Introduction
To date, stem/progenitor cell-based therapies are
promising therapeutic approaches for the treatment of various heart
diseases, including ischemic heart disease and heart failure. These
cells promote angiogenesis and endogenous stem cell activation, and
exert anti-apoptotic effects on surviving cardiomyocytes through
paracrine signaling, resulting in improved cardiac function
(1–3). Two potential adult stem cell sources
are cardiac stem cells (CSCs) isolated from the heart itself and
bone marrow-derived mesenchymal stem cells (BM-MSCs), and each has
been implicated in contributing to the restoration of cardiac
function (4,5). Previous evidence has suggested that
CSCs have greater cardioprotective potential in ischemic heart
failure compared to BM-MSCs (6)
and are necessary and sufficient for anatomical and functional
adult myocardial regeneration following severe diffuse myocardial
damage (7). Nonetheless, aging,
ischemic myocardial injury, cardiac hypertrophy and metabolic
disorders, together with environmental factors, can markedly affect
resident human CSC (hCSC) growth and differentiation (8–11).
Macrophage migration inhibitory factor (MIF) is a
pro-inflammatory factor that participates in the pathogenesis of
various inflammatory diseases, including sepsis, atherosclerosis
and rheumatoid arthritis (12–14). It is produced and stored in
various cell types, including immune cells, endothelial cells and
cardiomyocytes. It is then rapidly released from intracellular
stores in response to various noxious stimuli, such as infection,
inflammation and hypoxia (15–17). Increasing evidence has
demonstrated that MIF protects the heart following
ischemia/reperfusion (I/R) by activating adenosine
monophosphate-activated protein kinase (AMPK), inhibiting c-Jun
N-terminal kinase (JNK)-mediated apoptosis, and attenuating
cardiomyocyte oxidative stress, thereby reducing the infarct size
and preserving cardiac function (18,19). However, it is unknown whether MIF
protects the injured heart in other ways, such as by activating
resident CSCs and promoting their survival, proliferation and/or
differentiation into myocytes and coronary vessels.
CD74 was initially considered a major
histocompatability complex class II chaperone (20). However, it was later found to be
an MIF receptor, and it can form a complex with CD44 (21). In studies on other types of stem
cells, MIF was shown to promote stem/progenitor cell survival and
proliferation by increasing Akt, extracellular signal-regulated
kinase ERK) and AMPK phosphorylation, and to regulate
CD74-dependent cell migration (22–24). Recently, some researchers reported
that MIF protected BM-MSCs from senescence through Akt signaling
(25,26). The phosphoinositide 3-kinase
(PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway
has been shown to play a central role in several cellular
functions, including survival, proliferation, adhesion, migration,
differentiation and metabolism (27–29). However, whether MIF can affect
CSCs through CD74 and the PI3K/Akt/mTOR signaling pathway and AMPK
activition remains unknown.
In this study, we addressed two questions: i)
whether MIF promotes CSC survival, proliferation and endothelial
differentiation, and ii) whether the effects of MIF on CSCs are
mediated through the PI3K/Akt/mTOR and AMPK signaling pathways and
interactions with the CD74 receptor. Our results demonstrate that
MIF may be a potential therapeutic factor in the treatment of
degenerative heart disorders, as it is able to activate CSCs.
Materials and methods
Animals
BALB/c mice, weighing 18–25 g, were used for the
cell isolation and culture experiments. All BALB/c mice were
purchased from the Laboratory Animal Science Department, the Second
Affiliated Hospital of Harbin Medical University, Heilongjiang,
China. All experiments were carried out in accordance with the
Local Ethics Committee of Harbin Medical University Animal Care and
Use.
CSC isolation and identification
Mouse CSCs were obtained as described previously,
with a minor modification (30,31). Briefly, mice were sacrificed by
cervical dislocation, the heart tissue was chopped, followed by
enzymatic dissociation. The hybrid cells were then isolated using
an FITC rat anti-mouse CD117/c-kit antibody (561680; BD
Biosciences, Franklin Lakes, NJ, USA) and MACS anti-FITC microbeads
(130-048-701; Miltenyi Biotec, Bergisch Gladbach, Germany) for
positive sorting. These obtained cells were subjected to negative
selection with PE rat anti-mouse CD45 antibody (561087; BD
Biosciences, Franklin Lakes, NJ, USAs) and MACS anti-PE microbeads
(130-048-801; Miltenyi Biotec). The cells were cultured in HyClone
Dulbecco's modified Eagle's medium (DMEM)/F12 (Thermo Fisher
Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine
serum (FBS), basic fibroblast growth factor (bFGF; 10 ng/ml;
100-18B), insulin-like growth factor (IGF; 10 ng/ml; 250-19),
epidermal growth factor (EGF; 10 ng/ml; 100-15) and leukemia
inhibitory factor (LIF; 10 ng/ml; 300-05) (all from Peprotech,
Rocky Hill, NJ, USA), at 37°C in 5% CO2. All cells used
in the subsequent experiments were between passages 3 and 5.
To identify surface markers, the cells at passages
3–5 were harvested, washed with phosphate-buffered saline (PBS),
and labeled with the following conjugated antibodies: FITC-labeled
anti-CD CD117 (561680; BD Biosciences, Franklin Lakes, NJ, USA),
anti-CD45 (11-0451-82; eBioscience, San Diego, CA, USA), anti-CD29
(561796), anti-CD34 (560238), and phycoerythrin-labeled anti-Sca-1
(561076), APC-labeled anti-CD90 (561974), PE-labeled anti-CD31
(561073) and anti-KDR (561259) antibodies (all from BD Biosciences,
Franklin Lakes, NJ, USA). The labeled cells were analyzed by flow
cytometry using FACSDiva Pro Software (Becton-Dickinson, San Jose,
CA, USA).
In addition, the thymus and liver samples of the
mice sacrificed to obtain the CSCs were preserved in liquid
nitrogen as a positive control for the PCR experiment for examining
MIF and CD74. BM-MSCs were purchased from the Americal Type Culture
Collection (ATCC; Manassas, VA, USA) and also used as a positive
control. BM-MSCs were cultured in LG-DMEM (HyClone, Logan, UT,
USA), supplemented with 10% fetal calf serum (FCS; HyClone), at
37°C in 5% CO2.
Cell treatment
In preliminary experiments, a time course analysis
was performed in which the cells were incubated with various
concentrations (0, 50, 100 and 200 ng/ml) of MIF (1978-MF-025;
R&D Systems, Chantilly, VA, USA) for 0, 24 or 48 h. In
subsequent experiments, the optimal MIF concentration (200 ng/ml)
was incubated with the mCSCs for the indicated period of time (0,
30, 60, 90, 120 and 180 min) in complete medium without growth
factors. In blockade assays, ISO-1 (an MIF inhibitor; 100
µg/ml; 475837; Calbiochem, San Diego, CA, USA), LY294002 (a
PI3K inhibitor; 25 µM; L9908; Sigma, St. Louis, MO, USA),
MK-2206 (an Akt inhibitor; 3 µM; S1078; Selleckchem,
Houston, TX, USA), or compound C (an AMPK inhibitor; 40 µM;
171260; Merck, Darmstadt, Germany) was added to the medium. The
inhibitors were pre-incubated with the cells in complete medium for
90 min prior to exposure to MIF as previously described (32). For determining whether the
isolated cells can differentiate into 3 main cardiac lineages, the
CSCs at passage 3–5 were placed in differentiation medium
containing DMEM, 10% FBS and 10−8 M dexamethasone or 10
M 5-Azacytidine for 24 h. Following this procedure, the cells were
maintained in 2% FBS medium with or without dexamethasone for 21
days.
