Introduction
Hemorrhagic shock (HS) significantly contributes to
trauma-related mortality. HS and subsequent resuscitation are also
responsible for the most common clinical complications in patients
with traumatic injuries or who have undergone major surgery. Acute
hypovolemia/hemodynamic disorders and global ischemia/reperfusion
result in multiple organ failure mediated by priming the innate
immune system (1–3). In this regard, an exaggerated
inflammatory reaction prevails in distant vital organs,
characterized by the activation and the infiltration of phagocytes
(neutrophils and macrophages). It has been suggested that the liver
and lungs are the most commonly affected organs following
hemorrhage/resuscitation in clinical practice (4). As one of the first organs affected
by HS, the liver suffers adenosine triphosphate (ATP) depletion due
to ischemia, which results in the induction of inflammatory
signaling and necrosis thereafter (5). On the other hand, the reperfusion
phase in the liver consists of neutrophil activation mediated by
inducible nitric oxide synthase, and ultimately leads to hepatocyte
apoptosis (6,7). By priming excess reactive oxygen
species (ROS) production, ischemia/reperfusion activates
neutrophils sequestered in the lung and alveolar macrophages, thus
subsequently augmenting lung injury and dysfunction (8,9).
However, the precise mechanisms through which HS activates the
inflammatory response have not yet been fully clarified.
Nuclear factor-erythroid 2 (NF-E2) p45-related
factor-2 (Nrf2), an essential leucine zipper redox-susceptible
transcription factor, is abundantly expressed in most tissues and
is involved in regulating the induction of cytoprotective and
antioxidant genes. Under conditions of oxidative stress, Nrf2
dissociates from its cytosolic inhibitor, Kelch-like ECH-associated
protein 1 in the cytosol and translocates to the nucleus, where it
promotes the production of antioxidants and transactivates related
cytoprotective pathways (10).
Accordingly, Nrf2 has been identified as a pivotal mediator in
redox homeostasis and inflammatory disorders, including pulmonary
fibrosis, asthma, cigarette smoke-induced emphysema, colonic
inflammatory injury and experimental sepsis (11–15). Clinical studies have also
concluded that there is a correlation between Nrf2
activity/signaling and the pathogenesis of inflammatory diseases,
such as chronic obstructive pulmonary disease and chronic kidney
disorder (16,17).
Taking the above-mentioned findings into account, we
hypothesized that Nrf2 exerts protective effects against
inflammation-associated injury induced by HS, which may reveal a
novel regulatory mechanism therein. To confirm this hypothesis, in
this study, we examined the role of Nrf2 in the dysregulated
inflammatory response following HS. We examined the correlation
between Nrf2 expression and clinical HS. We also investigated the
inflammatory response and subsequent injury due to HS in wild-type
(WT; Nrf2+/+) and Nrf2-deficient [Nrf2−/− or
Nrf2-knockout (KO)] mice in vivo and ex vivo.
Materials and methods
Clinical specimens, processing and
RT-qPCR
The protocols for blood collection and analysis were
approved by the Ethics Review Committee of Zhejiang University
School of Medicine (Hangzhou, China). Additionally, written
informed consent was obtained from all patients prior to blood
collection. Whole blood was obtained from 6 blood donors without
any disease or from 6 patients with surgery-associated hemorrhage
subjected to resuscitation treatment (termed HS patients), of which
3 were undergoing hepatectomy, 2 splenectomy and 1 abdominal aorta
replacement. All 6 patients suffered blood loss >15% of total
blood volume with the duration of HS varying from 8–30 min. All the
surgeries were performed at the First Affiliated Hospital of
Zhejiang University School of Medicine. We collected the blood
samples at 1 h and 5 days post-operation. Briefly, blood samples
were obtained from a forearm vein and coded without patient
identifiers. All specimens were collected in Vacutainer tubes
containing the anticoagulant, ethylenediamine tetraacetic acid
(EDTA; Kang Shi Medical Equipment Co., Ltd., Hangzhou, China) and
were processed within 2 h. During sample collection and processing,
whole blood specimens were stored at 4°C. For blood specimen
processing, whole blood was treated with 5X BL solution [from a
Blood gDNA mini kit (Biomiga, San Diego, CA, USA)] for erythrocyte
depletion and then centrifuged at 3,000 x g for 10 min at 4°C in
order to collect the leukocytes.
Total RNA was extracted using the Total RNA kit I
(Omega Bio-Tek, Inc., Doraville, GA, USA) after the cells were
lysed using 500 µl TRIzol reagent (Invitrogen, Carlsbad, CA,
USA). First-strand cDNA was synthesized from 200 ng of total mRNA
using the ReverTra Ace qPCR RT Master Mix with a gDNA Remover kit
(Toyobo, Osaka, Japan). Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) was performed using the
All-in-One qPCR Mix kit (GeneCopoeia, Rockville, MD, USA) and
analyzed using the ΔΔCt method on a CFX Connect Real-Time PCR
system (Bio-Rad, Hercules, CA, USA). The reaction was initiated at
95°C for 10 min, followed by 40 cycles of denaturation at 95°C for
15 sec, annealing at 55°C for 20 sec and extension at 72°C for 20
sec. A primer pair for the detection of human GAPDH was used as an
internal control. The primers used for RT-qPCR in the present study
were as follows: Nrf2 (human) sense, 5′-TCAGCGACGGAAAGAGTATGA-3′
and antisense, 5′-CCACTGGTTTCTGACTGGATGT-3′; and GAPDH (human)
sense, 5′-CATTGCCCTCAACGACCACTTTGT-3′ and antisense,
5′-TCTCTCTCTTCCTCTTGTGCTCTTGC-3′.
Mice
All animal experimental protocols were performed in
accordance with the Policy and Procedures Manual of Zhejiang
University Animal Care and Use Committee (Hangzhou, China). Nrf2-KO
mice were generated as described in our previous study (18). The Nrf2-KO mice in the current
study were provided by Dr Rajesh K. Thimmulappa and Dr Shyam Biswal
who obtained the mice as a gift from a Japanese group (18). Briefly, the b-Zip region of mouse
nrf2 cDNA was replaced with SV40 nuclear localization
signal-β-galactosidase (lacZ) gene by homologous recombination. The
linearized targeting construct was electroporated into embryonic
stem cells (ES) and positive colonies were identified by PCR and
Southern blot analysis. Positive ES colonies were then introduced
into C57BL/6J blastocysts by microinjection to obtaine chimeric
mice, which were mated with ICR females and BALB/cA females for
germline transmission. Afterwards, F1 and F2 offspring were
identified by genotyping. Nrf2-KO mice were then backcrossed to the
C57BL/6 strain over 12 generations to obtain WT mice of the same
origin.
Induction of HS
Twenty mice were subjected to hemorrhage and
rescusitation (termed the HS group) and another 20 mice were
subjected to sham-operation according to previously described
protocols (19). Briefly, the
mice were anesthetized with isoflurane (Minrad, Inc., Orchard Park,
NY, USA) after being fasted overnight with only water ad
libitum. Polyethylene-10 tubing (BD Biosciences, San Jose, CA,
USA) was placed within both femoral arteries and the right femoral
vein. Upon awakening, the mice were bled rapidly through the other
arterial catheter to a mean arterial blood pressure of 35±5 mmHg
within 10 min, which was then maintained for the remaining 90 min.
