Introduction
Since the successful isolation of endothelial
progenitor cells (EPCs) by Asahara et al in 1997 (1), EPCs have received increasing
attention in the field of research on endothelial injury and
repair. As the origins of vascular endothelial cells (ECs), EPCs
repair damaged endothelium by directly differentiating into mature
ECs and indirectly secreting paracrine cytokines that promote the
proliferation of ECs (2,3). To date, EPC transplantation has been
used to treat atherosclerosis and coronary heart disease caused by
endothelial injuries (4–6). However, due to the limited
proliferative capacity of EPCs, this strategy often leads to
insufficient efficacy (7,8). Therefore, enhancing the
proliferative ability of EPCs is a key step in improving the
effects of EPC transplantation therapy.
The transcription factor E2-2, also referred to as
transcription factor 4 (TCF4) or class A basic helix-loop-helix
(bHLH) transcription factor, is a member of the E-protein family
that is universally expressed in mammalian cells. E2-2 regulates
transcription by binding to the E-box binding site in the promoter
and enhancer regions of various genes, including nerve-, pancreas-
and tumor-specific genes (9,10).
It also promotes the proliferation of human liver cancer cells and
dermal papilla cells through activating the Wnt/β-catenin signaling
pathway (11,12). By contrast, another study has
shown that knockout of E2-2 increased the proliferative and
vessel-forming capacities of mature ECs in mice (13). However, the role and mechanism by
which E2-2 regulates the proliferation of EPCs remains unclear.
Autophagy is a highly conserved metabolic process in
eukaryotes, and one which is essential for maintaining cellular
homeostasis. It has been previously reported that regulation of
autophagy affects the proliferation of tumor cells (14), nerve cells (15) and vascular smooth muscle cells
(16). A recent study has
revealed that inhibition of basal autophagy by 3-methyl-adenine
(3-MA) decreases the growth ability of human EPCs and prevents
their differentiation into mature ECs, while hypoxia-induced
autophagy increased the growth and differentiation of human EPCs,
which led to an increased survival rate of EPCs under hypoxic
conditions (17). In recent
years, E2-2 has been shown to suppress autophagy by activating the
Wnt/β-catenin signaling pathway in tumor cells (18). However, whether E2-2 regulates
levels of autophagy in EPCs and whether levels of autophagy affect
the proliferation of EPCs have not been previously reported.
In the present study, we firstly demonstrated that
inhibition of E2-2 expression significantly increased the
proliferative ability of EPCs through its effects on autophagy.
Moreover, we found that E2-2 downregulated the autophagy level by
decreasing the expression of AGT7. These findings provide a new
insight into EPC proliferation, as we have targeted the
'E2-2/autophagy related 7 (ATG7)/autophagy' pathway, which will
benefit EPC transplantation therapy used to treat atherosclerosis
and coronary heart disease caused by endothelial injury.
Materials and methods
Isolation and characterization of
EPCs
All animal experiments were approved by the Center
of Experimental Animals Committee of Xinqiao Hospital (Chongqing,
China). The male C57BL/6J mice (6–8 weeks, 18–20 g, from Xinqiao
Hospital Experimental Animal Center, Chongqing, China) were
sacrificed by cervical dislocation, and spleens were explanted and
thoroughly minced. Spleen-derived mononuclear cells were isolated
by density gradient centrifugation (Histopaque 1083; Sigma-Aldrich,
St. Louis, MO, USA) at 400 × g for 20 min in 4°C. The cells were
collected, washed in phosphate-buffered saline (PBS) three times,
and then resuspended in Dulbecco's modified Eagle's medium/nutrient
mixture F12 (DMEM/F12; Gibco BRL, Gaithersburg, MD, USA)
supplemented with 20% fetal calf serum (FCS; HyClone, Los Angeles,
CA, USA), 20 ng/ml vascular endothelial growth factor (VEGF;
R&D Systems, Inc., Minneapolis, MN, USA), 100 U/ml penicillin
and 100 g/ml streptomycin. Cells were seeded into gelatin-coated
cell culture flasks and incubated at 37°C under an atmosphere with
5% CO2. Forty-eight hours later, non-attached cells were
removed and adherent cells were cultured continuously. Only
adherent cells were used in further experiments. The medium was
refreshed with complete medium every 2 or 3 days.
To verify the phenotype of EPCs, cells were
incubated with Dil-ac-LDL (Biomedical Technologies, Inc.,
Stoughton, MA, USA) for 3 h, fixed with 4% paraformaldehyde and
then incubated with fluorescein isothiocyanate (FITC)-labeled
lectin (UEA-I; Sigma-Aldrich) for 1 h, then washed with PBS three
times. The cells were then observed under an immunofluorescence
laser scanning confocal microscope (Leica TCS; Leica, Mannheim,
Germany). Dual-stained cells positive for Dil-ac-LDL and UEA-I were
identified as EPCs. Additionally, fluorescence-activated cell
sorting (FACS) analysis was performed using FITC-conjugated
antibodies against mouse Sca-1 (ab25031) and vascular endothelial
growth factor receptor 2 (VEGFR2; ab11939), and the corresponding
isotype control antibodies (ab18446, ab171870; all from Abcam,
Cambridge, UK).
Small interfering RNA (siRNA)-mediated
silencing of genes
Transient silencing of E2-2 and ATG7 was induced by
transfection with siRNAs (siRNA-E2-2 and siRNA-ATG7, respectively)
(both from GenePharma, Shanghai, China). The selected siRNA duplex
sequences specifically targeted mouse E2-2 and ATG7, and showed no
homology to any other sequences during a BLAST search. The
siRNA-E2-2 efficient sequence was: sense, 5′-CAAGAAGGAUAUCAAAUCA-3′
and antisense, 5′-UGAUUUGAUAUCCUUCUUG-3′. The siRNA-ATG7 sequence
was: sense, 5′-GGAGUCACAGCUCUUCCUUTT-3′ and antisense,
5′-AAGGAAGAGCUGUGACUCCTT-3′. The corresponding non-silencing
control (siRNA-con) sequences (GenePharma) were designed according
to the sequence of a negative control. Transfection with siRNA-E2-2
or siRNA-ATG7 was carried out using Lipofectamine 2000 reagent
(Invitrogen, Carlsbad, CA, USA) with a molar ratio between DNA and
lipid of approximately 1:3. Forty-eight hours after transfection,
cells were collected and used for subsequent assays.
