Introduction
Chronic hypoxia (CH) occurs in individuals living in
high altitude areas as well as in individuals with cyanotic
congenital heart disease (such as Tetralogy of Fallot) and coronary
artery disease. A previous epidemiological study has revealed that
populations living at high altitude are less prone to chronic
ischemic heart disease and have lower rates of mortality from heart
disease (1). Clinical observation
has noted long-term survival with rare occurrences of heart failure
in children with cyanotic congenital heart defects. Furthermore, in
our experience these children are able to tolerate hypoxic-ischemic
injury caused by cardiac surgery that would likely cause myocardial
necrosis in children non-cyanotic heart defects (2). Moreover, several animal experimental
studies have demonstrated that myocardial chronic intermittent
hypoxia (CIH) is associated with more efficient metabolism,
increased coronary vasculature and tolerance to acute
hypoxic-ischemic injury, such as limitation of infarct size
(3), improved recovery of
ventricular systolic function (4,5)
and protection against ischemia/reperfusion (I/R)-induced
ventricular arrhythmia (6).
Although CIH induces a variety of adaptive changes in the
myocardium that play a crucial role in this protective phenomenon,
including the regulation of adenosine triphosphate (ATP)-sensitive
K+ channels (7,8),
reactive oxygen species (9),
nitric oxide (10), various
protein kinases (11,12), apoptosis (13) and pathways mediated by hypoxia
inducible factor (14), details
of the underlying molecular mechanisms remain to be elucidated.
Activating transcription factor 6 (ATF6), a key
transcriptional activator involved in maintaining cellular
homeostasis, is a 670-amino acid endoplasmic reticulum (ER)
transmembrane protein that is cleaved in response to ER stress. The
cleaved N-terminal fragment translocates to the nucleus and
activates ER stress response genes. A microarray study using mouse
hearts showed that ATF6 activation induces the expression of almost
400 genes (15), many of which
may serve protective roles. A previous study has revealed that
ATF6α modulates ER function to protect cells against chronic stress
(16), which suggests that ATF6
mediates cytoprotective and pro-survival functions. A previous
study (17) using a transgenic
mouse model indicated that ATF6 activation prior to ischemia exerts
protective effects against ischemia and/or reperfusion damage.
Furthermore, we have previously demonstrated that the cardiac ER
stress response is activated by CIH stress and that ATF6α is
continuously activated in primary human cardiomyocytes and rat
myoblast H9c2 cells (18).
Several studies have also demonstrated the importance of the
serine/threonine-specific protein kinase, Akt (also known as
protein kinase B), in cardiomyocyte survival (19), which is effected through Ser473
phosphorylation (20), thus
contributing to cardioprotection (21,22). Activation of ATF6 positively
regulates mammalian target of rapamycin (mTOR)C2-mediated
phosphorylation of Akt at Ser473, which is required for full
activity of Akt (23); however,
the involvement of Akt in the cardioprotective effects exerted by
ATF6 against myocardial I/R injury following CIH remain to be
elucidated. Binding immunoglobulin protein (BiP), also known as
heat shock protein 70, has been shown to be downstream of ATF6 in
the response to chronic stress in the ER (24). Analysis of BiP should thus provide
evidence that ATF6 has been activated.
In the present study, the role of ATF6 in the
mechanism by which CIH increases tolerance to myocardial I/R was
investigated using both in vivo and ex vivo models as
well as a cultured cardiomyocyte model system. The role of Akt in
this process was also investigated in a cultured cardiomyocyte
model system.
Materials and methods
Animals and experimental protocol
Male Sprague-Dawley rats (3 weeks old, 140–180 g)
were obtained from the Animal Center of Xinqiao Hospital at the
Third Military Medical University (Chongqing, China).
Animals were used in order to investigate the
following: i) the effects of global I/R injury simulated in ex
vivo rat hearts subjected to normoxic perfusion (n=6) and
exposed to CIH (n=6) using the Langendorff-perfusion system; ii)
determination of regional I/R injury induced by coronary ligation
and subsequent reperfusion of rat heart tissues subjected to
normoxic perfusion (n=6) and exposed to CIH (n=7); iii)
determination of the expression of ATF6 in heart tissues of rats
subjected to normoxic perfusion (n=5) and exposed to CIH (n=5).
All experiments involving the use of animals
performed as part of this study were conducted with the approval of
the Third Military Medical University Animal Care and Ethics
Committee.
Exposure to chronic hypoxia
Rats were randomly assigned to either the normoxia
group or the CIH group. The initial body weight was measured using
an electronic scale (DST673; SuHang Co. Ltd., Suzhou, China)
immediately before exposure to hypoxia. Rats in the CIH group were
housed for 4 weeks in a hypobaric chamber (equivalent to an
altitude of 5,000 m, with a barometric pressure of 404 mmHg,
PO2=84.98 mmHg). Barometric pressure in the chamber was
adjusted weekly (equivalent to an altitude of 3,000 m over a period
of 1 h) for cage maintenance. Age-matched rats in the normoxia
group were housed under normoxic conditions for the duration of the
experiments. All animals had free access to water and a standard
laboratory diet. At the end of the fourth week, the body weight of
the animals was measured and a blood sample was collected from the
abdominal aorta to determine the blood gas parameters using a blood
gas analyzer (I-STAT 300; Abbott Laboratories, Abbott Park, IL,
USA).
Examining I/R using isolated rat
hearts
Rats were anesthetized with pentobarbital sodium [50
mg/kg administered intraperitoneally (i.p.)]. Following a
laparotomy and thoracotomy, heparin (1,000 IU/kg body weight) was
injected intravenously. Hearts were rapidly excised and transferred
into cold (4°C), heparinized Krebs-Henseleit (K-H) perfusate [NaCl
(118 mmol/l), NaHCO3 (25 mmol/l), KCl (4.7 mmol/l),
MgSO4 (1.2 mmol/l), KH2PO4 (1.2
mmol/l), glucose (11 mmol/l), CaCl2 (2.5 mmol/l)]. The
hearts were connected to Langendorff apparatus via the aorta within
30 sec of excision and subsequently perfused with K-H perfusate in
a retrograde manner at constant pressure (80 cm H2O).
The perfusate was bubbled with gas (95% oxygen, 5% carbon dioxide)
to yield a pH of 7.4 at 37°C throughout the experiment. A
water-filled latex balloon connected to a pressure transducer was
inserted into the left ventricle (LV) through the left atrium and
the mitral annulus. Left ventricular developed pressure (LVDP) was
monitored by an amplifier. During the period of measurement, the
balloon volume was adjusted to produce a left ventricular end
diastolic pressure (LVEDP) of 6 mmHg. After a stabilization period
(approximately 20 min), the isolated heart was subjected to global
normothermic ischemia for 30 min and then perfused for 1 h. The
effluent was collected for lactate dehydrogenase (LDH) activity
assays at 60 min after reperfusion. At the end of the reperfusion,
the hearts were removed from the perfusion cannula and stored at
−80°C for triphenyl tetrazolium chloride (TTC) staining,
immunofluorescence staining and western blot analysis.
In vivo rat model of I/R injury
Animals were anesthetized with pentobarbital sodium
[50 mg/kg i.p.] and mechanically ventilated with air (10 ml/100 g
body weight; 80 breaths/min). The barometric pressure was changed
using a face mask; the breath ratio was 1:2. A left thoracotomy was
undertaken between the third and fourth ribs. The heart was
exposed, and a 6-0 Prolene suture was passed under the left
anterior descending coronary artery (LAD) 2–3 mm below the left
atrium using a needle. A piece of polyethylene tubing was placed
between the LAD and the suture before the LAD was ligated for 30
min, followed by coronary reperfusion for 24 h by releasing the
ligature. The chest cavity was closed and the animal was gradually
detached from the respirator. At the end of the experiment, the
rats were sacrificed by injection of 3 ml 2.6% potassium chloride
and re-thoracotomized to allow for the collection of blood samples
into ethylenediaminetetraacetic acid (EDTA) tubes via the inferior
vena cava (IVC) for creatine kinase (CK)-cardiac isoenzyme CK-MB
and LDH analysis. The hearts were excised and frozen at −20°C for
myocardial infarct size assessment.
