Introduction
Spontaneous subarachnoid hemorrhage (SAH) is a very
common cerebrovascular condition and affects 9/100,000 in the
Western world. Although it accounts for only 5% of all strokes, it
is a devastating neurological condition with generally poor outcome
and high levels of mortality and morbidity (1–3).
Early brain injury (EBI), acute hydrocephalus, delayed cerebral
vasospasm (CVS) and cerebral infraction play an important role in
poor prognosis after SAH. Even though a large number of previous
studies on SAH have focused mainly on delayed CVS (7,10,12,13,18,22), Laskowitz et al (4) found that the functional outcome was
not improved even if the angiographic vasospasm was reversed.
MacDonald et al (5)
confirmed that clazosentan, an endothelial receptor antagonist,
ameliorated CVS after SAH significantly, but it did not improve the
outcome.
EBI occurs within 72 h after SAH, Sehba et al
(6) have reported that it is more
common than CVS in cases with poor outcomes. The pathophysiological
mechanisims of EBI after SAH include increased intracranial
pressure, oxidative stress leading to inflammation, blood-brain
barrier (BBB) disruption and cerebral ischemia. All of these
factors lead to neuronal cell death and brain edema, and brain
edema also increases intracranial pressure and aggravates EBI
(7–9). Certain studies have shown that EBI,
which manifests as early cortical feedback depolarization waves,
cortical spreading depression and impaired neurovascular coupling,
plays a crucial important role in neurological deterioration after
SAH (10–12).
Statins, inhibitors of the
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, are
widely used in cardiovascular medicine as cholesterol-lowering
drugs and it has also been suggested to exert pleiotropic effects,
including exerting anti-inflammatory (13), anti-oxidative stress (14), and anti-CVS effects (7,15),
as well as inhibiting platelet aggregation (16,17). However, the full effect and
specific pathophysiological mechanisms of statins on EBI after SAH
remain not fully understood. Atorvastatin has been shown to act as
a potent statin and inhibitor of HMG-CoA, and Cheng et al
(7) have reported that
atorvastatin exerted neuroprotective- and anti-CVS effects in
experimental rat models of SAH.
The aim of the present study was to evaluate the
effect of atorvastatin on SAH-induced EBI, and then to demonstrate
that atorvastatin attenuates EBI and brain edema in experimentally
induced SAH via the inhibition of aquaporin 4 (AQP4)
expression.
Materials and methods
Animals and drugs
The animals used and the care protocols were
approved by the Animal Care and Use Committee of Anhui Medical
University (Wuxi, China) and all experiments conformed to the Guide
for the Care and Use of Laboratory Animals by the National
Institute of Health. All 72 New Zealand white rabbits used in the
present study were purchased from the Animal Central of Taihu
Hospital (Wuxi, China). They were raised in a comfortable room with
normal levels of atmospheric moisture and fed with a standard diet
at the Animal Center of Taihu Hospital (Wuxi, China) for 10 days
before the experiments. The temperature of the feeding room and
operation room was maintained at approximately 22°C. We chose a
dosage of 20 mg/kg/day atorvastatin. Atorvastatin was orally
administered by gastric gavage once daily for 3 days before and
also at 22 h after SAH to maintain drug levels. We assessed the
neurologic deficits at 24 h after SAH and rabbits were sacrificed
immediately after the neurological evaluation. Atorvastatin was
obtained from Pfizer (Jiangsu, China).
Experimental design
All adult male New Zealand white rabbits weighing
between 2.5 and 3.2 kg were assigned randomly to three groups: i)
Sham-operated group (n=24), ii) SAH group (n=24), and the SAH +
atorvastatin group (n=24). For the rabbits in the SAH +
atorvastatin group (n=24), atorvastatin (20 mg/kg/day) (7) was administered immediately after the
first blood injection and was continued every 24 for 72 h. The
sham-operated rabbits underwent the same operation to induce SAH as
the SAH group, but the sham-operated group was injected with saline
solution. All rabbits were sacrificed on day 3. Eight rabbits in
each group were sacrificed using the perfusion-fixation method. The
hippocampus was collected for terminal deoxynucleotidyl
transferase-mediated dUTP nick end labelling (TUNEL) staining.
Eight rabbits were sacrificed in order to evaluate the brain water
content. The other eight rabbits were exsanguinated and decollated
in each group. The brain tissue was removed and frozen in a deep
cryogenic refrigerator for biochemical studies. Before rabbits were
sacrificed, serums were sampled in two copies to detect
endothelin-1 (ET-1).
SAH model
Experimental SAH was induced according to the
two-hemorrhage model, as previously described, using rabbits
(7). The rabbits were
anesthetized via auricular marginal vein injection of 10% chloral
hydrate (2.5 ml/kg). Life signs were maintained at a stable level,
and we inserted a 23-gauge butterfly needle into the cisterna
magna. After 1 ml cerebrospinal fluid (CSF) was released, then 2 ml
non-heparinized fresh autologous auricular artery blood was
injected into the cisterna magna for 1 min under strict aseptic
conditions. To make blood flow easily from the cisterna magna to
the basilar cistern, all rabbits were kept in a 30° head-down
position for 30 min. They were returned to the feeding room after
recovering from anesthesia. The second injection was administered
after 48 h in the same manner as the first.
Behavior scoring
All rabbits behavior scores were recorded by the
same independent observer who was blinded to the study. We used a
previously modified scoring table (Table I) to evaluate the neurological
function every day, as previously described (18).
 | Table IBehavior scores. |
Table I
Behavior scores.
| Category | Behavior | Score |
|---|
| Appetite | Finished meal | 0 |
| Left meal
unfinished | 1 |
| Scarcely ate | 2 |
| Activity | Active, squeaking
or standing | 0 |
| Lying down, will
stand and walk with some stimulation | 1 |
| Almost always lying
down | 2 |
| Deficits | No deficits | 0 |
| Unable to walk due
to ataxia or paresis | 1 |
| Impossible to walk
and stand due to ataxia and paresis | 2 |
Measuring brain water content
The entire brain was removed at 72 h and weighed
immediately (wet weight) and again after being dried in an oven at
100°C for 24 h (dry weight). The percentage of brain water content
was calculated as follows: (wet weight-dry weight)/wet weight ×
100%. The number of rabbits used for this in each group was SAH +
atorvastatin group (n=8), SAH group (n=8) and sham-operated group
(n=8).
Perfusion-fixation
Eight rabbits in each group scheduled for sacrifice
were anesthetized with an injection of 10% chloral hydrate (4
ml/kg) to the uricular marginal veins. The chests of the rabbits
were quickly opened for intubation with a cannula in the left
ventricle, and the right atrium was opened. Perfusion began with
1,500 ml physiological phosphate buffer solution (0.01 M = PBS, pH
7.3) at 37°C, and was then followed by 1,000 ml 10% buffered
formaldehyde under 120 cm H2O perfusion pressure. After
perfusion, the whole brain and the brain tissue were removed and
stored in formalin.
TUNEL staining and cell counting
A TUNEL staining kit (Roche Inc., Basel,
Switzerland) was used to stain brain sections; the TUNEL-positive
cells were indicated by fluorescein-dUTP with dNTP or peroxidase
(POD) with 3–3′ diaminobenzidine (DAB). This was undertaken
according to the manufacturer's instructions for the in situ
Apoptosis Detection kit (Roche Inc., Mannheim, Germany) as
previously described (18). The
negative control was similarly performed but TUNEL reaction mixture
was omitted. Cells exhibiting nuclear condensation/fragmentation
and apoptotic bodies in the absence of cytoplasmic TUNEL reactivity
and brown staining of nuclei were considered apoptotic cells.