Adenovirus transfection
Adenovirus vector expressing E2-2 (Ad-E2-2) and
adenovirus-carrying blank vector (Ad-vector) were both constructed
by HanBio Technology Co., Ltd. (Shanghai, China). Before
transfection, EPCs were incubated in DMEM/F12 medium without FCS
and antibiotics for 6 h. Adenovirus vector was then added to the
cells at a multiplicity of infection (MOI) of 100. The transfection
medium was removed 2 h later, and the cells were maintained in the
complete medium for 48 h. Ad-vector was used as a control. The
mRFP-GFP-LC3 adenovirus vector for detecting autophagy was also
purchased from HanBio Technology Co., Ltd. The process of
mRFP-GFP-LC3 adenovirus transfection was similar to that of
Ad-E2-2. Autophagy was observed under an immunofluorescence laser
scanning confocal microscope (Leica TCS). Autophagy was detected by
counting the number of green fluorescent protein (GFP) and red
fluorescent protein (RFP) dots (dots/cell).
Cell proliferation assay
EPCs were collected from the cultures and replated
into a 96-well plate (1×105 cells/ml) and underwent
different treatments. Cell proliferation was measured using a Cell
Counting Kit-8 (CCK-8; Beyotime Biotech, Jiangsu, China) according
to the manufacturer's instructions. Before reading the absorbance
at 450 nm, 10 µl CCK-8 solution and 100 µl complete
medium were sequentially added to each well.
In addition, the cell cycle was analyzed by flow
cytometric analysis. EPCs were harvested and resuspended in PBS
solution at 1×106 cells/ml. The cells were then stored
in 70% ethanol at 4°C overnight. Before being analyzed, the cells
were mixed with 1 ml propidium iodide (PI; 1 mg/ml) (BD
Biosciences, Bedford, MA, USA) and then incubated at 4°C for 30
min. After centrifugation, the cells were resuspended and the cell
cycle was analyzed immediately using a flow cytometer (MoFloTMXDP;
Beckman Coulter, Co. Fullerton, CA, USA). The proliferation index
was defined as the total percentage of cells in S and G2/M phases.
All groups of experiments were undertaken in triplicate.
Western blot analysis
EPCs were lysed in ice-cold lysis buffer (RIPA)
containing a protease inhibitor (0.5 mM PMSF) (both from Beyotime
Biotech). The cell lysates were further centrifuged at 15,000 × g
for 15 min at 4°C. Proteins from EPC lysates were measured using
the Bradford method. The same amount of protein was loaded in each
lane, separated by 10–15% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE), and transferred to 0.22 µm
polyvinylidene difluoride (Millipore Corp., Boston, MA, USA)
membranes. Membranes were blocked with 5% non-fat milk 2 h followed
by incubated with primary antibodies overnight at 4°C. The primary
antibodies included: rabbit anti-mouse LC3 polyclonal antibody
(1:1,000; Cat. no. 2775), rabbit anti-mouse p62 monoclonal antibody
(1:1,000; Cat. no. 5114), rabbit anti-mouse ATG7 polyclonal
antibody (1:1,000; Cat. no. 8558) (all from Cell Signaling
Technology, Inc., Boston, MA, USA) and rat anti-mouse β-actin
monoclonal antibody (1:1,000; Cat. no. AA128; Beyotime Biotech).
The membranes were then incubated with horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:5,000; A0208, A0216;
Beyotime Biotech) for 1 h at 37°C. Protein bands were visualized by
enhanced chemiluminescence (ECL) (Thermo Fisher Scientific,
Waltham, MA, USA), and densitometric signals were quantified using
ImageQuant TL software (General Electric, Co., Fairfield, CT,
USA).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from EPCs using RNAiso Plus
(Takara Bio, Dalian, China) according to the manufacturer's
instructions. Single cDNAs was synthesized from RNA using a
PrimeScript™ RT reagent kit (Takara Bio). The following primer
sequences were used for ATG7 (sense, 5′-ACCCAGAAGAAGCTGAACGA-3′,
and antisense, 5′-CTCATTTGCTGCTTGTTCCA-3′); β-actin (sense,
5′-TTCTACAATGAGCTGCGTGTGG-3′ and anti-sense,
5′-GTGTTGAAGGTCTCAAACATGAT-3′). SYBR® Premix Ex Taq™ II
(Takara Bio) was used according to the manufacturer's instructions,
and RT-qPCR was performed using a StepOnePlus™ Real-Time PCR system
(Applied Biosystems Life Technologies, Waltham, MA, USA). The
reaction conditions were as follows: 95°C for 30 sec, 40 cycles of
95°C for 5 sec, and 60°C for 30 sec. Relative expression of target
mRNA was normalized to β-actin using the ΔΔCt method, as previously
described (19).
Statistical analysis
Data from at least three independent experiments are
expressed as the means ± SD. SPSS 16.0 software was used for
statistical analysis. Student's t-tests were used for comparisons
between groups. A P-value <0.01, or P<0.05 was considered to
indicate a statistically significant difference.
