusions from studies which did not discriminate between
the two compounds (11). In the
1970s amygdalin/leatrile became one of the most popular,
non-conventional anticancer treatments. By 1978, approximately
70,000 US cancer patients had used amygdalin (12). The National Cancer Institute (NCI)
initiated several studies (13–16) with sobering results. In a summary
of the only phase II trial, no substantive benefit was ascribed to
amygdalin, whereas several patients with symptoms of cyanide (HCN)
poisoning were described (13).
However, the quality of this study is questionable since a
heterogeneous patient cohort was used, no control groups were
included, and a racemate instead of pure amygdalin was used for the
i.v. therapy. All official judgement concerning amygdalin has been
based on this trial, since no other clinical trial with amygdalin
is available. The German Federal Institute for Drugs and Medical
Devices (BfArM) (http://www.bfarm.de/DE/Home/home_node.html), has also
classified amygdalin as a questionable drug, as have counterparts
in other countries. Despite the controversy and lack of
scientifically sound data on the benefits and risks of amygdalin,
many cancer patients use amygdalin (11,17). Thus, to clarify the many questions
which remain to be answered on the impact of amygdalin on tumor
growth and proliferation, the cell cycle progression and underlying
molecular action modes in RCC cells were determined in the present
study.
Materials and methods
Cell cultures and amygdalin
treatment
The kidney carcinoma cell lines, Caki-1, KTC-26 and
A498 were purchased from LGC Promochem (Wesel, Germany). The cells
were grown and sub-cultured in RPMI-1640 medium (obtained from
Seromed, Berlin, Germany) supplemented with 10% FCS, 20 mM
HEPES-buffer, 100 IU/ml penicillin and 100 µg/ml
streptomycin at 37°C in a humidified atmosphere with 5%
CO2 in an incubator. Sub-cultures from passages 5–24
were selected for use in the experiments. Amygdalin from apricot
kernels (Sigma-Aldrich, Taufkirchen, Germany) was freshly dissolved
in cell culture medium and was then added to the tumor cells at a
concentration of 10 mg/ml [previously evaluated as optimal
concentration (18)] for either
24 h or 2 weeks (three times a week) to evaluate acute versus
chronic treatment. The controls remained untreated. In all
experiments in the present study, treated tumor cell cultures were
compared to non-treated tumor cell cultures. In order to examine
the toxic effects of amygdalin, cell viability was determined by
Trypan blue (Gibco/ Invitrogen, Darmstadt, Germany).
Measuring tumor cell growth,
proliferation and apoptosis
Cell growth was assessed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
dye reduction assay (Roche Diagnostics, Penzberg, Germany). Caki-1
cells (50 µl, 1×105 cells/ml) were seeded onto
96-well tissue culture plates. After 24, 48 and 72 h, 10 µl
MTT (0.5 mg/ml) were added for an additional 4 h. Thereafter, cells
were lysed in a buffer containing 10% SDS in 0.01 M HCl. The plates
were incubated overnight at 37°C, 5% CO2. Absorbance at
550 nm was determined for each well using a microplate
enzyme-linked immunosorbent assay (ELISA) reader. After subtracting
background absorbance, results were expressed as the mean number of
cells.
Cell proliferation was measured using a BrdU cell
proliferation ELISA kit (Calbiochem/Merck Biosciences, Darmstadt,
Germany). Tumor cells, seeded onto 96-well plates, were incubated
with 20 µl BrdU-labeling solution per well for 8 h, and
fixed and detected using anti-BrdU mAb according to the
manufacturer's instructions. Absorbance was measured at 450 nm
using a microplate ELISA reader.
In order to evaluate whether tumor cell growth was
impaired or reduced due to apoptosis, the expression of Annexin
V/propidium iodide (PI) was evaluated using an Annexin V-FITC
Apoptosis Detection kit (BD Pharmingen, Heidelberg, Germany). Tumor
cells were washed twice with PBS and subsequently incubated with 5
µl Annexin V-FITC and 5 µl of PI in the dark for 15
min at room temperature. Cells were analyzed by flow cytometry
using FACSCalibur (BD Biosciences, Heidelberg, Germany). The
percentage of apoptotic cells (early and late) in each quadrant was
calculated using CellQuest software (BD Biosciences).
Percentage of cells in different cell
cycle phases
Cell cycle analysis was carried out on subconfluent
cell cultures. Tumor cell populations were stained with PI, using a
CycleTEST PLUS DNA Reagent Kit and then subjected to flow cytometry
using FACScan (both from Becton-Dickinson, Heidelberg, Germany).
From each sample 10,000 events were collected. Data acquisition was
carried out using CellQuest software, and cell cycle distribution
was calculated using ModFit software (Becton-Dickinson). The number
of gated cells in G1, G2/M or S phases is expressed in percentage
form.
Expression of cell cycle regulating
proteins
Cell cycle regulating proteins were investigated by
western blot analysis. Tumor cell lysates were applied to a 7-15%
polyacrylamide gel (depending on protein size) and electrophoresed
for 90 min at 100 V. The protein was subsequently transferred to
nitrocellulose membranes (1 h, 100 V). After blocking with non-fat
dry milk for 1 h, the membranes were incubated overnight with
monoclonal antibodies directed against the following cell cycle
proteins, which were all from BD Biosciences: cdk1 (IgG1, clone 1,
dilution 1:2,500; #610038), cdk2 (IgG2a, clone 55, dilution
1:2,500; #610146), cdk4 (IgG1, clone 97, dilution 1:250; #610148),
cyclin A (IgG1, clone 25, dilution 1:250; #611269), cyclin B (IgG1,
clone 18, dilution 1:1,000; #610220), cyclin D1 (IgG1, clone
G124-326, dilution 1:250; #554181), cyclin D3 (IgG2b, clone 1,
dilution 1:1,000; #610280), p19 (IgG1, clone 52/p19, dilution
1:5,000; #610530), p27 (IgG1, clone 57, dilution 1:500; #610244).