Apoptotic cells were confirmed with the help of a pathologist
blinded to the grouping. The number of TUNEL-positive cells in each
region was counted in a high-powered field (×400) by an
investigator who was blinded to the studies, and expressed as
number/mm2. The number of animals used in each group was
as follows: SAH + atorvastatin (n=8), SAH (n=8), and sham-operated
(n=8).
Enzyme-linked immunosorbent assay
(ELISA)
At day 3 after surgical intervention, blood samples
were collected from anesthetized animals, and analyzed for ET-1
expression levels using an ELISA kit (Abcam, Cambridge, UK)
specific for rabbits. To collect the plasma, the homogenates were
centrifuged at 3,000 g/min for 15 min, and the supernatant was
assayed for the protein concentration of ET-1 (Abcam), in
accordance with the manufacturer's instructions. The concentrations
(pg/ml) were determined based on a standard curve, prepared using a
known set of serial dilutions of standard proteins by BCA assay.
The number of animals used in the ELISA and histological study are
as follows: sham-operated group (n=8), SAH group (n=8), and SAH +
atorvastatin group (n=8).
Western blot analysis
The method of western blot analysis for evaluating
AQP4 and caspase-3 has been described previously (19). The samples (20 μg total
protein) were separated by sodium dodecyl sulfate polyacrylamide
gel for electrophoresis with 10% polyacrylamide gel. The following
primary antibodies were used: rabbit anti-AQP4 (1:2,000; ab46182)
and anti-caspase-3 (1:500; ab2171) antibody (Abcam). The
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (diluted in
1:6,000; Sigma-Aldrich, Inc., St. Louis, MO, USA) was used as a
loading control. After incubation with the primary antibodies, the
nitrocellulose membranes were washed with 0.01 M TBST dilution and
incubated with appropriate horseradish peroxidase-labeled secondary
antibodies (1:1,000; Santa Cruz Biotechnology Inc., Santa Cruz, CA,
USA) using 1% non-fat milk in TBST for 1 h at room temperature.
After two rinses and four washes with TBST, the membranes were
incubated in ECL (Amersham, Little Chalfont, UK) reagent for HRP
(60 sec) and exposed to autoradiography film for visualization of
the bands. The results were quantified using Quantity One Software
(Bio-Rad Laboratories, Hercules, CA, USA). The number of animals
used in each group was SAH + atorvastatin (n=8), SAH (n=8) and Sham
(n=8).
Statistical analysis
All data are presented as the means ± SD. SPSS 14.0
(Anhui Medical University, SPSS Inc., Anhui, China) was used for
statistical analysis of the data. Differences between the two
groups were analyzed using a two-tailed unpaired Student's t-test.
The differences among multiple groups were assessed using one-way
analysis of variance (one way ANOVA). Ranked data between the two
groups were evaluated using the rank sum test. A P-value <0.05
was considered to indicate a statistically significant
difference.
Results
General observations
In the interval and by the end of the experiment,
there was no obvious difference in blood pressure, injected
arterial blood gas data, body weight and blood pressure. The
mortality of the SAH group was 33.3% (8/24), 20.8% (5/24) in the
SAH + atorvastatin treated group and none in the sham-operated
group (0/24). The mortality of SAH + atorvastatin treated group was
significantly lower than in the SAH group (P<0.05). In the
process of constructing the model, we removed the rabbits which
died, or the rabbits which dif not meet the requirements, and added
new rabbits randomly to ensure the number of animals was maintained
in each group (all data not shown).
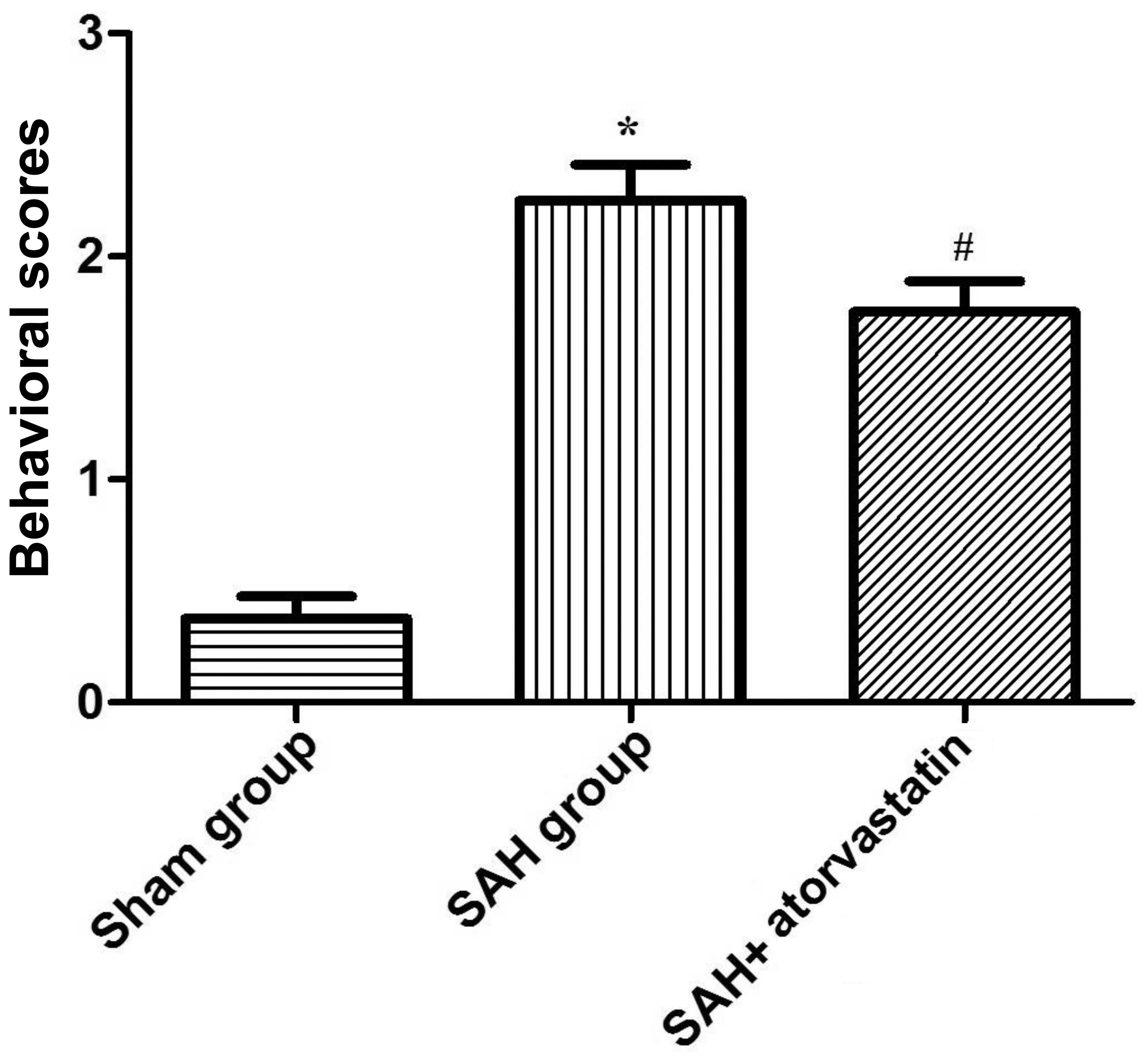
Behavior scoring
The behavior scores of rabbits in the SAH group and
SAH + atorvastatin group were both significantly higher than the
sham-operated group (P<0.01; according to ANOVA), but the
behavior scores in the SAH + atorvastatin group was significantly
lower (P<0.05; ANOVA) (Fig. 1)
than that in the SAH group after 72 h. Thus, our results showed
that atorvastatin improves neurological functional after SAH in
experimental rabbits.