Results
Biological properties of EPCs
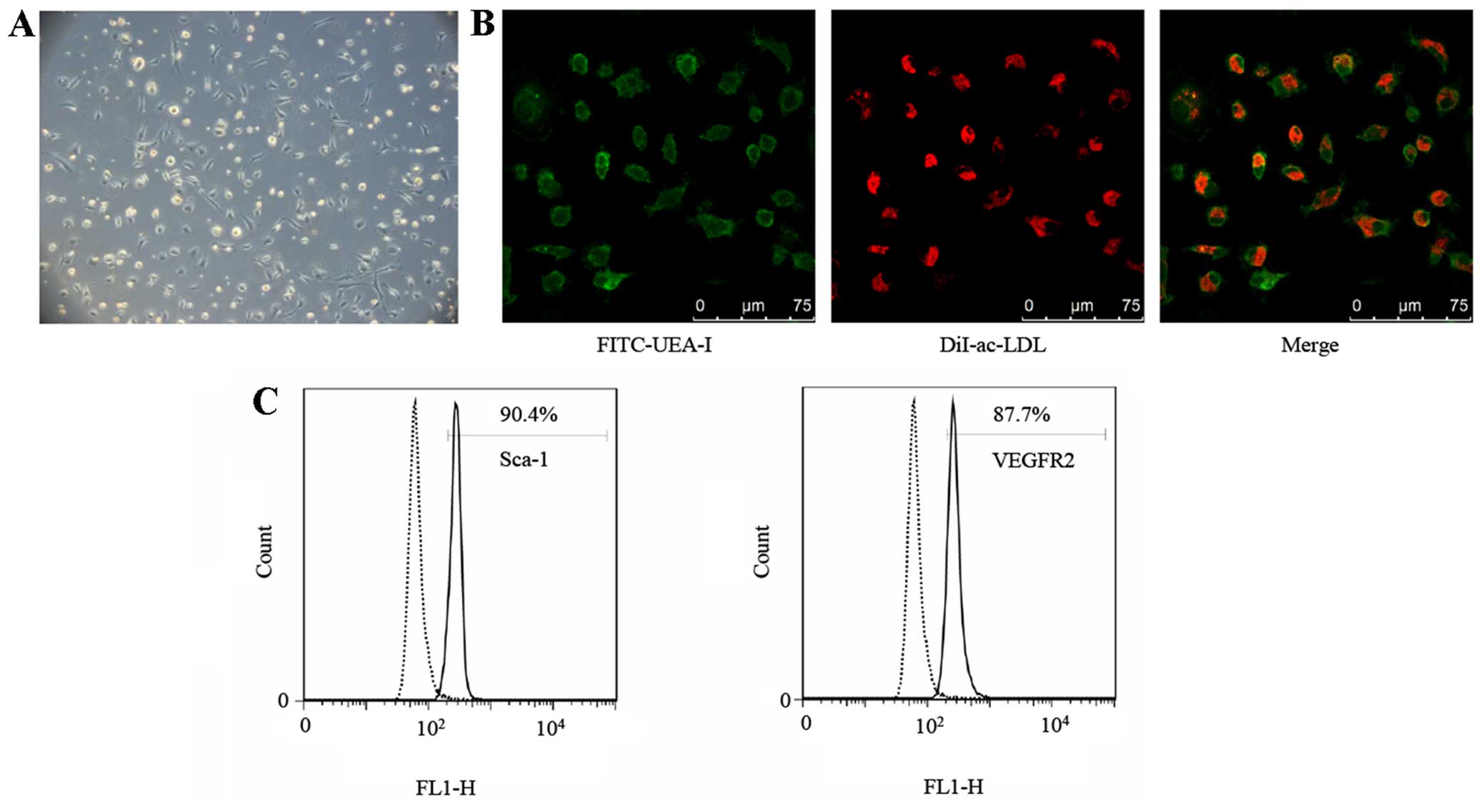
After 7 days of being cultured, round, oval and
spindle shapes of attached spleen-derived mono-nuclear cells were
noted under the microscope (Fig.
1A), similar to morphologies which have been previously
described (20).
Immunocytochemical (ICC) staining demonstrated that 92.33±2.63%
(n=3) of attached cells were stained positively for FITC-UEA-I and
Dil-ac-LDL (Fig. 1B), validating
the theory that the FITC-UEA-I/Dil-ac-LDL double-positive cells
possessed the characteristics of ECs. Furthermore, the attached
cells were identified by analyzing the expression of the mouse
stem-cell marker Sca-1 and EC marker VEGFR2 using flow cytometry.
The percentage of positive cells was 88.93±1.31 and 85.13±2.83%
(n=3), respectively (Fig. 1C).
Taking these results into account, the attached cells were
considered EPCs.
E2-2 inhibits the proliferation of
EPCs
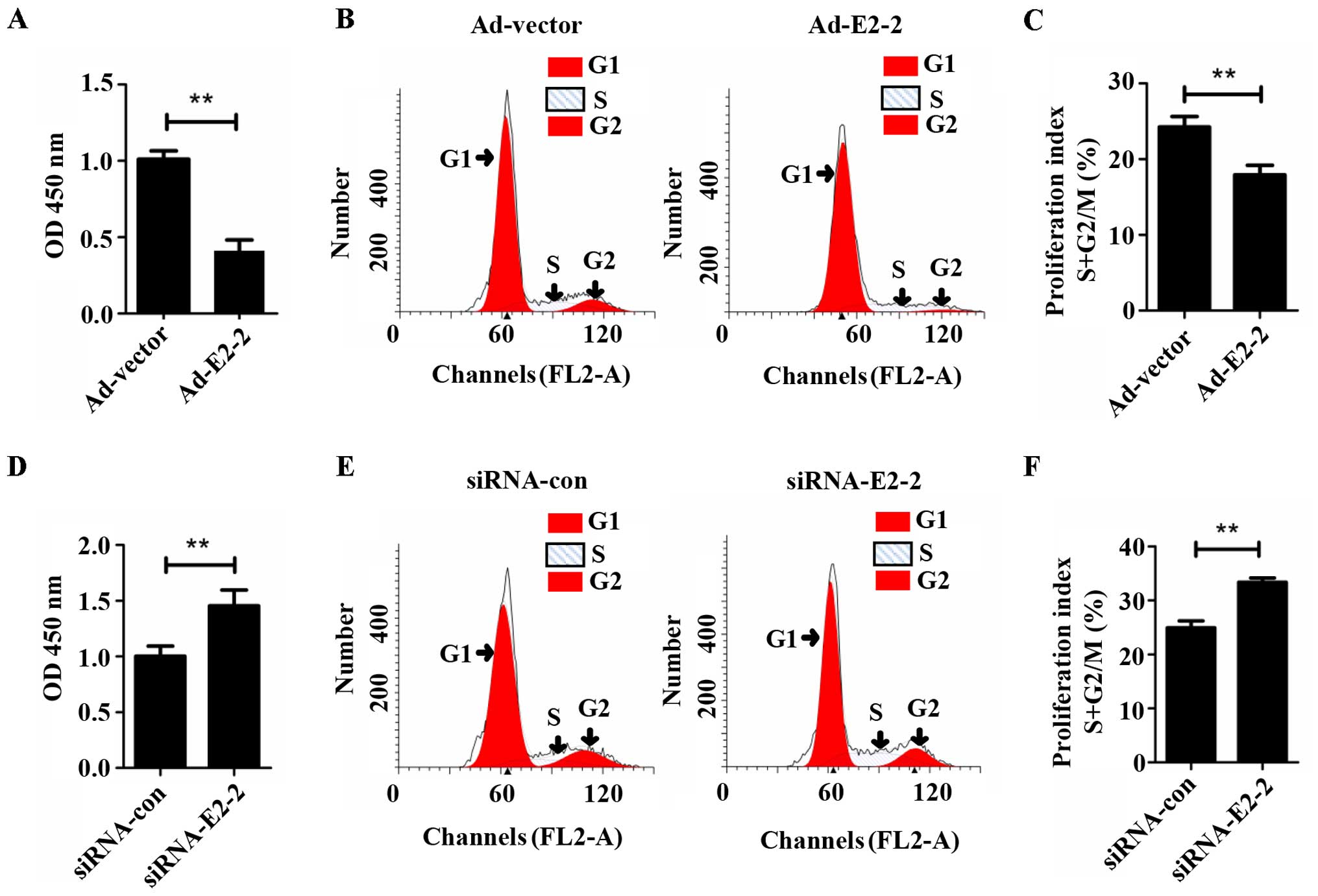
To determine whether E2-2 regulated the
proliferation of EPCs, we overexpressed or silenced E2-2 by
transfecting Ad-E2-2 or siRNA-E2-2 into the cells, and
proliferation was then detected by CCK-8 and cell cycle assays
in vitro. The CCK-8 assay demonstrated that the
proliferation of EPCs significantly decreased in the E2-2
overexpression (Ad-E2-2) group compared with the adenoviral control
vector (Ad-vector) group (Fig.