HRP-conjugated goat-anti-mouse IgG (dilution 1:5,000; #12-349;
Merck Millipore, Temecula, CA, USA) served as the secondary
antibody. The membranes were briefly incubated with ECL detection
reagent (ECL™; Amersham/GE Healthcare, München, Germany) to
visualize the proteins and then analyzed with the Fusion FX7 system
(Peqlab, Erlangen, Germany). β-actin (dilution 1:1,000; #A5441;
Sigma, Taufenkirchen, Germany) served as the internal control.
Surface expression of E- and
N-cadherin
Tumor cells were washed in blocking solution (PBS,
0.5% BSA) and then incubated for 60 min at 4°C with phycoerythrin
(PE)-conjugated monoclonal antibodies (mAB) directed against the
following: anti-human E-cadherin-PE (mouse IgG2b, clone 180224;
#FAB18381P) and anti-human N-cadherin-PE (rat IgG2a, clone 401408;
#IC1388P) (both from R&D Systems, Wiesbaden, Germany). E- and
N-cadherin surface expression of the RCC cells was then measured by
flow cytometry using FACscan [FL-2H (log) channel histogram
analysis; 1×104 cells/ scan;BD Biosciences] and
expressed as mean fluorescence units. Rat IgG2a-PE (clone RG7/1.30;
#558067) or mouse IgG2b-PE (clone 27-35; #555743) (both from BD
Biosciences) served as isotype controls.
Blocking experiments
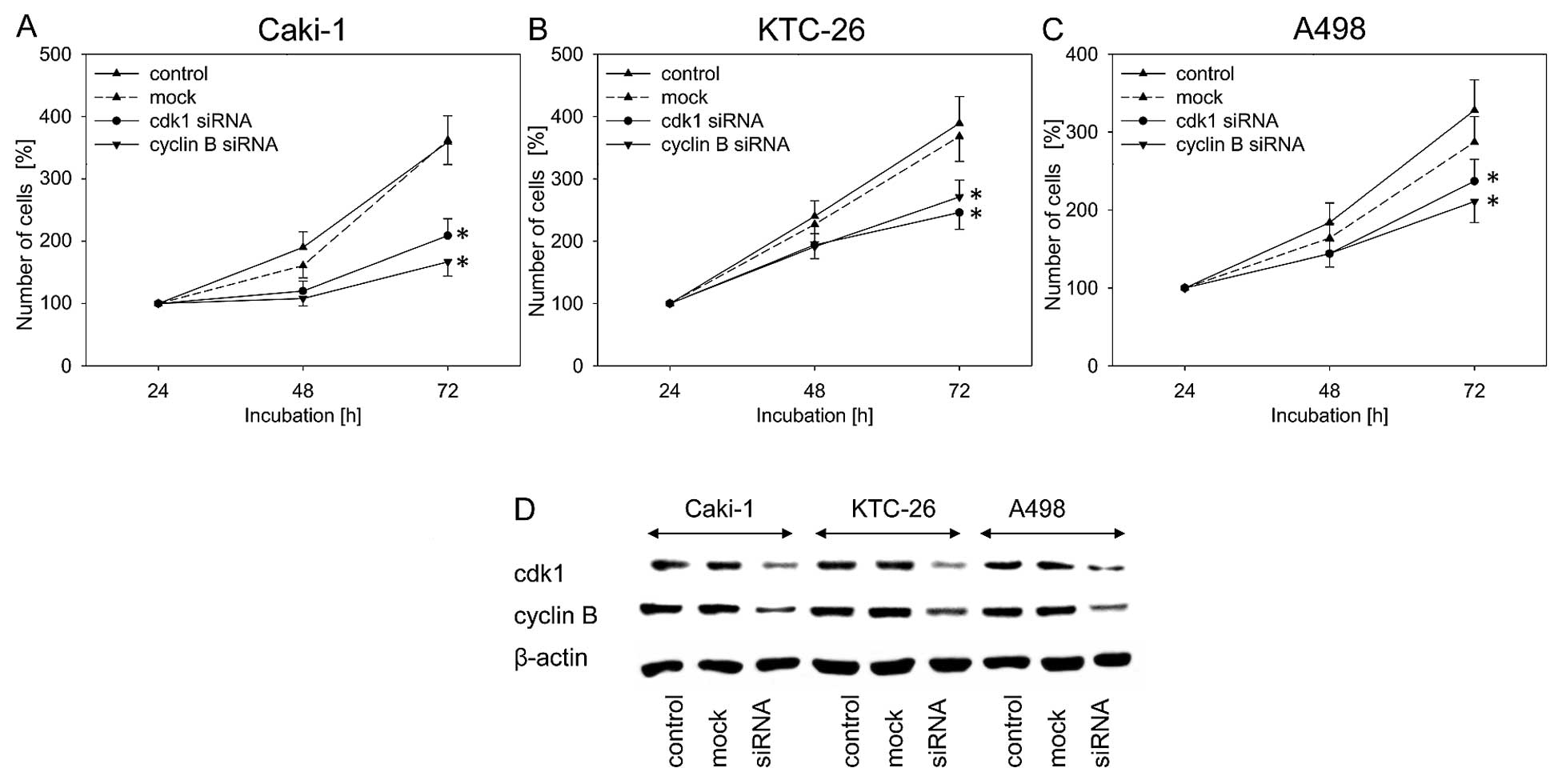
To determine whether cdk1 and cyclin B impacted
tumor cell growth in Caki-1, KTC-26 and A498 cell lines, cells were
transfected. Tumor cells (3×105/6-well) were transfected
with small interfering RNA (siRNA) directed against cdk1
(Hs_CDC2_10, gene ID: 983, target sequence: AAGGGGTTCCTAGTACTGCAA)
or cyclin B (Hs_CCNB1_6, gene ID: 891, target sequence:
AATGTAGTCATGGTAAATCAA) (both from Qiagen, Hilden, Germany), with
siRNA/transfection reagent (HiPerFect transfection reagent; Qiagen)
at a ratio of 1:6. Non-treated cells and cells treated with 5 nM
control siRNA (AllStars Negative Control siRNA; Qiagen) served as
controls. Subsequently, tumor cell growth was determined as
indicated above.
Statistical analysis
All experiments were performed 3–6 times.
Statistical significance was determined by the
Wilcoxon-Mann-Whitney U test. A p-value <0.05 was considered to
indicate a statistically significant difference.