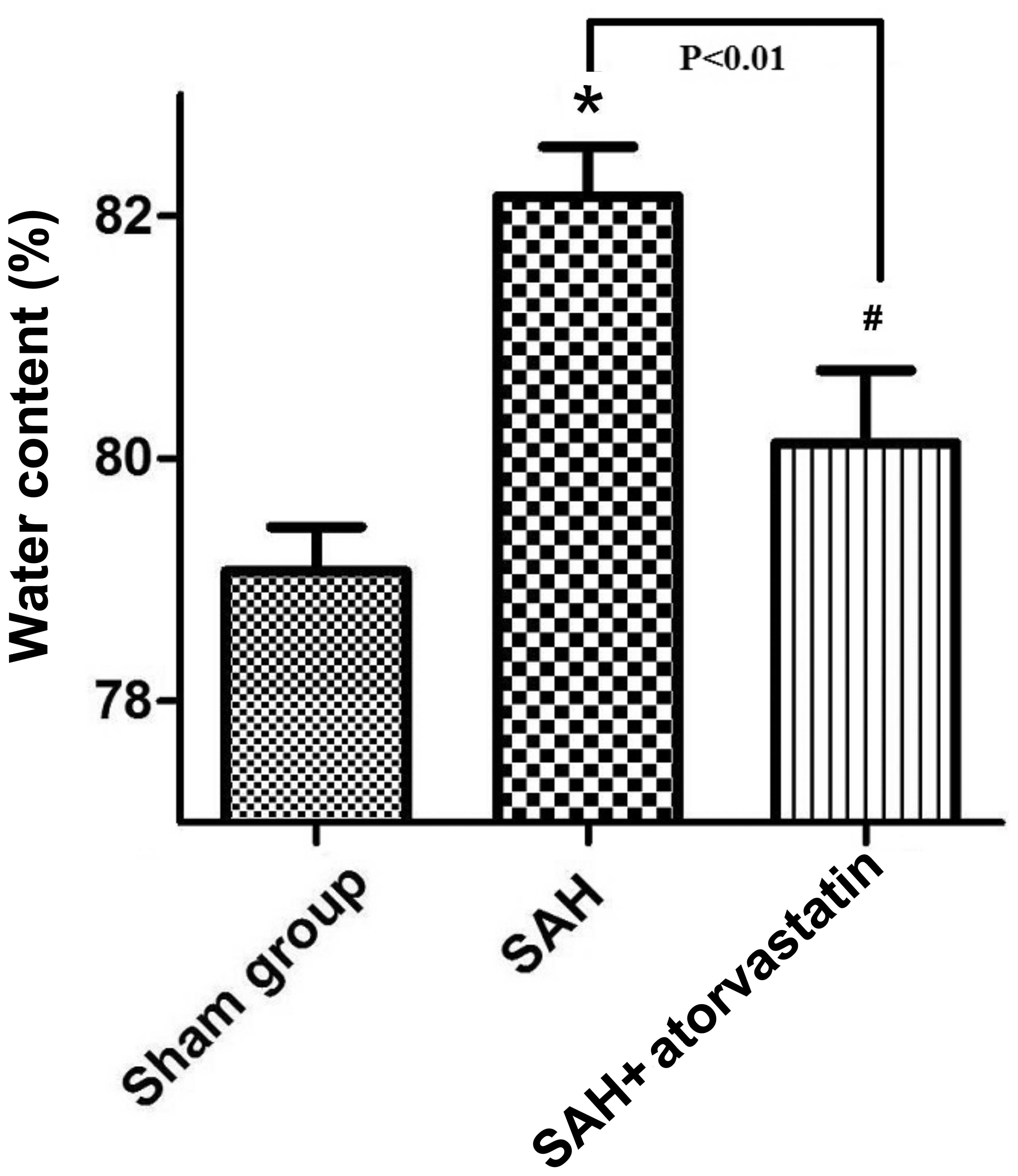
Brain water content
The brain water content of the SAH + atorvastatin
group and SAH group all significantly increased (80.130.60 vs.
79.080.36, P<0.05; 82.160.41 vs. 79.080.36, P<0.05) compared
to the sham-operated group at 72 h after SAH. Brain water content
was decreased significantly by atorvastatin treatment as compared
with that of the SAH group (80.130.60 vs. 82.160.41, P<0.05)
(Fig. 2).
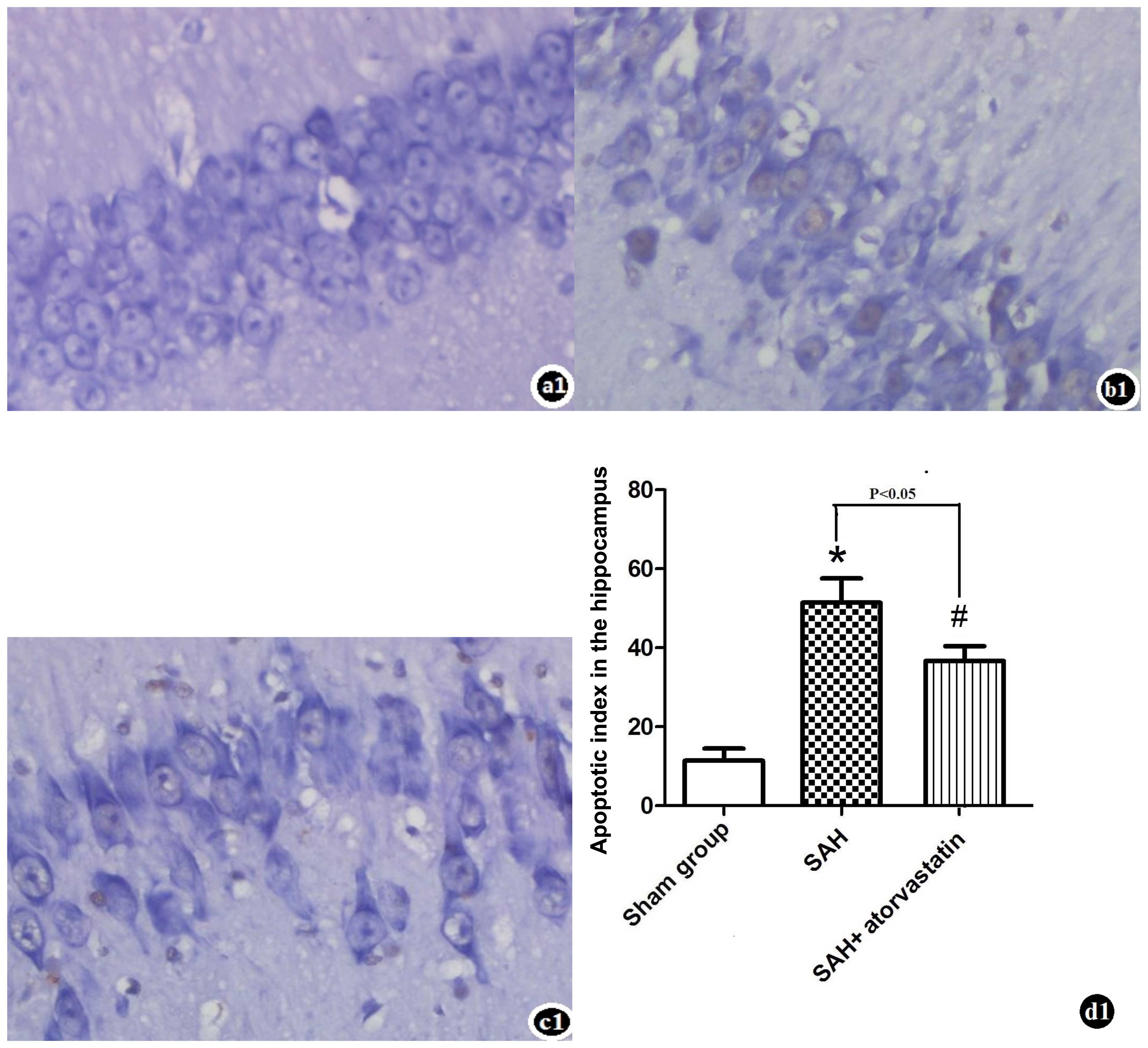
TUNEL staining and cell death assay
There were almost no TUNEL-positive cells detected
in the sham-operated group animals. TUNEL-positive cells were
significantly increased in the hippocampus of rabbits at 72 h after
SAH. TUNEL-positive cells significantly decreased in the SAH +
atorvastatin treatment group (P<0.05) (Fig. 4).
