Introduction
The early repolarization (ER) pattern on the
electrocardiogram (ECG) is defined as ≥0.1 mV J-point elevation of
either notched or slurred morphology in at least 2 contiguous
inferior and/or lateral leads. It was first described as a common
ECG variant in healthy human subjects in 1936 by Shipley and
Hallaran (1). For a long time,
the ECG pattern of ER has been regarded as a benign phenomenon,
which is observed predominantly in young healthy men, male
athletes, and African Americans (2,3).
However, over the past few decades, using evidence from various
studies, the pattern of ER has been considered as associated with
an increased risk of arrhythmogenic sudden death (44–7). Thus,
early repolarization syndrome (ERS), which is also termed inherited
J wave syndrome, was proposed in order to describe the ER pattern
with arrhythmic phenotypes, which is diagnosed only in patients who
have been resuscitated after cardiac arrest, those with documented
ventricular fibrillation (VF) or polymorphic ventricular
tachycardia (VT), and possibly in the relatives of the syndrome
carriers in whom a genetic mutation is documentable (8). There is increasing evidence
suggesting that ERS is related to mutations in ion channel genes.
Haïssaguerre et al have reported that mutations in the
adenosine triphosphate (ATP)-sensitive gene potassium channel,
inwardly rectifying subfamily J, member 8 (KCNJ8) was
responsible for idiopathic VF associated with ER (9). Mutations in the cardiac L-type
calcium channel, including calcium channel, voltage-dependent, L
type, alpha 1C subunit (CACNA1C), calcium channel,
voltage-dependent, beta 2 subunit (CACNB2B) and calcium
channel, voltage-dependent, alpha 2/delta subunit 1
(CACNA2D1), have also been associated with ERS and sudden
cardiac death (10).
In the present study, in a proband with ERS, we
identified a novel mutation in the sodium channel, voltage gated,
type V alpha subunit (SCN5A) gene and investigated the
functional consequences of the novel mutation.
Patients and methods
Clinical examination
For the purposes of the present study, the proband
and his family members who participated in the study provided
written informed consent before genetic and clinical
investigations, and the study conformed to the Declaration of
Helsinki and local ethics committees. The proband underwent
clinical evaluations, including laboratory tests, a 12-lead ECG,
echocardiography, chest roentgenogram, magnetic resonance imaging
(MRI) and coronary angiography in Fuwai Hospital (Beijing,
China).
Genetic analysis
Genomic DNA was extracted from peripheral blood
leukocytes using a TIANamp Blood DNA isolation kit (Tiangen,
Beijing, China) according to the manufacturer's instructions.
Genetic testing was performed in order to identify mutations in ion
channel genes including ATP-binding cassette, sub-family C
(CFTR/MRP), member 9 (ABCC9), KCNQ1, KCNH2, KCND3, KCNE1,
KCNE2, KCNJ8, CACNA1C, CACNB2, CACNA2D1, SCN1B and
SCN5A. All exons of these genes were amplified and screened
by direct sequencing. To be considered a mutation, the variant had
to have changed or disrupted the open reading frame and be absent
in 500 unrelated, healthy individuals (1,000 reference alleles) of
similar ethnicity [derived from our own database as previously
described (11,23)].
Mutagenesis and transfections
The wild-type (WT)-SCN5A cDNA (GenBank ID: NM198056;
Geneway, Beijing, China) was subcloned into a pcDNA3.1 expression
vector (Geneway). The missense mutation consistent with the variant
detected in the proband (A1055G-SCN5A) was created in the
pcDNA3.1 WT-SCN5A plasmid by a site-directed mutagenesis strategy
as previously described (12).
The mutations were verified by sequencing.
293 cells (from the Cell Resource Center, IBMS,
CAMS/PUMC, Beijing, China) were first transiently co-transfected
with 0.6 µg WT or an equal amount of mutant SCN5A
constructs using Effectene transfection reagent (Qiagen, Hilden,
Germany) according to the manufacturer's instructions. Green
fluorescent protein (GFP) plasmids (0.2 µg; Geneway) were
then co-transfected for use as a reporter gene. The sodium current
(INa) was recorded after the transfected cells were
cultured for 36 h. More than two independent transfection
experiments were conducted for further patch-clamp and confocal
experiments to confirm the reproducibility of the results.
Functional analysis
In the present study, INa was measured
using a whole-cell configuration of the patch-clamp technique with
Axon Patch 700B amplifiers (Axon Instruments, Foster City, CA,
USA). All of the patch-clamp experiments were performed at room
temperature (20–22°C). The transfected cells were bathed in a
solution that contained 140 mmol/l NaCl, 4 mmol/l KCl, 1.8 mmol/l
CaCl2, 0.75 mmol/l MgCl2 and 5 mmol/l HEPES,
at pH 7.4, which was adjusted with NaOH. The glass pipettes were
filled with a solution of 120 mmol/l CsF, 20 mmol/l CsCl, 5 mmol/l
ethylene glycol tetraacetic acid and 5 mmol/l HEPES, at pH 7.4,
which was adjusted with CsOH. The resistance of pipettes in the
bathing solution ranged from 1.5 to 2 MΩ. No leak subtraction was
applied during the recording, and the membrane capacitance was
measured for each of the cells. Both steady-state inactivation
curves and steady-state activation curves were calculated using
Boltzmann distribution.