2A). Cell cycle analysis showed that the proliferation index (S
+ G2/M) of the EPCs was lower in the Ad-E2-2 group than in the
Ad-vector group (Fig. 2B and C).
The trend of the cell cycle was similar to the results of the CCK-8
assay. However, in the knockdown E2-2 (siRNA-E2-2) group, the
proliferative capacity of EPCs significantly increased compared
with that in the negative control (siRNA-con) group (Fig. 2D). The EPC proliferation index
(S+G2/M) of the siRNA-E2-2 group was higher than that of the
siRNA-con group (Fig. 2E and F).
These results suggest that E2-2 suppressed the proliferation of
EPCs in vitro.
E2-2 negatively regulates the autophagy
of EPCs
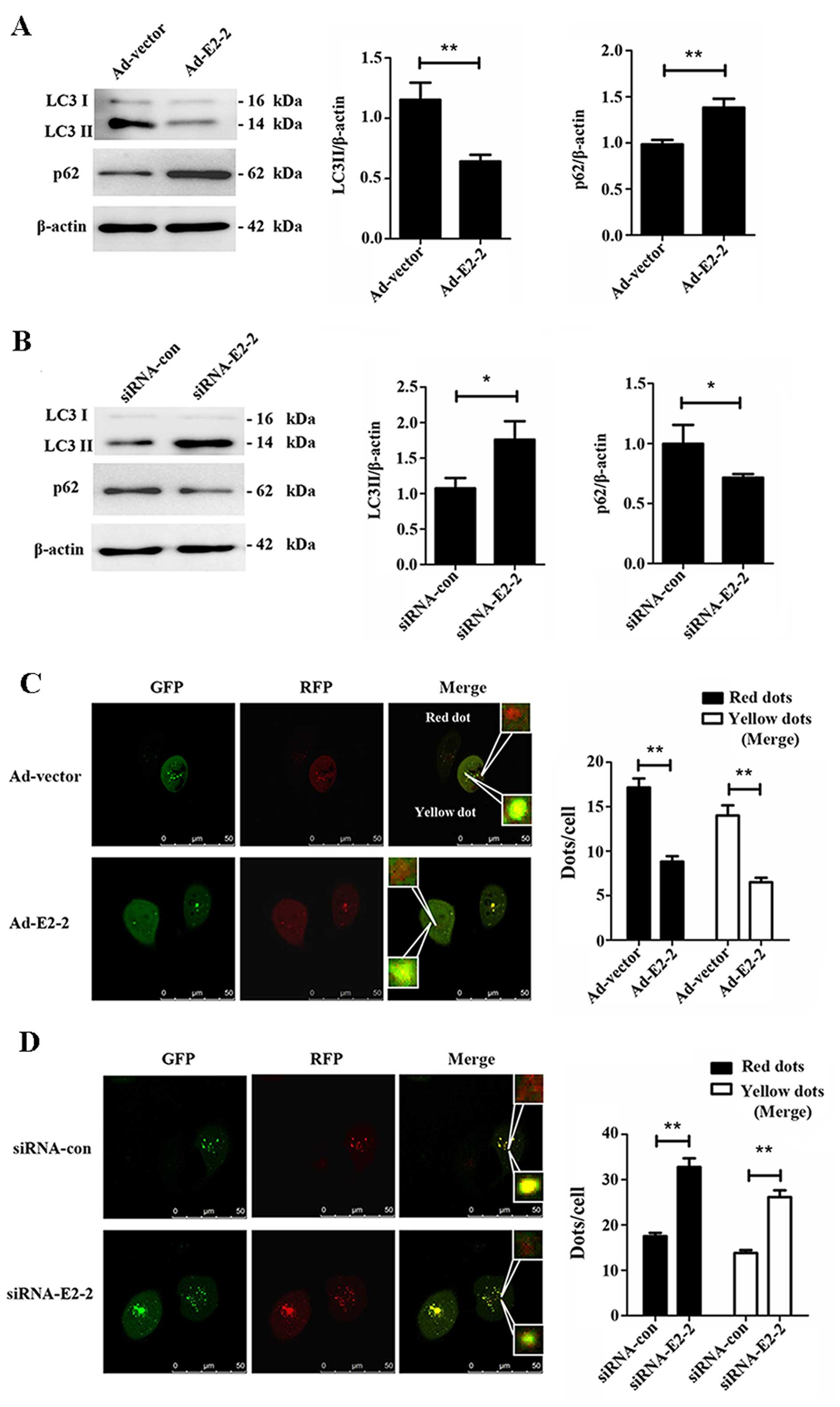
To investigate whether E2-2 regulates autophagy in
EPCs, we firstly studied the expression of LC3II and p62 while
overexpressing or knocking down E2-2. We noted a significant
decrease in LC3II expression, but a marked increase in the bands of
p62, in the Ad-E2-2 group compared with the Ad-vector group
(Fig. 3A), suggesting that
overexpression of E2-2 induced suppression of autophagy in EPCs. By
contrast, LC3II expression increased and the protein level of p62
decreased in the knockdown E2-2 group (Fig. 3B). Thus, knockdown of E2-2
upregulated autophagy of EPCs. In addition, we used the
mRFP-GFP-LC3 adenovirus to confirm the regulatory effects of E2-2
on autophagy. This assay is used to monitor progression from
autophagosome to autolysosome based on the pH difference between
the acidic autolysosome and the neutral autophagosome and the pH
sensitivity differences displayed by GFP and RFP. When
autolysosomes are formed by fusing autophagosome with a lysosome,
the GFP moiety degrades from the tandem protein, but RFP-LC3
maintains the dots (21,22). The red dots indicate
autophagosomes, and yellow dots (merge of RFP and GFP) represent
autolysosomes. When we co-transfected mRFP-GFP-LC3 adenovirus with
Ad-E2-2 or siRNA-E2-2 in EPCs for 48 h, we observed that
overexpression of E2-2 reduced the numbers of autophagosomes and
autolysosomes (Fig. 3C).