At the end of the above-mentioned period, the mice were
resuscitated via the venous line using a volume of Ringer's lactate
solution equal to 4 times the volume of blood lost. Following blood
vessel ligation, all catheters were removed and the incisions were
closed with sutures. The mice in the sham-operated group were
subjected to the same surgical procedures as those in the HS group,
but were not subjected to hemorrhage or resuscitation.
Histological analysis
To perform histological analysis, the livers and
lungs were harvested from the mice as indicated above. Briefly, the
mice were anaesthetized with ketamine and xylazine and sacrificed
by cervical dislocation 24 h following HS or sham-operation. After
removing the hair, the abdomens and chest of mice were cut
vertically and opened, revealing the liver and lungs. Half of the
two tissues were then isolated, respectively. The tissues were then
fixed in 4% paraformaldehyde and embedded in paraffin. The paraffin
blocks were then sectioned at 5-µm thick using a microtome
(Leica Biosystems, Heidelberger, Germany). After floating on a 40°C
water bath containing distilled water, the sections were
transferred onto the surface of clean glass slides, which were
placed in 37°C oven to dry overnight and stored in light-proof
boxes until ready for use. Sections (5-µm-thick) were
prepared and stained with hematoxylin and eosin. Two independent
investigators performed the quantitative analysis of liver and lung
injuries by assessing the histological scores. The extent of lung
injury was determined by grading 4 histological findings:
congestion, edema, inflammation and hemorrhage. The degree of lung
injury was scored on a scale of 0–4 (0, normal; 1, mild; 2,
moderate; 3, severe; and 4, very severe) for each feature, with a
cumulative maximum score of 16. For liver injury, all sections were
examined for the following 6 parameters: cytoplasmic color fading,
vacuolization, nuclear condensation, nuclear fragmentation, nuclear
fading and erythrocyte stasis. Each parameter was scored according
to the percentage of cells showing the particular parameter per 10
microscopic fields: 0, 0%, 1, 0–10%; 2, 10–50%; and 3, 50–100%. The
histological scores are reported as the sum of the individual
values.
Measurement of cytokine levels by
ELISA
The levels of interleukin (IL)-1β and tumor necrosis
factor (TNF)-α in plasma and protein lysis were measured using
ELISA kits (eBioscience, San Diego, CA, USA) according to the
manufacturer's instructions. The IL-6 levels were also measured
using an ELISA kit (R&D Systems, Minneapolis, MN, USA). The
plasma was obtained with the following protocol: 2 h following HS
or sham-operation, the mice were anaesthetized with ketamine and
xylazine and fixed in a supine position. The mouse left chest
between the 3 to 4 intercostal space was felt using an index finger
of the left hand to feel the heartbeat. Afterwards, a 23G needle of
a syringe was carefully punctured into the mouse chest cavity until
it entered the heart. When the heart was punctured, the blood
rushed into the syringe automatically, and 0.5–0.6 ml blood was
collected in vacutainer tubes containing the anticoagulant, EDTA.
The plasma was obtained by removing blood cells and platelets by
centrifugation for 15 min at 2,000 × g. Protein lysis of the lung
and liver tissue was carried out as follows: the isolated liver and
lung tissues were placed in liquid nitrogen and then smashed with a
wooden hammer. Afterwards, the tissue powders were resuspended in
RIPA buffer (1% Triton X-100, 1% deoxycholate and 0.1% SDS), which
was centrifuged at 15,000 × g for 15 min at 4°C. The supernatant
was carefully isolated as tissue protein lysis.
Immunoblot analysis
Protein was extracted from the serum in lysis buffer
(1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 5 mM EDTA, 1 mM
sodium orthovanadate, 0.1% aprotinin and 1 mM PMSF) and boiled in
SDS sample buffer for 5 min. Equal amounts of protein per sample
were separated by SDS-PAGE and transferred electrophoretically onto
a polyvinylidene fluoride membrane (Bio-Rad Laboratories). After
blocking in 5% milk, the membrane was incubated with a primary
antibody followed by an HRP-conjugated secondary antibodies
(115-475-062, goat anti-mouse IgG and 111-475-003, goat anti-rabbit
IgG; Jackson ImmunoResearch, West Grove, PA, USA).
Chemiluminescence was then detected using an ECL kit according to
the manufacturer's instructions (Amersham Life Science, Arlington
Heights, IL, USA). The primary antibodies, anti-high-mobility group
box 1 protein (HMGB1) (ab18256, rabbit anti-mouse/human,
polyclonal) and anti-β-actin (ab8226, mouse anti-mouse/human,
monoclonal), were purchased from Abcam (Cambridge, UK).
Myeloperoxidase (MPO) activity assay
MPO activity was assayed using techniques described
previously (20). Briefly, the
tissue samples were sonicated using an Ultrasonic Cell Disrupter
(Kontes, Vineland, NJ, USA) with 10 intervals for 9 times, then
incubated in a 60°C water bath for 1.5 h, and centrifuged at 10,500
× g for 15 min. For each assay, 100 µl of collected
supernatant were mixed with 2.9 ml assay solution (50 mM potassium
phosphate buffer, containing 5×10−4%
H2O2 and 0.167 mg/ml o-dianisidine, pH
6.0). MPO activity was then determined by measuring the absorbance
of 460 nm visible light using a spectrophotometer (Beckman DU7;
Beckman Coulter, Brea, CA, USA) and finally calculated in units per
microgram of wet tissue.
Culture and treatment of mouse peritoneal
macrophages
To isolate peritoneal macrophages, the mice were
injected with 2 ml 4% thioglycollate broth (Sigma-Aldrich, St.
Louis, MO, USA) and incubated for 3 days prior to being subjected
to HS or sham operation. At 24 h after resuscitation, they were
anesthetized and sacrificed by cervical dislocation. For the
harvesting of peritoneal macrophages 5 ml ice-cold PBS was injected
into the peritoneal cavity and peritoneal exudates were aspirated
after 2 min under sterile conditions. The isolated peritoneal
macrophages were cultured at a density of 2.5×105
cells/ml in RPMI-1640 medium (Gibco-Invitrogen Cell Culture,
Carlsbad, CA, USA) containing 10% fetal bovine serum under 5%
CO2 at 37°C. The medium was replaced after 1 h in order
to remove the unattached cells. Lipopolysaccharide (LPS; 100 ng/ml)
was then added to the medium. The supernatant was collected after 6
h for measuring the concentration of IL-1β, IL-6 and TNF-α.
Statistical analysis
All data were analyzed using the unpaired Student's
t-test or one-way ANOVA. A p-value <0.05 was considered to
indicate a statistically significant difference.