Confocal imaging
293 cells were fixed with 4% paraformaldehyde for 15
min at room temperature, and they were then permeabilized with 0.2%
Triton X-100 for 10 min. Non-specific sites were blocked by
incubation with 5% bovine serum albumin (BSA) for 1 h at room
temperature. Subsequently, the cells were incubated with mouse
monoclonal to Nav1.5 IgM primary antibody (1:40 dilution; ab62388)
(Abcam, Cambridge, UK) overnight at 4°C. The following day, the
cells were incubated with the anti-mouse FITC-conjugated donkey
secondary antibody (AB10081-100ul; Jackson ImmunoResearch, West
Grove, PA, USA, 1:500 dilution) for 30 min in the dark at room
temperature. Finally, the coverslips were then mounted using
mounting medium containing DAPI (Beyotime Biotechnology Co., Ltd.,
Shanghai, China). Confocal images were obtained using a confocal
laser scanning microscope (Leica TCS SP2, Leica, Leica
Microsystems, Wetzlar, Germany) and were analyzed using NIH image
software (ImageJ).
Statistical analysis
INa data for patch-clamp analysis were
analyzed using pClamp9 software (Molecular Device). Continuous
variables are expressed as the means ± SE and were compared using
the unpaired Student's t-test or one-way ANOVA when appropriate. A
P-value <0.05 was considered to indicate a statistically
significant difference.
Results
Clinical manifestation and
genotyping
The 67-year-old male proband was hospitalized due to
recurrent syncope, accompanied by palpitations, which occurred over
a period of 1 month prior to hospitalization. Baseline 12-lead ECG
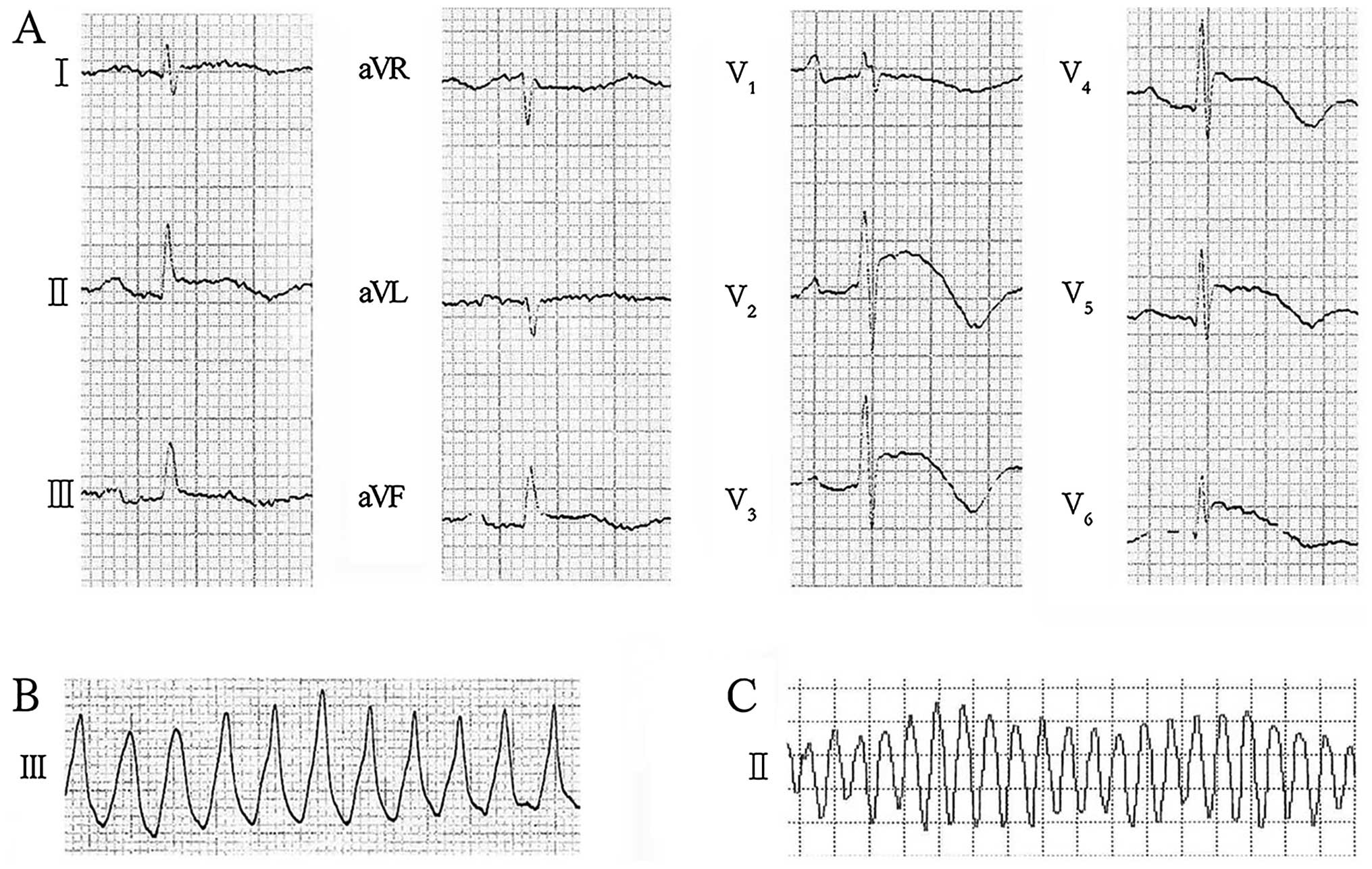
exhibited coved ST-segment elevation which mimicked acute
myocardial ischemia in lead V2–V6, and the
ECG also revealed J waves in lead II, III, aVF and
V2–V6 (Fig.