Conversely, knockdown of E2-2 increased not only the number of
autophagosomes but also that of autolysosomes (Fig. 3D). The results further confirmed
that E2-2 negatively regulated autophagy of EPCs.
E2-2 inhibits the proliferation of EPCs
via suppressing autophagy
To validate the role autophagy plays in EPC
proliferation, we treated EPCs with chloroquine (CQ) at different
concentrations and different periods of time. It was observed that
CQ inhibited the proliferation of EPCs in a concentration-dependent
manner. After treatment with CQ for 24 h at 20 µM, the
proliferation of EPCs as measured by CCK-8 decreased markedly
compared with the group which was not treated with CQ, and the
degree of decrease changed slightly with the increasing periods of
time (Fig. 4B). Subsequently, to
determine whether autophagy was involved in E2-2-mediated EPC
proliferation, we added 20 µM CQ to EPCs after transfection
with siRNA-E2-2 for 24 h. After transfection for 48 h, the CCK-8
assay showed that the increase in proliferation in the siRNA-E2-2
group was partially reversed by the administration of CQ (Fig. 4C). The alteration of the
proliferation index, as measured by flow cytometry, was similar to
the results of the CCK-8 assay (Fig.
4D). Furthermore, we transfected the different groups of EPCs
with siRNA-con, siRNA-E2-2 or siRNA-E2-2 and CQ and found that CQ
inhibited the increase in autophagosomes and autolysosomes
(Fig. 4E), suggesting that EPC
autophagy induced by E2-2 knockdown was reversed by CQ. These
results demonstrated that inhibition of basal autophagy reduced the
proliferation of EPCs, and E2-2 inhibited the proliferation of EPCs
through its effects on autophagy.
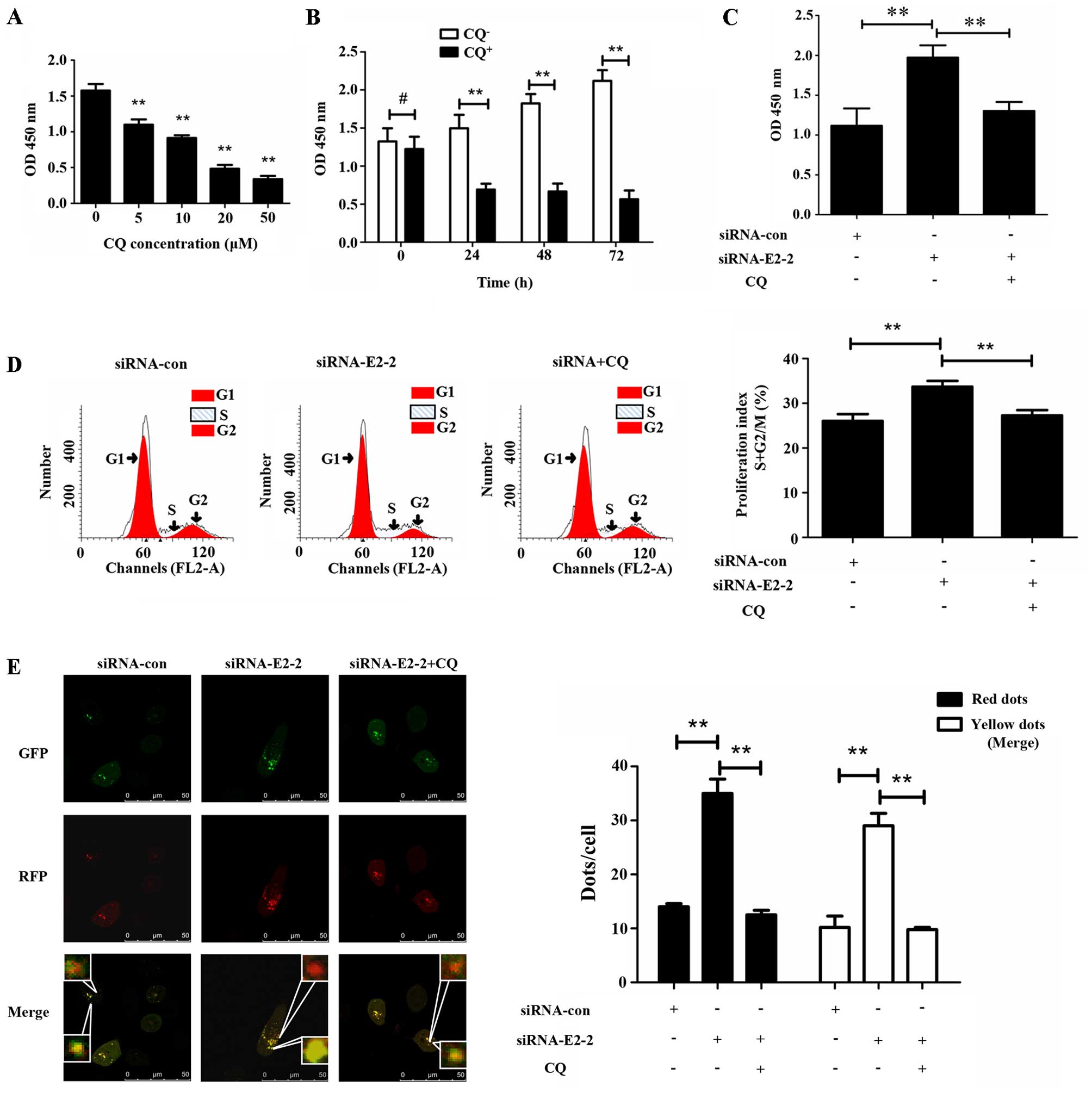 | Figure 4E2-2 inhibits the proliferation of
endothelial progenitor cells (EPCs) via suppressing autophagy. (A)
Chloroquine (CQ) was used to treat EPCs for 24 h. A CCK-8 assay
showed that the OD value significantly decreased at 5, 10, 20 and
50 µM (**P<0.01, n=3). (B) CQ-treated EPCs at
20 µM for different times. In the groups treated with CQ for
24, 48 and 72 h, the absorbance of EPCs was significantly reduced
compared with that in CQ-free group (**P<0.01, n=3).