Results
Tumor cell growth and proliferation is
blocked by amygdalin
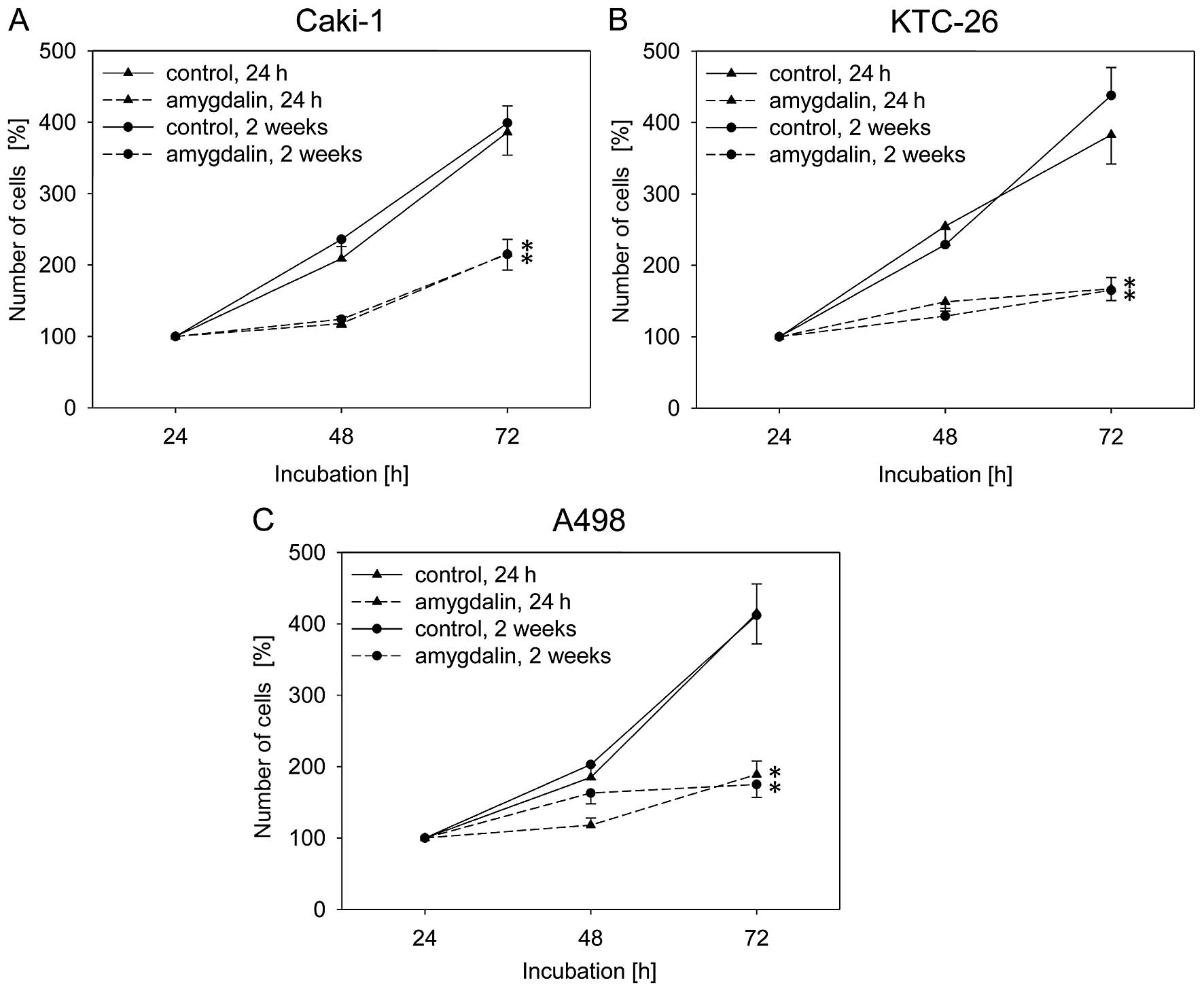
Exposure to amygdalin (10 mg/ml) for 24 h or 2 weeks
resulted in significant and similar degrees of growth inhibition
over 72 h in all three RCC cell lines, Caki-1, KTC-26 and A498,
compared to the untreated control cells (Fig. 1). Caki-1, KTC-26 and A498 cell
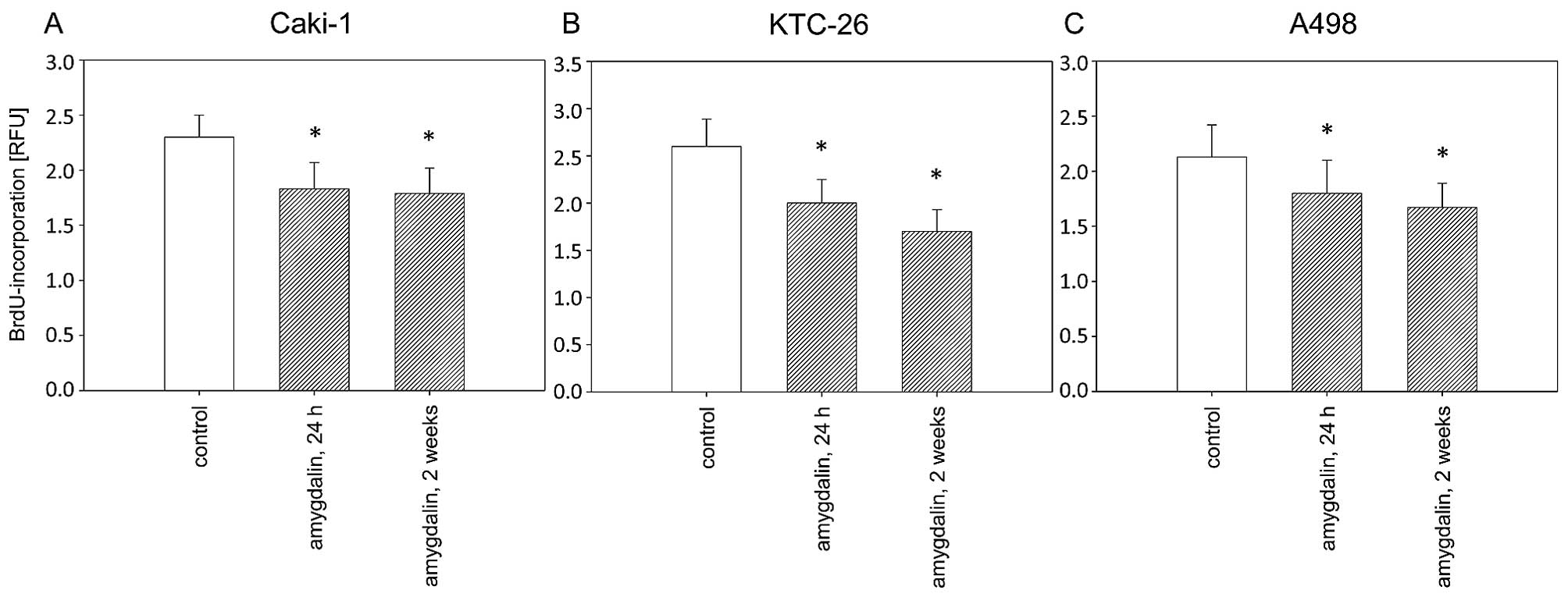
proliferation was also significantly reduced after 24 h or 2 weeks
of amygdalin exposure, compared to the controls (Fig. 2). Antitumor effects in the RCC
cells were comparable after 24 h and 2 weeks of amygdalin treatment
(Fig. 2).