1A). Polymorphic VT (Fig. 1B and
C), which were terminated successfully by direct current
cardioversion, were documented on the ECG obtained during his
syncope at the emergency department and in the ward during the
admission process. Coronary angiog-raphy exhibited no stenosis in
the coronary arteries. Structural abnormalities were excluded by
echocardiography, an MRI and a chest roentgenogram, and electrolyte
disturbances were excluded by laboratory tests. His medical history
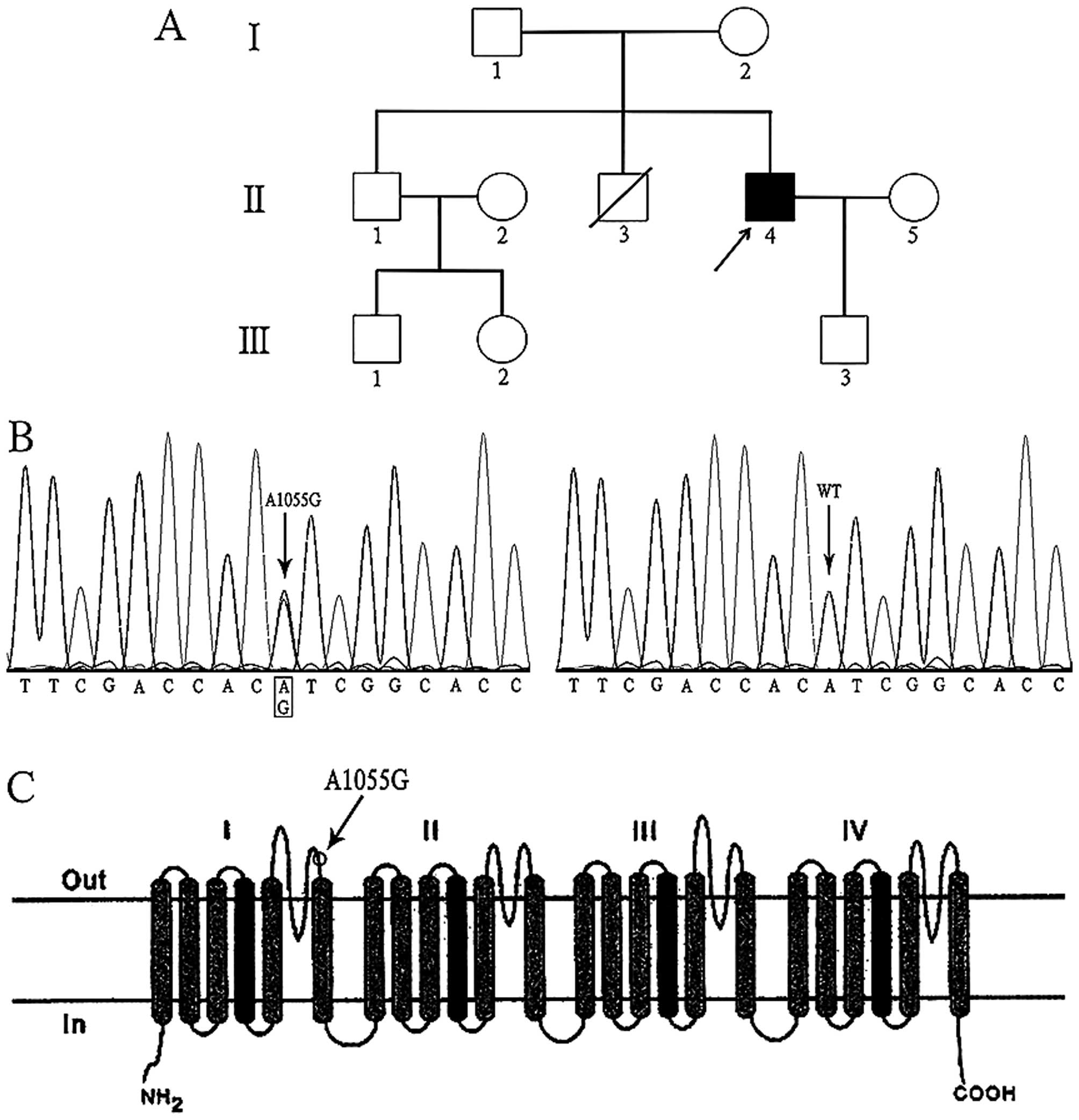
revealed that the proband had suffered from paroxysmal palpitations
for two years and that one of his brothers had died from sudden
death syndrome (Fig. 2A).
According to the clinical characteristics, the proband was
diagnosed with ERS and was provided with an implantable
cardioverter defibrillator (ICD). The other family members of the
proband were asymptomatic, and the son of the proband had a normal
ECG pattern.