No significant difference was observed in absorbance between
CQ-treated and CQ-free groups at 0 h (#P>0.05, n=3).
(C) Absorbance at 450 nm was detected by CCK-8 assay after 48 h
treatment of EPCs with siRNA-con, siRNA-E2-2, and siRNA-E2-2+CQ.
The OD value was significantly higher in the siRNA-E2-2 group than
in siRNA-con and siRNA+CQ groups (**P<0.01, n=3). (D)
Representative images of flow cytometry and statistical data were
indicative of distinct increases in proliferation index (S + G2/M)
in the siRNA-E2-2 group compared with the siRNA-con and siRNA+CQ
groups (**P<0.01, n=3). (E) Representative images of
immunofluorescence laser scanning using a confocal microscope and
statistical data from different groups. The numbers of red and
yellow dots per cell were significantly higher in the siRNA-E2-2
group compared to the siRNA-con and siRNA-E2-2+CQ groups
(**P<0.01, n=3; scale bar, 50 µm). |
E2-2 regulates autophagy of EPCs via
mediating the expression of ATG7
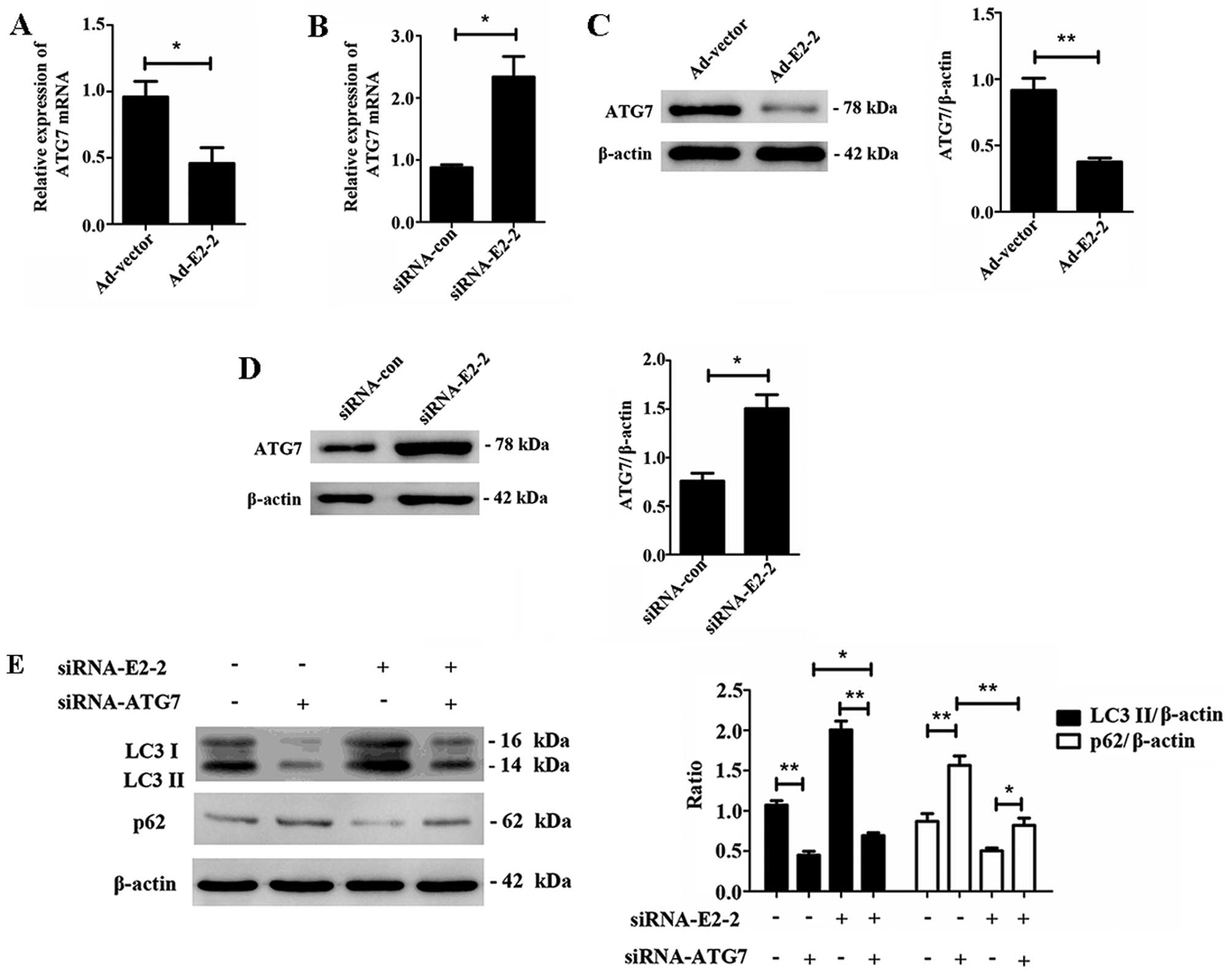
To further explore the mechanism by which E2-2
regulates autophagy in EPCs, we detected the expression of the
autophagy-related gene ATG7 after E2-2 overexpression or knockdown.
We found that overexpression of E2-2 decreased the expression level
of ATG7 mRNA (Fig. 5A), and
knockdown of E2-2 upregulated the expression of ATG7 mRNA (Fig. 5B). Subsequently, we noted that
overexpression of E2-2 downregulated ATG7 protein expression, and
knockdown of E2-2 upregulated ATG7 protein expression (Fig. 5C and D). Furthermore, to
demonstrate whether ATG7 was involved in E2-2-mediated autophagy,
we transfected the cells with siRNA-E2-2 and siRNA-ATG7
simultaneously or respectively. The transfection revealed that
autophagy of EPCs was downregulated by ATG7 knockdown alone, and
the increased autophagy induced by siRNA-E2-2 was reversed by ATG7
knockdown. Interestingly, the level of autophagy was still higher
in the co-transfected siRNA-E2-2 and siRNA-ATG7 group than in
siRNA-ATG7 group (Fig. 5E). We
demonstrated that E2-2 regulated autophagy of EPCs via partially
mediating the expression of ATG7.
