Introduction
Reactive oxygen species (ROS) and other free
radicals are produced during normal cell metabolism, a necessary
and normal process, and they play important physiological roles
(1,2). However, under pathological
conditions, the uncontrolled generation of ROS results in oxidative
stress in cells, which subsequently leads to damage to different
cellular structures, such as proteins, DNA and lipids (3). A proper balance between the
formation of ROS and the antioxidant network is known to be
essential for the regulation of biological processes. Clinical and
experimental studies have suggested that oxidative stress is
involved in the pathogenesis of a number of diseases (4,5).
The liver, due to its high metabolic activity and its anatomical
positioning to receive blood from the gastrointestinal tract, is
vulnerable to toxicity from a variety of drugs and environmental
contaminants. ROS-induced mechanisms have, for instance, been
related to different chronic liver diseases and hepatocellular
carcinoma (HCC), and are induced by various risk factors for liver
cancer, such as hepatitis B and C or aflatoxin-B, and thus may be a
possible driving force in hepatocarcinogenesis (6,7).
Previous research has demonstrated that antioxidants are
efficacious in preventing oxidative stress-related liver diseases
and protecting cells from toxic insults (8). Therefore, there is increasing
interest in the potential preventive/protective effects of
exogenous antioxidants on oxidative stress-related hepatocellular
disorders.
Carthamus tinctorius L (safflower), which
belongs to the Compositae family, has long been used in Chinese
medicine in clinical settings for the treatment of various diseases
due to its antioxidant properties. The chemical constituents in
safflower have been reported to include lignans, flavonoids,
triterpene alcohols and polysaccharides (9–12).
Previous research has indicated that the main effective constituent
of safflower is safflower yellow (SY), which is a flavonoid. SY
consists of hydroxysafflower yellow A (HSYA), safflower yellow A
(SYA) and safflower yellow B (SYB), as well as other chemicals
(13).
Safflower has long been used in the treatment of
cardiovascular diseases, and its anti-myocardial ischemic effects
are well known. Safflower also exerts other pharmacological
effects, including anticoagulant, antioxidant, neuroprotective and
calcium antagonist effects (14).
However, which component is responsible for these protective
effects remains largely unknown. As one of the main components of
safflower, SYB has been extensively used in the treatment of
cardiocerebro-vascular diseases in traditional Chinese medicine and
has been shown to exert neuroprotective effects following permanent
middle cerebral artery occlusion in rats (15). However, to the best of our
knowledge, little research on the effects of SYB on liver
preservation has been undertaken as of yet. Thus, the aim of the
present study was to determine whether SYB is an effective
component of safflower, and whether it can protect hepatocytes from
oxidative damage. In our experiments, we used a control group
treated with N-acetylcysteine (NAC, 200 µM, a frequently
used antioxidant in clinical settings), to establish whether SYB
has antioxidant potential.
Materials and methods
Materials
SYB (purity >98%) was purchased from the Chinese
National Institute for the Control of Pharmaceutical and Biological
Products (Beijing, China). Dulbecco's modified Eagle's medium
(DMEM) was obtained from Gibco Life Technologies (Grand Island, NY,
USA) and the fetal bovine serum (FBS) we used was provided by
Hangzhou Sijiqing Biological Engineering Materials Co., Ltd.
(Hangzhou, China). NAC,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
type I collagenase and western blot reagents were purchased from
Sigma (St. Louis, MO, USA). The kits for the determination of
lactate dehydrogenase (LDH), malondialdehyde (MDA), glutathione
peroxidase (GSH-Px), superoxide dismutase (SOD) and caspase-3
activity were obtained from Nanjing Jiancheng Bioengineering
Institute (Nanjing, China). Anti-β-actin primary (sc-7210),
anti-AKT (sc-8312), anti-phosphorylated (p-)AKT (sc-33437),
anti-heme oxygenase 1 (HO-1; sc-10789), anti-nuclear factor
erythroid 2-related factor 2 (Nrf2; sc-7943) and anti-NAD(P)H
dehydrogenase, quinone 1 (NQO1; sc-25591) antibodies were obtained
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The
caspase-3 assay kit was purchased from Chemicon International, Inc.
(Temecula, CA, USA).
9-[4-[Bis[2-[(acetyloxy)methoxy]-2-oxoethyl]amino]-3-[2-[2-[bis[2-[(acetyloxy)methoxy]-2-oxoethyl]amino]phenoxy]
ethoxy]phenyl]-3,6-bis(dimethylamino)xanthylium bromide (Rhod-2
AM), DAPI, JC-1 and 2′,7′-dichlorodihydrofluorescein diacetate
(H2DCF-DA) were purchased from Molecular
Probes/Invitrogen (Carlsbad, CA, USA).
Cell lines and cell culture
The human hepatoma cell line, HepG2, was obtained
from the American Type Culture Collection (ATCC; Manassas, VA, USA)
and cultured in DMEM supplemented with 10% FBS, glucose, penicillin
and streptomycin. The HepG2 cells were grown in 10-cm cell culture
dishes and incubated in a humidified atmosphere containing 5%
CO2 at 37°C. The cells were seeded at a density of
4×104 cells/ml in 96-well microplates for MTT assay, or
3×105 cells/ml in 6-well microplates for MDA activity
assay, antioxidant enzyme assays and other biochemical indicator
determinations. The culture medium was changed every second
day.
Cell treatments
The HepG2 cells were pre-treated with SYB (50, 100
and 150 nmol/l) for 24 h and then exposed to hydrogen peroxide
(H2O2; 200 µmol/l) for a further 6 h.
Cells not exposed to H2O2 served as the
controls, and cells exposed to H2O2 and not
subjected to any treatments, served as the model group. In order to
determine the ability of SYB to protect the cells from damage, the
cells were also treated with NAC (200 µM), a
well-characterized chemoprotective compound. In addition, in order
to determine the role of AKT in the effects of SYB, the cells were
treated with the AKT inhibitor, LY294002 (10 µM; purchased
from Sigma).
Cell viability and cytotoxicity
assays
Cell viability was determined by MTT assay. The
cells were seeded at a density of 1×104 cells/well in
96-well plates. After the cells were subjected to the different
treatments, 20 µl MTT solution (5 mg/ml) was added to each
well and the final concentration at 5 mg/ml was maintained for 4 h
at 37°C. Subsequently, the medium was removed and DMSO (150 ml) was
added to each well. The optical density (OD) was determined
spectrophotometrically at 490 nm using a microplate reader
(Infinite M200 PRO; Tecan, Männedorf, Switzerland), and the cell
survival ratio was expressed as a percentage of the control.
Cytotoxicity was evaluated by LDH leakage assay
after collecting the culture medium, and the cells were scraped in
phosphate-buffered saline (PBS) after the cells were subjected to
the different treatments. The cells were first sonicated to ensure
the cell membrane broke down to release the total amount of LDH;
subsequently, centrifugation (1,000 × g for 15 min) to clear up the
cell sample was undertaken. LDH leakage was estimated from the
ratio between the LDH activity in the culture medium and that of
the whole cell content.
Determination of intracellular ROS
accumulation
Intracellular ROS accumulation in the HepG2 cells
was monitored using the fluorescent marker, H2DCF-DA.
Briefly, the HepG2 cells (1×105 cells/well) were seeded
into 6-well plates and pre-treated with or without NAC or
increasing concentrations of SYB (50, 100 and 150 nmol/l) for 24 h.