Genetic analysis revealed that the proband carried a
novel heterozygous missense mutation of c.1055 A>G in the
SCN5A gene (Fig. 2B),
which had led to the substitution of a tyrosine by a cysteine in
position 352 (Tyr352Cys). The gene encoded the predominant cardiac
sodium channel α subunit and the corresponding substitution located
in the extracellular loop between segment five and six of domain I
in sodium channel α subunit (Fig.
2C). This variant was predicted to have deleterious effect on
the protein by the bioinformatics tools SIFT (13). Moreover, the mutation was not
detected in the son of the proband, nor in ethnicity-matched 500
healthy unrelated controls.
Electrophysiology
In the present study, whole-cell configuration of
the patch-clamp technique was used to record INa from
293 cells, which were transiently transfected with WT-SCN5A or
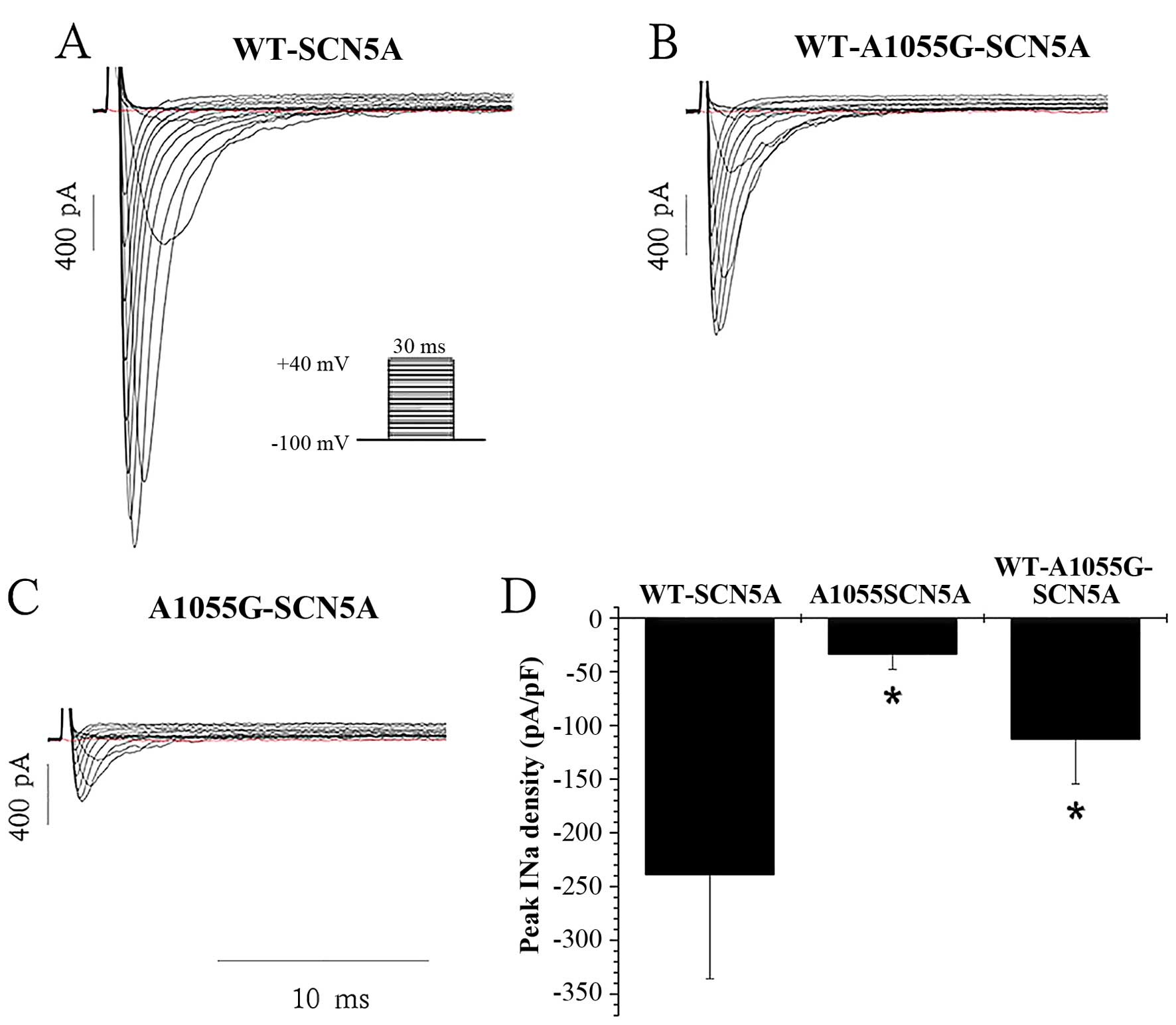
mutant (A1055G-SCN5A) sodium channels. The mean peak INa
density of A1055G-SCN5A channels (−33.39±14.29 pA/pF, n=7) was
markedly lower than that of WT-SCN5A channels (−238.68±97.13 pA/pF,
n=7, P<0.001). Additionally, heterozygous co-expression of
WT-SCN5A and A1055G-SCN5A channels (WT-A1055G-SCN5A), which mimic
the genotype of the proband, significantly reduced the peak
INa density (−112.69±41.66 pA/pF, n=6) to 47% of the
WT-SCN5A channels (−238.68±97.13 pA/pF, n=7, P<0.001). The
changes in peak INa density are summarized in Fig. 3.