Knockdown experiments using small
interfering RNA (siRNA)
The following sequences of mouse-specific siRNA
targeting CD74 (CD74-siRNA), 5′-CCAGGACCAUGUGAUGCAUTT-3′ and
negative control siRNA (NC-siRNA) 5′-UUCUCCGAACGUGUCACGUTT-3′ were
used for gene silencing experiments. Single cells were seeded in 6-
or 96-well plates, and siRNA constructs were transfected at a
concentration of 80 nM using X-tremeGENE siRNA Transfection Reagent
(Roche Applied Science, Penzberg, Germany) according to the
manufacturer's instructions. The siRNA-CD74 knockdown efficiency
was determined by western blot analysis.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from the cultured cells using
TRizol reagent (Invitrogen, Shanghai, China) and reverse
transcribed into cDNA using AccuPower® RocketScript™ RT
PreMix (Bioneer, Shanghai, China) according to the manufacturer's
instructions. RT-qPCR was performed using AcccuPower® 2X
Greenstar qPCR master mix (Bioneer) and the ABI fluorescence
quantitative PCR system (Bio-Rad, Hercules, CA, USA). The PCR
conditions were as follows: 1 cycle of 10 min at 95°C, followed by
40 cycles of 95°C for 30 sec, 60°C for 30 sec, and 72°C for 60 sec.
Relative gene expression levels were calculated using the
2−ΔΔCt method, as previously described (32). Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) mRNA levels were used as internal
normalization controls. For semi-quantitative PCR, the PCR products
were resolved on a 2% agarose gel stained with Dured nucleic acid
dye (Fanbo Biochemicals, Beijing, China). Primer pairs used to
detect target gene mRNA levels are listed in Table I.
 | Table IPrimer sequences used for
RT-qPCR. |
Table I
Primer sequences used for
RT-qPCR.
| Genes | Sequences |
|---|
| CD74 | F:
5′-ACGCGACCTCATCTCTAACC-3′ |
| R:
5′-GGTCATGTTGCCGTACTTGG-3′ |
| GAPDH | F:
5′-AGGTCGGTGTGAACGGATTTG-3′ |
| R:
5′-TGTAGACCATGTAGTTGAGGTCA-3′ |
Cell proliferation assay
Cell survival/proliferation was assessed using a
CCK-8 assay (Beyotime Institute of Biotechnology, Shanghai, China)
according to the manufacturer's instructions. Cells in a 96-well
plate were incubated with CCK-8 for 2 h at 37°C. Absorbance of each
well was measured at 450 nm.
In 5-ethynyl-2′-deoxyuridine (EdU) chase
experiments, the CSCs were seeded into 96-well plates at a density
of 1–2×103 cells/ml and cultured in serum-free medium
for 24 h. The cells were then treated with MIF for 24 h in complete
culture medium and EdU (RiboBio, Guangzhou, China) was added to a
final concentration of 50 µM for a further 2 h. The cells
were fixed with 4% paraformaldehyde (PFA) for 30 min, permeabilized
with PBS containing 0.5% Triton X-100 for 10 min and stained with
1X Apollo® reaction cocktail for 30 min, followed by
staining with Hoechst 33342 for 30 min at room temperature. At
least 10 different viewing fields were counted for analysis. All
images were obtained using a fluorescence microscope (Leica
DM4000B; Leica, Solms, Germany).
For cell cycle analysis, the mCSCs were treated with
MIF (200 ng/ml) for 48 h, then harvested and washed with PBS, and
fixed with 75% ethanol overnight. The cells were then washed with
PBS and incubated with RNase A (20 mg/ml) for 30 min at 37°C. This
was followed by a further incubation with propidium iodide (PI, 0.5
mg/ml) for 30 min at 4°C. Finally, the cells were washed and
resuspended in 500 µl of PBS, then analyzed using a
Becton-Dickinson flow cytometer (BD Biosciences, Franklin Lakes,
NJ, USA) to detect the DNA content.
Western blot analysis
Western blot analysis was performed as previously
described (33). Briefly, cells
were washed in ice-cold PBS and lysed in RIPA buffer (50 mM
Tris-HCl, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and
0.1% SDS, pH 7.6) (Beyotime Institute of Biotechnology) containing
protease and phosphatase inhibitors (cocktail tablet; Roche Applied
Science). The lysates were centrifuged at 12,000 × g for 15 min at
4°C, and supernatants were collected. The protein concentrations of
each sample were determined with a BCA protein assay kit (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructoins. Equal amounts of proteins were electrophoresed on 10%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) gels and transferred onto polyvinylidene fluoride (PVDF)
membranes (Beyotime Institute of Biotechnology). The membranes were
blocked with 5% skim milk in Tris-buffered saline containing 0.1%
Tween-20 (TBST) at room temperature for 1 h, then incubated
overnight at 4°C with the following primary antibodies: Akt
(1:1,000; 4691), phospho-Akt (Thr308; 1:750; 4056), mTOR (1:1,000;
2983), phospho-mTOR (Ser2448; 1:750; 2971), AMPK (1:1,000; 5831s),
phospho-AMPK (Thr172; 1:750; 4188) (all from Cell Signaling
Technology, Danvers, MA, USA), CD74 (1:100; sc-5438), vascular
endothelial growth factor (VEGF; 1:200; sc-507), von Willebrand
factor (vWF; 1:200; sc-14014) (all from Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) and GAPDH (1:1,000; AB-P-R 001; Good
Here Biochemicals, Hangzhou, China). The membranes were washed in
TBST and incubated with the appropriate horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:5,000, goat anti-rabbit;
ZDR-5306; Zhongshan Golden Bridge Biotechnology, Beijing, China)
for 1 h at room temperature. Signals were detected with an ECL-Plus
Substrate (P0018; Beyotime Institute of Biotechnology) and exposed
to Hyper film (Kodak, Rochester, NY, USA), and quantified and
analyzed using Quantity One software (Bio-Rad).
Immunofluorescence staining
For identifying the isolated cells, cells at
passages 3–5 were seeded onto 48-well plates and fixed with 4% PFA
for 30 min at room temperature, permeabilized with 0.5% Triton
X-100, blocked with 1% BSA and incubated with rabbit anti-c-kit
(stemness marker; sc-168), anti-NK2 homeobox 5 (Nkx2.5; sc-14033)
and anti-GATA binding protein 4 (GATA-4; sc-9053) (cardiac lineage
markers) (1:50; Santa Cruz Biotechnology, Inc.) antibodies at 4°C
overnight. After washing, the cells were incubated with TRITC- or
FITC-conjugated AffiniPure goat anti-rabbit IgG (H+L) antibodies,
TRITC-conjugated AffiniPure goat anti-mouse IgG (H+L) antibodies
(1:50; ZF-0316, ZF-0311, ZF-0313; Zhongshan Golden Bridge
Biotechnology) for 1 h at room temperature. The nuclei were stained
with 4′,6-diamidino-2-phenylindole (DAPI; D8417; Sigma).