Oxidative stress was induced by the addition of
H2O2 to the culture medium for 30 min. At the
end of the incubation period, the culture supernatant was removed
and the cells were washed twice with PBS. H2DCF-DA (10
µM) was mixed with 500 µl DMEM and added to the
culture plate. Following incubation for 30 min, the relative
fluorescence intensity was quantified using a fluorescence
spectrophotometer (Hidex Oy, Turku, Finland) at 485/535 nm
(A485/535). The percentage of ROS generation was calculated as
follows: (A485/535 of treated cells/A485/535 of untreated cells)
×100.
Measurement of GSH-Px and SOD
activities
The activities of antioxidant enzymes were
determined according to the instructions provided with the assay
kits (Nanjing Jiancheng Bioengineering Institute). Briefly, after
the cells were subjected to the different treatments, they were
washed twice with PBS, resuspended in 1 ml 0.1 M phosphate buffer
(pH 7.4) and homogenized. The homogenate was centrifuged at 2,200 ×
g for 10 min at 4°C, and the supernatants were collected following
centrifugation for the following analyses. SOD activity was assayed
at 560 nm on the basis of its ability to inhibit the oxidation of
hydroxylamine via the superoxide anion from the xanthine oxidase
system. GSH-Px activity was measured at 412 nm on the basis of the
rate of oxidation of the reduced glutathione to oxidized
glutathione by H2O2 under the catalyst,
GSH-Px. The protein content in the cell lysate was determined using
Coomassie blue staining solution (from Nanjing Jiancheng
Bioengineering Institute).
Apoptosis
Apoptosis was evaluated by Annexin V/propidium
iodide (PI) double staining assay. Briefly, the cells were
trypsinized following a wash in PBS and resuspended in 400
µl binding buffer. Subsequently, 5 µl Annexin V-FITC
and 5 µl PI (50 µM) working solution were added to
every 200 µl cell suspension. The cells were incubated at
room temperature for 20 min in the dark and analyzed by flow
cytometry. Annexin V and PI emissions were detected in the FL1-H
and FL2-H channels of a FACSCalibur flow cytometer (BD Biosciences,
San Jose, CA, USA) using emission filters of 525 and 575 nm,
respectively. The number of each type of cells was expressed as
percentages of the number of total stained cells. Data were
acquired using CellQuest software and analyzed by ModFit
software.
Apoptosis was also evaluated by examining the
activation of caspase-3. The cells were lysed in buffer containing
5 mM Tris, pH 8, 20 mM ethylenediaminetetraacetic acid (EDTA) and
0.5% Triton X-100 (both from Sigma). The reaction mixture contained
20 mM HEPES, pH 7, 10% glycerol, 2 mM dithiothreitol and 30
µg protein per well, as well as 20 µM Ac-DEVD-AMC as
substrate. The enzymatic activity was determined by measuring
fluorescence at an excitation wavelength of 380 nm and an emission
wavelength of 440 nm (BioTek Instruments, Inc., Winooski, VT,
USA).
Protein extraction and western blot
analysis
The cells were lysed with either mammalian protein
extraction reagent or nuclear and cytoplasmic extraction reagent
kits (Pierce Biotechnology, Rockford, IL, USA). Protein
concentrations were determined using Bio-Rad protein assay reagent
(Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein
samples (30 µg) were separated by 10% SDS-PAGE, and the
separated proteins were transferred onto polyvinylidene fluoride
(PVDF) membranes (Bio-Rad Laboratories) overnight. The transferred
protein membranes were blocked with 5% non-fat dried milk for 30
min, followed by incubation with specific primary anti-bodies
(β-actin, AKT, p-AKT, HO-1, Nfr2 and NQO1) overnight at 4°C, and
either horseradish peroxidase-conjugated goat anti-rabbit or
anti-mouse antibodies for 1 h at 37°C. The blots were detected
using VL Chemi-Smart 3000 (Viogene-Biotek, Sunnyvale, CA, USA) with
enhanced chemiluminescence (ECL) western blotting reagent
(Millipore, Billerica, MA, USA). β-actin was used as a loading
control.
Measurement of mitochondrial membrane
potential (ΔΨm)
ΔΨm in the HepG2 cells was evaluated by
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine
iodide (JC-1) staining. The fluorescent dye, JC-1, exhibits
potential dependent accumulation in mitochondria by a fluorescence
emission shift from green (monomeric form, indicating
depolarized/low ΔΨm) to red (aggregated form, indicating
polarized/normal ΔΨm). In this study, the HepG2 cells
(1×105 cells/well) were seeded in 35-mm dishes and
pre-treated with or without increasing concentrations of SYB or NAC
for 24 h. Oxidative stress was induced by the addition of
H2O2 into the culture medium. At the end of
the incubation period, the culture supernatant was removed and the
cells were washed twice with PBS. The cells were incubated with
JC-1 (Qcbio Science & Technologies Co., Ltd., Shanghai, China)
staining liquid for 20 min at 37°C, washed 3 times with JC-1
staining buffer and examined under a confocal microscope (SP5-FCS;
Leica Microsystems, Wetzlar, Germany).
Measurement of mitochondrial calcium
To follow H2O2-induced
intramitochondrial calcium trafficking, mitochondrial calcium was
estimated by co-incubating the cells with a mitochondria-permeant
calcium fluorophore, Rhod-2 AM (2 µM). For confocal images,
2×104 cells were seeded onto a glass coverslip,
pre-treated with SYB for 24 h, and then subjected to
H2O2. Subsequently, the cells were stained
with Rhod-2 AM (2 µM), fixed with 4% buffered
paraformaldehyde, washed 3 times with PBS and mounted with antifade
on a glass slide for image acquisition using a confocal microscope
(SP5-FCS; Leica Microsystems). The untreated unstained cells served
as the negative controls and untreated fluorophore-loaded cells
served as the controls for background correction for the confocal
experiments.
Statistical analysis
All values are expressed as the means ± SD. A
one-way analysis of variance test, followed by Bonferoni's
correction, was carried out to test for any differences between the
mean values of all groups. A P-value <0.05 was considered to
indicate a statistically significant difference.