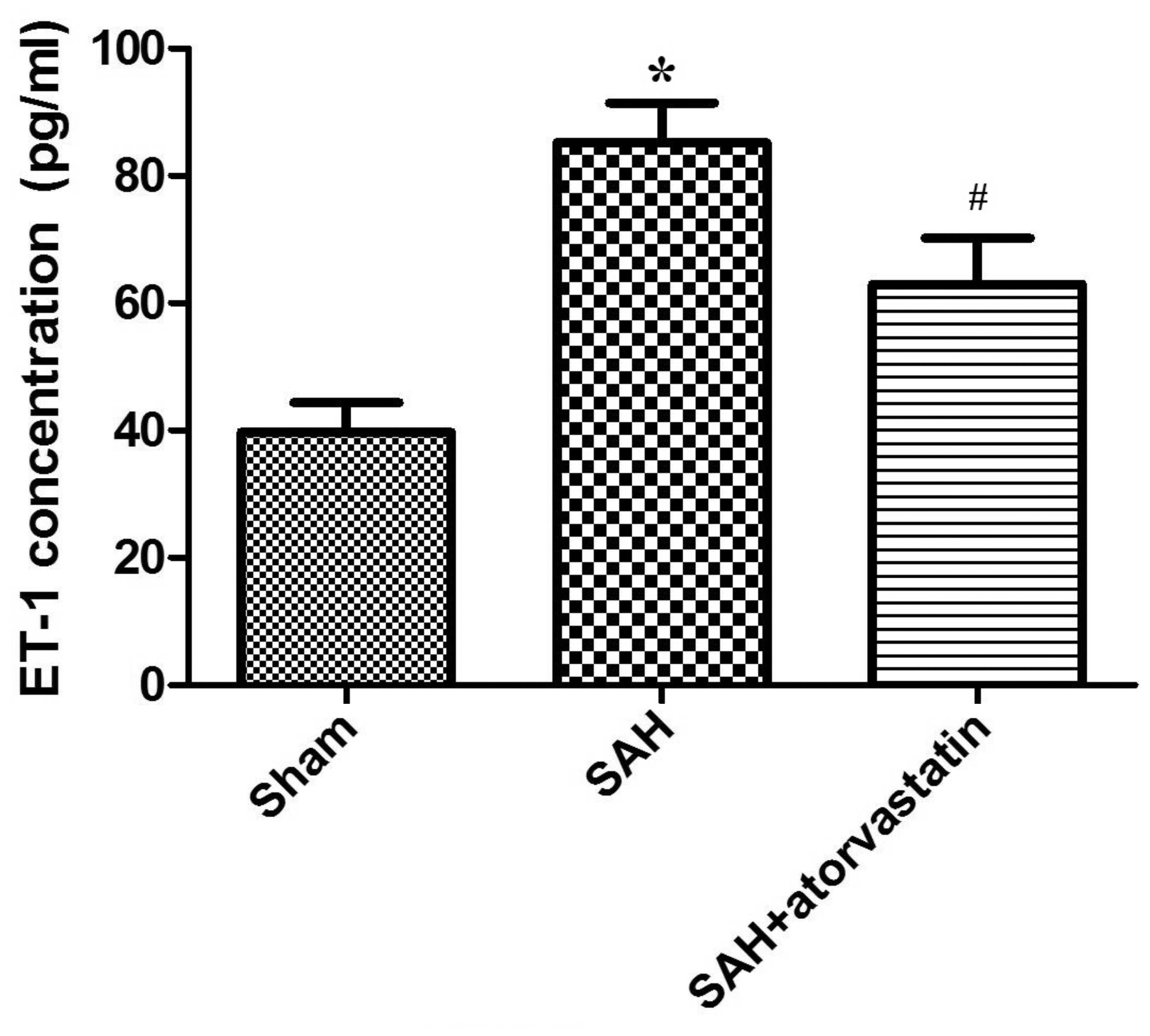
Protein ET-1 expression
In this study, we performed ELISA to examine the
changes in protein expression of the vasoconstrictor ET-1, to
determine the effect of atorvastatin on cerebral edema after SAH.
As is shown in Fig. 3, compared
with the sham-operated group, ET-1 expression was markedly
increased in all SAH rabbits (SAH group, 85.24+6.25 vs. 39.72+4.67
pg/ml, P<0.01; SAH + atorvastatin, 62.92±7.27 vs. 39.72±4.67
pg/ml, P<0.01). After atorvastatin treatment, the elevation of
plasma ET-1 concentration was significantly lower than the SAH
group (62.92±7.27 vs. 85.24±6.25 pg/ml, P<0.01) (Fig. 3).
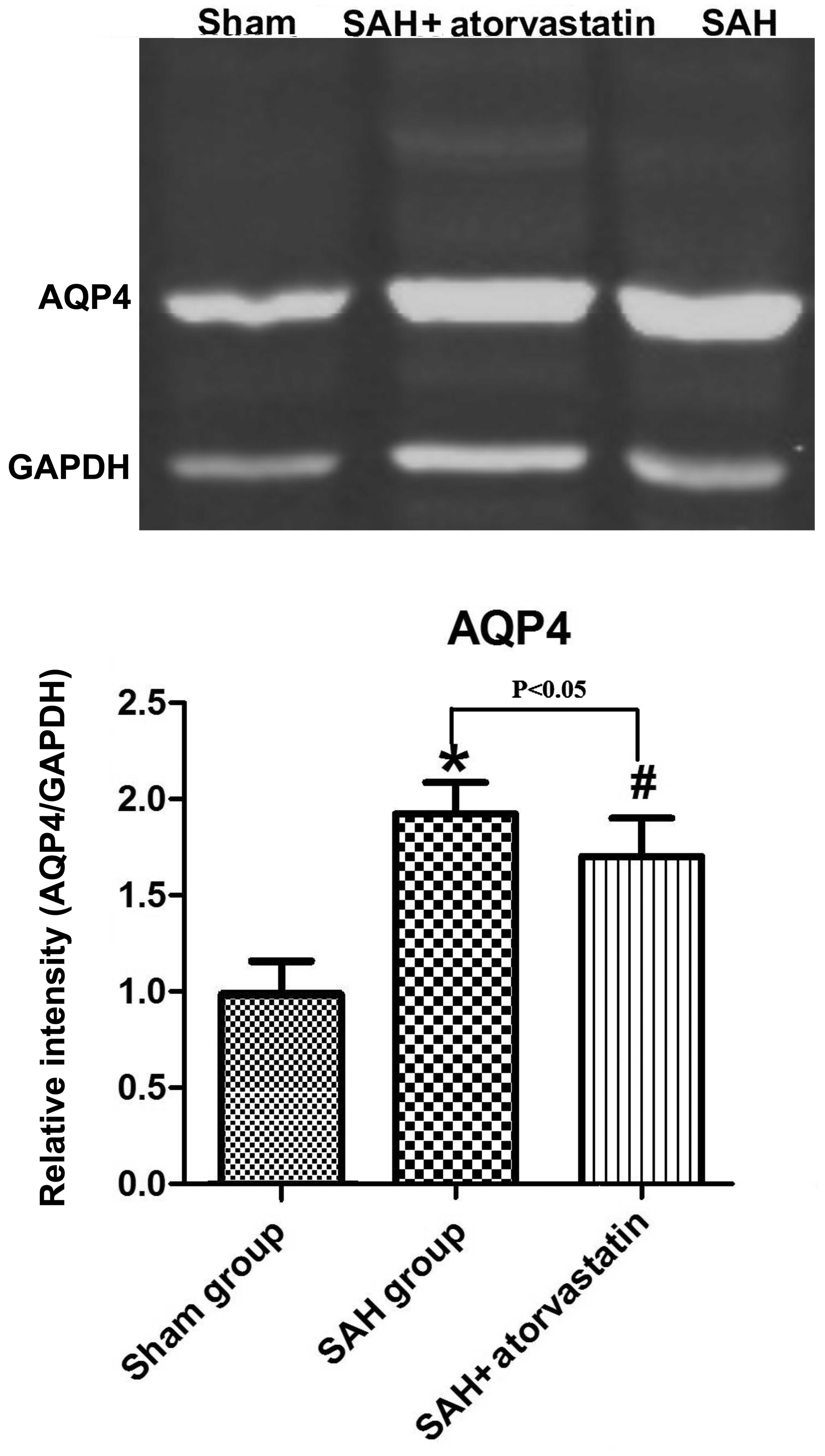
AQP4 protein expression
In the present study, AQP4 protein expression was
evaluated in the brain cortex; the level of cleaved AQP4 was
evaluated by western blot analysis after SAH (P<0.05 vs.