Fluorescent images were acquired using a fluorescence microscope
(Leica DMI4000B). To examine CD74 expression in the CSCs, rabbit
anti-CD74 antibody (1:50; sc-5438; Santa Cruz Biotechnology, Inc.)
was used. The CSCs used in the endothelial differentiation
experiments were stained with rabbit anti-VEGF (1:50; sc-507) and
rabbit anti-vWF (1:50; sc-14014; Santa Cruz Biotechnology, Inc.)
antibodies. To identify the differentiation of CSCs into myocardial
cells or smooth muscle cells, mouse anti-TnI (1:100; ab19615;
Abcam, Cambridge, MA, USA) and mouse anti-SMA (1:100, BM0002;
Boster, Wuhan, China) were used.
Tube formation assay
To measure tube formation, 48-well plates were
coated with Matrigel (BD Biosciences, Bedford, MA, USA) according
to the manufacturer's instructions. Human umbilical vein
endothelial cells (HUVECs) were purchased from ATCC and cultured in
DMEM-1640 and endothelial cells (ECs) differentiated from the CSCs
(CSC-ECs) in DMEM/F12 supplemented with 0.2% FBS were seeded on
Matrigel-coated plates (1.5×104 cells/well) and
incubated for 4 h at 37°C. Subsequently, capillary-like structures
were observed and were quantified by calculating the number of
junctions per field; at least 5 different viewing fields were
analyzed. All images were obtained using an inverted microscope
(Olympus IX73; Olympus, Tokyo, Japan).
MIF enzyme-linked immunosorbent assay
(ELISA)
A mouse MIF ELISA kit (BlueGene, Shanghai, China)
was used to measure the amount of secreted MIF in the culture
supernatants. The assays were conducted in 96-well microplates
according to the manufacturer's instructions.
Statistical analysis
All values are expressed as the means ± standard
deviation (SD). The statistical differences between 2 groups was
determined using a Student's t-test, and differences among groups
were examined by one-way ANOVA with the statistical software SPSS
package v19.0 (IBM Corp., Armonk, NY, USA). A value of p<0.05
was considered to indicate a statistically significant
difference.
Results
Cells isolated from mouse hearts exhibit
CSC characteristics
FACS analysis revealed that the majority of cells
from passages 3–5 expressed common surface markers for CSCs: they
were positive for c-kit (93.4±0.95%), Sca-1 (82.33±0.90%), CD90
(71.6±1.55%) and CD29 (93.7±1.2%), but negative for CD45