Results
SYB protects cells against
H2O2-induced cell damage
In order to elucidate the mechanisms responsible for
the protective effects of SYB against
H2O2-induced cell damage, the human hepatoma
HepG2 cells were pre-treated with SYB (50, 100 and 150 nmol/l) for
24 h and then exposed to H2O2 (200
µmol/l) for a further 6 h. Concentrations of SYB which did
not cause any measurable adverse effects to the cells (data not
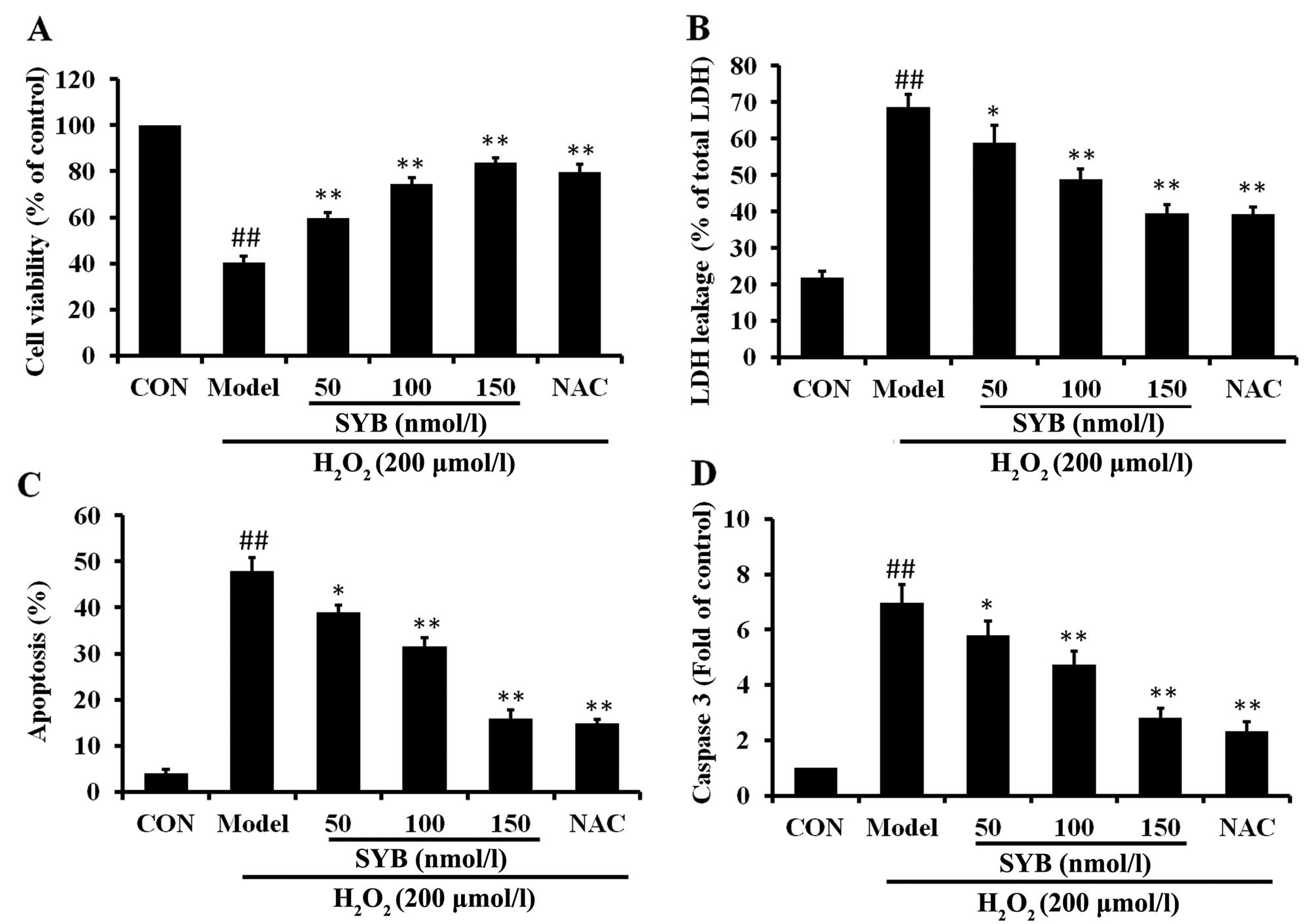
shown) were selected. The effects of pre-treatment with SYB on cell
viability and cell integrity following exposure to
H2O2 were determined by MTT assay and LDH
leakage assay, respectively, and the ability of SYB to protect the
cells from damage was compared to a well-characterized
chemoprotective compound, NAC (200 µM). The results revealed
that pre-treatment with SYB for 24 h protected the HepG2 cells from
subsequent H2O2-induced damage in a
concentration-dependent manner, as shown by the measurement of cell
viability by MTT assay (Fig. 1A).
Similarly, the LDH leakage assay revealed that treatment with SYB
for 24 h prior to exposure to H2O2 decreased
LDH leakage from the cells, indicating that SYB enabled the HepG2
cells to maintain cell integrity. The protective effects of SYB
against LDH leakage also occurred in dose-dependent manner
(Fig. 1B). The protective effects
of SYB (150 nmol/l) were equal or greater to those exerted by NAC
(200 µM).
To determine whether SYB exerts anti-apoptotic
effects, Annexin V-FITC/PI double staining and caspase-3 assay were
performed. The results of flow cytometric detection revealed that
H2O2 induced a significant increase in the
apoptosis of the HepG2 cells compared with the control group, and
that apoptosis was markedly decreased by pre-treatment with SYB
(Fig. 1C). As the sequential
activation of caspases plays a central role in the execution phase
of cell apoptosis (16), the
activation of caspase-3 was examined in this study. Exposure of the
HepG2 cells to H2O2 induced a significant
increase in the level of caspase-3 compared to the control group;
however, pre-treatment with SYB or NAC significantly decreased the
level of caspase-3 (Fig. 1D).
These results suggested that SYB protected the HepG2 cells against
apoptosis induced by H2O2.
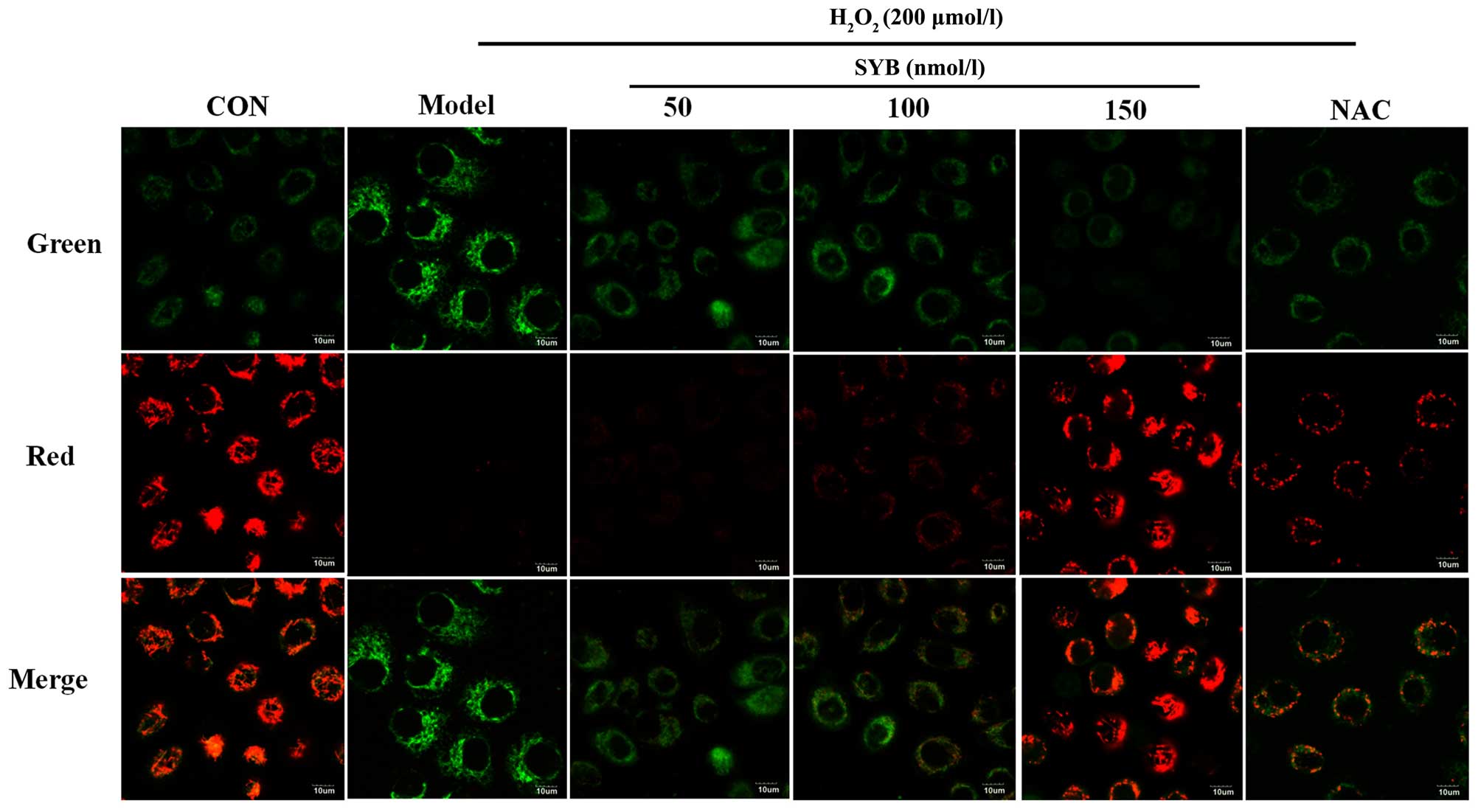
Effects of SYB on ΔΨm
In many systems, apoptosis is associated with the
loss of ΔΨm, which may be regarded as a limiting factor in the
apoptotic pathway (17).