Results
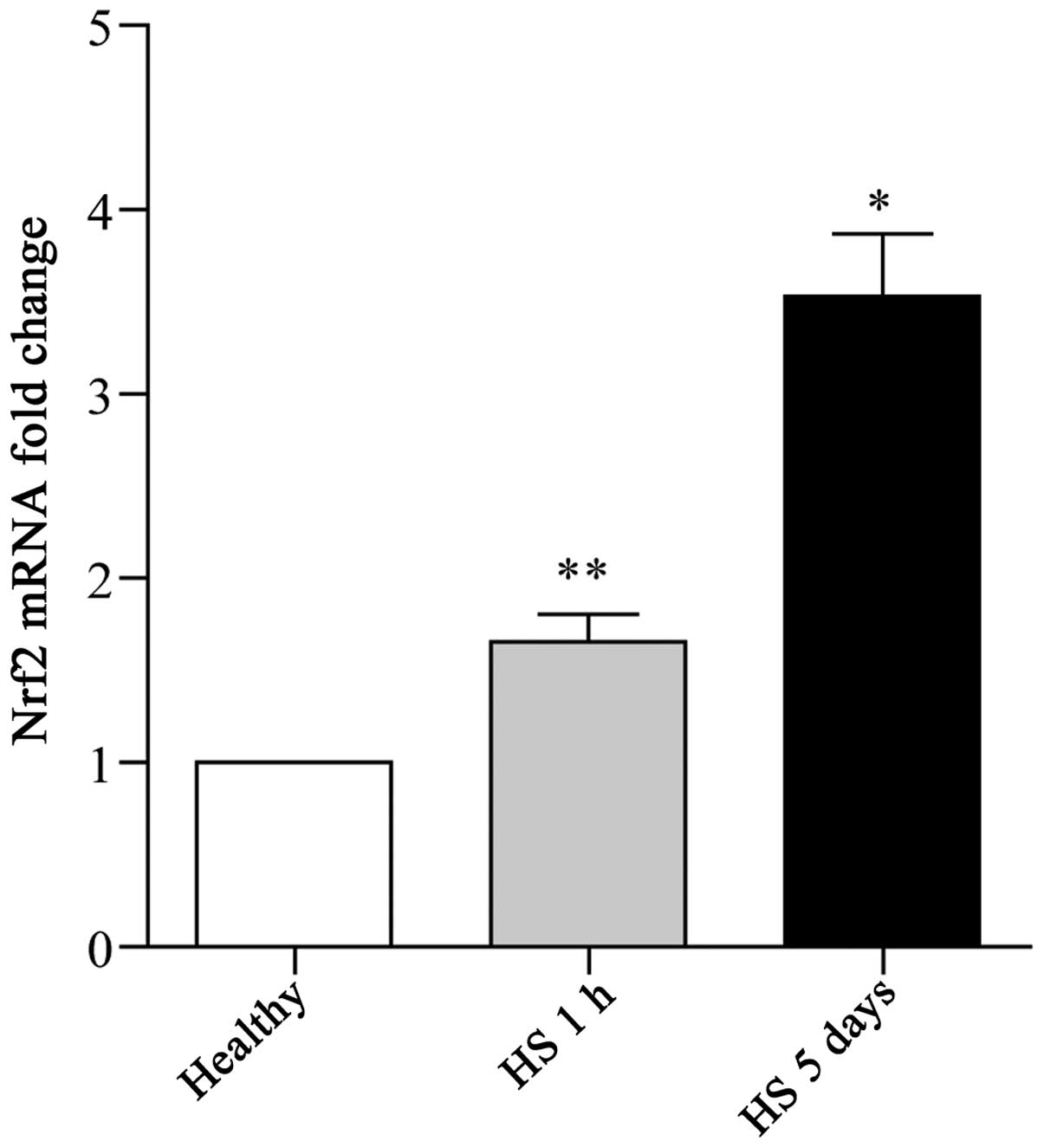
Marked induction of Nrf2 expression in
leukocytes is associated with clinical HS
Nrf2 expression in leukocytes has been described in
a few in vitro studies (21,22). In this study, to identify the
potential role of Nrf2 in the HS-induced inflammatory response, the
expression pattern of Nrf2 in leukocytes was investigated. To this
end, whole blood samples were collected from patients with
surgery-associated hemorrhage subjected to resuscitation treatment,
or from healthy donors. Isolated leukocytes from these clinical
samples were then subjected to RT-qPCR for Nrf2. A normal
expression of Nrf2 was detected in the whole blood samples from the
healthy donors, whereas a significant increase in the Nrf2
expression levels was observed in the samples collected from the HS
patients (Fig. 1). Furthermore,
the trend in the induction of Nrf2 expression was synchronized with
the duration of HS (Fig. 1).
Thus, these findings indicate that there is a close correlation
between Nrf2 expression and the development of HS.
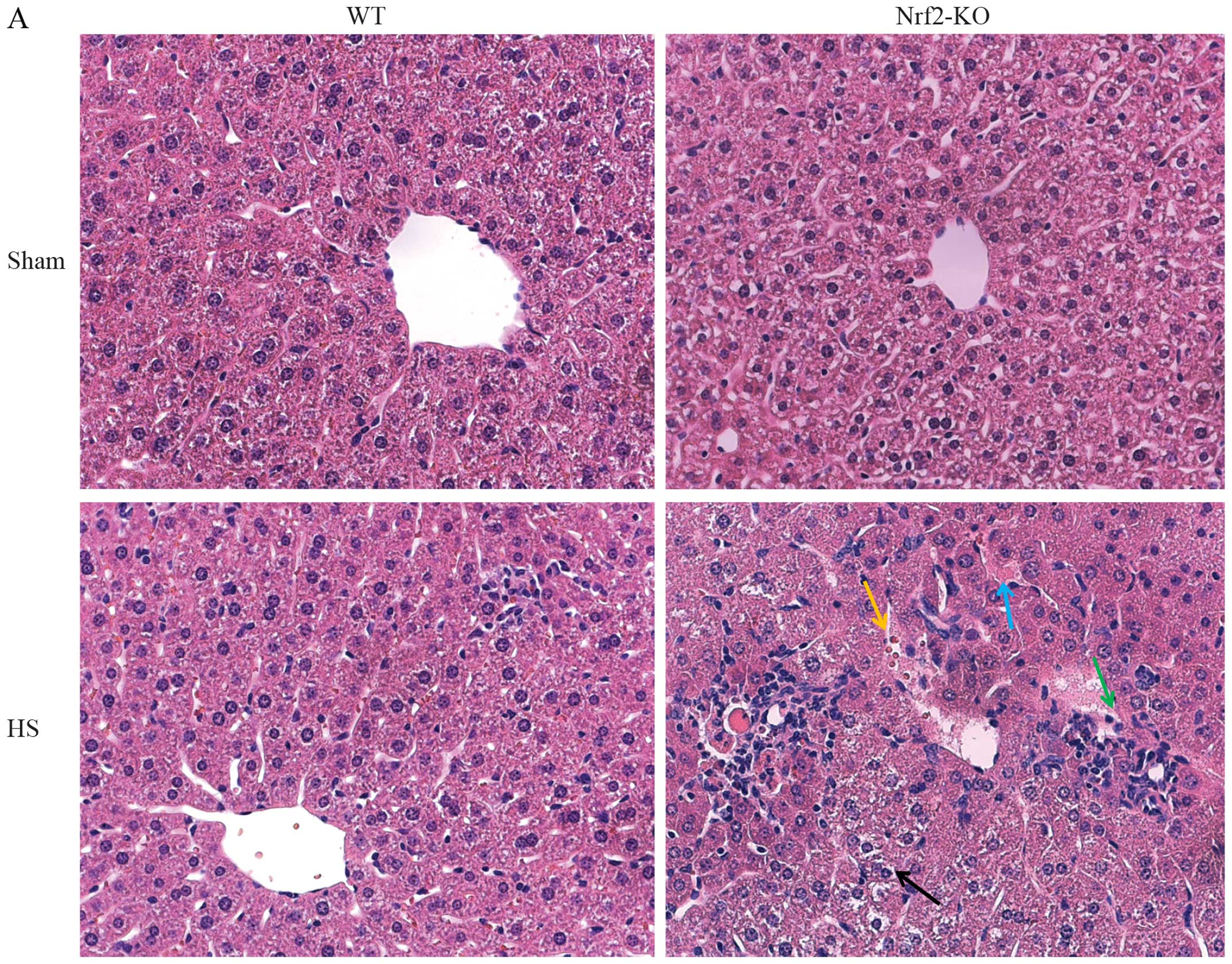
Nrf2 deficiency aggravates lung and liver
injury in a mouse model of HS
In order to further explore the function of Nrf2 in
the pathological setting of HS, we established a mouse model of HS,
which mimicked clinical HS, as indicated in the Materials and
methods. More severe lung and liver injury was observed in the
Nrf2-KO mice following HS. Histological analysis revealed more
interstitial edema, alveolar wall thickening and the interstitial
infiltration of leukocytes, as well as alveolar hemorrhage in the
lungs of the Nrf2-KO mice compared with the WT group (Fig. 2B, with the histological score
shown in Fig. 2D). Similarly, the
livers of the Nrf2-KO mice subjected to HS had far more
interstitial edema, leukocyte infiltration and interstitial
hemorrhage, as well as more severe hepatocellular injury with
necrotic areas (Fig. 2A, with the
histological score shown in Fig.