(2.1±0.26%), CD34 (3.4±0.5%), CD31 (1.67±0.35%) and KDR (2.8±0.42%)
(Fig. 1). In addition,
immunofluorescence staining revealed that these cells were positive
for c-kit, Nkx2.5 and GATA-4, and positive for troponin I (TnI),
smooth muscle actin (SMA) and vWF following treatment with
dexamethasone or 5-Azacytidine (Fig.
1). These data suggest that the cells used in our experiments
were stem cells of cardiac origin.
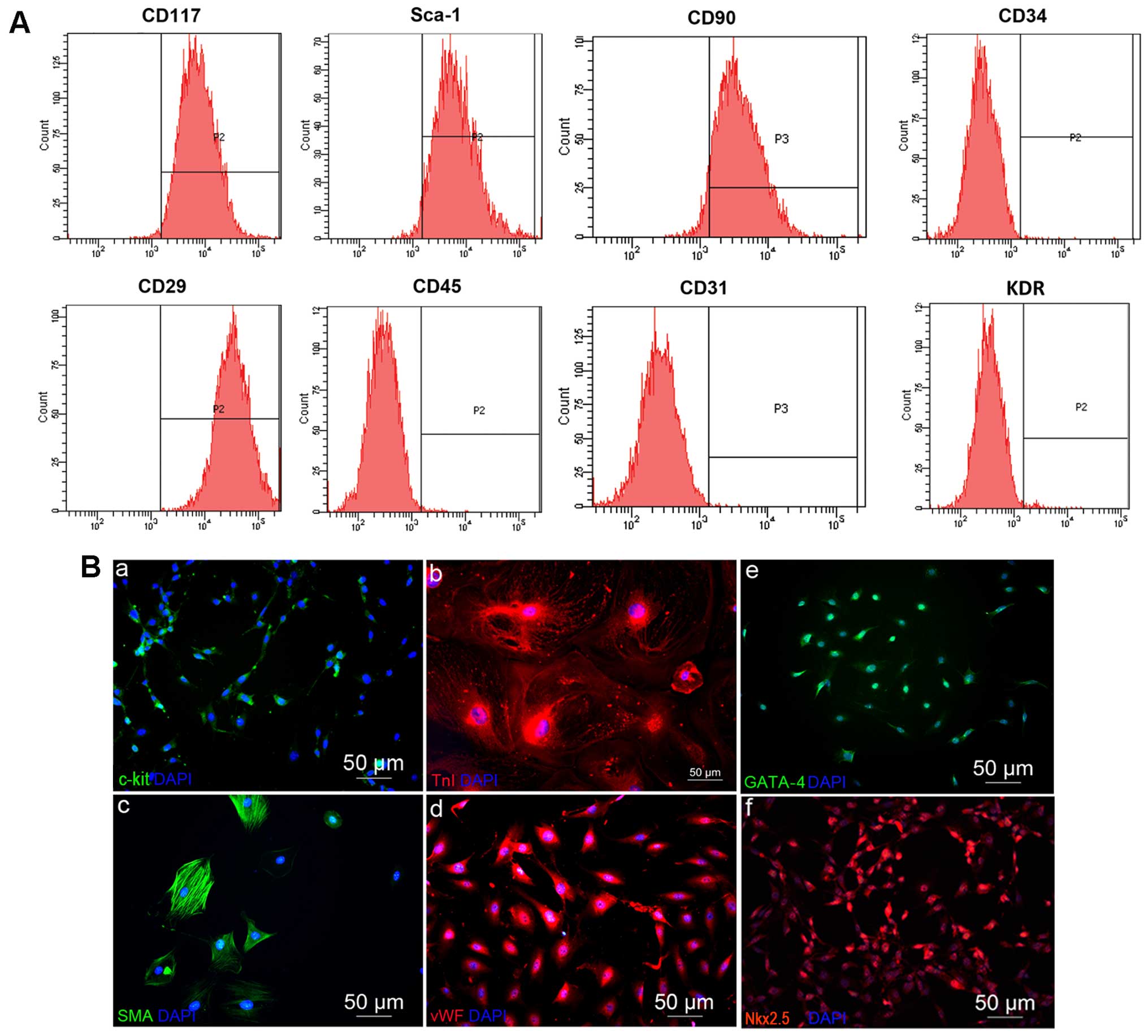 | Figure 1Identification of isolated cells. (A)
As shown by flow cytometry analysis, at passage 3 (P3) after
sorting, the c-kit-positive cells reached 93.4±0.95%, but were
negative for the hematopoietic and endothelial antigens, CD45
(2.1±0.26%), CD31 (1.67±0.35%), CD34 (3.4±0.5%) and KDR
(2.8±0.42%). As expected, they were positive for the control
mesenchymal markerS, CD90 (71.6±1.55%), CD29 (93.7±1.2%), and THYE
stemness marker Sca-1 (82.33±0.90%). (B) Immunofluorescence
staining of the main cardiac lineages differentiated from
c-kit+ cells, namely (panel b) the cardiomyocyte marker,
TnI; (panel c) the smooth muscle cell marker, SMA; and (panel d)
the endothelial cell marker, vWF. The expression of the cardiac
lineage markers (panel e) GATA-4; and (panel f) Nkx2.5 in the
c-kit+ cells was also analyzed. Nuclei were
counterstained with DAPI. |
MIF and MIF receptor CD74 expression in
CSCs
It has previously been demonstrated that MIF is a
ligand of CD74 which can activate various signaling pathways in
various cell types, including stem/progenitor cells (22). Another study demonstrated that
cardiac-derived MIF enhances post-ischemic injury myocardial
healing by protecting cardiomyocytes from apoptosis (34). Therefore, we hypothesized that
CSC-derived MIF can support CSC survival and proliferation through
its interactions with CD74, which is expressed in CSCs. Thus, we
first examined MIF and CD74 gene expression in CSCs using RT-PCR,
and then we confirmed CD74 protein expression by immunofluorescence
staining and the amount of secreted MIF from CSC supernatants by
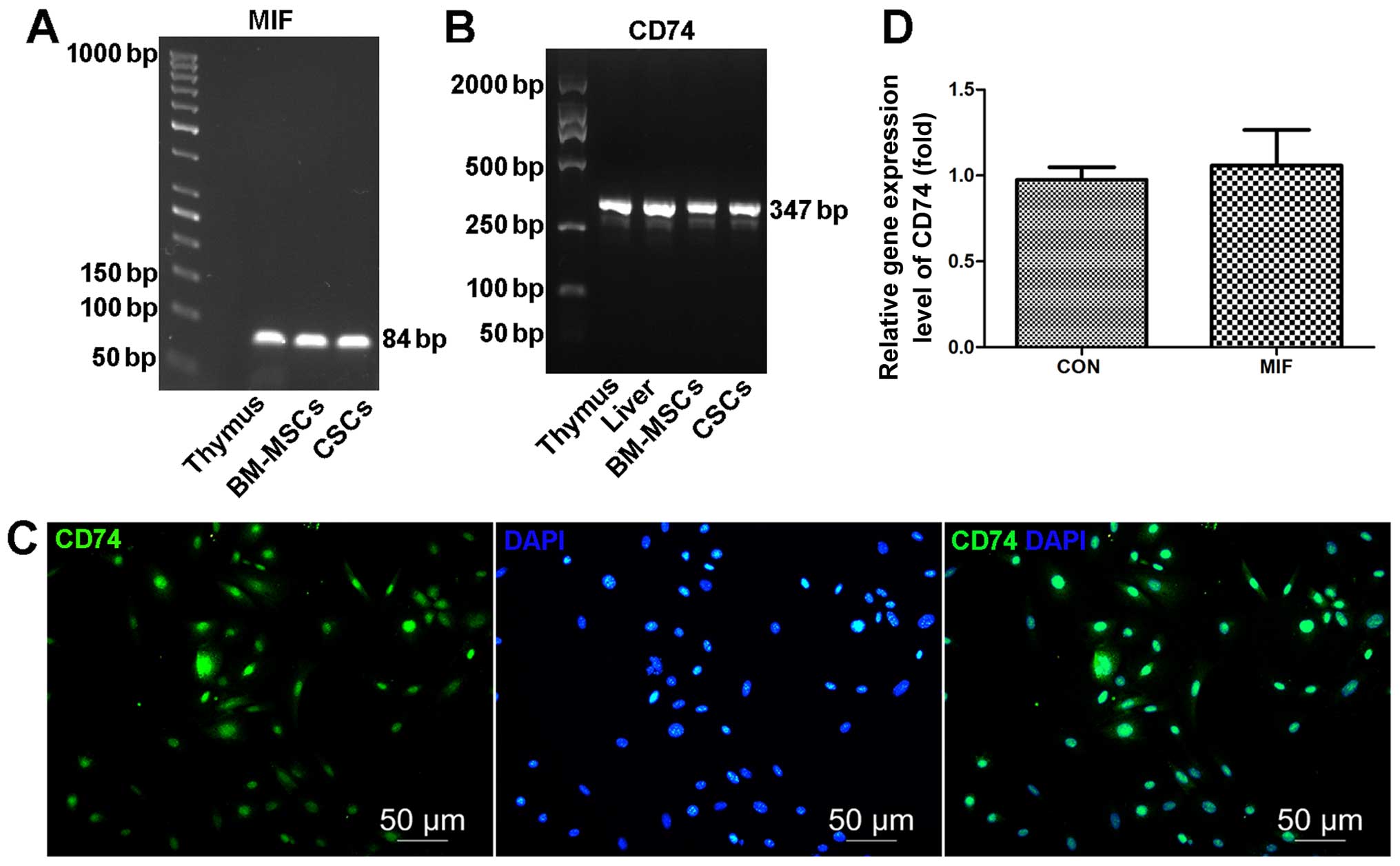
ELISA. We found that CSCs expressed MIF (Fig. 2A) and secreted MIF (317.07±5.56
pg/ml for 1–2×106 mCSCs cultured in 5 ml medium for 24
h) (Fig. 2A) and expressed CD74
(Fig. 2B and C). In addition,
co-culture with exogenous MIF had no effect on the CD74 mRNA levels
(Fig. 2D).