Excessive ROS production, which leads to a decrease in ΔΨm, may
also induce apoptotic cell death (18). In light of these facts, in this
study, we assessed whether there was any reduction in ΔΨm in the
H2O2-exposed cells using the
potential-sensitive dye, JC-1. The exposure of the HepG2 cells to
H2O2 for 6 h resulted in a decrease in the
ratio of red to green fluorescence, as detected by confocal
microscopy when compared to the control cells, and treatment with
SYB or NAC restored this ratio (Fig.
2).
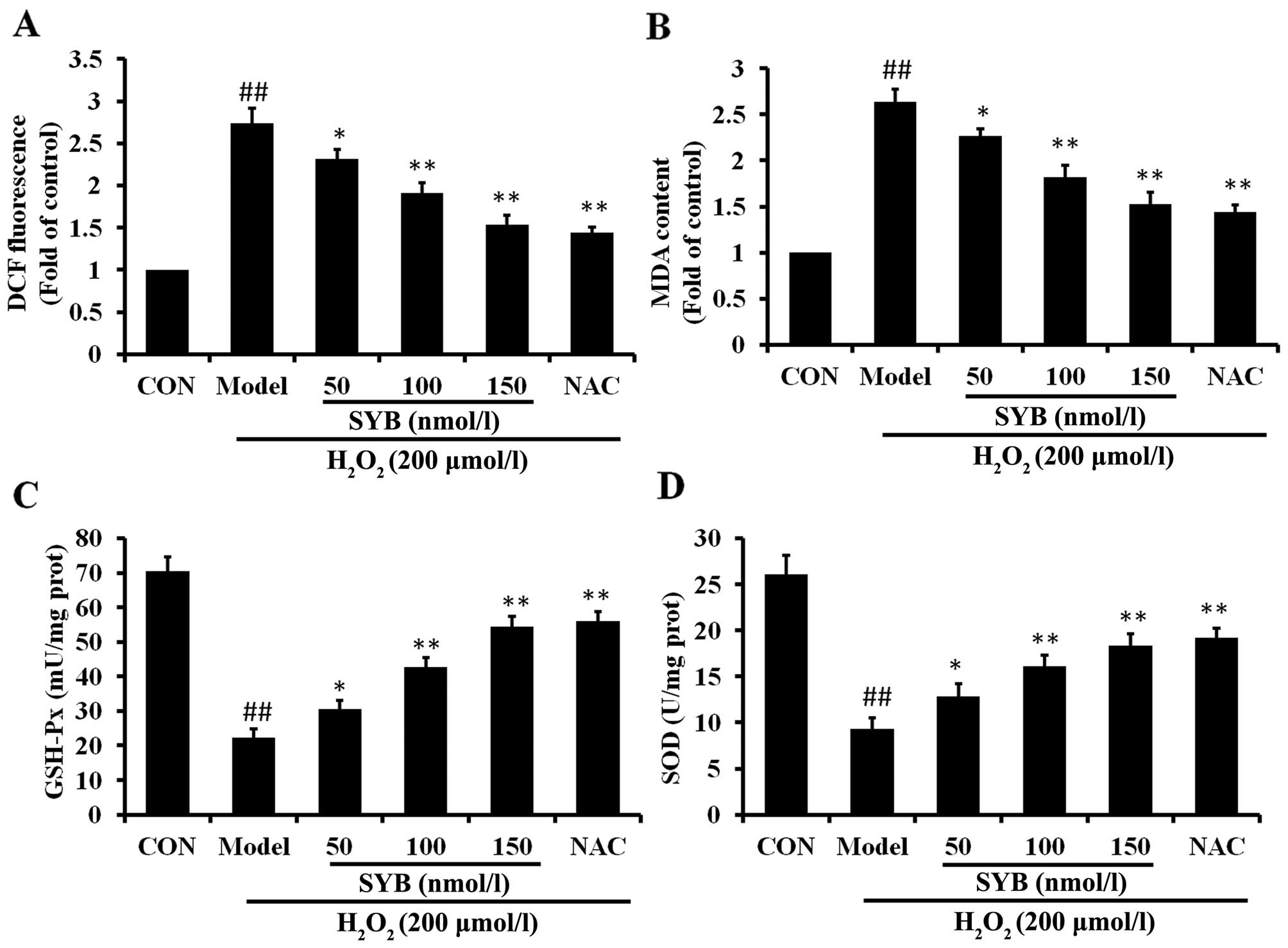
SYB reduces
H2O2-dependent intracellular ROS production
and MDA formation and increases antioxidant enzyme activity
To investigate the mechanisms through which SYB
protects cells against H2O2-induced damage,
the effects of pre-treatment with SYB on the intracellular ROS
levels were determined. The HepG2 cells were pre-treated with SYB
for 24 h, and then exposed to 200 µmol/l
H2O2 for 6 h, and the ROS levels were
measured by DCF fluorescence. The results revealed that exposure to
H2O2 induced a rapid and significant increase
in the intracellular ROS levels, when compared to the untreated
controls (Fig. 3A). Pre-treatment
of the cells with SYB for 24 h significantly decreased the
H2O2-induced production of intra-cellular ROS
compared with the H2O2-exposed cells not
treated with SYB (model group; P<0.05). To examine the effects
of SYB on H2O2-induced lipid peroxidation,
the level of MDA in the cells was measured. Exposure of the HepG2
cells to H2O2 markedly increased the
intracellular MDA levels (Fig.
3B). When the cells were pre-treated with SYB, the increase in
the MDA levels induced by H2O2 was
significantly prevented in a dose-dependent manner.
GSH-Px and SOD activity was measured as an index of
the enzymatic antioxidant defense system in HepG2 cells during
oxidative stress. The HepG2 cells were exposed to 200 µmol/l
H2O2 and either pre-treated or not with SYB
or NAC. The results revealed that exposure to
H2O2 markedly decreased the GSH-Px and SOD
levels compared to the control group. More significantly, the
results revealed that pre-treatment with SYB increased the GSH-Px
and SOD levels, which were decreased following exposure to
H2O2 (Fig. 3C
and D). These data suggest that SYB exerts its protective
effects by enhancing the the anti-oxidant capability of the
cells.