sham-operated group). Atorvastatin clearly reduced the expression
of cleaved AQP4 (P<0.05 vs. SAH rabbits) after SAH (Fig. 5).
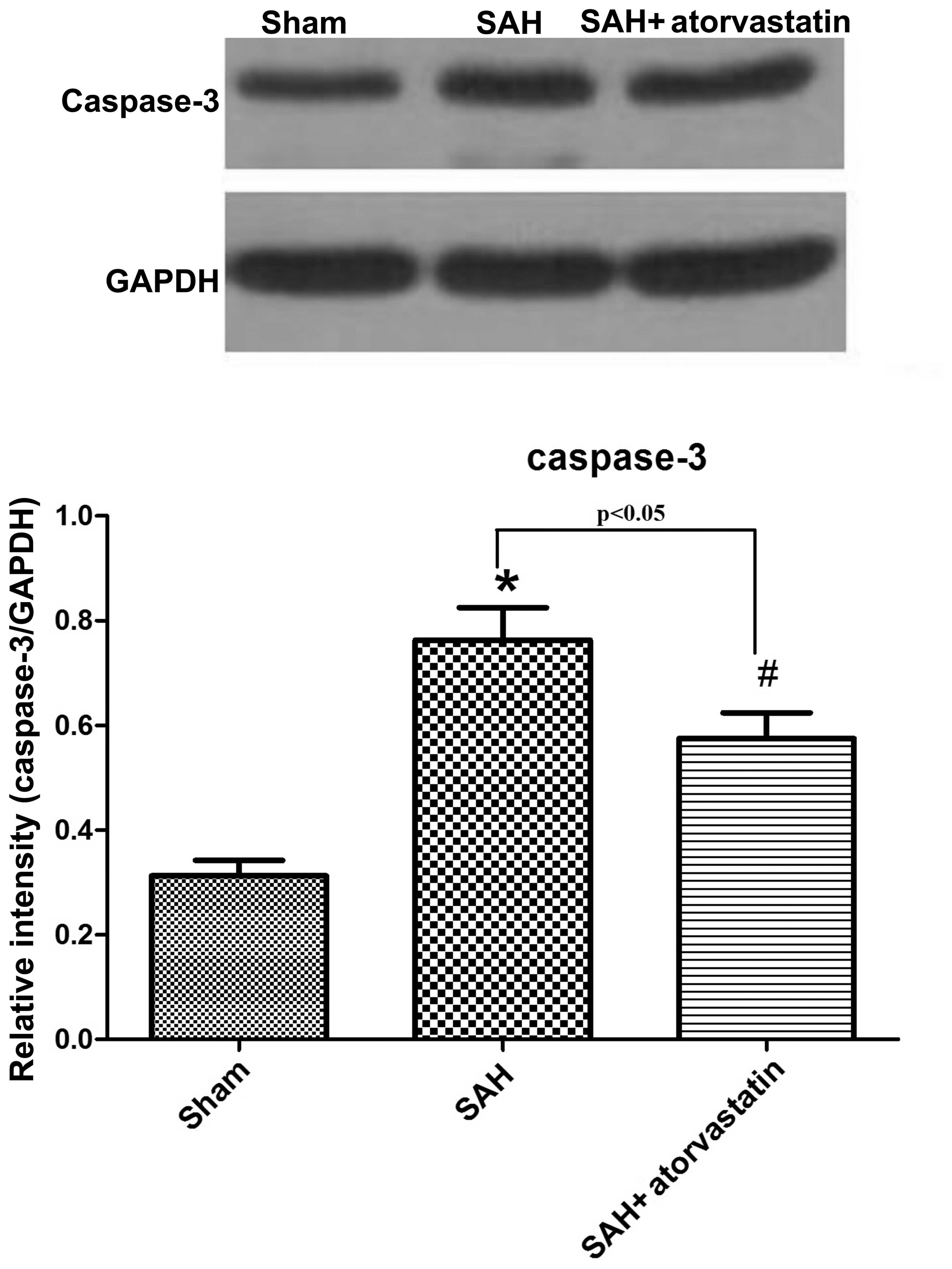
Caspase-3 protein expression
Caspase-3 expression in the hippocampus was detected
by western blot analysis in order to observe neuronal apoptosis at
72 h after SAH. SAH induced a marked increase in caspase-3 in the
hippocampus, whereas the level of caspase-3 decreased markedly in
the SAH + atorvastatin group (P<0.05) (Fig. 6).
Discussion
Certain randomized, controlled clinical trials
(15,20,21) have investigated the effects of
acute statin post-treatment on SAH, and mainly noted delayed CVS
and outcomes. By contrast, previous studies (22–24) have found that the beneficial
effects of statins on functional outcomes and delayed CVS are
controversial, or even invalid. Therefore, the effects of acute
statin post-treatment on SAH were questionable. However, a large
number of experiments (7,9,14,15,25,26) have confirmed that statins prevent
delay CVS, ameliorate EBI and improve the outcome of patients after
SAH. However, the mechanism of EBI remained not fully understood.
In the present study, we demonstrated that atorvastatin ameliorated
EBI and brain edema after experimental SAH. Inhibition of AQP4 was
also observed after treatment with atorvastatin.
EBI is one of the most important causes of delayed
cerebral dysfunction; Broderick et al (27) firstly proposed the concept of EBI
after SAH, and key pathological hallmarks of EBI after SAH were
thought to be neuronal cell death and brain edema. Claassen et
al (28) demonstrated that 8%
of patients had brain edema after bleeding, according to CT
examination, and 12% exhibited brain edema 6 days after bleeding.