Effects of MIF on CSC proliferation
To examine the effects of MIF on the CSCs, we
evaluated the proliferation of MIF-treated cells using CCK-8 and
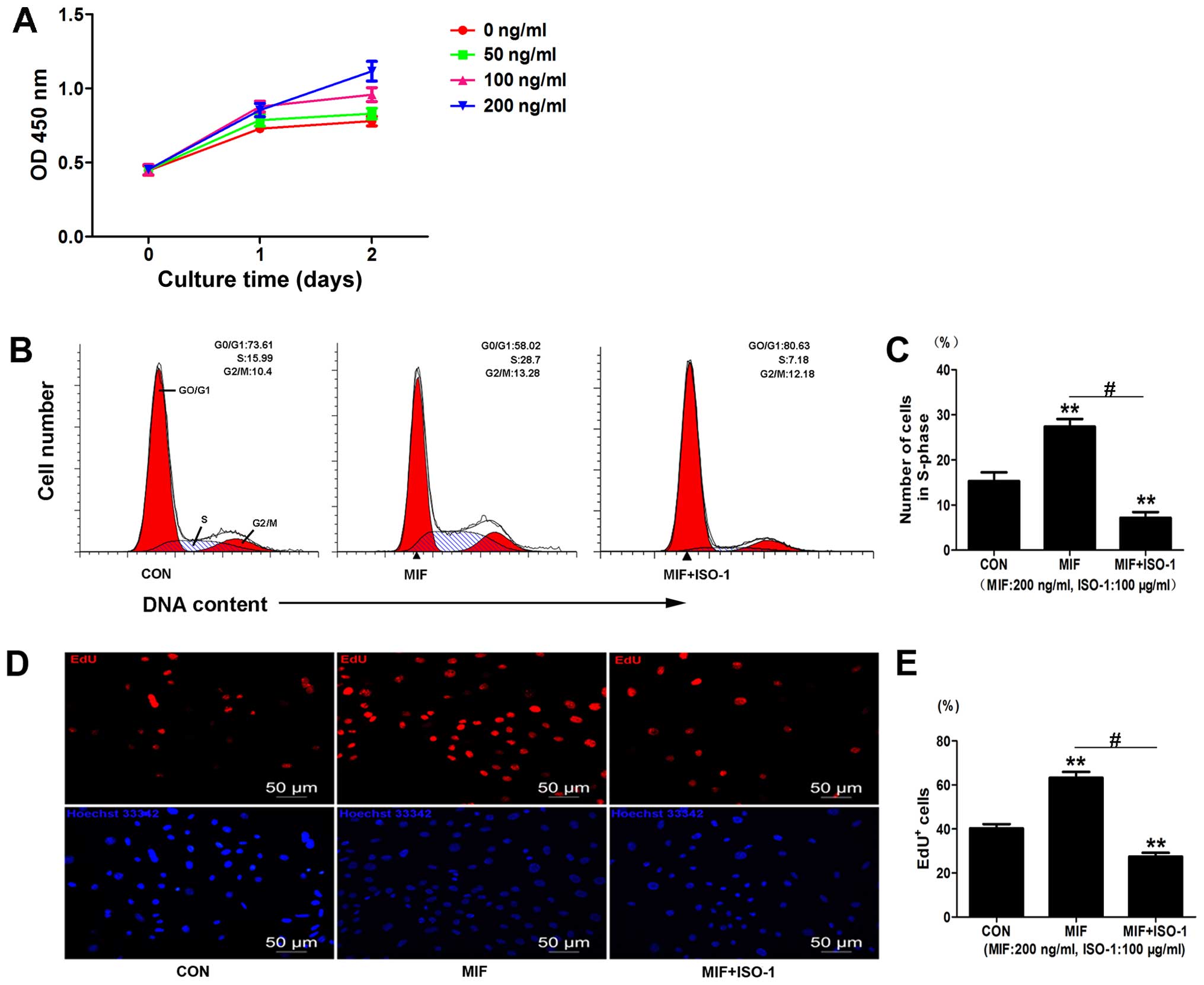
EdU assays. The CCK-8 assay indicated that MIF significantly
increased cell viability at day 1 (100 ng/ml, 1.206±0.089-fold
compared to control, p<0.01, n=3; 200 ng/ml, 1.173±0.098-fold
compared to control, p<0.05, n=3) (Fig. 3A) and at day 2 (100 ng/ml,
1.228±0.012-fold compared to control, p<0.01, n=3; 200 ng/ml,
1.431±0.036-fold compared to control, p<0.01, n=3) (Fig. 3A). MIF at 50 ng/ml had no
significant effect on cell viability (Fig. 3A). The optimal concentration of
MIF selected for use in the subsequent experiments was 200
ng/ml.
We further examined the effects of MIF on CSC
proliferation by EdU assay and found increased EdU incorporation
following treatment with MIF compared to the control
(1.57±0.13-fold compared to control, p<0.01; n=3) (Fig. 3D and E). We then examined the
effects of the MIF-specific inhibitor, ISO-1, on the CSCs and found
that treatment with ISO-1 (100 µg/ml) decreased cell
proliferation, as assessed by EdU assay (0.67±0.05-fold of control,
0.267±0.05-fold of MIF, p<0.01; n=3) (Fig. 3D and E).
Cell cycle analysis also revealed that MIF increased
the number of cells in the S-phase (1.82±0.04-fold of control,
p<0.01; n=3) (Fig. 3B and C),
and treatment with ISO-1 significantly decreased the population of
cells in the S-phase (0.49±0.09-fold of control, 0.43±0.04-fold of
MIF, p<0.01; n=3) (Fig. 3B and
C). Taken together, these data suggest that MIF promotes CSC
proliferation.
MIF promotes CD74-dependent CSC
proliferation
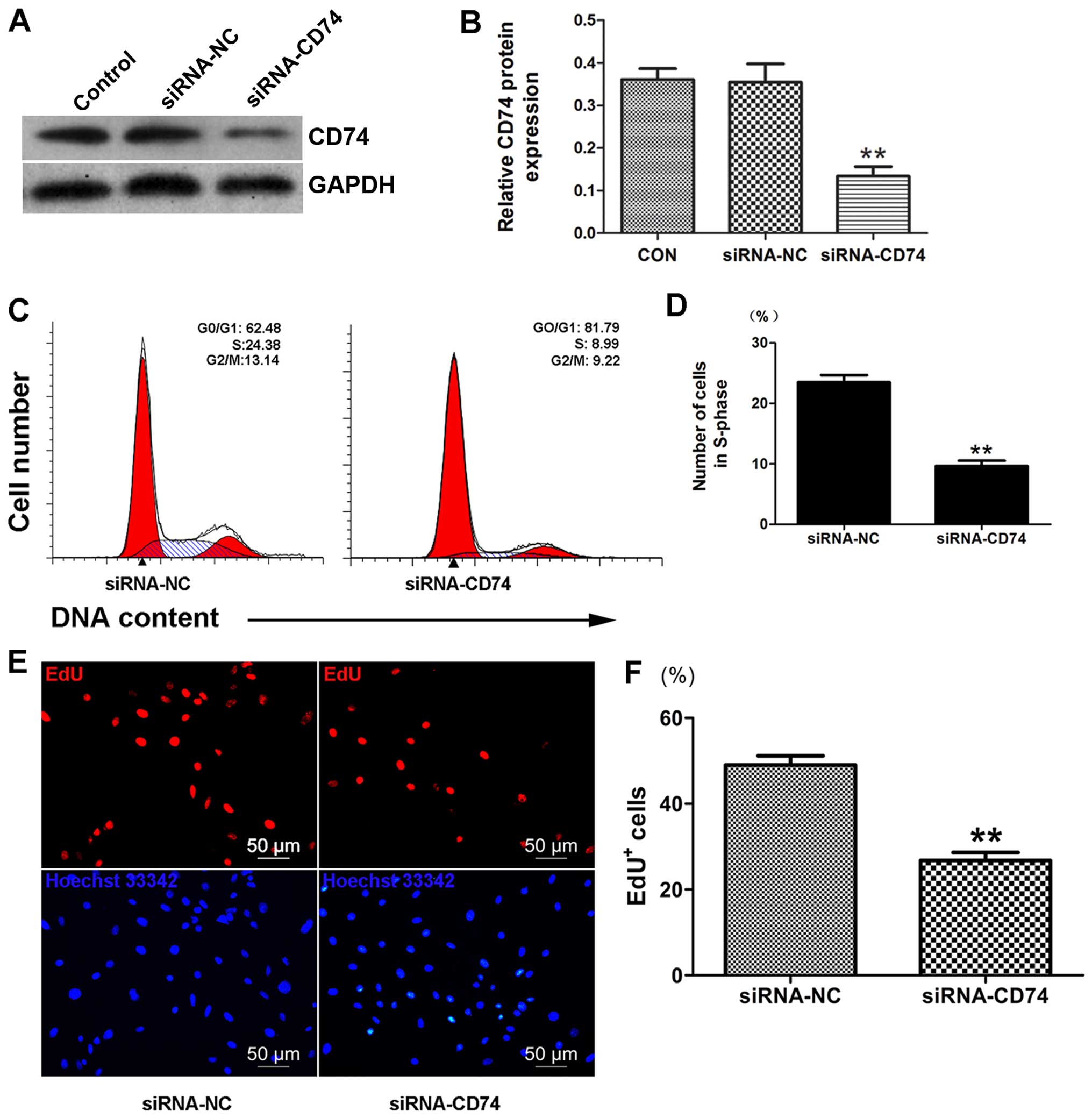
To determine whether CD74 is involved in the
pro-proliferative effects of MIF on CSCs, we examined CSC
proliferation following the knockdown of CD74 by siRNA. We
confirmed a decrease in CD74 protein expression (36.52±6.61%
compared to control, p<0.01; n=3) (Fig. 4A and B) by western blot analysis.