Neither apoptosis nor necrosis is induced
by amygdalin
Neither significant early or late apoptosis, nor
induction of necrosis, was detected after amygdalin administration
(data not shown).
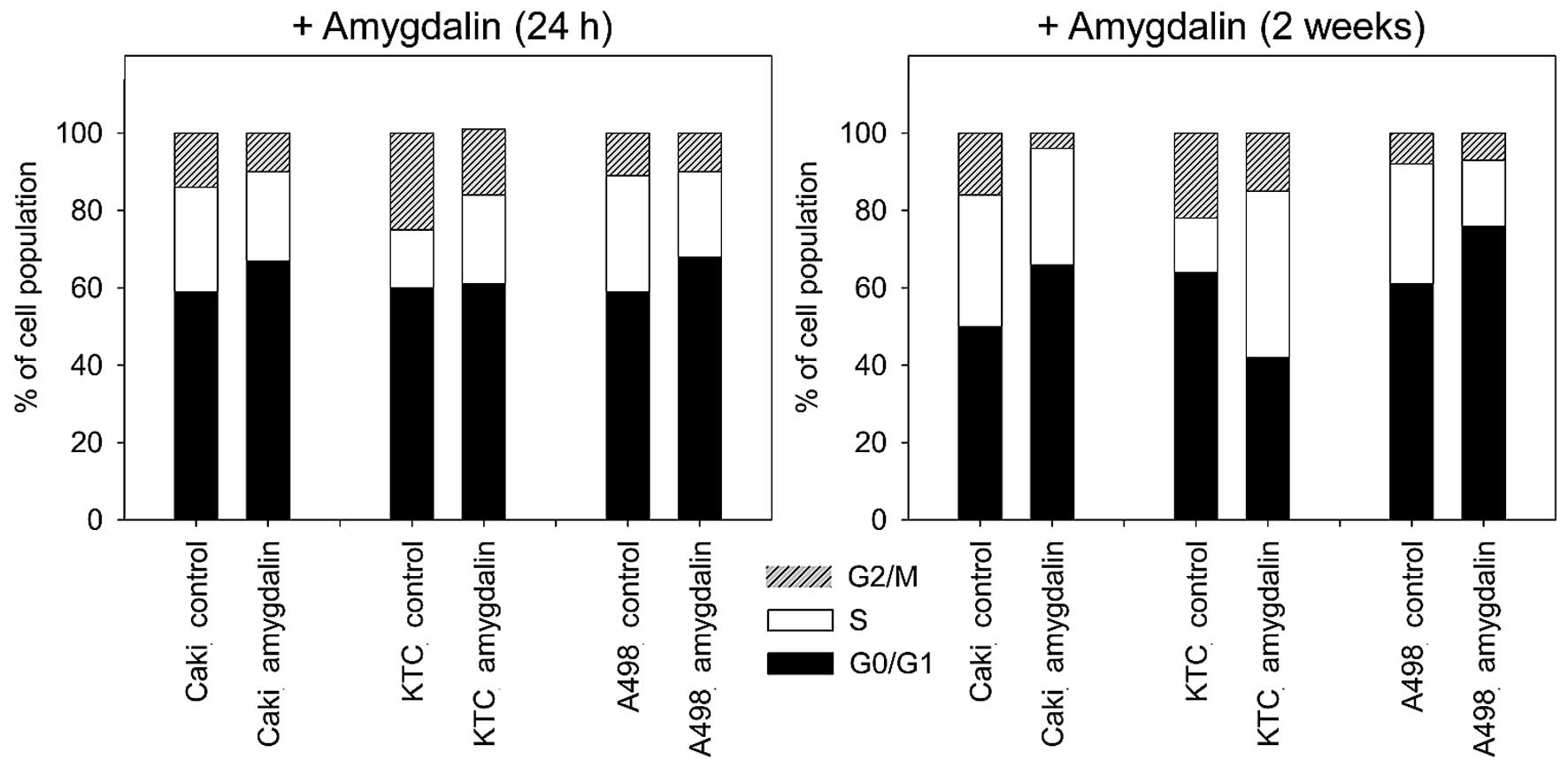
Amygdalin alters the percentage of RCC
cells in G0/1-, S- and G2/M-phases
Amygdalin significantly increased the percentage of
Caki-1 and A498 cells in the G0/G1-phase and reduced the amount of
S- and G2/M-phase cells after 24 h and 2 weeks (Fig. 3) of exposure, compared to
untreated controls. In KTC-26 cells, amygdalin caused the
percentage of G2/M-phase cells to significantly decrease, while
S-phase cells increased (24 h, <2 weeks). No significant
increase in the percentage of G0/G1-phase cells was measured after
24 h amygdalin treatment in KTC-26 cells. After 2 weeks of amydalin
treatment, in KTC-26 cells, concomitant with the S-phase increase,
the number of G0/G1-phase cells significantly decreased (Fig. 3), compared to the control.