Discussion
As a transcription factor, E2-2 binds to the
promoter regions and affects the expression of target proteins,
ultimately influencing several cellular biological functions,
including proliferation. In ECs, Bach1 binds to E2-2, which
represses the activation of Wnt/β-catenin signaling and decreases
the proliferative and vessel-forming abilities of the cells
(23). Tanaka et al
revealed that E2-2 suppressed the proliferation of vascular ECs via
binding to the promoter of VEGFR2. Knockdown of the E2-2 gene
significantly improved the proliferative and vessel-forming
abilities of ECs (13).
Consistent with these reports, our findings showed that E2-2
negatively regulated the proliferation of EPCs. However, another
study has shown that downregulation of E2-2 suppressed the
proliferation and survival of corneal epithelial stem cells
(24). Xiong et al
demonstrated that upregulation of E2-2 activated the Wnt/β-catenin
signaling pathway, initiating the proliferation of dermal papilla
cells (12). Therefore, we
consider that the mechanisms of E2-2 in the regulation of cell
proliferation are complicated, and may be associated with the
different cellular contents and microenvironment.
Autophagy is a highly conserved cell activity that
involves degradation of dysfunctional organelles and proteins. The
breakdown of cellular components promotes cellular survival by
maintaining cellular energy levels and homeostasis (25,26). Previously it has been confirmed
that autophagy plays an indispensable role in maintaining stem-cell
characteristics, self-renewal, and the capacity for differentiation
in various stem cells (27).
Moreover, previous research has demonstrated that upregulation of
autophagy promotes the proliferation of cancer cells (28), while downregulation of autophagy
decreases cell proliferation (29). In nerve cells, autophagy induced
by high mobility group box 1 protein (HMGB1) increased the growth
of neuroblasts (30). In immune
cells, inhibition of autophagy decreased cell growth and
differentiation and promoted apoptosis (31). By contrast, another study has
suggested that induction of autophagy suppressed cancer cell
proliferation (32). In the
present study, inhibition of autophagy at basal levels by the
inhibitor CQ decreased the proliferative ability of EPCs.
Therefore, we suggest that distinct cell types have different
levels of autophagy, thus generating various effects on cell growth
and survival. The underlying mechanism of action remains to be
explored.
In the present study, to the best of our knowledge,
we were the first to find that E2-2 inhibited the autophagy of EPCs
via decreasing the expression of ATG7. ATG7 is a gene that plays a
key role in autophagy and normal cell physiology (33,34). Previous studies showed that ATG7
is required for normal functioning of ECs via maintaining the basal
levels of autophagy (35). In
HeLa cells, inhibition of ATG7 suppressed the level of autophagy,
which decreased cell proliferation (36). Moreover, Lee et al
discovered that depletion of ATG7 suppressed autophagy and
inhibited the activation of p53 in mouse embryonic fibroblasts,
thus inducing cell cycle arrest and apoptosis (37). Our present study is thus the first
to elucidate that overexpression of E2-2 in mouse EPCs
downregulated ATG7 mRNA and protein levels, whereas knockdown of
E2-2 led to upregulation of ATG7 mRNA and protein levels. After
simultaneous or respective knockout of E2-2 and ATG7 with siRNAs,
we found that knockdown of ATG7 caused a decrease in autophagy
levels. Moreover, increased autophagy levels after knockdown of
E2-2 waspartially reversed by ATG7 knockdown. Our findings suggest
that in EPCs ATG7 mRNA and protein levels are regulated by E2-2.
Increased levels of autophagy after depletion of E2-2 may be
partially attributed to the enhanced transcription of ATG7.
Therefore, we noted the existence of the
E2-2/ATG7/autophagy signaling pathway and its regulatory effects on
EPC proliferation. However, the binding site of E2-2 with ATG7 and
the mechanism by which ATG7 transcription is regulated remain to be
explored in future research.
The present study attempted to clarify the mechanism
underlying EPC proliferation by investigating autophagy, and
provided new insights into research on EPCs and potential novel
therapies for repairing endothelial injury and treating
atherosclerosis and coronary heart disease.
Acknowledgments
The present study was supported by grants from the
National Natural and Science Foundation of China (NSFC) (no.
81270224). We thank Ms. Huali Kang, Ms. Chunxing Xu and Mr. Jie
Yang for their kind help (Department of Cardiology, Institute of
Cardiovascular Science of PLA, Xinqiao Hospital, Third Military
Medical University). Thanks to Professor Jihang Zhang (Department
of Cardiology, Institute of Cardiovascular Science of PLA, Xinqiao
Hospital, Third Military Medical University) for a critical reading
of the manuscript.
References
|
1
|
Asahara T, Murohara T, Sullivan A, Silver
M, van der Zee R, Li T, Witzenbichler B, Schatteman G and Isner JM:
Isolation of putative progenitor endothelial cells for
angiogenesis. Science. 275:964–967. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ii M, Nishimura H, Iwakura A, Wecker A,
Eaton E, Asahara T and Losordo DW: Endothelial progenitor cells are
rapidly recruited to myocardium and mediate protective effect of
ischemic preconditioning via 'imported' nitric oxide synthase
activity. Circulation. 111:1114–1120. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
George AL, Bangalore-Prakash P, Rajoria S,
Suriano R, Shanmugam A, Mittelman A and Tiwari RK: Endothelial
progenitor cell biology in disease and tissue regeneration. J
Hematol Oncol. 4(24)2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wassmann S, Werner N, Czech T and Nickenig
G: Improvement of endothelial function by systemic transfusion of
vascular progenitor cells. Circ Res. 99:e74–e83. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee PS and Poh KK: Endothelial progenitor
cells in cardiovascular diseases. World J Stem Cells. 6:355–366.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu JY, Lee YK, Wang Y and Tse HF:
Therapeutic application of endothelial progenitor cells for
treatment of cardiovascular diseases. Curr Stem Cell Res Ther.