Cerebral edema increased intracranial pressure and decreased
cerebral blood flow. The mechanism of brain edema may be related to
its actions: i) high levels of matrix metalloproteinases (MMPs)
play a major role in brain edema. Rosell et al (29) reported that MMP-9 was closely
related to BBB breakdown during reperfusion injury in cases of
human ischemic stroke. Rosenberg and Navratil (30) suggested that the reason for brain
edema was MMP-2 overexpression. Ramos-Fernandez et al
(31) confirmed that the MMP-9
level was associated with the volume of cerebral infarction, the
severity of stroke, hemorrhagic transformation and the functional
outcome after an acute stroke; ii) another mechanism was abnormal
transformation of water molecules, and water molecule channel
proteins: e.g., aquaporins (AQPs) play an important role in brain
edema. AQP4 is a two-way water transfer channel protein, and
distributes to astrocyte, capillary endothelial cells, ependymal
cells and choroid plexus epithelial cells. Papadopoulous and
Verkman (32) demonstrated that
brain edema in AQP4-null mice was significantly less severe than
that of the wild-type mice. Manley et al (33) also found that AQP4-null mice
survived more easily than wild-type mice in the experimental model
of acute water poisoning. Yang et al (34) found that glial cell AQP4
overexpression in transgenic mice accelerated brain edema and brain
swelling. In the present study, we noted that the expression of
AQP4 was higher, and brain edema was more severe in the SAH groups,
and that atorvastatin reduced AQP4 expression and water content of
the brain tissues. Our results indicated that AQP4 was the key to
brain edema, and thus our study provides a new therapeutic target
for brain edema.
BBB plays a vital role in the homeostasis of the
special internal environment in the central nervous system. The
tight junctions of brain microvascular endothelial cells (BMECs)
are important in terms of the core structure in the BBB (35). The tight junctions have many
important physiological functions and proteins, including occludin,
claudins, junction-associated molecules and zonula occludens
(36). Researchers have found
that occludin interacts closely with other tight junction proteins
and they maintain the structure and function of the tight junction
complex through the structural domain. Terry et al (37) confirmed that occludin was closely
related to the barrier function of BMECs. Huber et al
(38) and Persidsky et al
(39) confirmed that
overex-pression of occludin reduced the permeability of the blood
brain barrier. Therefore, the tight junction and BMECs maintain the
function and reduce the permeability of BBB, and affect cerebral
edema. Yi et al (40)
found that atorvastatin prevented angiotensin II (Ang II)-induced
hyperpermeability and dysregulation of ZO-1 by suppressing Rho
kinase (ROCK) signaling, and thus atorvastatin increases the tight
junction protein to protect the BBB. Garrido et al (41) also found that atorvastatin
protects the BBB and ameliorates brain edema by upregulating
claudin 5, tight junction protein 1. Kalayci et al (42) found that long-term nitric oxide
(NOS) inhibition with N omega-nitro-L-arginine methyl ester
(L-NAME) followed by Ang II markedly disrupted the BBB, which may
affect CNS homeostasis, and that treatment with atorvastatin
improved the perturbations in the BBB by increasing the expression
of tight junction proteins.
Atorvastatin inhibits the HMG-CoA reductase
(7,40,42). Several studies (30,43) have confirmed that statins
significantly inhibit the activity of MMP-2 and MMP-9 to maintain
the stability of the BBB. We suggest that atorvastatin ameliorates
early brain injury and improves patient prognosis after SAH. Chang
et al (26) found that
pitavastatin exerts its neuroprotective effect through the dual
action of inhibiting cJNK (p46/p55) activation and reducing cleaved
caspase-9a and MMP-9 expression. Zhu et al (44) also demonstrated that simvastatin
protected the cerebrum from neuronal excitotoxicity and cytotoxic
edema by downregulating the expression of phosphorylated-CaMK II
and AQP4 in an animal model of experimental ischemic stroke. Tseng
et al (15) first
demonstrated that acute treatment with pravastatin after a SAH was
safe and ameliorated CVS, improved cerebral autoregulation, and
reduced vasospasm-related delayed ischemic deficits; also,
unfavorable outcomes at time of discharge were reduced primarily
after aneurysmal SAH in a phase II randomized placebo-controlled
trial. Chou et al (20)
found that simvastatin for the prevention of delayed cerebral
ischemia was safe and feasible after SAH using a randomized
placebo-controlled trial containing 39 patients. In the present
study, we also observed brain edema and EBI, and demonstrated that
atorvastatin ameliorated cerebral edema in a rat SAH model, proving
its clinical potential as a treatment strategy to reverse brain
edema and EBI in patients suffering from SAH.
Certain studies have reported contradictory results,
where statins were not found to have a significant impact on brain
edema, delayed cerebral ischemia and brain injury after SAH. One
multicenter randomized, controlled, double-blind clinical trial
(45) confirmed that high-dose
simvastatin or lower doses of simvastatin had no long-term effect
on the incidence of delayed ischemic deficits or on the rate of
favorable outcomes after SAH. Kirkpatrick et al (23) also reached a similar conclusion:
they did not detect any benefit in the use of simvastatin either
for the long-term or short-term outcome in patients with SAH in an
aneurysmal subarachnoid haemorrhage (STASH) trial.
The mechanism underlying the physiological effects
of atorvastatin on sympathetic nerve activity or reversal of over
activity, irrespective of whether the sympathetic disorder causes a
critical reduction in EBI, can be explained by the relief of brain
edema and improvement of outcome. The molecular mechanisms
underlying atorvastatin may be related to changes in the expression
of two major factors that contribute to brain edema after SAH, AQP4
and ET-1. ET-1 binds to specific receptors on smooth muscle cells
and causes constriction of the blood vessels and proliferation of
endothelial cells; it exerts deleterious effects on water
homeostasis, cerebral edema, and BBB integrity (46). Wang et al (47) confirmed that high levels of ET-1
are closely associated with BBB disruption, and thus it was
suggested that it plays an important role in the pathogenesis of
secondary brain injury after intracerebral hemorrhage (ICH).
Michinaga et al (48)
examined the effects of ETB antagonists on brain edema formation
and disruption of the BBB in a mouse model of cold injury and found
that ETB receptor antagonists are important to the amelioration of
brain edema. Jo et al (49) also found that endothelial ET-1
expression contributes to epilepticus-induced vasogenic edema
formation via BBB disruption in an AQP4/monocyte chemotactic
protein 1 (MCP1)-independent manner.
Certain studies (49–51) indicate that the water channel
protein AQP4 plays an essential role in water homeostasis and is
implicated in the pathogenesis of brain edema. Wang et al
(52) found that increased
expression of hypoxia-inducible factor 1-α (HIF-1α) plays a central
role in brain edema and BBB disruption by regulating both AQP4 and
MMP-9 following experimental SAH. Wang et al (53) studied the functional roles of AQP4
in the different stages of cerebral edema by systematic review;
AQP4 may facilitate cerebral edema fluid formation and aggravate
the clinical symptoms in cytotoxic edema. It has been suggested
that AQP4 is involved in the mechanism of water removal from the
interstitial space, ameliorating the level of vasogenic edema as
well as intracranial pressure in models of vasogenic edema, such as
freezing injury and brain tumors (50).
In the present study, we found that atorvastatin
significantly decreased the expression of ET-1 and AQP4 after SAH,
possibly resulting in a molecular cascade favoring water removal.
This discovery suggests that atorvastatin alleviates brain edema by
regulating the plasma concentration of ET-1 and AQP4. However, the
precise mechanisms of atorvastatin action need to be studied
further.
It has previously been suggested that apoptosis
plays an important role in the long-term morbidity associated with
SAH. In the present study, we performed a TUNEL assay to identify
and quantify the number of positive cells in the hippocampus of our
SAH rabbit model with and without atorvastatin treatment. In our
study, we demonstrated that in response to atorvastatin,
TUNEL-positive cells were significantly decreased after SAH.
Caspases are a family of cysteine proteases that
play an essential role in programmed cell death (7,54).