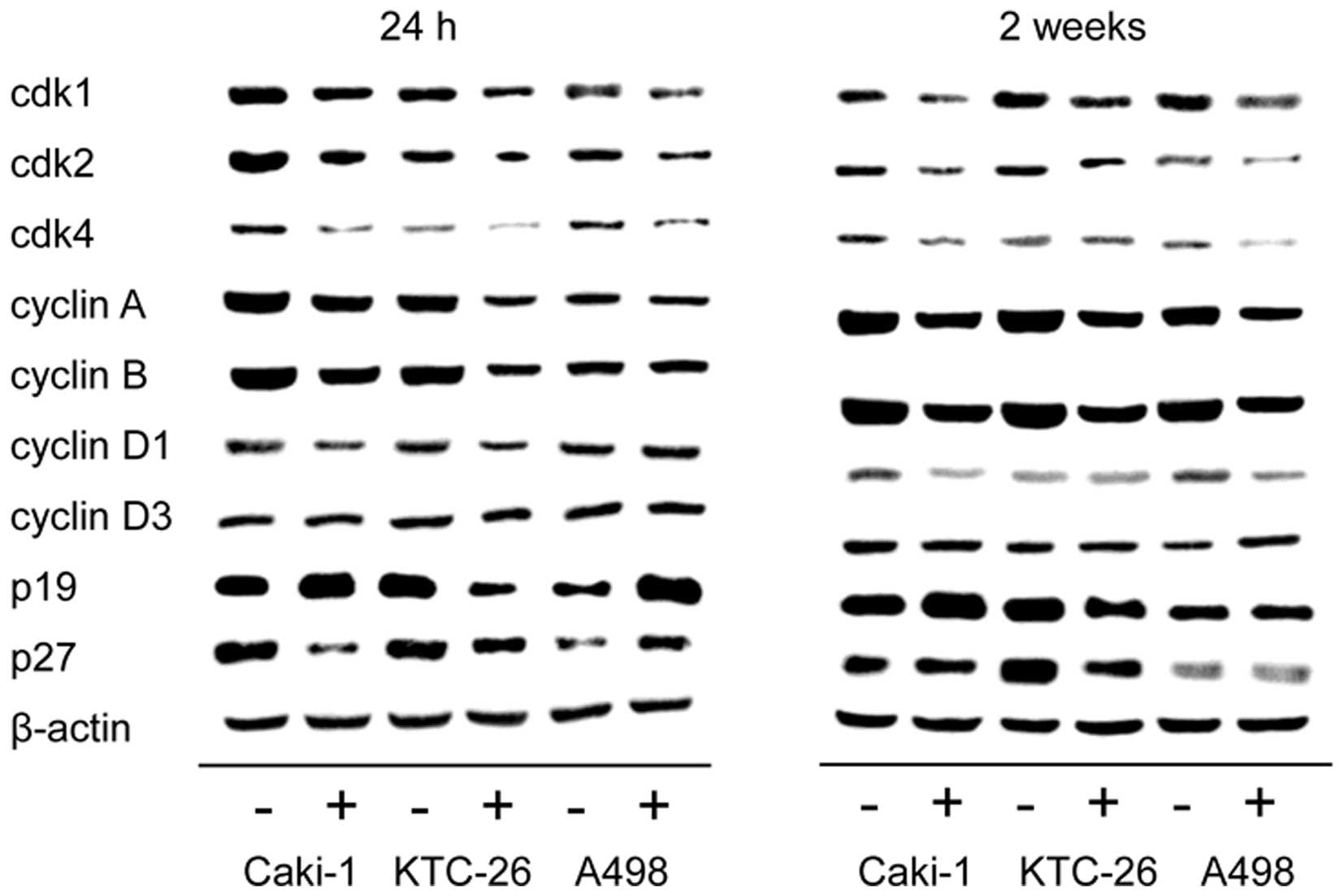
Amygdalin causes a reduction in cell
cycle activating protein expression
We noted that the alterations in cell cycle
progression were accompanied by modulation of cell cycle regulating
proteins (Fig. 4). In all three
cell lines, Caki-1, KTC-26 and A498, treatment with amygdalin for
24 h and 2 weeks contributed to downregulation of the cell cycle
activating proteins cdk1, cdk2 and cdk4 as well as cyclin A and B,
with the strongest effects being noted in relation to cdk1 and
cyclin B expression. Cyclin D1 was also diminished in Caki-1 and
KTC-26 after 24 h (Fig. 4, left
panel) and in Caki-1 and A498 after 2 weeks amygdalin application
(Fig. 4, right panel). No marked
changes were detectable for cyclin D3, in any cell line. By
contrast to the cell cycle activating proteins, expression of the
cell cycle inhibiting protein p19 was enhanced after amygdalin
exposure in Caki-1 (24 h and 2 weeks) and A498 (24 h) cell lines.
p27 was also elevated in A498 cells (24 h) (Fig. 4, left panel). However, p19 and p27
were reduced in KTC-26 cells, and diminished p27 was noted in
Caki-1 cells after 24 h.
A decrease in cdk1 and cyclin B is
involved in growth inhibition caused by amygdalin
Due to the fact that in the present study the most
striking inhibitory effect of amygdalin was noted in relation to
cdk1 and cyclin B expression, the impact of those two proteins on
tumor cell growth was evaluated by blocking their function using
siRNA. Knockdown of cdk1 and cyclin B resulted in significant
inhibition of cell growth in all three cell lines, compared to the
untreated and mock control (Fig.
5A–C). In all three RCC cell lines, blocking of cdk1 and cyclin
B protein expression was verified by western blot analysis
(Fig. 5D).