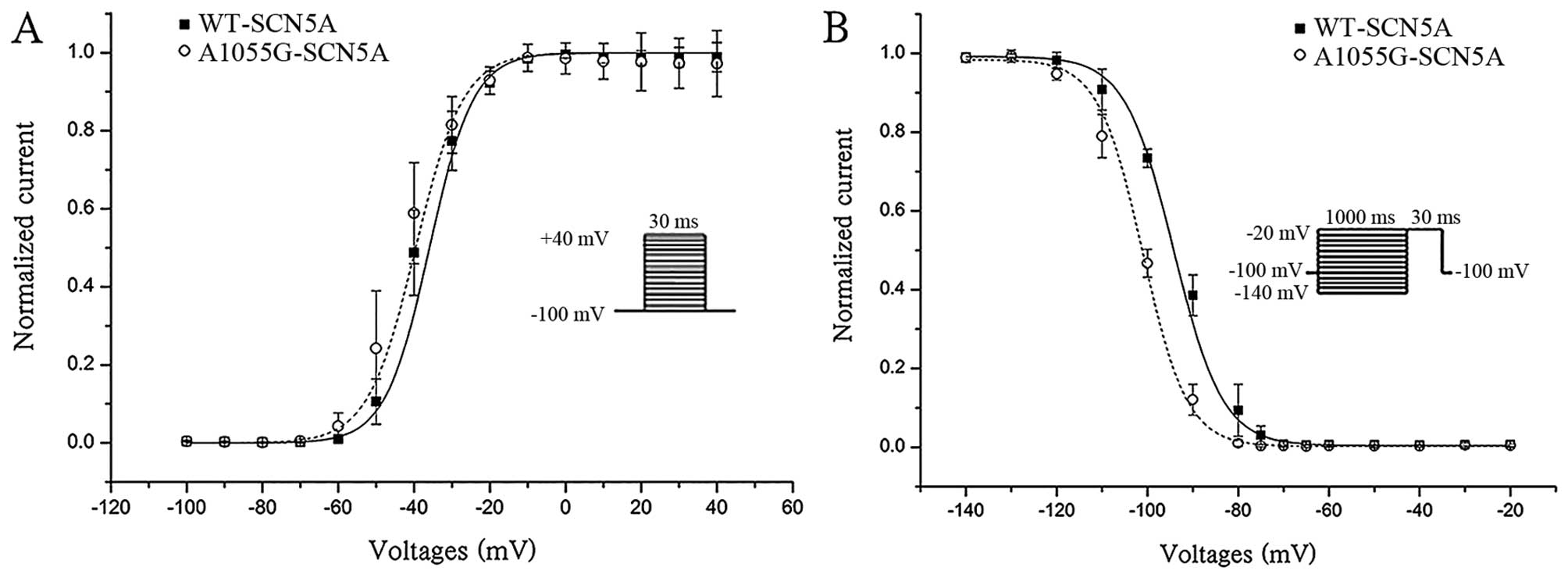
The steady-state activation curve of A1055G-SCN5A
channels exhibited no significant changes compared with that of
WT-SCN5A channels (P>0.05, Fig.
4A), while the steady-state inactivation curve of A1055G-SCN5A
channels was shifted to a more negative potential compared with
that of WT-SCN5A channels (P=0.003, Fig. 4B).
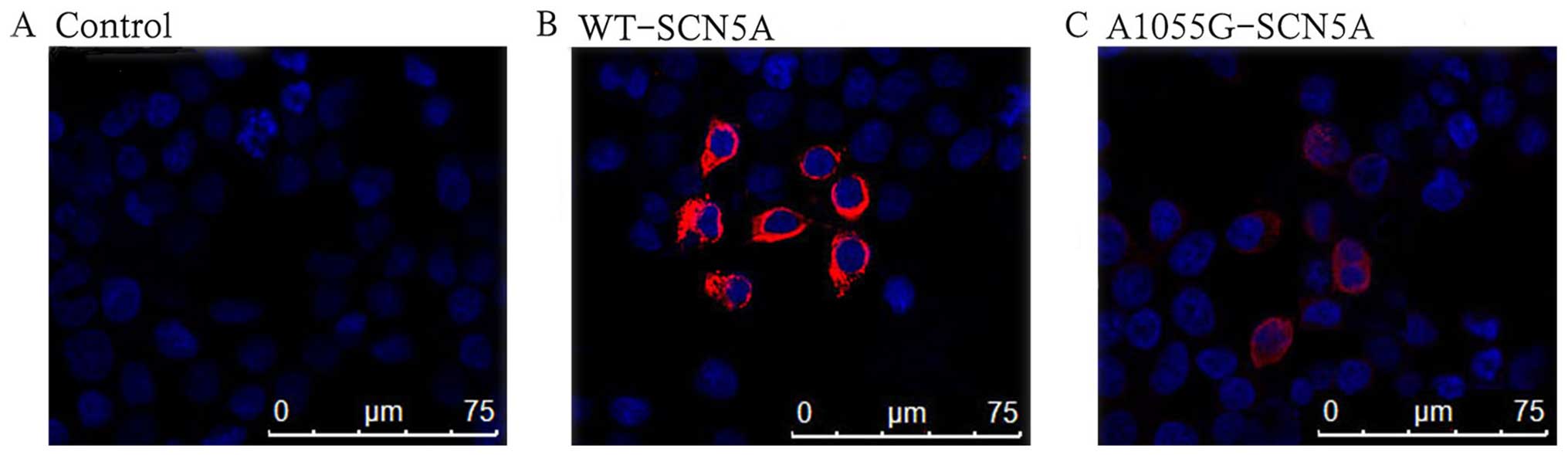
Confocal imaging
In order to understand the mechanisms underlying the
marked reduction in INa, immunocytochemical analysis was
performed to explore cellular expression and the localization of
the WT and mutant channels. The non-transfected cells had blue
background fluorescence (Fig.