2C). On the other hand, the extent of organ injury was
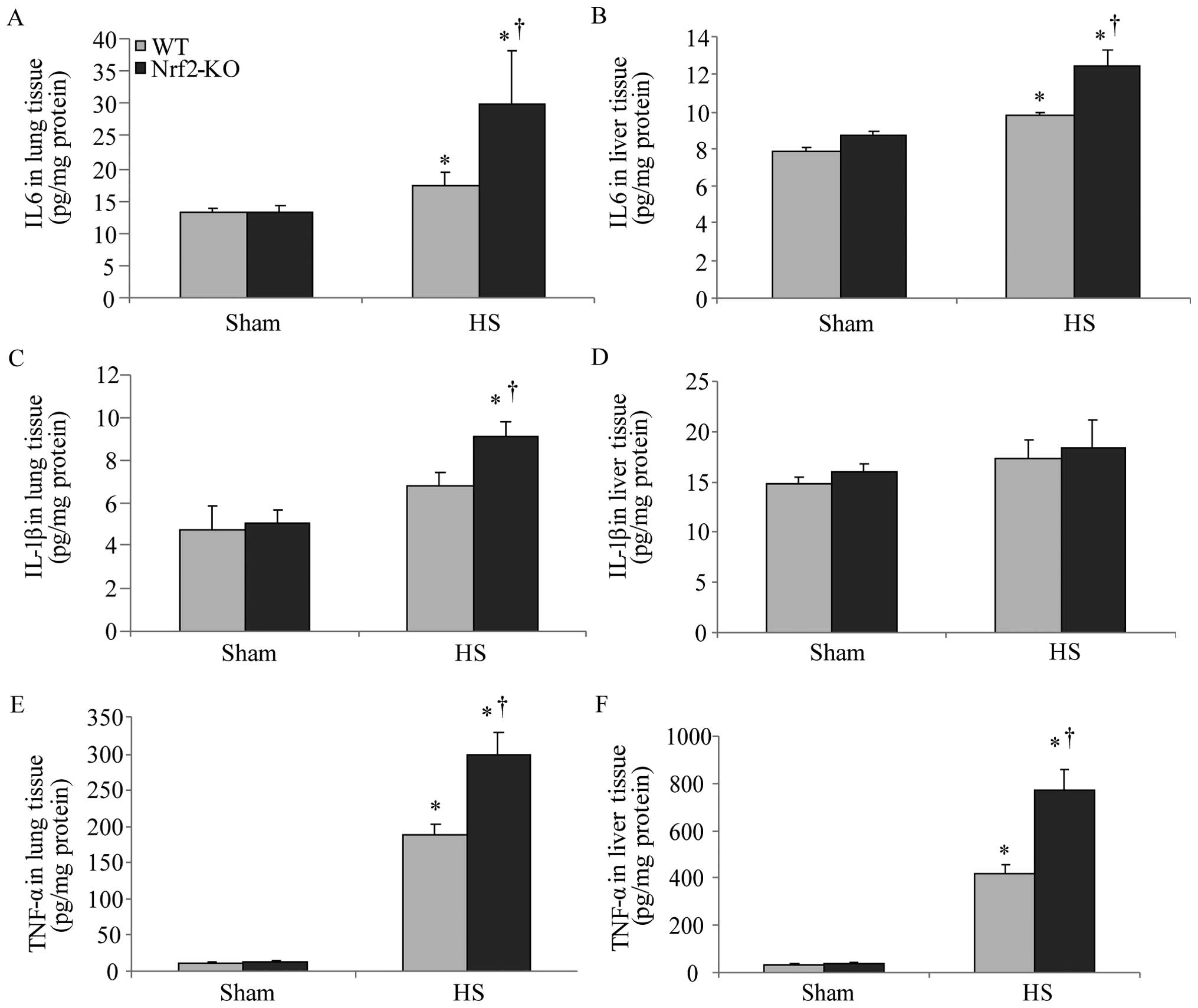
evaluated by determining the levels of pro-inflammatory cytokines
in the organ tissues. We measured the levels of these cytokines in
homogenized tissue protein by ELISA. In the lungs and livers of all
the mice, the levels of IL-6 and TNF-α were significantly increased
following HS; however, the Nrf2-KO mice exhibited significantly
higher levels of these cytokines compared with the WT mice
(Fig. 3A, B, E and F). The
Nrf2-KO mice exhibited much higher expression levels of IL-1β in
the lungs compared with the WT mice following HS, whereas there was
no significant difference observed in the IL-1β expression levels
in the livers of the Nrf2-KO and WT mice (Fig. 3C and D). The accumulation of
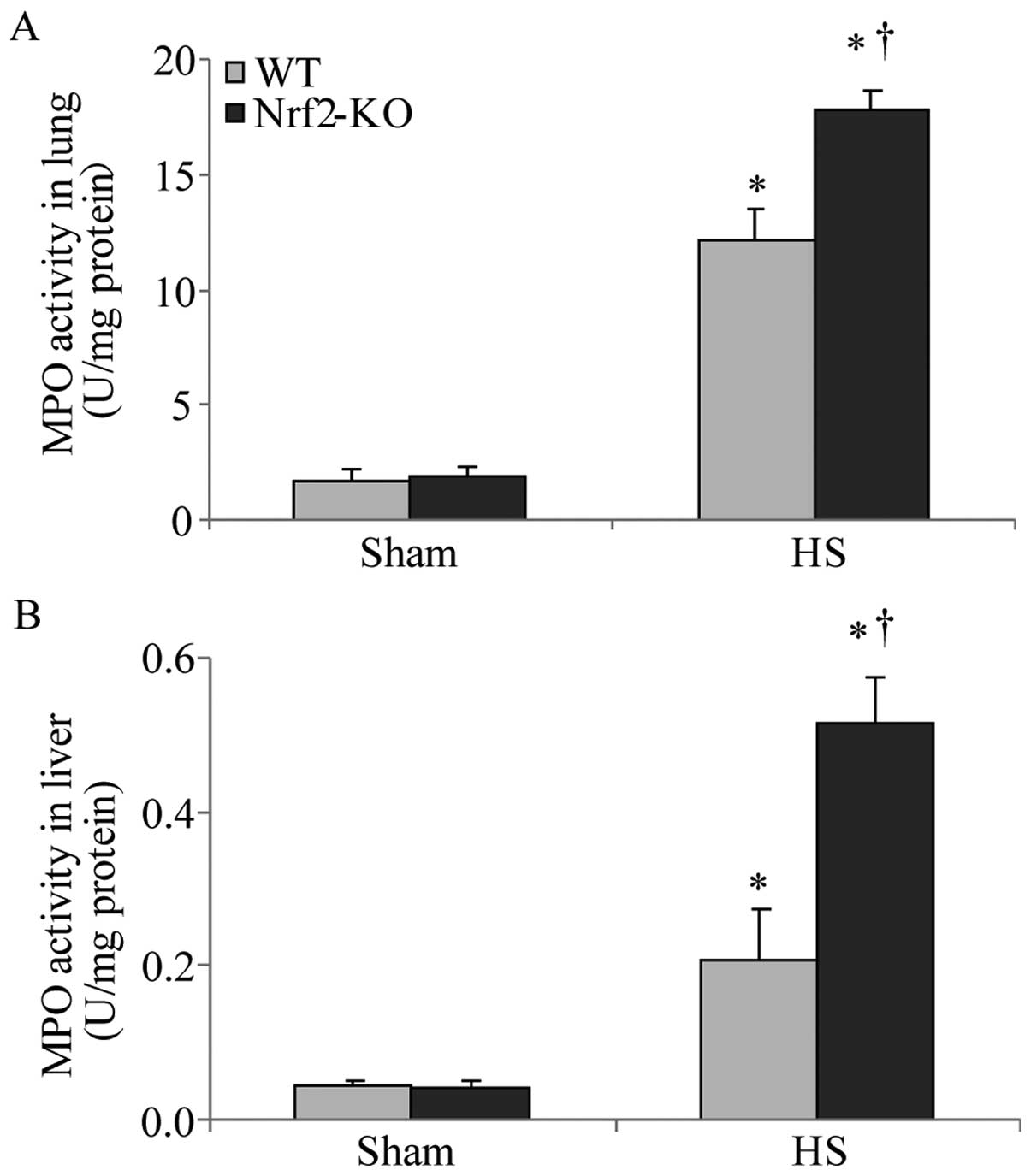
neutrophils in the lung and liver tissues was assessed by measuring
MPO activity, which is also indicative of the severity of
inflammation in the organs. Following HS, an increased MPO activity
in the lungs and livers was detected in both the WT and Nrf2-KO
mice; however, the Nrf2-KO mice had a significantly higher MPO
activity than the WT mice in both organs (Fig. 4). Taken together, these findings
indicate that Nrf2 deficiency induces a more severe inflammatory
response in the vital organs examined (lungs and liver) following