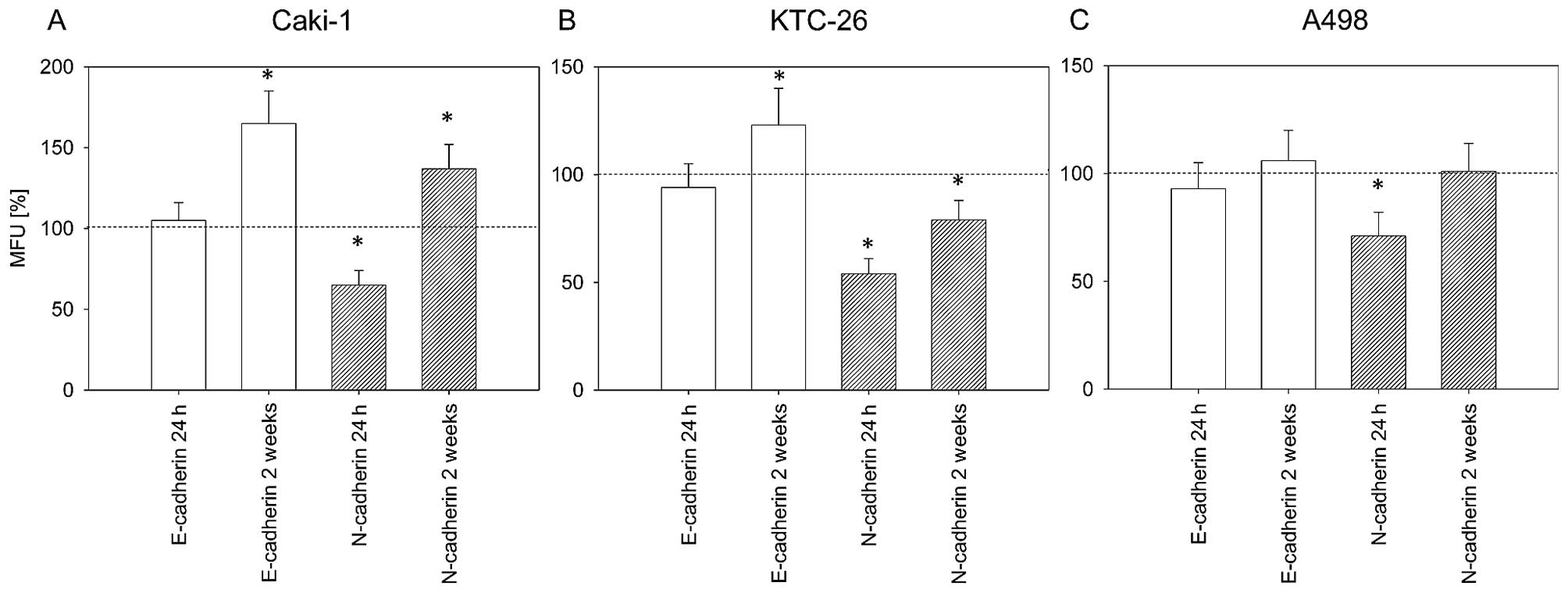
Differentiation markers are modulated by
amygdalin treatment
Dedifferentiation of tumor cells is accompanied by
loss of E-cadherin and increased N-cadherin expression. Expression
of these two differentiation markers was determined in order to
evaluate whether amygdalin influences tumor cell differentiation.
After 24 h of treatment with amygdalin, we noted a significant
decrease of surface N-cadherin in the three cell lines (Fig. 6). After 2 weeks of amygdalin
treatment, markedly increased E-cadherin expression on the surface
of Caki-1 and KTC-26 cells was noted (Fig. 6A and B). In KTC-26 cells,
E-cadherin elevation was associated with a significant reduction in
surface N-cadherin (Fig. 6B).
However, N-cadherin expression in Caki-1 cells significantly
increased after 2 weeks of amygdalin exposure, although the MFU was
still lower than that for E-cadherin (Fig. 6A). No significant effect on
E-cadherin was noted in the A498 cells after 2 weeks of amygdalin
application.
Discussion
In the present study, we noted that treatment of the
RCC cell lines Caki-1, KTC-26 and A498 with amygdalin caused
significant inhibition of cell growth and proliferation. Similar
growth reduction after amygdalin application has been noted in
non-small cell lung cancer (19)
and bladder cancer cells (18)
in vitro, as well as cervical cancer cells in vivo
(20). Based on our data we
conclude that the inhibition of growth induced by amygdalin is not
due to apoptosis or necrosis. Other cancer cells such as cervical,
bladder and prostate cancer cells react to amygdalin with
apoptosis, leading to growth inhibition (18,20,21). Thus, the mode of action of
amygdalin seems to depend on the type of cancer.
Although inhibition of growth in all three RCC cell
lines was accompanied by changes in the percentage of cells in
different cell cycle phases, the changes were not homogeneous.
Treatment of Caki-1 and A498 cells with amygdalin caused an
increase in G0/G1-phase cells by reducing the S- (Caki-1 and A498)
and G2/M-phases (Caki-1). Amygdalin treatment caused an increase of
S-phase cells in KTC-26 cells, while the G2/M- and G0/G1-phases
were reduced after 2 weeks. The elevation of S-phase cells in
KTC-26 after amygdalin application is likely indicative of cell
cycle arrest in the S-phase. In various bladder cancer cell lines,
amygdalin-induced growth blockade, effected by differently
influencing cell cycle progression, has also been noted, increasing
the percentage of G0/G1 phase cells in one cell line and elevating
S-phase cells in another (18).
Alteration of the percentage of cell cycle phases
was correlated with modulation of the expression of cell cycle
regulating proteins. In all three RCC cell lines, most cell cycle
activating proteins were reduced after amygdalin treatment, in
particular cdk1 and cyclin B. Cdk1 is known to be a key kinase for
mitotic entry (22). The
cdk1-cyclin B axis in tumor cells has been shown to be involved in
promoting mitosis and overcoming chemotherapy-dependent cell cycle
arrest (23). In all three RCC
cell lines, the decrease in cdk1 and cyclin B was related to the
inhibitory effect exerted by amygdalin, as proved by siRNA
knockdown. As well as the cdk1-cyclin B axis, the cdk2-cyclin A
axis was also distinctly altered in RCC cells. Cdk2/cyclin A
promotes G1/S-phase transition and has been shown to be important
to the inhibition of bladder cancer cells caused by amygdalin
(18). We suggest that the
accumulation of G0/G1-cells was due to the inhibitory effect which
amygdalin exerted on cdk2 and cyclin B. However, amygdalin treament
of the KTC-26 cell line did not result in G0/G1-, but rather
S-phase, arrest. Conceivably, this is due to the cell cycle
inhibiting protein p19, which was elevated in Caki-1 and A498 cells
after amygdalin application, but diminished in KTC-26 cells. p19 is
involved in G1 checkpoint activity, stopping the entry of cells
into the S-phase (24).