Transthoracic echocardiography
Echocardiographic measurements were performed prior
to ischemic injury and at 24 h after reperfusion. The rats were
anesthetized with pentobarbital (50 mg/kg i.p.) and heart rates
were maintained at 200–400 beats/min. Transthoracic 2D-guided
M-mode echocardiography was performed using a Vevo 2100 Imaging
System (VisualSonics Inc., Toronto, ON, Canada) with a 13–24 MHz
scan probe (MS250). Ejection fraction (EF) and fractional
shortening (FS) were calculated according to standard formulas as
indicators of ventricular function.
TTC staining
Frozen hearts were first partially thawed and then
frozen sectioned (thickness, 1 mm) perpendicular to the
longitudinal axis. The remaining heart tissues were used for
western blot analysis. Sections were incubated in 1% TTC in
phosphate-buffered saline (PBS) solution at 37°C for 15 min and
then fixed in 4% formalin solution overnight at 4°C. Sections were
then placed on glass slides and photographed. The infarction area
for each section was determined using ImageJ software and the
infarct size was expressed as a percentage of the total
cross-sectional area of the heart.
LDH and CK-MB activity
LDH and CK-MB activity in the collected blood
samples was assayed using a Lactate Dehydrogenase test kit, and a
Creatine Kinase MB test kit (both from Ruiyuan Biotechnology Co.,
Ltd., Ningbo, China), respectively. The results were analyzed using
a Beckman Coulter Chemistry Analyzer (AU-5800; Beckman Coulter
Inc., Tokyo, Japan).
Cell culture and experimental
protocols
H9c2 cells were obtained from the American Type
Culture Collection (ATCC, Rockville, MD, USA) and cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS) (both from Invitrogen, Carlsbad, CA,
USA).
After 24 h, the H9c2 cells were randomly divided
into the normoxia and mild hypoxia groups. The cells were cultured
in an incubator (Thermo 3131; Thermo Fisher Scientific, Waltham,
MA, USA) in which the fraction of inspired oxygen (FiO2)
levels were 21 and 13%, respectively, as has been previously
described (8). At 0, 12, 24, 48
and 72 h, ATF6 expression was assayed. The time point at which ATF6
expression reached a maximum was used in subsequent experiments to
investigate the protective effect of ATF6 on cells subjected to I/R
injury. The experimental groups were as follows: i) normoxia + I/R;
ii) hypoxia + I/R; iii) hypoxia + control-siRNA + I/R, iv) hypoxia
+ ATF6-siRNA + I/R. I/R was simulated by placing cells in ischemia
buffer [CaCl2 (1.13 mmol), KCl (5 mM),
KH2PO4 (0.3 mmol), MgCl2 (0.5
mmol), MgSO4 (0.4 mmol), NaCl (128 mmol),
NaHCO3 (4 mmol), HEPES (10 mmol); pH 6.8], which was
previously equilibrated in the anoxia chamber (95% N2,
5% CO2, 37°C) for 1 h. Subsequently, cells were
reperfused for 3 h under normal culture conditions. There were five
replicates in each group. Cell viability assays for all groups were
performed at 3 h after reperfusion.
Transfection of siRNA
H9c2 cells were seeded in 24-well plates
(4×104 cells/well) and transfected when the cells
reached approximately 50% confluence. Each well was transfected
with siRNA targeted against rat ATF6 (5′-ACCACAAGACCG AAGATG-3′) or
a negative control (5′-GCCTGCCGTCCA AAGTTGTAA-′3) (both from
Shanghai GenePharma Co., Ltd., Shanghai, China) with 5% FBS using
Lipofectamine 2000 Transfection reagent (Invitrogen) according to
the manufacturer's protocol. After 24 h, the cells were switched to
a medium containing 5% fetal calf serum for use in subsequent
experiments.
Cell viability assessment
Cell viability was assessed using a cell counting
kit-8 (CCK-8; Boster Biological Technology Ltd., Wuhan, China) and
studying LDH release into the culture medium. For the CCK-8 assay,
CCK-8 solution (10 µl) was added to each well and the plate
was incubated at 37°C for 4 h. Absorbance was measured at 450 nm
using the VICTOR™ X2 Multilabel Plate reader (PerkinElmer, Inc.,
Waltham, MA, USA) with a reference wavelength of 650 nm. LDH
activity was assayed using a Lactate Dehydrogenase test kit
(Ruiyuan Biotechnology Co., Ltd.,) and analyzed with a Beckman
Coulter Chemistry Analyzer (AU-5800; Beckman Coulter Inc.).
Immunofluorescence staining
Immunofluorescence assays for the detection of ATF6
were performed using DAPI on the frozen sections of the ventricular
myocardium. The sections were blocked with 1% bovine serum albumin
in PBS, and then incubated overnight at 4°C with a mouse monoclonal
antibody against ATF6α (ab11909, 1:50; Abcam, Cambridge, MA, USA).
Immunoreactivity was detected by incubating the sections for 40 min
at 37°C with a FITC-labeled goat anti-mouse IgG (A0568, 1:50;
Beyotime Institute of Biotechnology, Beijing, China) secondary
antibody, and images of immunofluorescence staining were obtained
by laser scanning confocal microscopy (TCS SP5; Leica, Mannheim,
Germany).
Western blot analysis
Total proteins were extracted from ventricular
myocardial tissues or cultured cells, which were homogenized in an
in ice-cold lysis buffer (T-TER Tissue Protein Extraction Reagent;
Thermo Fisher Scientific) containing phosphatase and protease
inhibitors. Extracts were centrifuged at 15,000 × g, at 4°C for 5
min, and protein concentration was measured using the Bradford
assay (Bio-Rad, Hercules, CA, USA). Lysates were separated by 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF)
membranes (Roche, Mannheim, Germany). The membranes were blocked
(SuperBlock blocking buffer in TBS, with 0.5% Tween-20) for 2 h at
37°C. Primary antibodies were used in western blot analysis
according to the manufacturer's instructions. The membranes were
incubated overnight with antibodies against ATF6α (Abcam), Akt
(Cat. no. 2966), phosphorylated (p)-Akt (Ser473; Cat. no. 8200),
BiP (Cat. no. 3177) (all from Cell Signaling Technology, Inc.,
Beverly, MA, USA), GAPDH (ab8245; Abcam) and β-actin (A1978; Sigma,
St. Louis, MO, USA) (all 1:500 in SuperBlock blocking buffer in
TBS, with 0.5% Tween-20) at 4°C overnight. After washing six times
with Tris-buffered saline (5 min/wash), the blots were incubated
with a horseradish peroxidase-conjugated goat anti-rat IgG
(sc-2041, 1:1,000; Santa Cruz Biotechnology, Inc. Santa Cruz, CA,
USA) for 1 h at 37°C. The blots were washed three times with
Tris-buffered saline and antibody binding was detected with an
imaging system (Kodak Gel Logic 212; Kodak, Rochester, NY, USA).
The blots were analyzed by Quantity One software (Bio-Rad).
Statistical analysis
All data are expressed as the means ± standard error
of the means (SEM). Data were analyzed by SPSS 17.0. Statistically
meaningful differences were determined using independent Student's
t-tests or paired t-tests. A P-value <0.05 was considered to
indicate a statistically significant difference.