5A). As anticipated, the cells transfected with WT-SCN5A
channels displayed strong red fluorescence on the cell membranes
(Fig. 5B). By contrast, the
mutation of A1055G considerably reduced the expression in both the
cell membrane and cytoplasm (Fig.
5C).
Discussion
In the present study, we discovered a novel
pathogenic variant in SCN5A of A1055G, which was correlated
with ERS. This heterozygous missense mutation was identified in a
male proband suffering from recurrent syncope, in whose ECG we
noted an ER pattern in inferior leads and predominantly elevated
ST-segment in mass precordia leads (V2–V6).
Polymorphic VT was documented, and the functional analysis
suggested the mutation had severely deleterious effects on the
sodium channel, which we suggest is the pathogenic mechanism for
the arrhythmogenic characteristics of ERS.
Moreover, in the baseline 12-lead ECG we noted coved
ST-segment elevation in lead V2–V6, which
mimicked type 1 Brugada syndrome (BrS). The ECG diagnostic criteria
of type 1 BrS indicate that a coved pattern should be present in
lead V1–V2 (14). However, it should be noted that
lead V1 in the ECG of the proband did not exhibit any
such coved pattern. Furthermore, according to research by Zorzi
et al, the ST-segment elevation at J point (STJ)
and at 80 msec after J point (ST80) was useful for distinguishing
ER from BrS (15). An upsloping
ST-segment configuration (STJ/ST80 <1) had
a high diagnostic accuracy for the diagnosis of ER, which presented
in the ECG of the proband. Thus, the proband was diagnosed with
ERS. However, the absence of a sodium channel blockade test to
further confirm the diagnosis was one of limitations.
In 2011, Noseworthy et al found that the ER
pattern had a heritable basis in the general population (13). ERS has been associated with
mutations in six genes, namely KCNJ8, ABCC9,
CACNA1C, CACNB2, CACNA2D1 and SCN5A
(16). Medeiros-Domingo et
al put forward the theory that gain-of-function in the cardiac
IK-ATP channel secondary to the missense mutation S422L
was the pathogenic mechanism for the phenotypic expression of both
BrS and ERS (17). Previously, Hu
et al found mutations in ABCC9, which encoded
ATP-binding cassette transporter of IK-ATP (SUR2A)
(18). The mutations also caused
a gain-of-function in IK-ATP and contributed to both BrS
and ERS (18). Loss-of-function
mutations were discovered in CACNA1C, CACNB2 and
CACNA2D1, which respectively encode α1, β2 and α2δ subunits
of the cardiac L-type calcium channel, as well as in SCN5A
(10,19). Loss-of-function mutations in
SCN5A are related to a wide range of inherited arrhythmia
syndromes, such as BrS, progressive cardiac conduction disease, and
sick sinus syndrome (20–22). In one of our previous studies, a
mutation of G4297C in SCN5A was found to be responsible for
ERS. It decreased INa density due to abnormal
translation processes, which were rescued by a synonymous
polymorphism of T5457C on the same allele of the gene (23).
In the present study, another novel loss-of-function
mutation of A1055G in SCN5A was identified in the patient
with ERS. After excluding other gene variants (KCNJ8,
ABCC9, CACNA1C, CACNB2 and CACNA2D1)
related to ERS, a variant of A1055G was speculated to be a
causative mutation. The SCN5A mutation was considered to be
pathologic based on the following reasons: firstly, the variant was
heterozygous; secondly, the variant was not detected in
ethnicity-matched 500 healthy unrelated controls or in the son of
the proband, whose ECG was normal. However, the genetic material
and ECG from the living brother of the proband was not available.
Additionally, the variant resulted in a substitution of a tyrosine
by a cysteine in the extracellular loop between segments five and
six of domain I, which located in highly conserved region across
mammals. Furthermore, the functional analysis demonstrated
remarkably decreased INa and obviously altered biologic
features of sodium channel.