Caspase-3 is an important member of the caspase family and plays a
central role in the execution phase of cell apoptosis. It has
previously been reported that the expression of caspase-3 was
increased in brain neurons after SAH and promoted neuron apoptosis
(7). In the present study, we
analyzed the level of caspase-3 in the hippo-campus after SAH. Our
data showed that caspase-3 increased significantly in the SAH group
but decreased markedly in the SAH + atorvastatin group. This
indicates that atorvastatin activates the anti-apoptotic pathways
via the caspase-dependent apoptosis pathway and thus exerts a
neuroprotective effect on hippocampal neurons (7), in SAH rabbits.
In conclusion, EBI after SAH is a major contributor
to mortality and morbidity. The pathophysiology involves the
development of brain edema. Therapeutic options are still limited,
as the mechanisms are not fully understood. Our present study
provides evidence that atorvastatin may be used for the treatment
of cerebral edema and EBI.
References
|
1
|
Komotar RJ, Schmidt JM, Starke RM,
Claassen J, Wartenberg KE, Lee K, Badjatia N, Connolly ES Jr and
Mayer SA: Resuscitation and critical care of poor-grade
subarachnoid hemorrhage. Neurosurgery. 64:397–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosengart AJ, Schultheiss KE, Tolentino J
and Macdonald RL: Prognostic factors for outcome in patients with
aneurysmal subarachnoid hemorrhage. Stroke. 38:2315–2321. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Steiner T, Juvela S, Unterberg A, Jung C,
Forsting M and Rinkel G: European Stroke Organization guidelines
for the management of intracranial aneurysms and subarachnoid
haem-orrhage. Cerebrovasc Dis. 35:93–112. 2013. View Article : Google Scholar
|
|
4
|
Laskowitz DT and Kolls BJ: Neuroprotection
in subarachnoid hemorrhage. Stroke. 41(Suppl 10): S79–S84. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
MacDonald RL, Higashida RT, Keller E,
Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A,
et al: Clazosentan, an endothelin receptor antagonist, in patients
with aneurysmal subarachnoid haemorrhage undergoing surgical
clipping: a randomised, double-blind, placebo-controlled phase 3
trail (CONSCIOUS-2). Lancet Neurol. 10:618–625. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sehba FA, Hou J, Pluta RM and Zhang JH:
The importance of early brain injury after subarachnoid hemorrhage.
Prog Neurobiol. 97:14–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng G, Wei L, Zhi-Dan S, Shi-Guang Z and
Xiang-Zhen L: Atorvastatin ameliorates cerebral vasospasm and early
brain injury after subarachnoid hemorrhage and inhibits
caspase-dependent apoptosis pathway. BMC Neurosci. 10:72009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Budohoski KP, Czosnyka M, Kirkpatrick PJ,
Smielewski P, Steiner LA and Pickard JD: Clinical relevance of
cerebral autoregulation following subarachnoid haemorrhage. Nat Rev
Neurol. 9:152–163. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Uekawa K, Hasegawa Y, Ma M, Nakagawa T,
Katayama T, Sueta D, Toyama K, Kataoka K, Koibuchi N, Kawano T, et
al: Rosuvastatin ameliorates early brain injury after subarachnoid
hemorrhage via suppression of superoxide formation and nuclear
factor-kappa B activation in rats. J Stroke Cerebrovasc Dis.
23:1429–1439. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cahill J, Calvert JW, Solaroglu I and
Zhang JH: Vasospasm and p53-induced apoptosis in an experimental
model of subarachnoid hemorrhage. Stroke. 37:1868–1874. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Endo H, Nito C, Kamada H, Yu F and Chan
PH: Reduction in oxidative stress by superoxide dismutase
overexpression attenuates acute brain injury after subarachnoid
hemorrhage via activation of Akt/glycogen synthase kinase-3beta
survival signaling. J Cereb Blood Flow Metab. 27:975–982. 2007.
|
|
12
|
Aoki K, Zubkov AY, Ross IB and Zhang JH:
Therapeutic effect of caspase inhibitors in the prevention of
apoptosis and reversal of chronic cerebral vasospasm. J Clin
Neurosci. 9:672–677. 2002. View Article : Google Scholar
|
|
13
|
Sehba FA, Pluta RM and Zhang JH:
Metamorphosis of subarachnoid hemorrhage research: from delayed
vasospasm to early brain injury. Mol Neurobiol. 43:27–40. 201l.
View Article : Google Scholar
|
|
14
|
Tousoulis D, Antoniades C, Katsi V,
Bosinakou E, Kotsopoulou M, Tsioufis C and Stefanadis C: The impact
of early administration of low-dose atorvastatin treatment on
inflammatory process, in patients with unstable angina and low
cholesterol level. Int J Cardiol. 109:48–52. 2006. View Article : Google Scholar
|
|
15
|
Tseng MY, Czosnyka M, Richards H, Pickard
JD and Kirkpatrick PJ: Effects of acute treatment with pravastatin
on cerebral vasospasm, autoregulation, and delayed ischemic
deficits after aneurismal subarachnoid hemorrhage: a phase 2
randomized placebo-controlled trial. Stroke. 36:1627–1632. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee YM, Chen WF, Chou DS, Jayakumar T, Hou
SY, Lee JJ, Hsiao G and Sheu JR: Cyclic nucleotides and
mitogen-activated protein kinase: regulation of simvastatin in
platelet. J Biomed Sci. 17:452010. View Article : Google Scholar
|
|
17
|
Luzak B, Boncler M, Rywaniak J, Wilk R,
Stanczyk L, Czyz M, Rysz J and Watala C: The effect of a platelet
cholesterol modulation on the acetylsalicylic acid-madiated blood
platelet inhibition in hypercholesterolemic patients. Eur J
Pharmacol. 658:91–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou C, Yamaguchi M, Kusaka G, Schonholz
C, Nanda A and Zhang JH: Caspase inhibitors prevent endothelial
apoptosis and cerebral vasospasm in dog model of experimental
subarachnoid hemorrhage. J Cereb Blood Flow Metab. 24:419–431.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Z, Meng CJ, Shen XM, Shu Z, Ma C, Zhu
GQ, Liu HX, He WC, Sun XB, Huo L, et al: Potential contribution of
hypoxia-inducible factor-1α, aquaporin-4, and matrix
metalloproteinase-9 to blood-brain barrier disruption and brain
edema after experimental subarachnoid hemorrhage. J Mol Neurosci.
48:273–280. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chou SH, Smith EE, Badjatia N, Nogueira
RG, Sims JR, Ogilvy CS, Rordorf GA and Ayata C: A randomized,
double-blind, placebo-controlled pilot study of simvastatin in
aneurysmal subarachnoid hemorrhage. Stroke. 39:2891–2893. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garg K, Sinha S, Kale SS, Chandra PS, Suri
A, Singh MM, Kumar R, Sharma MS, Pandey RM, Sharma BS and Mahapatra
AK: Role of simvastatin in prevention of vasospasm and improving
functional outcome after aneurysmal subarachnoid hemorrhage: a
prospective, randomized, double-blind, placebo-controlled pilot
trial. Br J Neurosurg. 27:181–186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matthew J: Simvastatin for the prevention
of symptomatic cerebral vasospasm following aneurysmal subarachnoid
hemorrhage: a single-institution prospective cohort study. J
Neurosurg. 110:968–974. 2009. View Article : Google Scholar
|
|
23
|
Kirkpatrick PJ, Turner CL, Smith C,
Hutchinson PJ and Murray GD; STASH Collaborators: Simvastatin in
aneurysmal subarachnoid haemorrhage(STASH): a multicentre
randomised phase 3 trial. Lancet Neurol. 13:666–675. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vergouwen MD, Meijers JC, Geskus RB, Coert
BA, Horn J, Stroes ES, van der Poll T, Vermeulen M and Roos YB:
Biologic effects of simvastatin in patients with aneurysmal
subarachnoid hemorrhage: a double-blind, placebo-controlled
randomized trial. J Cereb Blood Flow Metab. 29:1444–1453. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen J, Chen G, Li J, Qian C, Mo H, Gu C,
Yan F, Yan W and Wang L: Melatonin attenuates inflammatory
response-induced brain edema in early brain injury following a
subarachnoid hemorrhage: a possible role for the regulation of
pro-inflammatory cytokines. J Pineal Res. 57:340–347. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chang CZ, Wu SC, Kwan AL and Lin CL:
Preconditioning with pitavastatin, an HMG-CoA reductase inhibitor,
attenuates C-Jun N-terminal kinase activation in experimental
subarachnoid hemorrhage-induced apoptosis. Acta Neurochi.