Inhibiting p19 increases the S-phase cell fraction (25). Thus, this likely explains why we
noted an increase in in the G0/G1-phase of Caki-1 and A498 cells,
while KTC-26 cells accumulated in the S-phase. Hence, we suggest
that amygdalin-induced alterations to cell cycle regulating protein
expression are responsible for different effects on cell cycle
progression in different cell lines.
During RCC tumor genesis and progression,
dedif-ferentiation accompanied by epithelial mesenchymal transition
(EMT) takes place (26,27). During transition the tumor cells
lose epithelial (E)-cadherin and gain neural (N)-cadherin (26,28). In all three RCC cell lines used in
this study, application of amygdalin for 24 h caused a significant
decrease in N-cadherin expression, which indicates
re-differentiation. N-cadherin has previously been associated with
aggressiveness and malignant potential of RCC (29). Hence, we suggest that impairing
N-cadherin expression with 24 h of amygdalin application results in
a less malignant tumor type. After 2 weeks of amygdalin exposure, a
switch in the mode of action of amygdalin became apparent, mainly
affecting E-cadherin expression. Caki-1 and KTC-26 E-cadherin
surface expression significantly increased. In various RCC cells
in vitro epithelial-mesenchymal transition, tumor growth and
an aggressive phenotype have been shown to be inversely linked to a
low level of E-cadherin (30,31). Poor prognosis and high-grade RCC
tumors have been associated with a lack of E-cadherin (32). In human RCC tumor tissue, a 3-fold
decrease of E-cadherin has been observed (33), and it has been postulated that
E-cadherin expression in RCC is an important predictor for disease
recurrence (34). Thus, we
suggest that the amygdalin-induced E-cadherin increase in Caki-1
and KTC-26 cells which we noted indicates re-differentiation back
to a less aggressive phenotype. The observed switch from N-cadherin
reduction to E-cadherin amplification indicates different modes of
amygdalin action. Since N-cadherin was no longer diminished in any
of the three cell lines after 2 weeks of amygdalin exposure, and
was even enhanced in Caki-1 cells, we hypothesize that the ratio
between E- and N-cadherin expression is crucial for differentiation
status. Indeed, it has been shown that the effect of N-cadherin
depends on E-cadherin expression (29).
In conclusion, amygdalin inhibits cell cycle
progression and tumor cell growth in RCC cells, at least partially,
by impairing the expression of cdk1 and cyclin B, thus exerting
antitumor effects in vitro. Although no necrotic effects
have been detected in vitro, toxic effects caused by the
degradation of amygdalin to HCN are possible, and this aspect
requires evaluation. Further investigation using animals is
necessary to verify the in vitro effects of amygdalin and to
evaluate whether HCN causes cytotoxicity in vivo.
Acknowledgments
This study was supported by the 'Brigitta und
Norbert Muth Stiftung' and 'Prof. Dr. Karl und Gerhard
Schiller-Stiftung'.
References
|
1
|
Maute L, Grünwald V, Weikert S, Kube U,
Gauler T, Kahl C, Burkholder I and Bergmann L: Therapy of mRCC
beyond mTOR-inhibition in clinical practice: results of a
retrospective analysis. J Cancer Res Clin Oncol. 140:823–827. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Najjar YG and Rini BI: Novel agents in
renal carcinoma: a reality check. Ther Adv Med Oncol. 4:183–194.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sun M, Thuret R, Abdollah F, Lughezzani G,
Schmitges J, Tian Z, Shariat SF, Montorsi F, Patard JJ, Perrotte P
and Karakiewicz PI: Age-adjusted incidence, mortality, and survival
rates of stage-specific renal cell carcinoma in North America: a
trend analysis. Eur Urol. 59:135–141. 2011. View Article : Google Scholar
|
|
4
|
Huebner J, Micke O, Muecke R, Buentzel J,
Prott FJ, Kleeberg U and Senf B: User rate of complementary and
alternative medicine (CAM) of patients visiting a counseling
facility for CAM of a German comprehensive cancer center.
Anticancer Res. 34:943–948. 2014.PubMed/NCBI
|
|
5
|
Saghatchian M, Bihan C, Chenailler C,
Mazouni C, Dauchy S and Delaloge S: Exploring frontiers: use of
complementary and alternative medicine among patients with
early-stage breast cancer. Breast. 23:279–285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bolarinwa IF, Orfila C and Morgan MR:
Determination of amygdalin in apple seeds, fresh apples and
processed apple juices. Food Chem. 170:437–442. 2015. View Article : Google Scholar
|
|
7
|
Tanaka R, Nitta A and Nagatsu A:
Application of a quantitative 1H-NMR method for the determination
of amygdalin in Persicae semen, Armeniacae semen, and Mume fructus.
J Nat Med. 68:225–230. 2014. View Article : Google Scholar
|
|
8
|
Lee J, Zhang G, Wood E, Rogel Castillo C
and Mitchell AE: Quantification of amygdalin in nonbitter,
semibitter, and bitter almonds (Prunus dulcis) by UHPLC-(ESI)QqQ
MS/MS. J Agric Food Chem. 61:7754–7759. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moss RW: The laetrile controversy. The
Cancer Industry. The Classic Expose on the Cancer Establishment.
First Equinox Press; Brooklyn, NY: pp. 131–152. 1996
|
|
10
|
Curt GA: Unsound methods of cancer
treatment. Princ Pract Oncol Updates. 4:1–10. 1990.
|
|
11
|
PDQ Cancer Complementary and Alternative
Medicine Editorial Board Laetrile/Amygdalin (PDQ®): Health
Professional Version PDQ Cancer Information Summaries [Internet].