None of the previously identified variations are
enough to distinguish benign from malignant ER variants (24). Based on the episodes of
life-threatening arrhythmia which occurred in the proband and based
on the results of the patch-clamp, the A1055G mutation studied here
is thought to be a variant indicated higher risk of malignant
arrhythmia. According to previous research, the estimated absolute
risk of sudden cardiac death in subjects with ER pattern was
70/100,000 every year (25). It
was recommended that ERS be divided into three subtypes: Type 1,
which has an ER pattern distributed predominantly in the lateral
precordial leads, was prevalent among healthy male athletes with
lower risk of arrhythmias; Type 2, which had an ER pattern
distributed predominantly in the inferior or inferolateral leads,
was associated with a higher level risk of arrhythmias; Type 3,
which had an ER pattern distributed globally in the inferior,
lateral and right precordial leads, was associated with the highest
level of risk of developing malignant arrhythmias (26). It was reported that mutations in
SCN5A were associated with Type 3 ERS under baseline
conditions or following a sodium block challenge (19). In the present study, the proband
who carried a mutation in SCN5A was diagnosed with Type 3
ERS according to the manifestation of the ECG, which was in
accordance with the information above.
In previous research, the mechanism underlying ERS
has been posited to be the increasing action potential (AP) notches
at either the left or right ventricular epicardium, which creates a
transmural voltage gradient that causes the appearance of J-waves
with or without ST segment elevation on the ECG (27,28). In this way, dispersion of
repolarization between the epicardium and endocardium as well as
within the epicardium was generated, leading to transmural
dispersion and a vulnerable window across the ventricular wall,
finally resulting in phase 2 reentry that triggers VT or VF
(24,28). The imbalanced repolarizing
currents caused by either decreased inward currents, such as
INa or ICa, or increased outward currents,
such as Ito, IK-ATP, IK-ACh, or
other outward currents, give rise to ERS (24,29,30). In the present study, the
patch-clamp analysis demonstrated the heterozygous expression of
A1055G reduced peak INa density to 47% of the WT.
Possible explanations for this phenomenon are the defects of
protein expression demonstrated by the immunocytochemical analysis,
which displayed reduced red fluorescence in both the cell membrane
and cytoplasm. Moreover, A1055G accelerated the inactivation of the
sodium channel, which possibly led to a relatively increased
outward current in ER of AP and finally caused arrhythmogenic
characteristic in the patient with ERS. The results of functional
tests, as well as the proposed mechanisms, are consistent with
findings in previous studies (23). Thus, we suggest that both the
defects of channel surface expression and altered kinetic features
of sodium channels, which resulted from the mutation of A1055G,
were critical for ERS in the proband.
In conclusion, in the present study we demonstrated
that a novel heterozygous missense mutation A1055G in SCN5A
led to a loss-of-function in the sodium channels, which likely
accounts for the clinical phenotype of the proband and contributed
to the arrhythmogenic characteristics of ERS.
Acknowledgments
The National Basic Research Program of China (973
program projects, program no. 2013CB531105) provided support to J.
Pu for this research.
References
|
1
|
Shipley RA and Hallaran WR: The four lead
electrocardiogram in two hundred normal men and women. Am Heart J.
11:32–45. 1936. View Article : Google Scholar
|
|
2
|
Wasserburger RH and Alt WJ: The normal
RS-T segment elevation variant. Am J Cardiol. 8:184–192. 1961.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Klatsky AL, Oehm R, Cooper RA, Udaltsova N
and Armstrong MA: The early repolarization normal variant
electrocardiogram: correlates and consequences. Am J Med.
115:171–177. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haïssaguerre M, Derval N, Sacher F, Jesel
L, Deisenhofer I, de Roy L, Pasquié JL, Nogami A, Babuty D,
Yli-Mayry S, et al: Sudden cardiac arrest associated with early
repolarization. N Engl J Med. 358:2016–2023. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rosso R, Kogan E, Belhassen B, Rozovski U,
Scheinman MM, Zeltser D, Halkin A, Steinvil A, Heller K, Glikson M,
et al: J-point elevation in survivors of primary ventricular
fibrillation and matched control subjects: incidence and clinical
significance. J Am Coll Cardiol. 52:1231–1238. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Derval N, Simpson CS, Birnie DH, Healey
JS, Chauhan V, Champagne J, Gardner M, Sanatani S, Yee R, Skanes
AC, et al: Prevalence and characteristics of early repolarization
in the CASPER registry: cardiac arrest survivors with preserved
ejection fraction registry. J Am Coll Cardiol. 58:722–728. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mahida S, Derval N, Sacher F, Berte B,
Yamashita S, Hooks DA, Denis A, Lim H, Amraoui S, Aljefairi N, et
al: History and clinical significance of early repolarization
syndrome. Heart Rhythm. 12:242–249. 2015. View Article : Google Scholar
|
|
8
|
Priori SG, Wilde AA, Horie M, Cho Y, Behr
ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, et al:
HRS/EHRA/APHRS expert consensus statement on the diagnosis and
management of patients with inherited primary arrhythmia syndromes:
document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF,
AHA, PACES, and AEPC in June 2013. Heart Rhythm. 10:1932–1963.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haïssaguerre M, Chatel S, Sacher F,
Weerasooriya R, Probst V, Loussouarn G, Horlitz M, Liersch R,
Schulze-Bahr E, Wilde A, et al: Ventricular fibrillation with
prominent early repolarization associated with a rare variant of
KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 20:93–98. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Burashnikov E, Pfeiffer R,
Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M,
Häissaguerre M, Kanter R, Pollevick GD, et al: Mutations in the
cardiac L-type calcium channel associated with inherited J-wave
syndromes and sudden cardiac death. Heart Rhythm. 7:1872–1882.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang RR, Li N, Zhang YH, Wang LL, Teng SY
and Pu JL: Novel compound heterozygous mutations T2C and 1149insT
in the KCNQ1 gene cause Jervell and Lange-Nielsen syndrome. Int J
Mol Med. 28:41–46. 2011.PubMed/NCBI
|
|
12
|
Teng S, Gao L, Paajanen V, Pu J and Fan Z:
Readthrough of nonsense mutation W822X in the SCN5A gene can
effectively restore expression of cardiac Na+ channels.