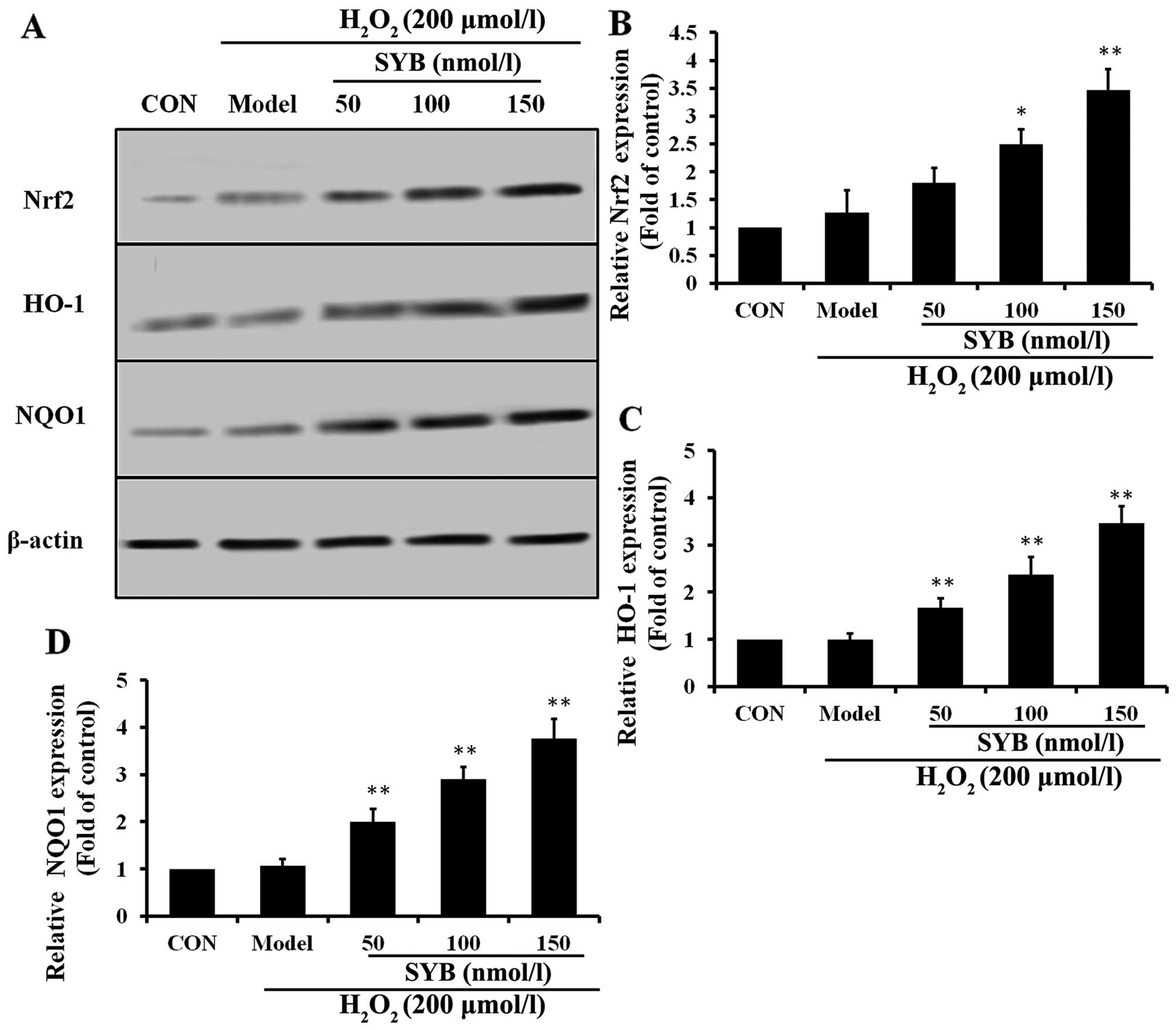
Effects of SYB on Nrf2-mediated
antioxidant defense system proteins in HepG2 cells
To examine the effects of SYB on Nrf2 activity, we
first treated the HepG2 cells with various concentrations of SYB
for 24 h and then examined the dose-response effects of SYB on the
expression of Nrf2. Pre-treatment with SYB gradually increased
cytosolic Nrf2 expression in a dose-dependent manner (Fig. 4A and B). Activated Nrf2 binds to
antioxidant response element (ARE) sites and causes the
upregulation of its target genes (19). It is well known that HO-1 and NQO1
are two major antioxidant enzymes that play an important role in
H2O2-induced antioxidant defense in hepatic
cells (20). Thus, we
hypothesized that the inhibitory effects of SYB on
H2O2-induced hepatic enzyme leakage and/or
the augmentation of GSH-Px and SOD levels result from the induction
of antioxidant genes, such as HO-1 and NQO1. As expected, we
observed that SYB significantly increased the HO-1 and NQO1
expression levels in the H2O2-exposed HepG2
cells in a dose-dependent manner (Fig. 4C and D). These data clearly
indicate that the cytoprotective effects of SYB are mediated
through Nrf2.
Role of AKT in the SYB-induced
upregulation of Nrf2 and HO-1
It has previously been noted that AKT is involved in
the induction of Nrf2/ARE-driven gene expression (21). Thus, in order to identify which
signaling cascade controls Nrf2 activation and the induction of
HO-1 expression in the SYB-treated HepG2 cells, the activation of
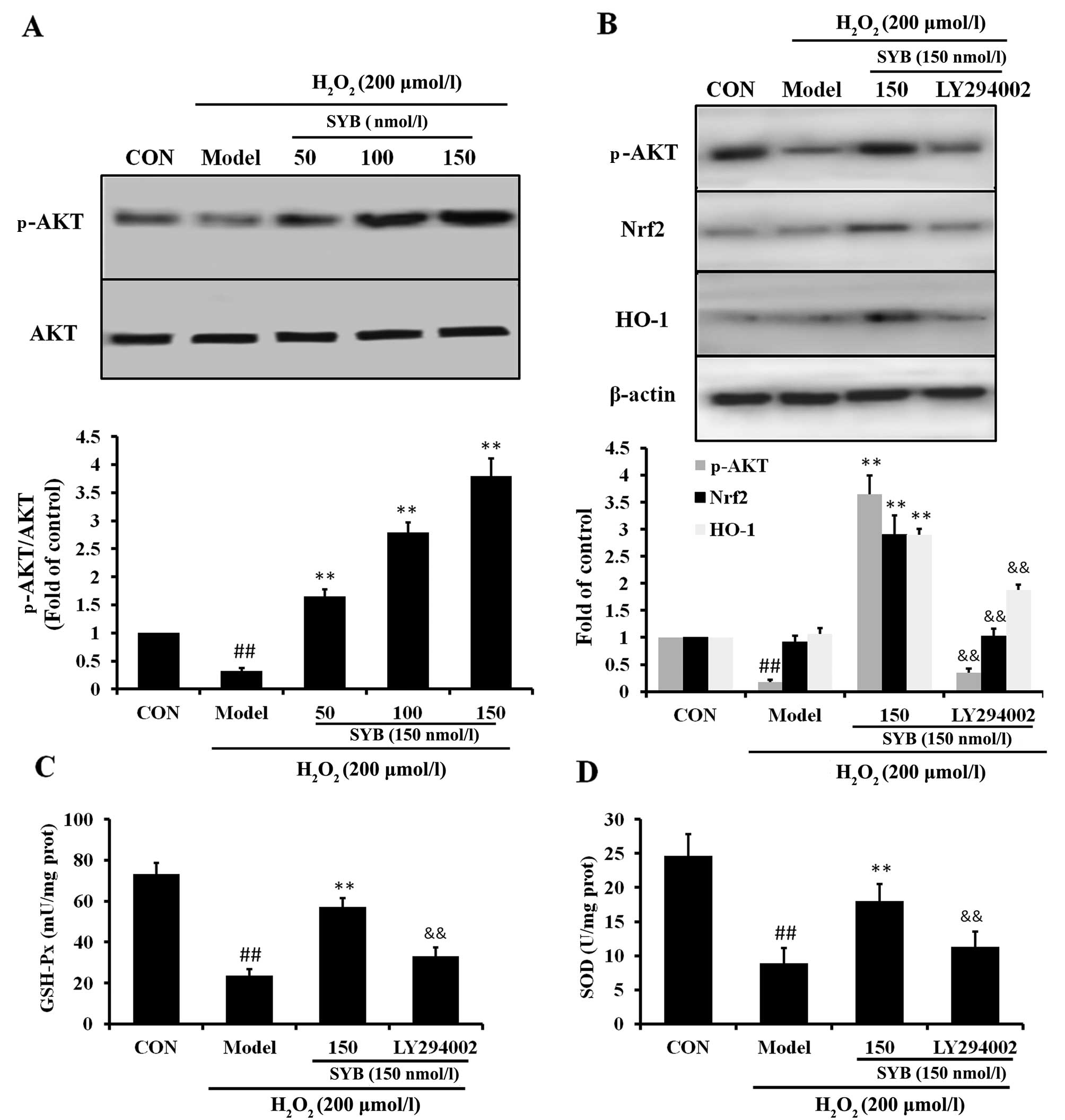
the AKT pathway was investigated. As shown in Fig. 5A, in the HepG2 cells exposed to
H2O2 (model group), the reduced activation of
AKT was observed; however, treatment with SYB significantly
increased the phosphorylation of AKT in a dose-dependent manner.