Results
CIH changes physical parameters
Although there was no significant difference in the
initial body weight of the rats prior to exposure to hypoxia, after
4 weeks of exposure the final body weight of the rats in the CIH
group was significantly lower than those in the normoxia group
(P<0.01) (Table I).
Furthermore, the blood hemoglobin levels, hematocrit values, and
the heart weight/body weight ratios of the CIH group were
significantly higher than those of the normoxia group (P<0.01).
In addition, arterial oxygen tension and oxygen saturation were
significantly higher in the normoxia group compared with those in
the CIH group (both P<0.01).
 | Table IPhysical parameters measured in the
animals at the end of 4 weeks of exposure to normoxic or chronic
hypoxic conditions. |
Table I
Physical parameters measured in the
animals at the end of 4 weeks of exposure to normoxic or chronic
hypoxic conditions.
| Parameter | Normoxia | Chronic
hypoxia |
|---|
| Number of rats
(n) | 17 | 18 |
| Initial body weight
(g) | 161.24±11.58 | 160.12±10.87 |
| Final body weight
(g) | 311.34±7.06 | 228.44±5.66a |
| Heart weight
(mg) | 781.80±180.50 |
924.40±384.30a |
| Heart/final body
weight ratio (mg/g) | 2.60±0.06 | 4.04±0.12a |
| Hematocrit value
(%) | 41.24±0.01 | 57.11±0.01a |
| Hemoglobin
(g/l) | 13.97±0.19 | 19.08±0.32a |
| Arterial oxygen
tension (mmHg) | 81.65±1.01 | 45.94±0.69a |
| Arterial oxygen
saturation (mmHg) | 93.53±0.61 | 66.50±13.04a |
CIH improves post-ischemic recovery of
myocardial performance in isolated rat hearts
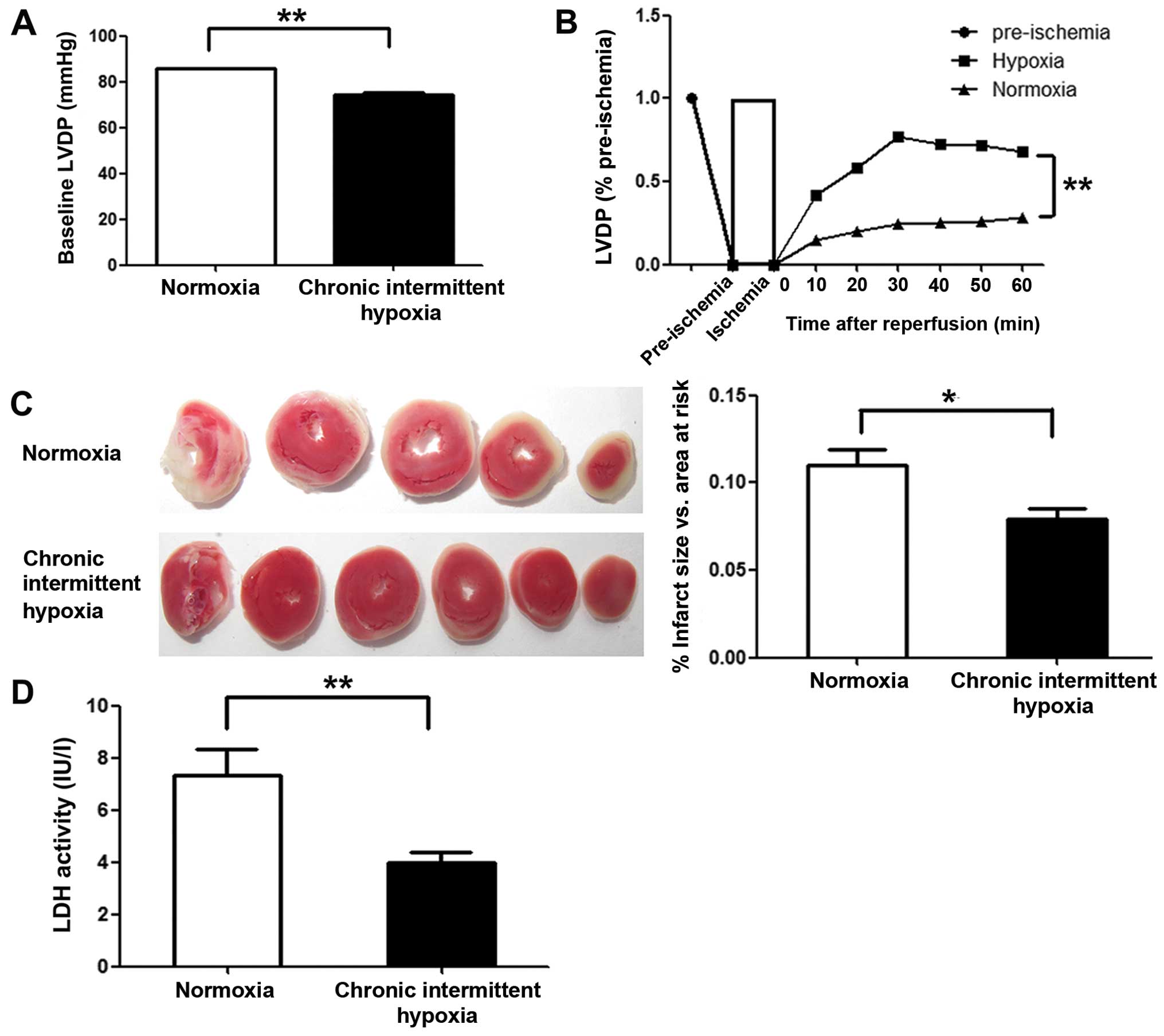
To examine the cardioprotective effects of CIH, we
monitored left ventricular function in Langendorff-perfused hearts
subjected to 30 min of global ischemia followed by 60 min of
reperfusion. The LVDP (pre-ischemia) in the CIH group was markedly
lower than that in normoxia group (P<0.01) (Fig. 1A). However, at all time points
investigated during the reperfusion period, greater recovery of
LVDP was observed in the CIH group compared with the normoxia group
(P<0.01) (Fig. 1B). To assess
the degree of myocardial injury following global ischemia, the
infarct size of all reperfused hearts was determined, and LDH
activity in the effluent at the end of reperfusion was assayed.
Markedly smaller infarcts (P<0.05) and lower LDH activity in the
effluent (P<0.01) were observed in the CIH group compared with
the normoxia group (Fig. 1C and
D).