Importantly, the results from EdU assay indicated that CD74
knockdown decreased the proliferation of the MIF-treated CSCs
(Fig. 4E and F). Cell cycle
analysis also revealed that CD74 knockdown in the CSCs markedly
decreased the number of cells in the S-phase (Fig. 4C and D), which further supports
our hypothesis that MIF promotes CSC proliferation through its
interaction with CD74.
MIF activates the PI3K/Akt/mTOR signaling
and AMPK pathways
The PI3K/Akt/mTOR axis intersects with a number of
intracellular signaling pathways, and thus regulates many cellular
events, including cell-cycle progression, proliferation/growth and
angiogenesis (27). Therefore, in
this study, we investigated whether these pathways mediate the
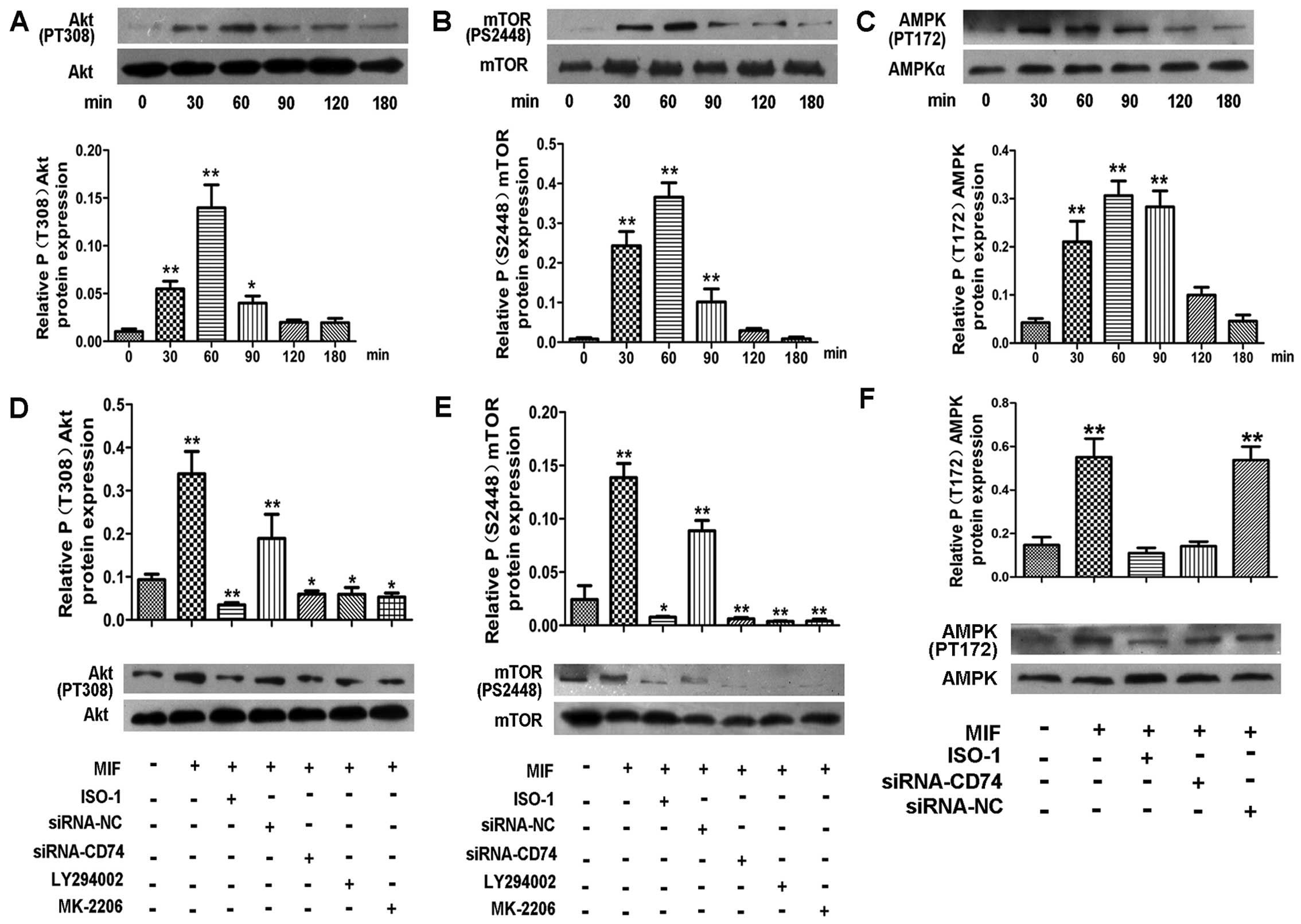
pro-proliferative effects of MIF on CSCs. The results of western
blot analysis revealed that MIF induced a pronounced increase in
the Akt and mTOR phosphorylation levels in a time-dependent manner,
with a peak at 60–90 min (Fig. 5A and
B). However, in the cells treated with a PI3K inhibitor
(LY294002) or the Akt inhibitor (MK-2206) prior to the addition of
MIF, the Akt and mTOR phosphorylation levels were significantly
decreased (Fig. 5D and E). These
data suggest that MIF regulates CSC proliferation through the
PI3K/Akt/mTOR pathway.
There is evidence to suggest that MIF also activates
AMPK to protect cardiomyocytes from ischemic heart disease
(18) and promotes the survival
and proliferation of neural stem/progenitor cells (22). Thus, in this study, we examined
the effects of MIF on the phosphorylation of AMPK in CSCs by
western blot analysis. The results revealed that MIF also induced a
marked increase in AMPK phosphorylation in a time-dependent manner,
with a peak at 30–90 min (Fig.