Subsequently, we examined whether SYB enhances Nrf2 expression
through the AKT pathway. Treatment of the cells with the AKT
inhibitor, LY294002, suppressed the SYB-induced phosphorylation of
AKT in the HepG2 cells exposed to H2O2.
Moreover, the inhibition of the AKT pathway by LY294002 markedly
reduced the capacity of SYB to increase Nrf2 and HO-1 expression
(Fig. 5B), as well as the GSH-Px
and SOD levels (Fig. 5C and D).
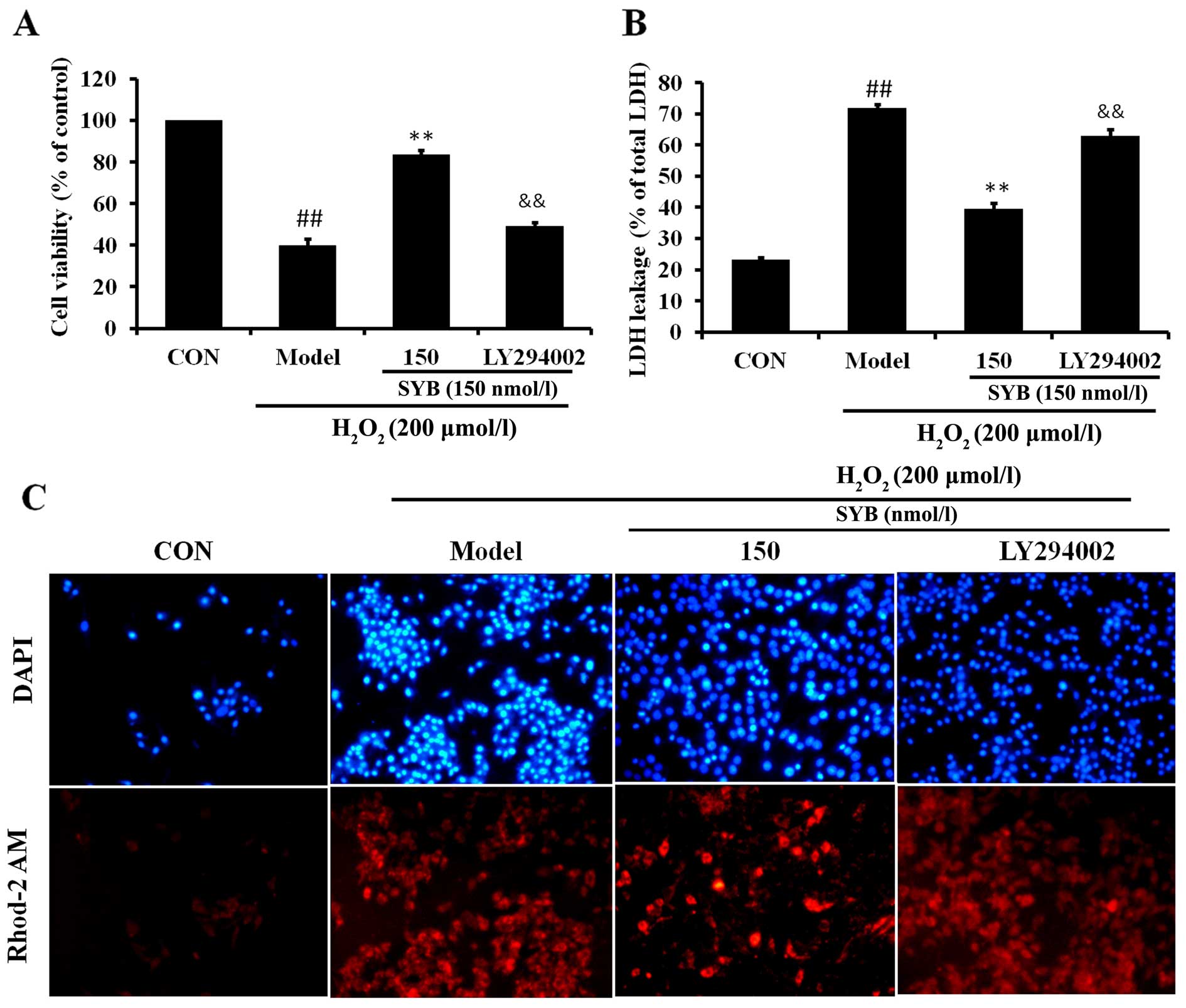
Consistently, the inhibition of AKT by LY294002 also eliminated the
SYB-induced cytoprotective effects against
H2O2-induced cell death; the cell viability
rate decreased, LDH leakage increased and calcium overload was
noted in the mitochondria following treatment with LY294002
(Fig. 6). These results suggest
that SYB induces the activation of Nrf2/HO-1 and the antioxidant
defense machinery by activating the AKT pathway.
Discussion
Oxidative stress is an abnormal phenomenon that
occurs inside the human body when the production of free radicals
exceeds the antioxidant capacity. The excessive production of free
radicals and other ROS damages essential macromolecules of the
cell, leading to the development of a number of human diseases,
including atherosclerosis, rheumatoid arthritis, inflammation,
cancer and neurodegenerative diseases (22). Oxidative stress associated with
the formation of ROS plays an important role in the pathogenesis of
hepatic ischaemia/reperfusion (I/R) injury. Previous research has
indicated that although ROS are generated mainly as by-products of
mitochondrial respiration (as the primary cellular consumer of
oxygen, together with multiple electron carriers and redox
enzymes), they are themselves extremely susceptible to oxidative
damage (23). The overproduction
of ROS leads to lipid peroxidation, damage to the mitochondrial
membrane, the release of mitochondrial apoptogenic factors into the
cytoplasm, followed by caspase activation and finally, cell
apoptosis (24). Natural
antioxidants that inhibit ROS generation are considered essential
in terms of protecting the liver from chemical- or I/R-induced
damage. Consequently, previous research has focused on natural
anti-oxidants that have chemopreventive properties and on their
mechanisms of action (25).