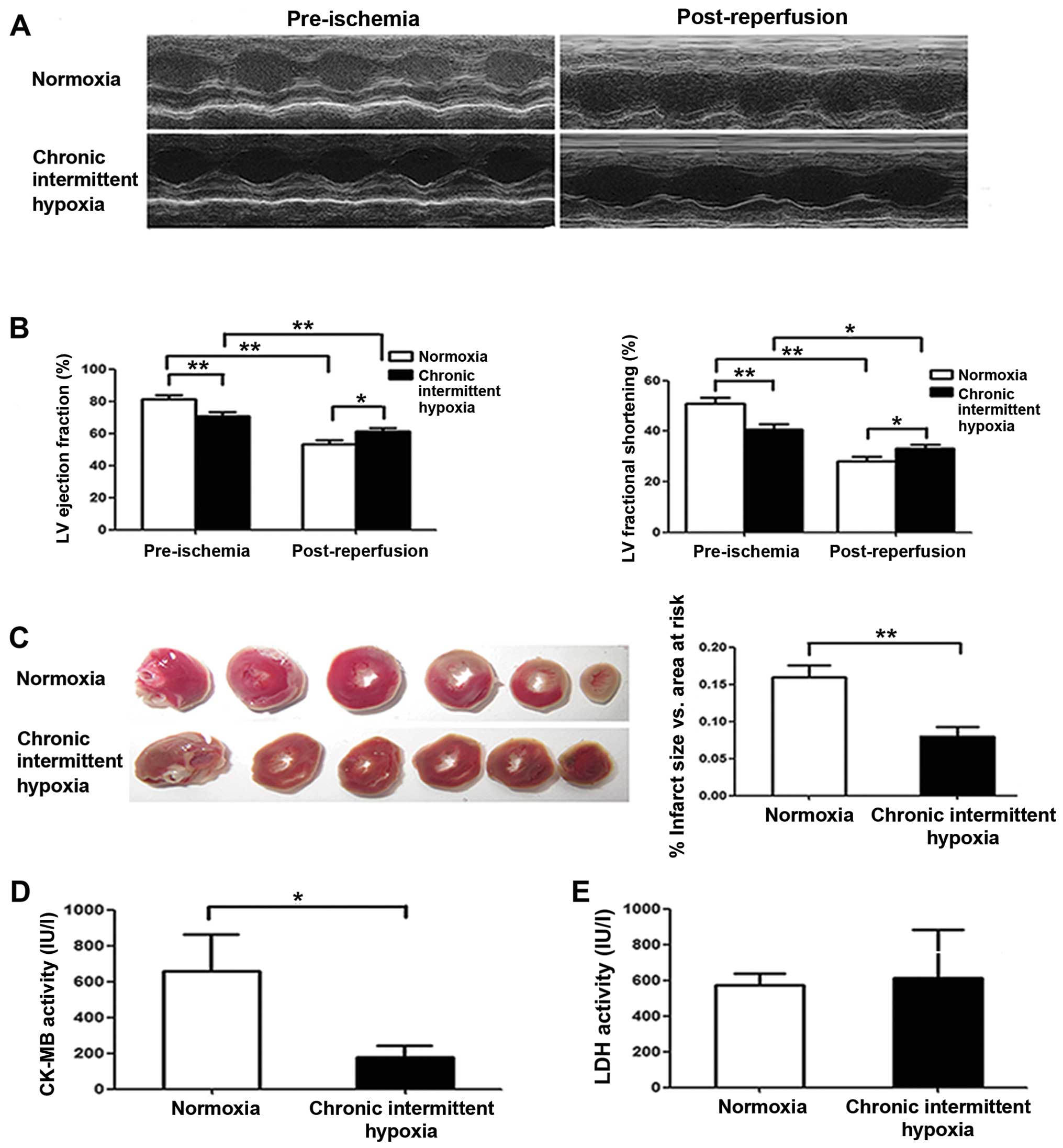
CIH preserves rat cardiac function after
I/R in vivo
Rats were subjected to I/R injury by 30 min LAD
ligation followed by 24 h coronary reperfusion, and cardiac
function was evaluated by echocardiography. We noted that
pre-ischemic cardiac function was markedly lower in the CIH group
than that in the normoxia group (Fig.
2A and B), although LV systolic function was markedly reduced
in both the normoxia and CIH groups at 24 h after reperfusion
compared with that observed pre-ischemia (Fig. 2B). However, we noted that the
decrease in EF and FS in CIH rats was less than in the normoxia
group. Notably, both the EF and the FS after I/R in the CIH group
were higher than that in the normoxia group (P<0.01) (Fig. 2B), suggesting that CIH preserves
cardiac function after I/R. Similarly, the infarct size measured at
24 h after reperfusion was greater in the normoxia group than that
in the CIH group (P<0.01) (Fig.
2C). Furthermore, CK-MB activity following I/R was noted as
being significantly lower in the CIH group than in the normoxia
group (P<0.05) (Fig. 2D),
indicating less myocardial damage in the CIH group; however, there
was no significant difference in LDH activity between the two
groups (P>0.05) (Fig. 2E).
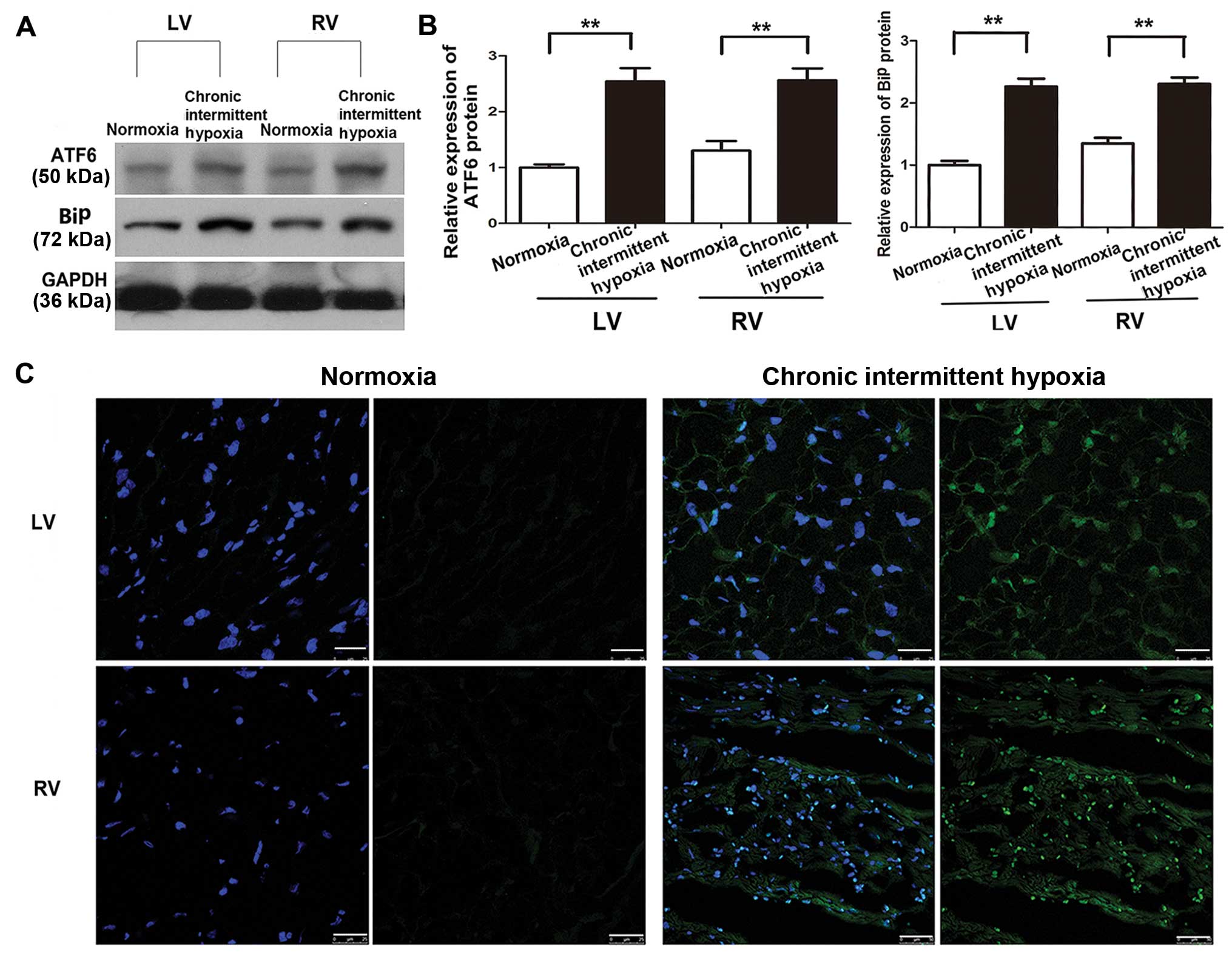
CIH induces ATF6 expression
Our previous study showed that ATF6 was
overexpressed in human specimens and H9c2 cells under anoxic
conditions (18). To confirm this
effect in the present study, cleaved ATF6α expression in tissue
samples taken from the right ventricle (RV) and LV of rat hearts
exposed to normoxic and chronic intermittent hypoxic conditions was
determined using western blot analysis. The samples were taken
immediately after removing the animals from the hypobaric chamber.