HS.
Nrf2 deficiency exacerbates systemic
inflammation induced by HS
In α previous study, Nrf2 was shown to be a critical
transcription factor in lethal septic shock (15). In this study, to determine the
role of Nrf2 in HS, we compared the severity of systemic
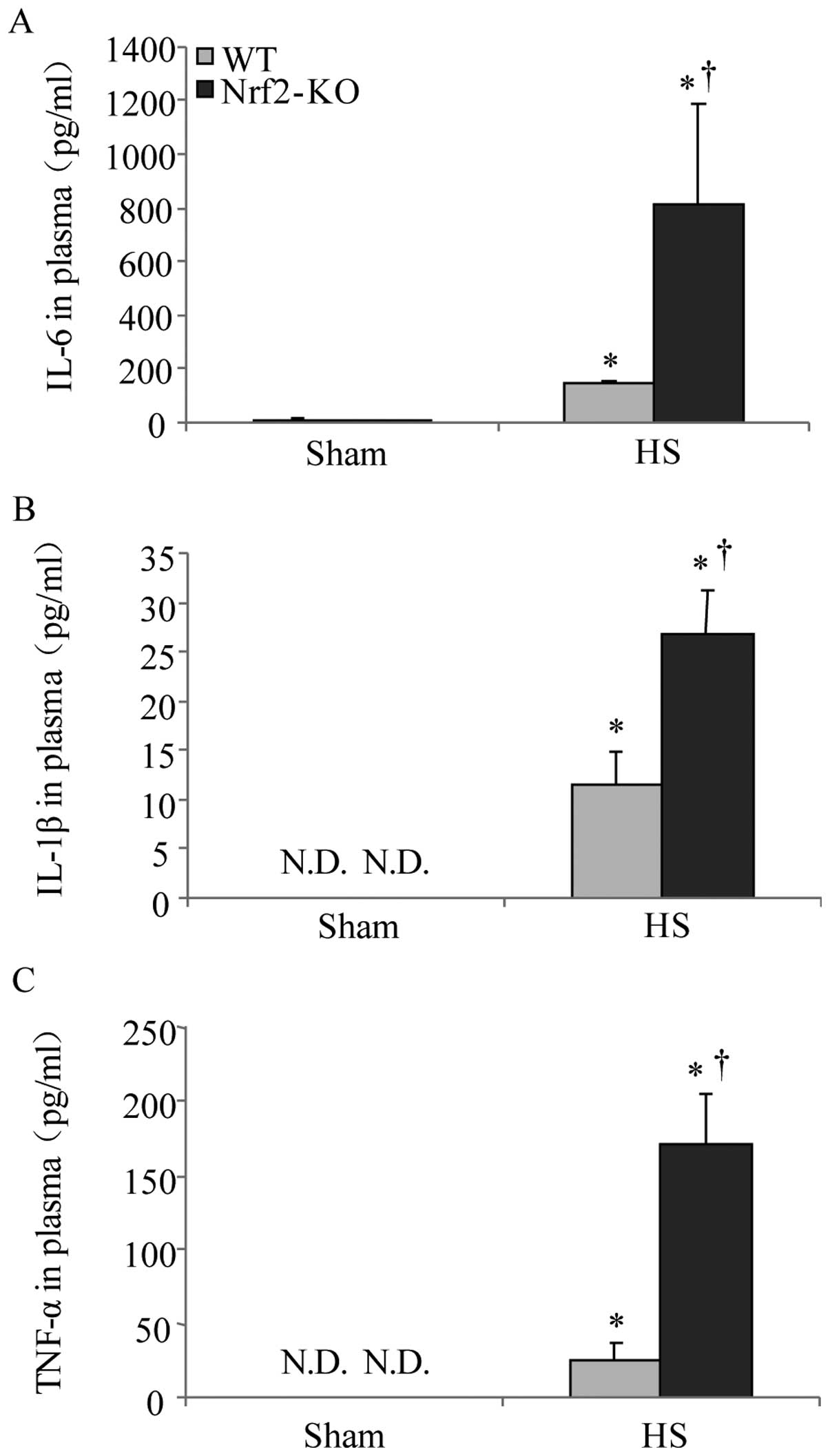
inflammation in the WT and Nrf2-KO mice. As IL-6, IL-1β and TNF-α
are well established as key cytokines involved in the mediation of
inflammation and associated mortality (23), the plasma concentrations of these
cytokines were thereby examined following HS. The results of ELISA
revealed an increasing trend in the levels of IL-6, IL-1β, and
TNF-α 2 h following HS. However, the levels of all 3 cytokines were
significantly higher in the Nrf2-KO mice compared with the WT mice
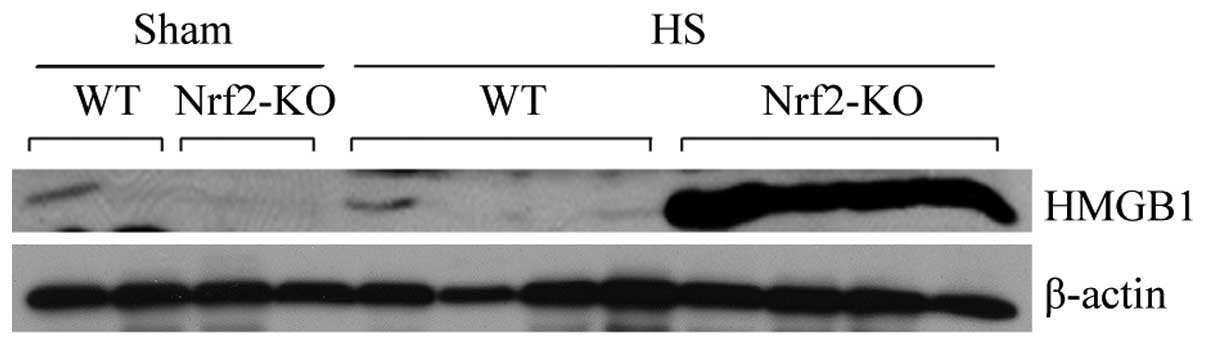
(Fig. 5). On the other hand,
HMGB1, a cytokine and alarmin molecule, has been characterized as a
crucial early mediator of inflammation after HS and organ
ischemia/reperfusion (24). Thus,
in the present study, we also examined the expression of HMGB1 in
mouse plasma following HS and a marked induction in HMGB1
expression in the Nrf2-KO group was detected by immunoblot analysis
(Fig. 6). These data suggest that
Nrf2 is a negative mediator for modulating systemic inflammation in
HS.