5C). Following the knockdown of CD74, the activation of AMPK
was significantly inhibited (Fig.
5F).
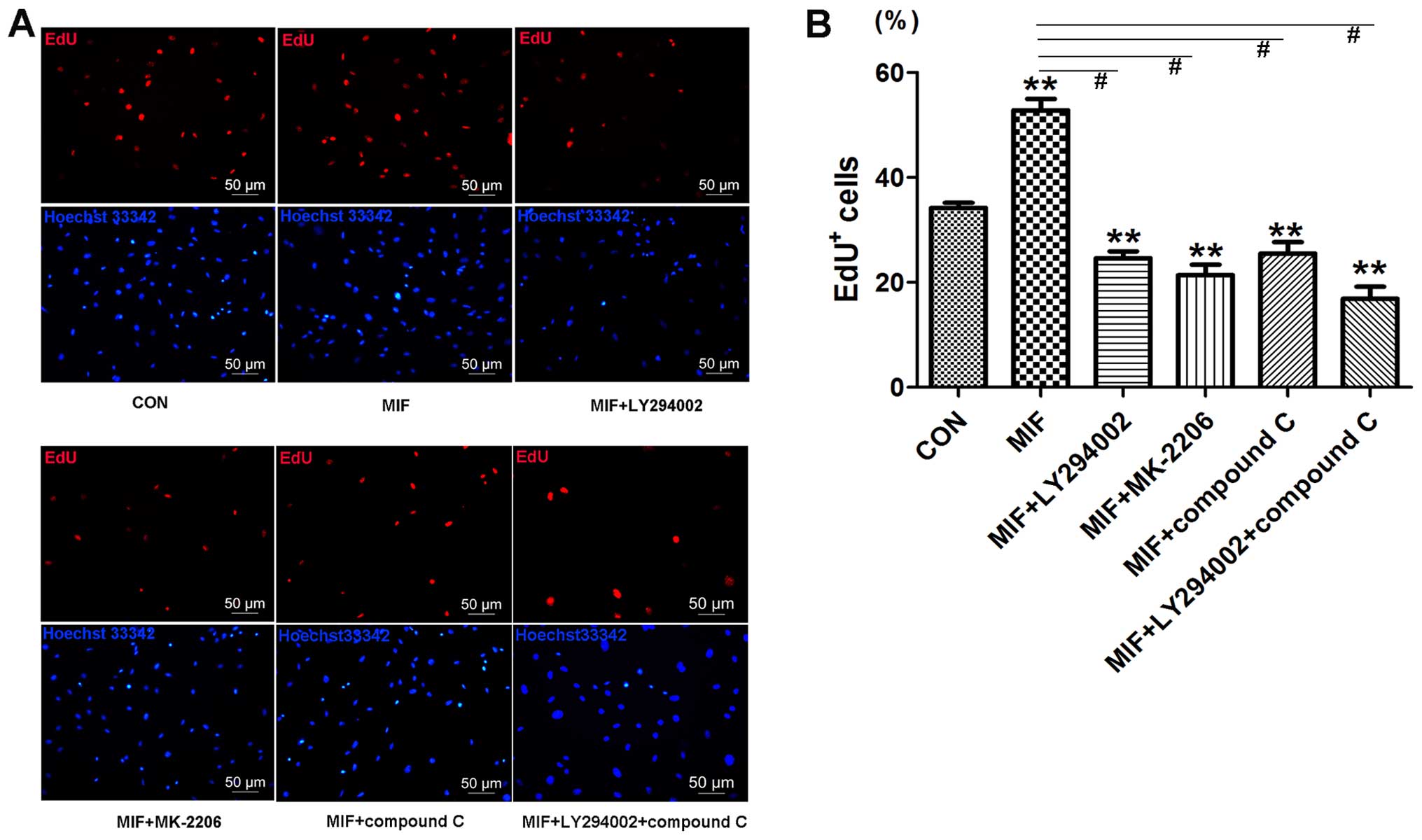
To further confirm that MIF acts through the
PI3K/Akt/mTOR and AMPK pathways to regulate CSC proliferation, the
cells were treated with a PI3K inhibitor (LY294002), Akt inhibitor
(MK-2206) and the AMPK inhibitor (compound C) prior to the addition
of MIF. EdU assay revealed that all the inhibitors markedly
inhibited MIF-induced cell proliferation (Fig. 7), which was consistent with the
effects of transient CD74 knockdown.
To determine whether CD74 mediates MIF-induced
PI3K/Akt/mTOR and AMPK activation in CSCs, we knocked down CD74 in
the CSCs. The results of western blot analysis revealed that CD74
knockdown significantly decreased the levels of phosphorylated Akt,
mTOR and AMPK compared to the control cells (Fig. 5D–F).
Effects of MIF on endothelial
differentiation
CSCs can differentiate into cardiomyocytes, smooth
muscle cells and endothelial cells under certain conditions
(35). Previous studies have
indicated that MIF can promote angiogenesis in teratoma and corneal
tissues (36,37). Akt phosphorylation plays an
important role in BM-MSC differentiation into endothelial cells
(38). Thus, we examined whether
MIF regulates CSC differentiation into endothelial cells. We
cultured the CSCs with MIF (200 ng/ml) for 7 days, and then removed
the growth factors and MIF, and allowed the cells to grow for an
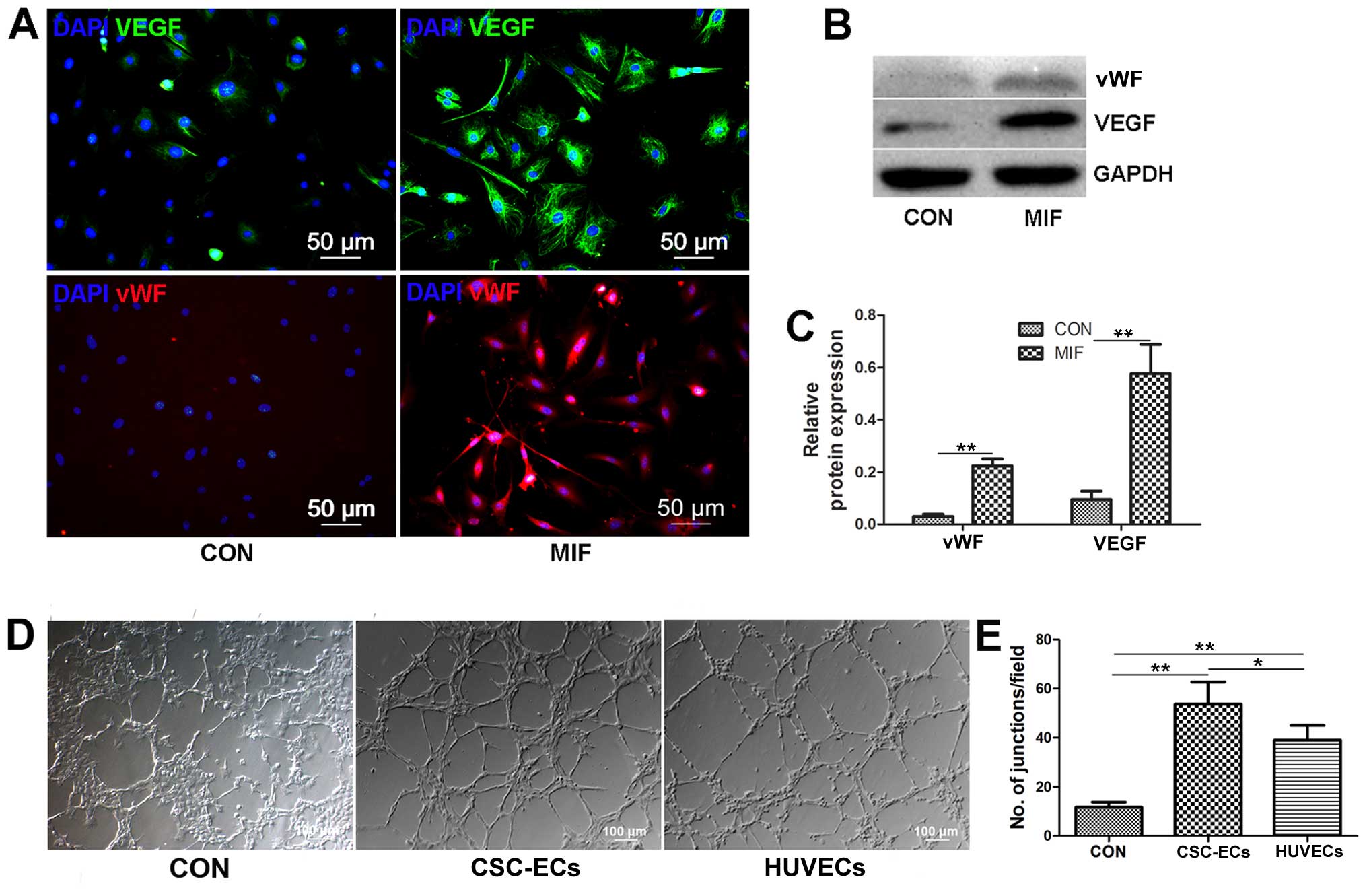
additional 7 days. We found that the MIF-treated CSCs expressed
significant amounts of VEGF and vWF compared to the controls
(Fig. 6A–C), and the MIF-treated
CSCs formed tubes in Matrigel paralleled with the positive control
cells, the HUVECs (Fig. 6D and
F); these results suggest that MIF-treated CSCs can
differentiate into endothelial cells. However, the CSCs in which
CD74 was knocked down, or the CSCs treated with MK-2206 (Akt
inhibitor) could cannot differentiate into endothelial cells (data
not shown). Thus, MIF may promote CSC differentiation into
endothelial cells through the activatwion of the Akt pathway.
Discussion
In the present study, we firstly isolated and
identify cardiac stem cells and we then demonstrated that CSCs
secrete the pleiotropic cytokine, MIF, to promote their survival
and proliferation. MIF also promoted CSC differentiation into
endothelial cells. We further found that MIF exerted its effects on
the CSCs through the MIF receptor CD74 expressed in CSCs,
suggesting that MIF can maintain CSC self-renewal and
differentiation capacity through autocrine and/or paracrine
mechanisms. These findings suggest that treatment with MIF may be
prove to be an effective strategy for the treatment of heart
diseases, including myocardial infarction and heart failure, by
activating native resident CSCs.
In our experiments, the used cells were positive for
thye stem cell marker, CD117, and negative for CD45, CD34 and KDR,
thus excluding contaminated cells, mainly hematopoietic lineage
cells containing blood cell lineage precursors, mast cells,
endothelial cells and endothelial progenitor cells (7,39).
The results of immunofluorescence staining revealed that our
isolated cells expressed early cardiac transcription factors and
can differentiate into 3 main cardiac lineages. These results
suggested that the cells were indeed stem cells of cardiac
origin.
Myocardial infarction and related heart failure are