Myocardial cleaved ATF6α (50 kDa) protein expression was
significantly higher in the CIH group compared with the normoxia
group (P<0.01) (Fig. 3A and
B), with no statistically significant difference noted between
the expression in the LV and RV in the CIH group (P>0.05)
(Fig. 3B). Similar results were
noted for BiP expression, which was increased in the CIH group in
both the LV and RV in comparison with the normoxia group.
Immunofluorescence staining also revealed a significant increase in
ATF6 expression both in the LV and the RV induced by CIH (Fig. 3C).
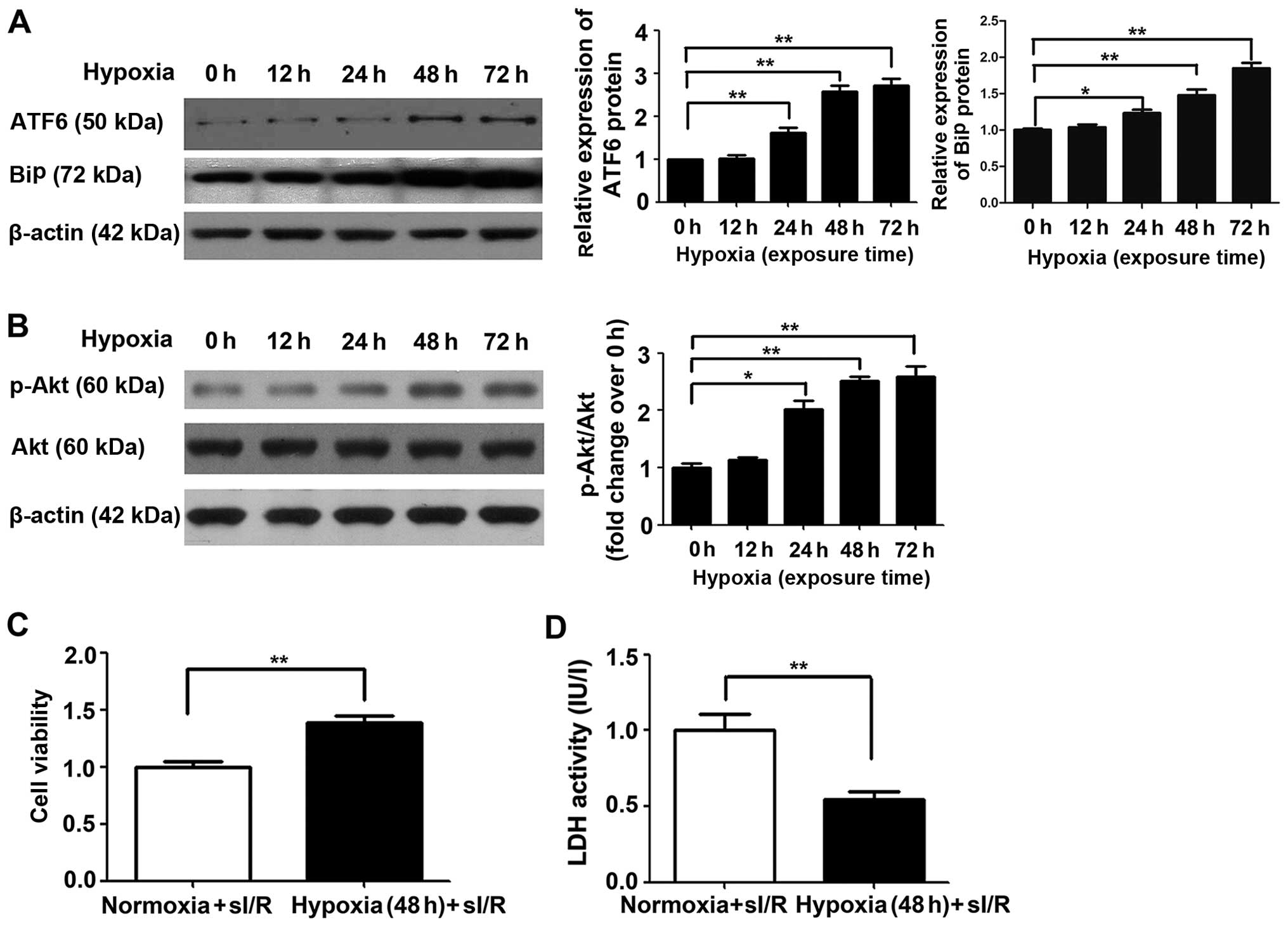
Cardioprotective effects of ATF6 against
myocardial I/R injury are induced by mild hypoxia
The cardioprotective effects of ATF6 against
myocardial I/R injury caused by CH were first investigated by
western blot analysis of cleaved ATF6 protein expression in rat
myocardial H9c2 cells subjected to chronic mild hypoxia, in a
previous study (8). As shown in
Fig. 4A, endogenous cleaved ATF6
protein expression was significantly upregulated in H9c2 cells
after 24 h exposure to mild hypoxia (P<0.05), with continued
upregulation after 48 h and a high level of expression maintained
up to 72 h (P<0.01). Similar results were also noted in relation
to BiP expression, which increased up to 72 h.
Subsequently, the effect of chronic mild hypoxia on
sI/R-induced cellular injury was evaluated. H9c2 cells exposed to
hypoxia for 48 h (the time point determined for high ATF6
expression) were subjected to ischemia for 1 h followed by
reperfusion for 3 h, as previously described (25). H9c2 cells cultured under normoxic
conditions served as a control. Chronic mild hypoxia increased H9c2
cell viability by 38% and reduced LDH activity compared with that
detected in cells cultured under normoxic conditions (both
P<0.01) (Fig. 4C and D,
respectively).