Nrf2 deficiency augments cytokine
production induced by LPS in peritoneal macrophages following
HS
As the major component of innate immunity,
macrophages play an essential role in the inflammatory response
(23). In this study, to evaluate
the potential anti-inflammatory effects of Nrf2 in HS, mouse
peritoneal macrophages were harvested 24 h after resuscitation and
then cultured with LPS for a further 6 h. The levels of cytokine
production in the supernatant were subsequently determined using
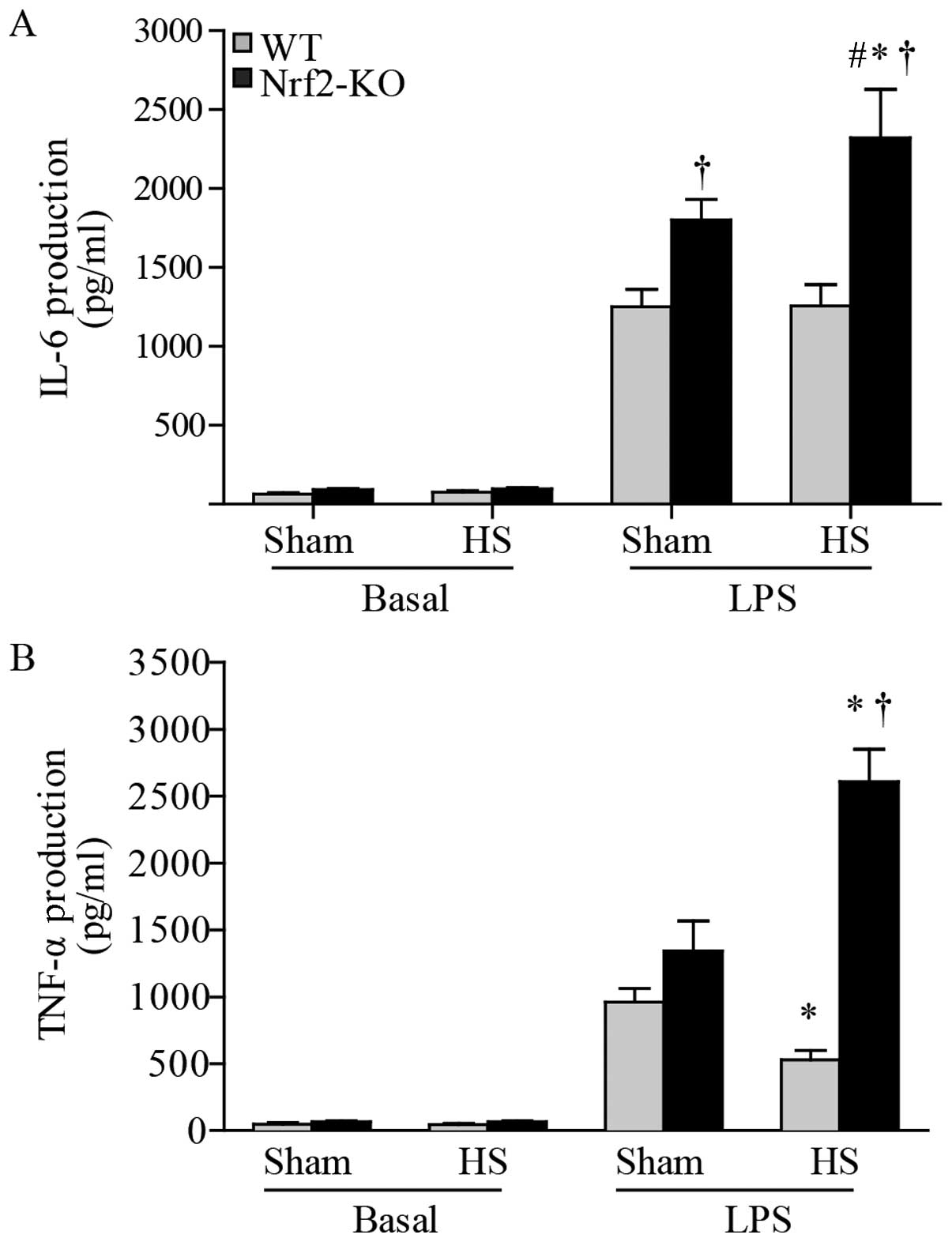
ELISA. The results revealed an undetectable level of IL-1β (data
not shown), whereas there was an abundant level of IL-6 and TNF-α
in the peritoneal macrophages. At the basal level, the macrophages
produced similarly low levels of IL-6 and TNF-α in the cells from
both the WT and Nrf2-KO mice. Following exposure to LPS, the level
of IL-6 was significantly higher in the macrophages from the
Nrf2-KO mice compared with those from the WT mice, even in the
sham-operated groups. However, when making comparisons between the
same mouse strains in the 2 groups (HS group and sham-operated
group), we found that IL-6 production was significantly increased
only in the macrophages from the Nrf2-KO mice with HS compared to
those from the Nrf2-KO mice in the sham-operated group; there were
no significant differences observed in the IL-6 levels between the
WT mice in the HS and sham-operated groups (Fig. 7A). Notably, a decrease in TNF-α
levels induced by LPS was detected in the macrophages from the WT
mice subjected to HS, whereas there was an increase in these levels
in the cells from the Nrf2-KO mice (Fig. 7B). Thus, Nrf2 deficiency resulted
in the increased production of inflammatory cytokines in the
LPS-exposed macrophages from mice subjected to HS.