Bethesda (MD): National Cancer Institute (US); 2002–2015
|
|
12
|
Moss RW: Patient perspectives: Tijuana
cancer clinics in the post-NAFTA era. Integr Cancer Ther. 4:65–86.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moertel CG, Fleming TR, Rubin J, Kvols LK,
Sarna G, Koch R, Currie VE, Young CW, Jones SE and Davignon JP: A
clinical trial of amygdalin (Laetrile) in the treatment of human
cancer. N Engl J Med. 306:201–206. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moertel CG, Ames MM, Kovach JS, Moyer TP,
Rubin JR and Tinker JH: A pharmacologic and toxicological study of
amygdalin. JAMA. 245:591–594. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ames MM, Moyer TP, Kovach JS, Moertel CG
and Rubin J: Pharmacology of amygdalin (laetrile) in cancer
patients. Cancer Chemother Pharmacol. 6:51–57. 1981.PubMed/NCBI
|
|
16
|
Newell GR and Ellison NM: Ethics and
designs: laetrile trials as an example. Cancer Treat Rep.
64:363–365. 1980.PubMed/NCBI
|
|
17
|
Wahab MF, Breitbach ZS, Armstrong DW,
Strattan R and Berthod A: Problems and pitfalls in the analysis of
amygdalin and its epimer. J Agric Food Chem. 63:8966–8973. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Makarević J, Rutz J, Juengel E, Kaulfuss
S, Reiter M, Tsaur I, Bartsch G, Haferkamp A and Blaheta RA:
Amygdalin blocks bladder cancer cell growth in vitro by diminishing
cyclin A and cdk2. PLoS One. 9:e1055902014. View Article : Google Scholar
|
|
19
|
Qian L, Xie B, Wang Y and Qian J:
Amygdalin-mediated inhibition of non-small cell lung cancer cell
invasion in vitro. Int J Clin Exp Pathol. 8:5363–5370.
2015.PubMed/NCBI
|
|
20
|
Chen Y, Ma J, Wang F, Hu J, Cui A, Wei C,
Yang Q and Li F: Amygdalin induces apoptosis in human cervical
cancer cell line HeLa cells. Immunopharmacol Immunotoxicol.
35:43–51. 2013. View Article : Google Scholar
|
|
21
|
Chang HK, Shin MS, Yang HY, Lee JW, Kim
YS, Lee MH, Kim J, Kim KH and Kim CJ: Amygdalin induces apoptosis
through regulation of Bax and Bcl-2 expressions in human DU145 and
LNCaP prostate cancer cells. Biol Pharm Bull. 29:1597–1602. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang WL, Yu CC, Chen CS and Guh JH:
Tubulin-binding agents down-regulate matrix metalloproteinase-2 and
-9 in human hormone-refractory prostate cancer cells - a critical
role of Cdk1 in mitotic entry. Biochem Pharmacol. 94:12–21. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Visconti R, Della Monica R, Palazzo L,
D'Alessio F, Raia M, Improta S, Villa MR, Del Vecchio L and Grieco
D: The Fcp1-Wee1-Cdk1 axis affects spindle assembly checkpoint
robustness and sensitivity to antimicrotubule cancer drugs. Cell
Death Differ. 22:1551–1560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang WT, Catto JW and Meuth M:
Differential response of normal and malignant urothelial cells to
CHK1 and ATM inhibitors. Oncogene. 34:2887–2896. 2015. View Article : Google Scholar
|
|
25
|
Carcagno AL, Marazita MC, Ogara MF, Ceruti
JM, Sonzogni SV, Scassa ME, Giono LE and Cánepa ET: E2F1-mediated
upregulation of p19INK4d determines its periodic expression during
cell cycle and regulates cellular proliferation. PLoS One.
6:e219382011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yuan H, Meng X, Guo W, Cai P, Li W, Li Q,
Wang W, Sun Y, Xu Q and Gu Y: Transmembrane-bound IL-15-promoted
epithelial-mesenchymal transition in renal cancer cells requires
the Src-dependent Akt/GSK-3β/β-catenin pathway. Neoplasia.
17:410–420. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
He H and Magi-Galluzzi C:
Epithelial-to-mesenchymal transition in renal neoplasms. Adv Anat
Pathol. 21:174–180. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Ren J, Yan L, Tang Y, Zhang W, Li
D, Zang Y, Kong F and Xu Z: Cytoplasmic expression of pontin in
renal cell carcinoma correlates with tumor invasion, metastasis and
patients' survival. PLoS One. 10:e01186592015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shimazui T, Kojima T, Onozawa M, Suzuki M,
Asano T and Akaza H: Expression profile of N-cadherin differs from
other classical cadherins as a prognostic marker in renal cell
carcinoma. Oncol Rep. 15:1181–1184. 2006.PubMed/NCBI
|
|
30
|
Cheng C, Wan F, Liu L, Zeng F, Xing S, Wu
X, Chen X and Zhu Z: Overexpression of SATB1 is associated with
biologic behavior in human renal cell carcinoma. PLoS One.
9:e974062014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang J, Yao X, Zhang J, Dong B, Chen Q,
Xue W, Liu D and Huang Y: Hypoxia-induced downregulation of miR-30c
promotes epithelial-mesenchymal transition in human renal cell
carcinoma. Cancer Sci. 104:1609–1617. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gervais ML, Henry PC, Saravanan A, Burry
TN, Gallie BL, Jewett MA, Hill RP, Evans AJ and Ohh M: Nuclear
E-cadherin and VHL immunoreactivity are prognostic indicators of
clear-cell renal cell carcinoma. Lab Invest. 87:1252–1264. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ho MY, Tang SJ, Chuang MJ, Cha TL, Li JY,
Sun GH and Sun KH: TNF-α induces epithelial-mesenchymal transition
of renal cell carcinoma cells via a GSK3β-dependent mechanism. Mol
Cancer Res. 10:1109–1119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Harada K, Miyake H, Kusuda Y and Fujisawa
M: Expression of epithelial-mesenchymal transition markers in renal
cell carcinoma: impact on prognostic outcomes in patients
undergoing radical nephrectomy. BJU Int. 110:E1131–E1137. 2012.
View Article : Google Scholar : PubMed/NCBI
|