To further determine the cardioprotective effects of
ATF6 induced by chronic mild hypoxia against subsequent I/R damage,
cleaved ATF6 expression was blocked in H9c2 cells by transfection
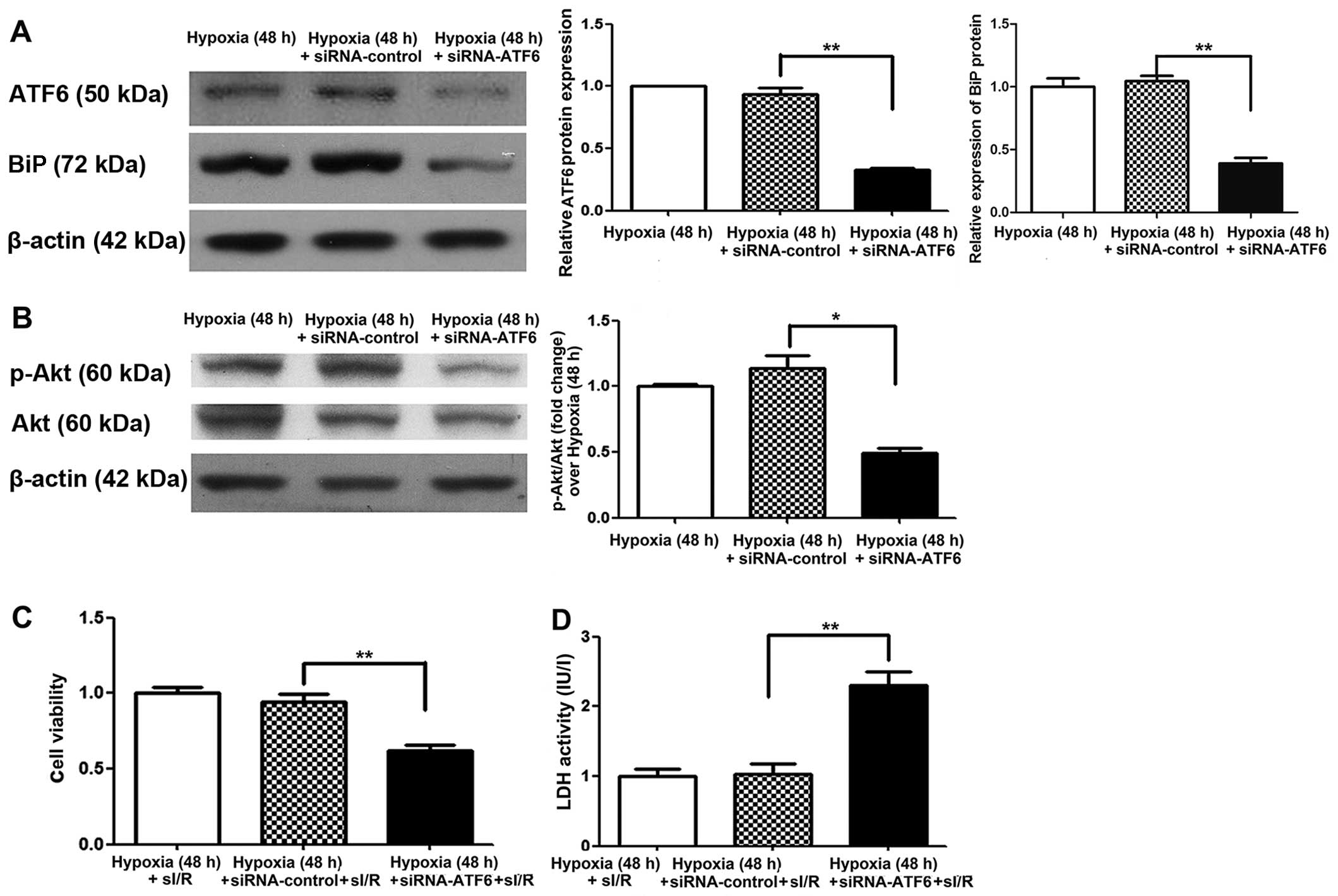
with siRNA-ATF6 prior to hypoxic exposure. Suppression of cleaved
ATF6 expression was confirmed, at 48 h after exposure to mild
hypoxia, by western blot analysis. We also noted a decrease in BiP
expression in comparison to the siRNA and hypoxic controls
(P<0.01) (Fig. 5A). Following
I/R, the viability of siRNA-ATF6 transfected H9c2 cells was also
significantly reduced (by 35%) (P<0.01) (Fig. 5C), while LDH activity was
significantly increased (by 1.2-fold) compared with siRNA-control
transfected cells (P<0.01) (Fig.
5D).
ATF6 exerts cardioprotective effects
against I/R injury through upregulation of p-Akt
To elucidate the molecular mechanism by which ATF6
mediates cardioprotection against I/R injury induced by CH, the Akt
signaling pathway was examined. Firstly, Akt expression was
investigated in H9c2 cells cultured under chronic mild hypoxia
conditions. After 24 h of hypoxia, p-Akt expression was observed,
with expression peaking after 48 h (Fig. 4B). Of note, increased Akt
expression was consistent with the changes observed in cleaved ATF6
protein levels. Subsequently, the influence of siRNA-ATF6-mediated
suppression of cleaved ATF6 on Akt expression in H9c2 cells was
investigated. Western blot analysis showed that transfection of
H9c2 cells with ATF6-siRNA suppressed hypoxia-induced p-Akt
expression compared with siRNA-control transfected cells (Fig. 5B).
Discussion
In the present study, CIH induced high expression of
cleaved ATF6, which was associated with significantly attenuated
myocardial infarct size and increased cardiac tolerance to acute
I/R injury both in vivo and ex vivo. Furthermore,
mild hypoxia-mediated cytoprotection against simulated I/R in H9c2
cells was demonstrated by improved cellular viability and increased
p-Akt and BiP levels. Thus, the results of the present study
demonstrated that exposing rats to CIH conferred resistance to
subsequent I/R injury of the myocardium by upregulating cleaved
ATF6 and increased Akt pathway signaling.
Previous studies have demonstrated that CIH confers
long-lasting cardioprotection against acute I/R injury (7,10,26). Furthermore, clinical observations
and epidemiological studies have implicated CIH in cardioprotection
(2). These observations are,
however, contradicted by opposing studies stating that CIH inhibits
the recovery of left ventricular function and cardioprotection
(27,28). To investigate this phenomenon in
the present study, rats and H9c2 cells were exposed to CIH and
their resistance to acute I/R injury was evaluated both in
vivo and ex vivo. Using these models, CIH was shown to
decrease the cardiac infarct size after I/R. Furthermore, CIH was
found to improve the post-ischemic recovery of LV function in
Langendorff rat hearts, as shown by increased LVDP (Fig. 1) and decreased LDH release into
the perfusate (Fig. 2), and also
to preserve post-I/R cardiac function as assessed by EF or FS and
CK-MB activity in an in vivo rat model (Fig. 2). These data suggest that CIH
confers resistance to I/R injury, although the precise mechanism
remains to be determined.
CIH is known to induce local and systemic adaptive
responses, including increased blood hemoglobin concentrations,
mitochondrial metabolic adaptation (29) and ER stress (30). It has also been shown that chronic
hypoxia without reoxygenation upregulates the unfolded protein
response in the mouse myocardium (31). ATF6 is activated in response to ER
stress, and although some studies demonstrate that ATF6 plays a
potentially cardioprotective role (17), other studies have concentrated on
the protective effects exerted in acute ischemia (23,32) (such as myocardial infarction) or
by mimicking ATF6 activation in transgenic mouse hearts (17). Studies of ATF6 in cardiomyocytes
cultured under hypoxic conditions are rare (30,33), and the status of ATF6 activation
by CIH has not been examined either in animals. ATF6α has been
shown to optimize long-term ER function to protect cells against
chronic stress (16), and our
previous study revealed that hypoxia induced ATF6 and GRP78
expression in primary human cardiomyocytes and H9c2 cells (18). In the present study, cleaved ATF6
protein expression levels were shown to increase nearly 3-fold in
rat hearts after exposure to CH, with no significant differences
being noted between the LV and RV (Fig. 3). Therefore, we hypothesized that
ATF6 exerts cardioprotective effects against myocardial I/R injury
caused by CIH. To test this hypothesis, we examined cleaved ATF6
expression in H9c2 cells subjected to mild hypoxia, as previously
described (8). Endogenous cleaved
ATF6 expression was induced by chronic mild hypoxia in a
time-dependent manner, with peak levels detected after 48 h and
maintained up to 72 h (Fig. 4).
Subsequently, we used RNA interference (RNAi) technology to
suppress cleaved ATF6 expression in transfected H9c2 cells. After
48 h of exposure to mild hypoxia, cleaved ATF6 was significantly
decreased in siRNA-ATF6 transfected H9c2 cells. Furthermore, after
1-h simulated ischemia followed by 3-h reperfusion (25), CCK-8 analysis showed a significant
decrease in siRNA-ATF6-transfected H9c2 cell survival compared with
that of siRNA-control-transfected cells, with a concomitant
increase in LDH activity as a marker of cellular necrosis. These
observations confirmed that endogenous ATF6 plays a vital,
protective role against I/R injury induced by CIH.