In a previous study, it was demonstrated that SYB is
able to significantly attenuate brain injury induced by ischemia,
and it was pointed out that SYB exerts neuroprotective effects
against brain injury induced by ischemia by increasing the
activities of antioxidant enzymes in the brain tissue, thus
decreasing free radical generation and improving mitochondrial
function (15). However, to the
best of our knowledge, there are no studies available to date on
the effects of SYB on I/R- or H2O2-induced
oxidative stress in liver cells. Thus, in the present study, we
examined the inhibitory effects of SYB on
H2O2-induced injury to HepG2 cells. The
results revealed that the exposure of HepG2 cells to
H2O2 significantly decreased cell viability
and increased the release of LDH, and pre-treatment of the cells
with SYB increased cell viability and reduced the release of LDH,
implying that SYB has the ability to protect HepG2 cells from
H2O2-induced injury. In addition, we
established that the ability of SYB to modulate important proteins
of the cell signaling cascades is directly involved in its
protective effects.
Oxidative stress-induced cell injury is associated
with increases in ROS, such as H2O2, hydroxyl
radicals, superoxide anion, singlet oxygen, nitric oxide and
peroxynitrites (26). Lipid
peroxidation of polyunsaturated fatty acid produces ROS and toxic
aldehydes, such as 4-hydroxy-2-nonenal (4-HNE) and MDA (27). Therefore, the concentration of MDA
in cells or tissue lysates is considered to be a major cause of
lipid peroxidation. In this study, we demonstrated that exposure to
H2O2 induced the overproduction of ROS in
hepatic cells and led to oxidative stress; however, treatment with
SYB significantly suppressed H2O2-induced ROS
generation and lipid peroxidation, possibly due to the powerful
antioxidant and free radical scavenging activity of SYB.
It has previously been noted that oxidants not only
stimulate inflammatory cytokines in HepG2 cells and cause cellular
senescence, but also induce the apoptosis of HepG2 cells, and
certain apoptotic agents increase the production of ROS in
mitochondria (28), and
antioxidants such as NAC and vitamin E prevent cell apoptosis.
Apoptosis is programmed cell death and it plays an important role
in embryogenesis, metamorphosis and cellular homeostasis (29). In this study, we discovered that
H2O2 induced the apoptosis of HepG2 cells
using flow cytometry. SYB markedly decreased the apoptosis induced
by H2O2 in a dose-dependent manner, and these
results were verified by determining the caspase-3 levels.
Mitochondria are an important source of ROS within
the majority of mammalian cells. Excessive ROS production
contributes to mitochondrial damage in a wide range of pathologies
(30). Damaged mitochondria can
then cause defects in lipid homeostasis, bioenergetics and ROS
production, leading to lipid accumulation and oxidative stress
(31). Increasing evidence
suggests that ΔΨm assay can be used as a more specific test for
early mitochondrial injury (32).
In the present study, using JC-1, we revealed that
H2O2 induced the loss of ΔΨm in HepG2 cells.
Pre-treatment with SYB for 24 h restored the ΔΨm to the basal
level. These findings implied that the ability of SYB to attenuate
oxidative stress partly depends on inhibiting mitochondrial-related
apoptosis.
Oxidative stress caused by ROS is responsible for a
wide variety of cellular damage and is the most validated mechanism
of secondary injury (33).
Following oxidative stress, the overproduction of ROS, and
subsequently the depletion of antioxidants, results in the total
breakdown of the endogenous antioxidant defense mechanisms,
culminating in failure to protect cells from damage induced by
oxidative stress. SOD is an oxygen radical scavenger that scavenges
superoxide radicals by converting them into
H2O2, which is then converted to water by
catalase and GSH-Px (34). In our
study, exposure of the HepG2 cells to H2O2
induced a marked decrease in the concentration of SOD and GSH-Px,
which was reversed by pre-treatment with SYB for 24 h.
Nrf2, a basic leucine zipper (bZIP) transcription
factor that belongs to the cap'n'collar (CNC) family, plays an
important role in the transcriptional regulation of phase II
enzymes (35). The
Nrf2-activating pathway provides a rapid feedback triggered
mechanism through which the cell is challenged by oxidative stress
(36). Upon stimulation, Nrf2
disassociates from its cytosolic inhibitor, Keap-1, and
translocates into the nucleus, then binds to the ARE in the
promoter regions of many phase II enzymes, including HO-1 and NQO1
(37). As the Nrf2/ARE pathway
has been shown to provide protection against several oxidative
impacts in different organs, the induction of HO-1 and NQO1 under
the regulation of Nrf2/ARE may provide a therapeutic option for
liver diseases in cases of severe oxidative stress (38). We hypothesized that the increased
protein expression level of HO-1 and NQO1 was due to activation of
the Nrf-2 signaling pathway. As expected, SYB had the effect of
activating Nrf2, which led to the increased protein expression of
HO-1 and NQO1.
Previous research has demonstrated that a wide
variety of phytochemicals from natural products, such as but in and
3′,4′-dide-methylnobiletin protect against oxidative stress-induced
cell damage through the PI3K/AKT/Nrf2-dependent pathway (39). Therefore, we investigated whether
that pathway contributes to the protective effects of SYB against
H2O2-induced oxidative stress. Importantly,
the phosphorylation of AKT was significantly increased in the
SYB-treated cells, and the inhibition of AKT signaling by the AKT
inhibitor, LY294002, blocked the SYB-induced protein expression of
Nrf2 and HO-1 and reduced the expression level of GSK-Px and SOD.
Further analysis also indicated that LY294002 abolished the ability
of SYB to control the calcium levels which were significantly
increased by H2O2. These results indicate
that AKT/Nrf2 signaling is involved in the cytoprotective effects
of SYB.
In conclusion, the findings of this study
demonstrate that pre-treatment with SYB leads to the protection of
hepatic cells against H2O2-induced oxidative
stress through a mechanism that involves Nrf2 activation and the
upregulation of the expression of its downstream antioxidant genes,
mediated by AKT. These results provide a scientific basis for the
hepatoprotective effects of pure compound SYB derived from
Safflower and suggest that it may be of therapeutic value in the
treatment of various liver diseases associated with oxidative
stress.
Acknowledgments
This study was supported by the National Science
Foundation of China (nos. 81173514 and 81302695).
References
|
1
|
Willcox JK, Ash SL and Catignani GL:
Antioxidants and prevention of chronic disease. Crit Rev Food Sci
Nutr. 44:275–295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cui K, Luo X, Xu K and Ven Murthy MR: Role
of oxidative stress in neurodegeneration: Recent developments in
assay methods for oxidative stress and nutraceutical antioxidants.
Prog Neuropsychopharmacol Biol Psychiatry. 28:771–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reuter S, Gupta SC, Chaturvedi MM and
Aggarwal BB: Oxidative stress, inflammation, and cancer: how are
they linked? Free Radic Biol Med. 49:1603–1616. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bitar MS, Al-Saleh E and Al-Mulla F:
Oxidative stress - mediated alterations in glucose dynamics in a
genetic animal model of type II diabetes. Life Sci. 77:2552–2573.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coskun O, Ocakci A, Bayraktaroglu T and
Kanter M: Exercise training prevents and protects
streptozotocin-induced oxidative stress and beta-cell damage in rat
pancreas. Tohoku J Exp Med. 203:145–154. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tien Kuo M and Savaraj N: Roles of
reactive oxygen species in hepatocarcinogenesis and drug resistance
gene expression in liver cancers. Mol Carcinog. 45:701–709. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu R, Wang Y, Zhang L and Guo Q:
Oxidative stress and liver disease. Hepatol Res. 42:741–749. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Palter R, Lundin RE and Haddon WF: A
cathartic lignan glycoside isolated from Carthamus tinctorius.