157:1031–1041. 2015. View Article : Google Scholar
|
|
27
|
Broderick JP, Brott TG, Duldner JE,
Tomsick T and Leach A: Initial and recurrent bleeding are the major
causes of death following subarachnoid hemorrhage. Stroke.
25:1342–1347. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Claassen J, Carhuapoma JR, Kreiter KT, Du
EY, Connolly ES and Mayer SA: Global cerebral edema after
subarachnoid hemorrhage: frequency, prediction, and impact on
outcome. Stroke. 33:1225–1232. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rosell A, Cuadrado E, Ortega-Aznar A,
Hernandez-Guillamon M, Lo EH and Montaner J: MMP-9-positive
neutrophil infiltration is associated to blood-brain barrier
breakdown and basal lamina type IV collagen degradation during
hemorrhagic transformation after human ischemic stroke. Stroke.
39:1121–1126. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rosenberg GA and Navratil M:
Metalloproteinase inhibition blocks edema in intracerebral
hemorrhage in the rat. Neurology. 48:921–926. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ramos-Fernandez M, Fernanda Bellolio M and
Stead LG: Matrix metalloproteinase-9 as a marker for acute ischemic
stroke:a systematic review. J Stroke Cerebrovasc Dis. 20:47–54.
2011. View Article : Google Scholar
|
|
32
|
Papadopoulous MC and Verkman AS:
Aquaporin-4 gene disruption in mice reduces brain swelling and
mortality in pneumococcal meningitis. J Biol Chem. 280:13906–13912.
2005. View Article : Google Scholar
|
|
33
|
Manley GT, Fujimura M, Ma T, Noshita N,
Filiz F, Bollen AW, Chan P and Verkman AS: Aquaporin-4 deletion in
mice reduces brain edema after acute water intoxication and
ischemic stroke. Nat Med. 6:159–163. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang B, Zador Z and Verkman AS: G1ial cell
aquaporin-4 overexpression in transgenic mice accelerates cytotoxic
brain swelling. J Biol Chem. 283:15280–15286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Abbott NJ and Patabendige AA: Structure
and function of the blood-brain barrier. Neurobiol Dis. 37:13–25.
2010. View Article : Google Scholar
|
|
36
|
Hawkins BT and Davis TP: The blood-brain
barrier/neurovascular unit in health and disease. Pharmacol Rev.
57:173–185. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Terry S, Nie M, Matter K and Balda MS: Rho
signaling and tight junction functions. Physiology (Bethesda).
25:16–26. 2010. View Article : Google Scholar
|
|
38
|
Huber JD, Egletun RD and Davis TP:
Molecular physiology and pathophysiology of tight junctions in the
blood-brain barrier. Trends Neurosci. 24:719–725. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Persidsky Y, Ramirez SH, Haorah J and
Kanmogne GD: Blood-brain barrier: structural components and
function under physiologic and pathologic conditions. J Neuroimmune
Pharmacol. 1:223–236. 2006. View Article : Google Scholar
|
|
40
|
Yi R, Gao X-P and Hui L: Atorvastatin
prevents angiotensin II-induced high permeability of human arterial
endothelial cell monolayers via ROCK signaling pathway. Biochem
Biophys Res Commun. 459:94–99. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Garrido JM, Esteban M, Roda O, Alaminos M
and Sánchez-Montesinos I: Lysophosphatidic acid pretreatment
prevents micromolar atorvastatin-induced endothelial cell death and
ensures the beneficial effects of high-concentration statin therapy
on endothelial gene expression. Ann Vasc Surg. 26:549–558. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kalayci R, Kaya M, Elmas I, Arican N,
Ahishali B, Uzun H, Bilgic B, Kucuk M and Kudat H: Effects of
atorvastatin on blood-brain barrier permeability during l-NAME
hypertension followed by angiotensin-II in rats. Brain Res.
1042:184–193. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Seo JH1, Guo S, Lok J, Navaratna D, Whalen
MJ, Kim KW and Lo EH: Neurovascular matrix metalloproteinases and
the blood-brain barrier. Curr Pharm Des. 18:3645–3648. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu MX, Lu C, Xia CM, Qiao Z and Zhu D:
Simvastatin pretreatment protects cerebrum from neuronal injury by
decreasing the expressions of phosphor-CaMK II and AQP4 in ischemic
stroke rats. J Mol Neurosci. 54:591–601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wong GK, Chan DY, Siu DY, Zee BCY, Poon
WS, Chan MTV, Gin T and Leung M: High-dose simvastatin for
aneurysmal subarachnoid hemorrhage: multicenter randomized
controlled double-blinded clinical trial. Stroke. 46:382–388. 2015.
View Article : Google Scholar
|
|
46
|
Chow M, Dumont AS and Kasselletal NF:
Endothelin receptor antagonists and cerebral vasospasm: an update.
Neurosurgery. 51:1333–1342. 2002.PubMed/NCBI
|
|
47
|
Wang LK, Hong Z, Wu GF and Li C:
Perihematomal endothelin-1 level is associated with an increase in
blood-brain barrier permeability in a rabbit model of intracerebral
hematoma. Chin Med J (Engl). 126:3433–3438. 2013.
|
|
48
|
Michinaga S, Nagase M, Matsuyama E,
Yamanaka D, Seno N, Fuka M, Yamamoto Y and Koyama Y: Amelioration
of cold injury-induced cortical brain edema formation by selective
endothelin ETB receptor antagonists in mice. PLoS One.
9:e1020092014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jo SM, Ryu HJ, Kim JE, Yeo SI, Kim MJ,
Choi HC, Song HK and Kang TC: Up-regulation of endothelial
endothelin-1 expression prior to vasogenic edema formation in the
rat piriform cortex following status epilepticus. Neurosci Lett.
501:25–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Manley GT, Binder DK, Papadopoulos MC and
Verkman AS: New insights into water transport and edema in the
central nervous system from phenotype analysis of aquaporin-4 null
mice. Neuroscience. 129:983–991. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Migliati E, Meurice N, DuBois P, Fang JS,
Somasekharan S, Beckett E, Flynn G and Yool AJ: Inhibition of
aquaporin-1 and aquaporin-4 water permeability by a derivative of
the loop diuretic bumetanide acting at an internal pore-occluding
binding site. Mol Pharmacol. 76:105–112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang Z, Meng CJ, Shen XM, Shu Z, Ma C, Zhu
GQ, Liu HX, He WC, Sun XB, Huo L, et al: Potential contribution of
hypoxia-inducible factor-1α, aquaporin-4, and matrix
metalloproteinase-9 to blood-brain barrier disruption and brain
edema after experimental subarachnoid hemorrhage. J Mol Neurosci.
48:273–280. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang WW, Xie C, Zhou LL and Wang GS: The
function of aquaporin4 in ischemic brain edema. Clin Neurol
Neurosurg. 127:5–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Alnemri ES, Livingston DJ, Nicholson DW,
Salvesen G, Thomberry NA, Wong WW and Yuan J: Human ICE/CED-3
protease nomenclature. Cell. 87:1711996. View Article : Google Scholar : PubMed/NCBI
|