The results of the present study in relation to ATF6
expression were supported by investigations into the expression of
BiP. BiP is involved in the action of ATF6 and plays an important
role in initiating adaptive ER stress and inhibiting apoptotic ER
stress (34). Androgen induces
BiP to dissociate from ATF6 and act as an androgen
receptor-interacting protein, suggesting that BiP is a regulator of
androgen receptor protein quality control (35). Thus, we would expect BiP
expression levels to increase with the 50-kDa cleaved form of ATF6.
We found that the expression of BiP in this study was similar to
the expression pattern of cleaved ATF6, supporting our hypothesis
on the role of ATF6.
The Akt signaling pathway is involved in the
regulation of growth, proliferation, protein translation and cell
survival in various cell types (36). It has been reported that the ER
stress transducer, protein kinase RNA-like ER kinase (PERK) is
regulated by phosphorylation mediated by the Akt signaling pathway
(37,38). Lin et al (39) demonstrated that Akt is the
downstream target of BiP in mediating cisplatin resistance in ER
stress-tolerant human lung cancer cells. Furthermore, a previous
study using RNAi technology suggested that Akt activation is
associated with the IRE1α and ATF6 pathways of the ER stress
response (40). The activation of
ATF6 and PERK contributes to the pro-survival effects of vascular
endothelial growth factor on endothelial cells by positively
regulating mTORC2-mediated phosphorylation of Akt (23). Collectively, these observations
indicate that Akt is regulated by ATF6 in crosstalk between the two
signaling pathways; however, there is little reported evidence in
relation to the heart. Therefore, based on the results of the
present study, we hypothesized that ATF6 is involved in alleviating
cardiac I/R damage mediated by CIH partly through regulation of the
Akt pathway. To test this hypothesis, we also examined Akt
expression in H9c2 cell lines subjected to mild hypoxia. We found
that endogenous Akt was activated, and the expression of p-Akt
increased, which is consistent with previous findings (30). Similarly, after 48 h of exposure
to mild hypoxia, p-Akt levels were significantly suppressed in
siRNA-ATF6-transfected H9c2 cells compared with those transfected
with the siRNA-control. These data indicate that ATF6 contributes
to cardioprotection by regulating p-Akt. Akt is activated by the
phosphorylation of Thr308 and Ser473. Activated Akt exerts
cardioprotective effects by phosphorylating multiple targets in the
cytoplasm, nucleus, mitochondria and on the surface of the ER
membrane, including glycogen synthase kinase-3β (41), cardiac mTOR (3), nuclear factor-κB (42), the pro-apoptotic Bcl-2 family
member BAD (43) and caspase-9
(44). However, the detailed
mechanism by which ATF6 regulates Akt remains to be clarified.
In conclusion, using in vivo and ex
vivo models, we have demonstrated for the first time to the
best of our knowledge, that CIH protects the myocardium from I/R
injury by upregulating ATF6 through a mechanism potentially
involving the Akt pathway. These observations likely explain the
mechanism underlying the low morbidity rate of patients with
chronic ischemic heart disease and the lower rate of mortality due
to myocardial infarction in populations living at high altitudes
(1), which is consistent with our
clinical observations. Furthermore, ATF6 has been implicated as a
molecular target in interventions aimed at alleviating I/R injury
in patients experiencing a cardiovascular event.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (nos. 81270228 and 81370004).
References
|
1
|
Faeh D, Gutzwiller F and Bopp M; Swiss
National Cohort Study Group: Lower mortality from coronary heart
disease and stroke at higher altitudes in Switzerland. Circulation.
120:495–501. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hu Y, Sun Q, Li Z, Chen J, Shen C, Song Y
and Zhong Q: High basal level of autophagy in high-altitude
residents attenuates myocardial ischemia-reperfusion injury. J
Thorac Cardiovasc Surg. 148:1674–1680. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neckár J, Papousek F, Nováková O, Ost'ádal
B and Kolár F: Cardioprotective effects of chronic hypoxia and
ischaemic preconditioning are not additive. Basic Res Cardiol.
97:161–167. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Naghshin J, McGaffin KR, Witham WG,
Mathier MA, Romano LC, Smith SH, Janczewski AM, Kirk JA, Shroff SG
and O'Donnell CP: Chronic intermittent hypoxia increases left
ventricular contractility in C57BL/6J mice. J Appl Physiol.
107:787–793. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Naghshin J, Rodriguez RH, Davis EM, Romano
LC, McGaffin KR and O'Donnell CP: Chronic intermittent hypoxia
exposure improves left ventricular contractility in transgenic mice
with heart failure. J Appl Physiol. 113:791–798. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Asemu G, Papousek F, Ostádal B and Kolár
F: Adaptation to high altitude hypoxia protects the rat heart
against ischemia-induced arrhythmias. Involvement of mitochondrial
K(ATP) channel. J Mol Cell Cardiol. 31:1821–1831. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Borchert GH, Yang C and Kolár F:
Mitochondrial BKCa channels contribute to protection of
cardiomyocytes isolated from chronically hypoxic rats. Am J Physiol
Heart Circ Physiol. 300:H507–H513. 2011. View Article : Google Scholar :
|
|
8
|
Crawford RM, Jovanović S, Budas GR, Davies
AM, Lad H, Wenger RH, Robertson KA, Roy DJ, Ranki HJ and Jovanović
A: Chronic mild hypoxia protects heart-derived H9c2 cells against
acute hypoxia/reoxygenation by regulating expression of the SUR2A
subunit of the ATP-sensitive K+ channel. J Biol Chem.
278:31444–31455. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kolár F, Jezková J, Balková P, Breh J,
Neckár J, Novák F, Nováková O, Tomásová H, Srbová M, Ostádal B, et
al: Role of oxidative stress in PKC-delta upregulation and
cardioprotection induced by chronic intermittent hypoxia. Am J
Physiol Heart Circ Physiol. 292:H224–H230. 2007. View Article : Google Scholar
|
|
10
|
Baker JE, Holman P, Kalyanaraman B,
Griffith OW and Pritchard KA Jr: Adaptation to chronic hypoxia
confers tolerance to subsequent myocardial ischemia by increased
nitric oxide production. Ann NY Acad Sci. 874:236–253. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morel S, Milano G, Ludunge KM, Corno AF,
Samaja M, Fleury S, Bonny C, Kappenberger L, von Segesser LK and
Vassalli G: Brief reoxygenation episodes during chronic hypoxia
enhance posthypoxic recovery of LV function: role of
mitogen-activated protein kinase signaling pathways. Basic Res
Cardiol. 101:336–345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Strnisková M, Ravingerová T, Neckár J,
Kolár F, Pastoreková S and Barancík M: Changes in the expression
and/or activation of regulatory proteins in rat hearts adapted to
chronic hypoxia. Gen Physiol Biophys. 25:25–41. 2006.PubMed/NCBI
|
|
13
|
Bianciardi P, Fantacci M, Caretti A,
Ronchi R, Milano G, Morel S, von Segesser L, Corno A and Samaja M:
Chronic in vivo hypoxia in various organs: hypoxia-inducible
factor-1alpha and apoptosis. Biochem Biophys Res Commun.