Discussion
HS is a medical emergency, characterized by the
sudden major loss of intravascular volume, which may cause death
due to massive blood loss. Even though fluid resuscitation may
restore circulatory failure, it also increases the severity of
ischemia/reperfusion injury, which induces multiple organ
dysfunction and progressive multiple organ failure (25,26). Under these circumstances,
overwhelming oxidative stress is the major consideration, resulting
from failed mitochondrial aerobic metabolism and excessive NADPH
oxidase-dependent ROS generation in the cytoplasm (27–29). However, the precise regulatory
mechanisms responsible for oxidative stress-mediated HS injury
remain largely unknown. In the present study, clinical analysis
demonstrated significantly increased Nrf2 levels in the patients
with surgery-associated hemorrhage subjected to resuscitation
treatment, and the Nrf2 levels increased in a time-dependent manner
in these patients. These results suggest that there is a
correlation between Nrf2 expression and the development of HS.
Based on these findings, as well as the results of a previous study
investigating the role of Nrf2 in the pathogenesis of sepsis
(15), we hypothesized that Nrf2,
the critical transcriptional factor mediating antioxidant
signaling, plays an important role in inflammatory injury following
HS. To this end, Nrf2-KO mice with WT mice being used as controls
were employed in order to establish an animal model of HS.
Histological analysis revealed that Nrf2 deficiency resulted in
more severe liver and lung injury; the liver and lungs are major
organs that are very responsive to ischemia/reperfusion during HS
(30,31). Thus, these findings indicate that
Nrf2 potentially exerts protective effects against organ injury
following HS.
It is well known that the ROS-mediated activation of
the innate immune response is the dominant modulator of multiple
organ failure and even mortality due to HS (32,33). Excess levels of intracellular and
extracellular ROS induce the hyper-activation of innate immune
cells (34,35). In this study, to determine whether
Nrf2 is involved in this process, we further investigated the
activation of neutrophils and macrophages in Nrf2-KO mice compared
with WT mice; both were subjected to HS. Nrf2 deficiency enhanced
MPO-1-labeled neutrophil activation in vivo, as well as
LPS-induced macrophage activation ex vivo. Additionally, the
production of classical innate inflammatory cytokines, namely IL-6,
IL-1β and TNF-α, was highly elevated in the Nrf2-KO group, both in
the blood and in local organs. Under these circumstances,
neutrophils and macrophages are the major sources of these
pro-inflammatory cytokines, as we confirmed. Such results further
support the negative effects of Nrf2 on the augmentation of the
innate immune response. On the other hand, the development of
systemic inflammatory response syndrome during HS is characterized
by the excessive production of these pro-inflammatory cytokines,
which results in end-organ injury. Animal and clinical studies have
demonstrated the key pathological event of the imbalance between
pro-inflammatory and anti-inflammatory cytokine production for the
progression of organ failure in HS (36,37). Of note, the downregulation of
pro-inflammatory cytokines in the early pathological process of HS
is of utmost importance. Concerning our results, the constitutive
protective function of Nrf2 in inflammation following HS may
therefore be attributed to the suppression of pro-inflammatory
cytokine production at the initial stage of the immune
response.
Notably, the expression of circulatory HMGB1 was
markedly upregulated in the absence of Nrf2 in mice. HMGB1 was
originally identified as a nuclear DNA-binding protein and
subsequently as a novel late-acting inflammatory cytokine, which
mediates lethality in sepsis (38,39). HMGB1 has been identified as an
inducible alarmin molecule for cell injury at the early stages of
inflammation. During HS ischemia/reperfusion, HMGB1 has been shown
to be highly activated and released in neutrophils and macrophages,
acting as an inflammatory mediator responsible for acute lung and
liver injury (40,41). In a clinical setting, increased
concentrations of HMGB1 have been detected in the serum of patients
with HS and its neutralization may improve related clinical
outcomes (42,43). Therefore, the inhibitory effects
of Nrf2 on HMGB1 expression may contribute to another regulatory
mechanism of Nrf2 in organ injury following HS.
In conclusion, the present study demonstrated that
Nrf2 played a negative role in the activation of the innate immune
response during HS by targeting pro-inflammatory cytokine
production and HMGB1 expression, thereby inhibiting
inflammation-associated organ injury. Thus, Nrf2 is an important
regulator in protecting major organs from ischemia/reperfusion
damage in HS. The modulation of systemic Nrf2 expression and
activity may represent a novel strategy for the treatment of
HS-related disease.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81000138, 81270357,
81422005 and 31470057), the Zhejiang Provincial Natural Science
Foundation of China (grant no. LR14H020002), the Medical Science
Research Foundation of Zhejiang Province (grant no. 2012KYB102),
the Traditional Chinese Medicine Foundation of Zhejiang Province
(grant no. 2012ZA083) and the Fundamental Research Funds for the
Central Universities. We would also like to thank Dr Rajesh K.
Thimmulappa and Dr Shyam Biswal (Johns Hopkins University) for
providing extensive assistance with the current study.
References
|
1
|
Jarrar D, Chaudry IH and Wang P: Organ
dysfunction following hemorrhage and sepsis: mechanisms and
therapeutic approaches (Review). Int J Mol Med. 4:575–583.
1999.PubMed/NCBI
|
|
2
|
McGhan LJ and Jaroszewski DE: The role of
toll-like receptor-4 in the development of multi-organ failure
following traumatic haemorrhagic shock and resuscitation. Injury.
43:129–136. 2012. View Article : Google Scholar
|
|
3
|
Visser T, Pillay J, Koenderman L and
Leenen LP: Postinjury immune monitoring: can multiple organ failure
be predicted? Curr Opin Crit Care. 14:666–672. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heckbert SR, Vedder NB, Hoffman W, Winn
RK, Hudson LD, Jurkovich GJ, Copass MK, Harlan JM, Rice CL and
Maier RV: Outcome after hemorrhagic shock in trauma patients. J
Trauma. 45:545–549. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hurt RT, Zakaria R, Matheson PJ, Cobb ME,
Parker JR and Garrison RN: Hemorrhage-induced hepatic injury and
hypoperfusion can be prevented by direct peritoneal resuscitation.
J Gastrointest Surg. 13:587–594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kan WH, Hsu JT, Schwacha MG, Choudhry MA,
Raju R, Bland KI and Chaudry IH: Selective inhibition of iNOS
attenuates trauma-hemorrhage/resuscitation-induced hepatic injury.
J Appl Physiol 1985. 105:1076–1082. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsutani T, Kang SC, Miyashita M,
Sasajima K, Choudhry MA, Bland KI and Chaudry IH: Liver cytokine
production and ICAM-1 expression following bone fracture, tissue
trauma, and hemorrhage in middle-aged mice. Am J Physiol
Gastrointest Liver Physiol. 292:G268–G274. 2007. View Article : Google Scholar
|
|
8
|
Botha AJ, Moore FA, Moore EE, Kim FJ,
Banerjee A and Peterson VM: Post injury neutrophil priming and
activation: an early vulnerable window. Surgery. 118:358–364. 1995.
View Article : Google Scholar
|
|
9
|
Hogg JC: Neutrophil kinetics and lung
injury. Physiol Rev. 67:1249–1295. 1987.PubMed/NCBI
|
|
10
|
Kensler TW, Wakabayashi N and Biswal S:
Cell survival responses to environmental stresses via the
Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 47:89–116.
2007. View Article : Google Scholar
|
|
11
|
Cho HY, Reddy SP, Yamamoto M and
Kleeberger SR: The transcription factor NRF2 protects against
pulmonary fibrosis. FASEB J. 18:1258–1260. 2004.PubMed/NCBI
|
|
12
|
Rangasamy T, Guo J, Mitzner WA, Roman J,
Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN and
Biswal S: Disruption of Nrf2 enhances susceptibility to severe
airway inflammation and asthma in mice. J Exp Med. 202:47–59. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rangasamy T, Cho CY, Thimmulappa RK, Zhen
L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM and
Biswal S: Genetic ablation of Nrf2 enhances susceptibility to
cigarette smoke-induced emphysema in mice. J Clin Invest.