9:401–414. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wöhrle J, Birkemeyer R, Markovic S, Nguyen
TV, Sinha A, Miljak T, Spiess J, Rottbauer W and Rittger H:
Prospective randomised trial evaluating a paclitaxel-coated balloon
in patients treated with endothelial progenitor cell capturing
stents for de novo coronary artery disease. Heart. 97:1338–1342.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Raval Z and Losordo DW: Cell therapy of
peripheral arterial disease: from experimental findings to clinical
trials. Circ Res. 112:1288–1302. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Massari ME and Murre C: Helix-loop-helix
proteins: regulators of transcription in eucaryotic organisms. Mol
Cell Biol. 20:429–440. 2000. View Article : Google Scholar
|
|
10
|
Sobrado VR, Moreno-Bueno G, Cubillo E,
Holt LJ, Nieto MA, Portillo F and Cano A: The class I bHLH factors
E2-2A and E2-2B regulate EMT. J Cell Sci. 122:1014–1024. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao DH, Hong JJ, Guo SY, Yang RL, Yuan J,
Wen CY, Zhou KY and Li CJ: Aberrant expression and function of TCF4
in the proliferation of hepatocellular carcinoma cell line
BEL-7402. Cell Res. 14:74–80. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiong Y, Liu Y, Song Z, Hao F and Yang X:
Identification of Wnt/β-catenin signaling pathway in dermal papilla
cells of human scalp hair follicles: TCF4 regulates the
proliferation and secretory activity of dermal papilla cell. J
Dermatol. 41:84–91. 2014. View Article : Google Scholar
|
|
13
|
Tanaka A, Itoh F, Nishiyama K, Takezawa T,
Kurihara H, Itoh S and Kato M: Inhibition of endothelial cell
activation by bHLH protein E2-2 and its impairment of angiogenesis.
Blood. 115:4138–4147. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo S, Bai R, Liu W, Zhao A, Zhao Z, Wang
Y, Wang Y, Zhao W and Wang W: miR-22 inhibits osteosarcoma cell
proliferation and migration by targeting HMGB1 and inhibiting
HMGB1-mediated autophagy. Tumour Biol. 35:7025–7034. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rubinsztein DC, Mariño G and Kroemer G:
Autophagy and aging. Cell. 146:682–695. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dong N, Zhu Q, Zhang P, Zhu C, Wang M, Li
W, Liu J, Liu Y, Ma B and Wu K: Autophagy downregulates
thrombin-induced VSMCs proliferation through lysosomal pathway. Int
J Cardiol. 159:156–158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang HJ, Zhang D, Tan YZ and Li T:
Autophagy in endothelial progenitor cells is cytoprotective in
hypoxic conditions. Am J Physiol Cell Physiol. 304:C617–C626. 2013.
View Article : Google Scholar
|
|
18
|
Petherick KJ, Williams AC, Lane JD,
Ordóñez-Morán P, Huelsken J, Collard TJ, Smartt HJ, Batson J, Malik
K, Paraskeva C and Greenhough A: Autolysosomal β-catenin
degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J.
32:1903–1916. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Yin Y, Huang L, Zhao X, Fang Y, Yu S, Zhao
J and Cui B: AMD3100 mobilizes endothelial progenitor cells in
mice, but inhibits its biological functions by blocking an
autocrine/paracrine regulatory loop of stromal cell derived
factor-1 in vitro. J Cardiovasc Pharmacol. 50:61–67. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Liu J, Zhen J, Zhang C, Wan Q, Liu
G, Wei X, Zhang Y, Wang Z, Han H, et al: Histone deacetylase 4
selectively contributes to podocyte injury in diabetic nephropathy.
Kidney Int. 86:712–725. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ikeda Y, Shirakabe A, Maejima Y, Zhai P,
Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, et
al: Endogenous Drp1 mediates mitochondrial autophagy and protects
the heart against energy stress. Circ Res. 116:264–278. 2015.
View Article : Google Scholar
|
|
23
|
Jiang L, Yin M, Wei X, Liu J, Wang X, Niu
C, Kang X, Xu J, Zhou Z and Sun S: Bach1 represses Wnt/β-catenin
signaling and angiogenesis. Circ Res. 117:364–375. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu R, Qu Y, Ge J, Zhang L, Su Z,
Pflugfelder SC and Li DQ: Transcription factor TCF4 maintains the
properties of human corneal epithelial stem cells. Stem Cells.
30:753–761. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Galluzzi L, Pietrocola F, Levine B and
Kroemer G: Metabolic control of autophagy. Cell. 159:1263–1276.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Guan JL, Simon AK, Prescott M, Menendez
JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A and Zhang
J: Autophagy in stem cells. Autophagy. 9:830–849. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li Y, Zhang L, Zhou J, Luo S, Huang R,
Zhao C and Diao A: Nedd4 E3 ubiquitin ligase promotes cell
proliferation and autophagy. Cell Prolif. 48:338–347. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding Y, Gao H, Zhao L, Wang X and Zheng M:
Mitofusin 2-deficiency suppresses cell proliferation through
disturbance of autophagy. PLoS One. 10:e01213282015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Y and Song L: HMGB1-induced autophagy
in Schwann cells promotes neuroblastoma proliferation. Int J Clin
Exp Pathol. 8:504–510. 2015.PubMed/NCBI
|
|
31
|
Pei B, Zhao M, Miller BC, Véla JL,
Bruinsma MW, Virgin HW and Kronenberg M: Invariant NKT cells
require autophagy to coordinate proliferation and survival signals
during differentiation. J Immunol. 194:5872–5884. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cianfanelli V, Fuoco C, Lorente M, Salazar
M, Quondamatteo F, Gherardini PF, De Zio D, Nazio F, Antonioli M,
D'Orazio M, et al: AMBRA1 links autophagy to cell proliferation and
tumorigenesis by promoting c-Myc dephosphorylation and degradation.
Nat Cell Biol. 17:20–30. 2015. View
Article : Google Scholar
|
|
33
|
Lee IH, Cao L, Mostoslavsky R, Lombard DB,
Liu J, Bruns NE, Tsokos M, Alt FW and Finkel T: A role for the
NAD-dependent deacetylase Sirt1 in the regulation of autophagy.
Proc Natl Acad Sci USA. 105:3374–3379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kageyama S and Komatsu M: Impaired
G1-arrest, autophagy, and apoptosis in Atg7-knockout mice. Circ
Res. 111:962–964. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Torisu T, Torisu K, Lee IH, Liu J, Malide
D, Combs CA, Wu XS, Rovira II, Fergusson MM, Weigert R, et al:
Autophagy regulates endothelial cell processing, maturation and
secretion of von Willebrand factor. Nat Med. 19:1281–1287. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Thorburn J, Andrysik Z, Staskiewicz L,
Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM and Thorburn A:
Autophagy controls the kinetics and extent of mitochondrial
apoptosis by regulating PUMA levels. Cell Reports. 7:45–52. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee IH, Kawai Y, Fergusson MM, Rovira II,
Bishop AJ, Motoyama N, Cao L and Finkel T: Atg7 modulates p53
activity to regulate cell cycle and survival during metabolic
stress. Science. 336:225–228. 2012. View Article : Google Scholar : PubMed/NCBI
|