Phytochemistry. 11:2871–2874. 1972. View Article : Google Scholar
|
|
10
|
Kazuma K, Takahashi T, Sato K, Takeuchi H,
Matsumoto T and Okuno T: Quinochalcones and flavonoids from fresh
florets in different cultivars of Carthamus tinctorius L. Biosci
Biotechnol Biochem. 64:1588–1599. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Akihisa T, Yasukawa K, Oinuma H, Kasahara
Y, Yamanouchi S, Takido M, Kumaki K and Tamura T: Triterpene
alcohols from the flowers of compositae and their anti-inflammatory
effects. Phytochemistry. 43:1255–1260. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wakabayashi T, Hirokawa S, Yamauchi N,
Kataoka T, Woo JT and Nagai K: Immunomodulating activities of
polysaccharide fractions from dried safflower petals.
Cytotechnology. 25:205–211. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duan JL, Wang JW, Guan Y, Yin Y, Wei G,
Cui J, Zhou D, Zhu YR, Quan W, Xi MM and Wen AD: Safflor yellow A
protects neonatal rat cardiomyocytes against anoxia/reoxygenation
injury in vitro. Acta Pharmacol Sin. 34:487–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zheng WC, Chen DB, Li B and Zhang L: The
preventive effects of safflor yellow on myocardium injury of
myocardium ischemic reperfusion in rats. Chin Pharmacol Bull.
19:1032–1034. 2003.
|
|
15
|
Wang C, Zhang D, Li G, Liu J, Tian J, Fu F
and Liu K: Neuroprotective effects of safflor yellow B on brain
ischemic injury. Exp Brain Res. 177:533–539. 2007. View Article : Google Scholar
|
|
16
|
Cohen GM: Caspases: the executioners of
apoptosis. Biochem J. 326(Pt 1): 1–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kwong JQ, Henning MS, Starkov AA and
Manfredi G: The mitochondrial respiratory chain is a modulator of
apoptosis. J Cell Biol. 179:1163–1177. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Adam-Vizi V and Chinopoulos C:
Bioenergetics and the formation of mitochondrial reactive oxygen
species. Trends Pharmacol Sci. 27:639–645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Campbell MR, Karaca M, Adamski KN, Chorley
BN, Wang X and Bell DA: Novel hematopoietic target genes in the
NRF2-mediated transcriptional pathway. Oxid Med Cell Longev.
2013:1203052013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu D, Hu L, Xia X, Song J, Li L, Song E
and Song Y: Tetrachloro-benzoquinone induces acute liver injury,
up-regulates HO-1 and NQO1 expression in mice model: the protective
role of chlorogenic acid. Environ Toxicol Pharmacol. 37:1212–1220.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee YJ, Jeong HY, Kim YB, Lee YJ, Won SY,
Shim JH, Cho MK, Nam HS and Lee SH: Reactive oxygen species and
PI3K/Akt signaling play key roles in the induction of Nrf2-driven
heme oxygenase-1 expression in sulforaphane-treated human
meso-thelioma MSTO-211H cells. Food Chem Toxicol. 50:116–123. 2012.
View Article : Google Scholar
|
|
22
|
Okezie IA: Free radicals, oxidative
stress, and antioxidants in human health and disease. J Am Oil Chem
Soc. 75:199–212. 1998. View Article : Google Scholar
|
|
23
|
Fiskum G, Rosenthal RE, Vereczki V, Martin
E, Hoffman GE, Chinopoulos C and Kowaltowski A: Protection against
ischemic brain injury by inhibition of mitochondrial oxidative
stress. J Bioenerg Biomembr. 36:347–352. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ghosh M, Manna P and Sil PC: Protective
role of a coumarin-derived schiff base scaffold against tertiary
butyl hydroperoxide (TBHP)-induced oxidative impairment and cell
death via MAPKs, NF-κB and mitochondria-dependent pathways. Free
Radic Res. 45:620–637. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Masella R, Di Benedetto R, Varì R, Filesi
C and Giovannini C: Novel mechanisms of natural antioxidant
compounds in biological systems: involvement of glutathione and
glutathione-related enzymes. J Nutr Biochem. 16:577–586. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Trachootham D, Lu W, Ogasawara MA, Nilsa
RD and Huang P: Redox regulation of cell survival. Antioxid Redox
Signal. 10:1343–1374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Negre-Salvayre A, Coatrieux C, Ingueneau C
and Salvayre R: Advanced lipid peroxidation end products in
oxidative damage to proteins. Potential role in diseases and
therapeutic prospects for the inhibitors. Br J Pharmacol. 153:6–20.
2008. View Article : Google Scholar
|
|
28
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Renehan AG, Booth C and Potten CS: What is
apoptosis, and why is it important? BMJ. 322:1536–1538. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar
|
|
31
|
Serviddio G, Bellanti F, Sastre J,
Vendemiale G and Altomare E: Targeting mitochondria: a new
promising approach for the treatment of liver diseases. Curr Med
Chem. 17:2325–2337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang J, Yu S, Jiang Z, Liang C, Yu W, Li
J, Du X, Wang H, Gao X and Wang X: N-acetyl-serotonin protects
HepG2 cells from oxidative stress injury induced by hydrogen
peroxide. Oxid Med Cell Longev. 2014:3105042014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang CC, Fang KM, Yang CS and Tzeng SF:
Reactive oxygen species-induced cell death of rat primary
astrocytes through mitochondria-mediated mechanism. J Cell Biochem.
107:933–943. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kondo T, Higashiyama Y, Goto S, Iida T,
Cho S, Iwanaga M, Mori K, Tani M and Urata Y: Regulation of
gamma-glutamyl-cysteine synthetase expression in response to
oxidative stress. Free Radic Res. 31:325–334. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ryter SW, Alam J and Choi AMK: Heme
oxygenase-1/carbon monoxide: from basic science to therapeutic
applications. Physiol Rev. 86:583–650. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Okawa H, Motohashi H, Kobayashi A,
Aburatani H, Kensler TW and Yamamoto M: Hepatocyte-specific
deletion of the keap1 gene activates Nrf2 and confers potent
resistance against acute drug toxicity. Biochem Biophys Res Commun.
339:79–88. 2006. View Article : Google Scholar
|
|
37
|
Kay HY, Won Yang J, Kim TH, Lee da Y, Kang
B, Ryu JH, Jeon R and Kim SG: Ajoene, a stable garlic by-product,
has an antioxidant effect through Nrf2-mediated glutamate-cysteine
ligase induction in HepG2 cells and primary hepatocytes. J Nutr.
140:1211–1219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Na HK, Kim EH, Jung JH, Lee HH, Hyun JW
and Surh YJ: (−)-Epigallocatechin gallate induces Nrf2-mediated
antioxidant enzyme expression via activation of PI3K and ERK in
human mammary epithelial cells. Arch Biochem Biophys. 476:171–177.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Su JD, Yen JH, Li S, Weng CY, Lin MH, Ho
CT and Wu MJ: 3′,4′-Didemethylnobiletin induces phase II
detoxification gene expression and modulates PI3K/Akt signaling in
PC12 cells. Free Radic Biol Med. 52:126–141. 2012. View Article : Google Scholar
|