114:1248–1259. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Osburn WO, Karim B, Dolan PM, Liu G,
Yamamoto M, Huso DL and Kensler TW: Increased colonic inflammatory
injury and formation of aberrant crypt foci in Nrf2-deficient mice
upon dextran sulfate treatment. Int J Cancer. 121:1883–1891. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thimmulappa RK, Lee H, Rangasamy T, Reddy
SP, Yamamoto M, Kensler TW and Biswal S: Nrf2 is a critical
regulator of the innate immune response and survival during
experimental sepsis. J Clin Invest. 116:984–995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Malhotra D, Thimmulappa R, Navas-Acien A,
Sandford A, Elliott M, Singh A, Chen L, Zhuang X, Hogg J, Pare P,
et al: Decline in NRF2-regulated antioxidants in chronic
obstructive pulmonary disease lungs due to loss of its positive
regulator, DJ-1. Am J Respir Crit Care Med. 178:592–604. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kumawat M, Sharma TK, Singh I, Singh N,
Ghalaut VS, Vardey SK and Shankar V: Antioxidant enzymes and lipid
peroxidation in type 2 diabetes mellitus patients with and without
nephropathy. N Am J Med Sci. 5:213–219. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Itoh K, Chiba T, Takahashi S, Ishii T,
Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et
al: An Nrf2/small Maf heterodimer mediates the induction of phase
II detoxifying enzyme genes through antioxidant response elements.
Biochem Biophys Res Commun. 236:313–322. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hsieh CH, Frink M, Hsieh YC, Kan WH, Hsu
JT, Schwacha MG, Choudhry MA and Chaudry IH: The role of MIP-1
alpha in the development of systemic inflammatory response and
organ injury following trauma hemorrhage. J Immunol. 181:2806–2812.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gray KD, Simovic MO, Blackwell TS,
Christman JW, May AK, Parman KS, Chapman WC and Stain SC:
Activation of nuclear factor kappa B and severe hepatic necrosis
may mediate systemic inflammation in
choline-deficient/ethionine-supplemented diet-induced pancreatitis.
Pancreas. 33:260–267. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vnukov VV, Gutsenko OI, Milutina NP,
Kornienko IV, Ananyan AA, Danilenko AO, Panina SB, Plotnikov AA and
Makarenko MS: Influence of SkQ1 on expression of Nrf2 gene,
ARE-controlled genes of antioxidant enzymes and their activity in
rat blood leukocytes. Biochemistry (Mosc). 80:1598–1605. 2015.
View Article : Google Scholar
|
|
22
|
Morzadec C, Macoch M, Sparfel L,
Kerdine-Römer S, Fardel O and Vernhet L: Nrf2 expression and
activity in human T lymphocytes: stimulation by T cell receptor
activation and priming by inorganic arsenic and
tert-butylhydroquinone. Free Radic Biol Med. 71:133–145. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rao DA and Pober JS: Endothelial injury,
alarmins, and allograft rejection. Crit Rev Immunol. 28:229–248.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang D, Tewary P, de la Rosa G, Wei F and
Oppenheim JJ: The alarmin functions of high-mobility group
proteins. Biochim Biophys Acta. 1799:157–163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Reynolds PS, Barbee RW, Skaflen MD and
Ward KR: Low-volume resuscitation cocktail extends survival after
severe hemorrhagic shock. Shock. 28:45–52. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barbee RW, Reynolds PS and Ward KR:
Assessing shock resuscitation strategies by oxygen debt repayment.
Shock. 33:113–122. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gutierrez G, Reines HD and Wulf-Gutierrez
ME: Clinical review: hemorrhagic shock. Crit Care. 8:373–381. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fan J, Frey RS and Malik AB: TLR4
signaling induces TLR2 expression in endothelial cells via
neutrophil NADPH oxidase. J Clin Invest. 112:1234–1243. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Imai Y, Kuba K, Neely GG,
Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen
R, Leung YH, Wang H, et al: Identification of oxidative stress and
Toll-like receptor 4 signaling as a key pathway of acute lung
injury. Cell. 133:235–249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gill R, Ruan X, Menzel CL, Namkoong S,
Loughran P, Hackam DJ and Billiar TR: Systemic inflammation and
liver injury following hemorrhagic shock and peripheral tissue
trauma involve functional TLR9 signaling on bone marrow-derived
cells and parenchymal cells. Shock. 35:164–170. 2011. View Article : Google Scholar
|
|
31
|
Song Z, Zhao X, Liu M, Jin H, Wang L, Hou
M and Gao Y: Recombinant human brain natriuretic peptide attenuates
trauma-/haemorrhagic shock-induced acute lung injury through
inhibiting oxidative stress and the NF-κB-dependent
inflammatory/MMP-9 pathway. Int J Exp Pathol. 96:406–413. 2015.
View Article : Google Scholar
|
|
32
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Krause KH and Bedard K: NOX enzymes in
immuno-inflammatory pathologies. Semin Immunopathol. 30:193–194.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lorne E, Zmijewski JW, Zhao X, Liu G,
Tsuruta Y, Park YJ, Dupont H and Abraham E: Role of extracellular
superoxide in neutrophil activation: interactions between xanthine
oxidase and TLR4 induce proinflammatory cytokine production. Am J
Physiol Cell Physiol. 294:C985–C993. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mitra S and Abraham E: Participation of
superoxide in neutrophil activation and cytokine production.
Biochim Biophys Acta. 1762:732–741. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu LM and Dubick MA: Hemorrhagic
shock-induced vascular hyporeactivity in the rat: relationship to
gene expression of nitric oxide synthase, endothelin-1, and select
cytokines in corresponding organs. J Surg Res. 125:128–136. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ertel W, Keel M, Neidhardt R, Steckholzer
U, Kremer JP, Ungethuem U and Trentz O: Inhibition of the defense
system stimulating interleukin-12 interferon-gamma pathway during
critical illness. Blood. 89:1612–1620. 1997.PubMed/NCBI
|
|
38
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMG-1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang H, Ochani M, Li J, Qiang X, Tanovic
M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, et al:
Reversing established sepsis with antagonists of endogenous
high-mobility group box 1. Proc Natl Acad Sci USA. 101:296–301.
2004. View Article : Google Scholar :
|
|
40
|
Tsung A, Sahai R, Tanaka H, Nakao A, Fink
MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA and Billiar TR:
The nuclear factor HMGB1 mediates hepatic injury after murine liver
ischemia-reperfusion. J Exp Med. 201:1135–1143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim JY, Park JS, Strassheim D, Douglas I,
Diaz del Valle F, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama
I, et al: HMGB1 contributes to the development of acute lung injury
after hemorrhage. Am J Physiol Lung Cell Mol Physiol.
288:L958–L965. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Andersson U, Erlandsson-Harris H, Yang H
and Tracey KJ: HMGB1 as a DNA-binding cytokine. J Leukoc Biol.
72:1084–1091. 2002.PubMed/NCBI
|
|
43
|
Ombrellino M, Wang H, Ajemian MS, Talhouk
A, Scher LA, Friedman SG and Tracey KJ: Increased serum
concentrations of high-mobility-group protein 1 in haemorrhagic
shock. Lancet. 354:1446–1447. 1999. View Article : Google Scholar : PubMed/NCBI
|