Cardiovasc Res. 83:473–480. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Noseworthy PA, Tikkanen JT, Porthan K,
Oikarinen L, Pietilä A, Harald K, Peloso GM, Merchant FM, Jula A,
Väänänen H, et al: The early repolarization pattern in the general
population: clinical correlates and heritability. J Am Coll
Cardiol. 57:2284–2289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Serra G, Baranchuk A, Bayés-De-Luna A,
Brugada J, Goldwasser D, Capulzini L, Arazo D, Boraita A, Heras ME,
Garcia-Niebla J, et al: New electrocardiographic criteria to
differentiate the Type-2 Brugada pattern from electrocardiogram of
healthy athletes with r'-wave in leads V1/V2. Europace.
16:1639–1645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zorzi A, Leoni L, Di Paolo FM, Rigato I,
Migliore F, Bauce B, Pelliccia A and Corrado D: Differential
diagnosis between early repolarization of athlete's heart and
coved-type Brugada electrocardiogram. Am J Cardiol. 115:529–532.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Antzelevitch C: Genetic, molecular and
cellular mechanisms underlying the J wave syndromes. Circ J.
76:1054–1065. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Medeiros-Domingo A, Tan BH, Crotti L,
Tester DJ, Eckhardt L, Cuoretti A, Kroboth SL, Song C, Zhou Q, Kopp
D, et al: Gain-of-function mutation S422L in the KCNJ8-encoded
cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave
syndromes. Heart Rhythm. 7:1466–1471. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu D, Barajas-Martínez H, Terzic A, Park
S, Pfeiffer R, Burashnikov E, Wu Y, Borggrefe M, Veltmann C,
Schimpf R, et al: ABCC9 is a novel Brugada and early repolarization
syndrome susceptibility gene. Int J Cardiol. 171:431–442. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Watanabe H, Nogami A, Ohkubo K, Kawata H,
Hayashi Y, Ishikawa T, Makiyama T, Nagao S, Yagihara N, Takehara N,
et al: Electrocardiographic characteristics and SCN5A mutations in
idiopathic ventricular fibrillation associated with early
repolarization. Circ Arrhythm Electrophysiol. 4:874–881. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schott JJ, Alshinawi C, Kyndt F, Probst V,
Hoorntje TM, Hulsbeek M, Wilde AA, Escande D, Mannens MM and Le
Marec H: Cardiac conduction defects associate with mutations in
SCN5A. Nat Genet. 23:20–21. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abe K, Machida T, Sumitomo N, Yamamoto H,
Ohkubo K, Watanabe I, Makiyama T, Fukae S, Kohno M, Harrell DT, et
al: Sodium channelopathy underlying familial sick sinus syndrome
with early onset and predominantly male characteristics. Circ
Arrhythm Electrophysiol. 7:511–517. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Adsit GS, Vaidyanathan R, Galler CM, Kyle
JW and Makielski JC: Channelopathies from mutations in the cardiac
sodium channel protein complex. J Mol Cell Cardiol. 61:34–43. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li N, Wang R, Hou C, Zhang Y, Teng S and
Pu J: A heterozygous missense SCN5A mutation associated with early
repolarization syndrome. Int J Mol Med. 32:661–667. 2013.PubMed/NCBI
|
|
24
|
Antzelevitch C: J wave syndromes:
molecular and cellular mechanisms. J Electrocardiol. 46:510–518.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu SH, Lin XX, Cheng YJ, Qiang CC and
Zhang J: Early repolarization pattern and risk for arrhythmia
death: a meta-analysis. J Am Coll Cardiol. 61:645–650. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Antzelevitch C and Yan GX: J wave
syndromes. Heart Rhythm. 7:549–558. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan GX and Antzelevitch C: Cellular basis
for the electrocardiographic J wave. Circulation. 93:372–379. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Koncz I, Gurabi Z, Patocskai B, Panama BK,
Szél T, Hu D, Barajas-Martínez H and Antzelevitch C: Mechanisms
underlying the development of the electrocardiographic and
arrhythmic manifestations of early repolarization syndrome. J Mol
Cell Cardiol. 68:20–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yan GX and Antzelevitch C: Cellular basis
for the Brugada syndrome and other mechanisms of arrhythmogenesis
associated with ST-segment elevation. Circulation. 100:1660–1666.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yan GX, Lankipalli RS, Burke JF, Musco S
and Kowey PR: Ventricular repolarization components on the
electrocardiogram: cellular basis and clinical significance. J Am
Coll Cardiol. 42:401–409. 2003. View Article : Google Scholar : PubMed/NCBI
|