342:875–880. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kakinuma Y, Tsuda M, Okazaki K, Akiyama T,
Arikawa M, Noguchi T and Sato T: Heart-specific overexpression of
choline acetyltransferase gene protects murine heart against
ischemia through hypoxia-inducible factor-1α-related defense
mechanisms. J Am Heart Assoc. 2:e0048872013. View Article : Google Scholar
|
|
15
|
Belmont PJ, Tadimalla A, Chen WJ,
Martindale JJ, Thuerauf DJ, Marcinko M, Gude N, Sussman MA and
Glembotski CC: Coordination of growth and endoplasmic reticulum
stress signaling by regulator of calcineurin 1 (RCAN1), a novel
ATF6-inducible gene. J Biol Chem. 283:14012–14021. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu J, Rutkowski DT, Dubois M, Swathirajan
J, Saunders T, Wang J, Song B, Yau GD and Kaufman RJ: ATF6alpha
optimizes long-term endoplasmic reticulum function to protect cells
from chronic stress. Dev Cell. 13:351–364. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martindale JJ, Fernandez R, Thuerauf D,
Whittaker R, Gude N, Sussman MA and Glembotski CC: Endoplasmic
reticulum stress gene induction and protection from
ischemia/reperfusion injury in the hearts of transgenic mice with a
tamoxifen-regulated form of ATF6. Circ Res. 98:1186–1193. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jian Z, Li JB, Ma RY, Chen L, Wang XF and
Xiao YB: Pivotal role of activating transcription factor 6α in
myocardial adaptation to chronic hypoxia. Int J Biochem Cell Biol.
44:972–979. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fujio Y, Nguyen T, Wencker D, Kitsis RN
and Walsh K: Akt promotes survival of cardiomyocytes in vitro and
protects against ischemia-reperfusion injury in mouse heart.
Circulation. 101:660–667. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jacinto E, Facchinetti V, Liu D, Soto N,
Wei S, Jung SY, Huang Q, Qin J and Su B: SIN1/MIP1 maintains
rictor-mTOR complex integrity and regulates Akt phosphorylation and
substrate specificity. Cell. 127:125–137. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cai Z and Semenza GL:
Phosphatidylinositol-3-kinase signaling is required for
erythropoietin-mediated acute protection against myocardial
ischemia/reperfusion injury. Circulation. 109:2050–2053. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matsui T, Tao J, del Monte F, Lee KH, Li
L, Picard M, Force TL, Franke TF, Hajjar RJ and Rosenzweig A: Akt
activation preserves cardiac function and prevents injury after
transient cardiac ischemia in vivo. Circulation. 104:330–335. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Karali E, Bellou S, Stellas D, Klinakis A,
Murphy C and Fotsis T: VEGF Signals through ATF6 and PERK to
promote endothelial cell survival and angiogenesis in the absence
of ER stress. Mol Cell. 54:559–572. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gardner BM, Pincus D, Gotthardt K,
Gallagher CM and Walter P: Endoplasmic reticulum stress sensing in
the unfolded protein response. Cold Spring Harb Perspect Biol.
5:a0131692013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ren XP, Wu J, Wang X, Sartor MA, Qian J,
Jones K, Nicolaou P, Pritchard TJ and Fan GC: MicroRNA-320 is
involved in the regulation of cardiac ischemia/reperfusion injury
by targeting heat-shock protein 20. Circulation. 119:2357–2366.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujii Y, Ishino K, Tomii T, Kanamitsu H,
Mitsui H and Sano S: Tolerance of the developing cyanotic heart to
ischemiareper-fusion injury in the rat. Gen Thorac Cardiovasc Surg.
58:174–181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Joyeux-Faure M, Stanke-Labesque F,
Lefebvre B, Beguin P, Godin-Ribuot D, Ribuot C, Launois SH, Bessard
G and Levy P: Chronic intermittent hypoxia increases infarction in
the isolated rat heart. J Appl Physiol. 98:1691–1696. 2005.
View Article : Google Scholar
|
|
28
|
Milano G, Corno AF, Samaja M, Morel S,
Vassalli G and von Segesser LK: Daily reoxygenation decreases
myocardial injury and improves post-ischaemic recovery after
chronic hypoxia. Eur J Cardiothorac Surg. 37:942–949. 2010.
View Article : Google Scholar
|
|
29
|
Heather LC, Cole MA, Tan JJ, Ambrose LJ,
Pope S, Abd-Jamil AH, Carter EE, Dodd MS, Yeoh KK, Schofield CJ and
Clarke K: Metabolic adaptation to chronic hypoxia in cardiac
mitochondria. Basic Res Cardiol. 107:2682012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thuerauf DJ, Marcinko M, Gude N, Rubio M,
Sussman MA and Glembotski CC: Activation of the unfolded protein
response in infarcted mouse heart and hypoxic cultured cardiac
myocytes. Circ Res. 99:275–282. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tagliavacca L, Caretti A, Bianciardi P and
Samaja M: In vivo up-regulation of the unfolded protein response
after hypoxia. Biochim Biophys Acta. 1820:900–906. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Doroudgar S, Thuerauf DJ, Marcinko MC,
Belmont PJ and Glembotski CC: Ischemia activates the ATF6 branch of
the endoplasmic reticulum stress response. J Biol Chem.
284:29735–29745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Terai K, Hiramoto Y, Masaki M, Sugiyama S,
Kuroda T, Hori M, Kawase I and Hirota H: AMP-activated protein
kinase protects cardiomyocytes against hypoxic injury through
attenuation of endoplasmic reticulum stress. Mol Cell Biol.
25:9554–9575. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang L, Tang W, Jiang T, Lu P, Li Y, Sun
A, Shen Y, Chen Y, Wang H, Zong Z, et al: Endoplasmic reticulum
stress is involved in the neuroprotective effect of propofol.
Neurochem Res. 39:1741–1752. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang YC, Fu HC, Hsiao BL, Sobue G, Adachi
H, Huang FJ, Hsuuw YD, Wei KT, Chang C, Huang KE and Kang HY:
Androgen receptor inclusions acquire GRP78/BiP to ameliorate
androgen-induced protein misfolding stress in embryonic stem cells.
Cell Death Dis. 4:e6072013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Blaustein M, Pérez-Munizaga D, Sánchez MA,
Urrutia C, Grande A, Risso G, Srebrow A, Alfaro J and Colman-Lerner
A: Modulation of the Akt pathway reveals a novel link with
PERK/eIF2α, which is relevant during hypoxia. PLoS One.
8:e696682013. View Article : Google Scholar
|
|
38
|
Hu P, Han Z, Couvillon AD and Exton JH:
Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in
counteracting endoplasmic reticulum stress-induced cell death. J
Biol Chem. 279:49420–49429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin Y, Wang Z, Liu L and Chen L: Akt is
the downstream target of GRP78 in mediating cisplatin resistance in
ER stress-tolerant human lung cancer cells. Lung Cancer.
71:291–297. 2011. View Article : Google Scholar
|
|
40
|
Jiang CC, Yang F, Thorne RF, Zhu BK,
Hersey P and Zhang XD: Human melanoma cells under endoplasmic
reticulum stress acquire resistance to microtubule-targeting drugs
through XBP-1-mediated activation of Akt. Neoplasia. 11:436–447.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang Y, Xia Z, La Cour KH and Ren J:
Activation of Akt rescues endoplasmic reticulum stress-impaired
murine cardiac contractile function via glycogen synthase
kinase-3β-mediated suppression of mitochondrial permeation pore
opening. Antioxid Redox Signal. 15:2407–2424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Romashkova JA and Makarov SS: NF-kappaB is
a target of AKT in anti-apoptotic PDGF signalling. Nature.
401:86–90. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Aikawa R, Nawano M, Gu Y, Katagiri H,
Asano T, Zhu W, Nagai R and Komuro I: Insulin prevents
cardiomyocytes from oxidative stress-induced apoptosis through
activation of PI3 kinase/Akt. Circulation. 102:2873–2879. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cardone MH, Roy N, Stennicke HR, Salvesen
GS, Franke TF, Stanbridge E, Frisch S and Reed JC: Regulation of
cell death protease caspase-9 by phosphorylation. Science.
282:1318–1321. 1998. View Article : Google Scholar : PubMed/NCBI
|