the leading cause of mortality worldwide. However, the ability of
stem cells to restore heart function is encouraging and inspiring
(40). Urbanek et al
previously reported that the number of CSCs in normal heart atria
is approximately 5-fold higher than in the left ventricle, but in
ischemic heart failure, the number of multipotent CSCs in the left
ventricle increases to become greater than that in the atria
(11). These data suggest that in
the injured heart, there must be a substance that promotes CSC
growth and/or migration. Previous studies have found that
cardiomyocytes secrete MIF, which exerts anti-senescence,
antioxidant and anti-apoptotic effects on cardiomyocytes (41,42). In this study, we found that CSCs
secreted MIF and MIF promoted CSC survival and proliferation. These
results suggest that MIF secreted by CSCs or injured cardiomyocytes
may contribute to the increased number of CSCs in the injured left
ventricle. Furthermore, we found that MIF regulated cell cycle
progression by promoting the G1/S-phase transition, thereby
controlling cell proliferation, thus improving the number of CSCs
in the injured heart. Our study also indicated that the inhibition
of MIF or CD74 inhibited or delayed the G1/S-phase transition.
Proangiogenic therapy was originally a promising
strategy for the treatment of acute myocardial infarction, although
clinical trials have failed to elicit the expected effects
(43,44). Hilfiker-Kleiner et al found
that the endothelial differentiation capacity of c-kit+
resident stem cells was severely impaired in models of heart
failure (45). However, little is
known about the regulatory factors within the cardiac
microenvironment, particularly during heart failure and myocardial
infarction. Certain studies have suggested that circulating MIF
levels and MIF levels within the local damaged myocardium are both
increased. A number of studies have shown that MIF can promote
angiogenesis in teratomas, corneal tissue and heart by recruiting
stem cells or disrupting macrophage polarization (36,37). In the present study, we found that
MIF promoted CSCs to express VEGF and differentiate into
endothelial cells. Treatment with ISO-1 or CD74 knockdown inhibited
the effects of MIF on CSCs. At the same time, we performed a tube
formation assay to examine the angiogenic effect of MIF in
vitro and found that the CSCs treated with MIF formed tube
structures in parallel with the HUVECs, suggesting that MIF may
promote neovascularization following myocardial infarction by
promoting CSC differentiation into endothelial cells.
Neovascularization can often provide enough oxygen to support cell
growth and function. This effect further illustrated that MIF may
contribute to reverse heart dysfunction and decrease infarct size.
Whether MIF promotes neovascularization by regulating other
progenitor cells or other mechanisms requires further study.
The PI3K/Akt/mTOR signaling pathway plays a central
role in numerous cellular functions, including proliferation,
adhesion, migration, invasion, metabolism and survival (27). It is activated by a number of
inflammatory cytokines and agents, including lipopolysaccharide
(LPS) and phorbolmyristate acetate (PMA) (46). Our results demonstrated that
exogenous MIF activates the PI3K/Akt/mTOR pathway through its
receptor CD74. It has been demonsgtrated that the activation of the
PI3K/Akt pathway in cancer cells can also modulate the expression
of hypoxia-inducible factor-1 (HIF-1) and other angiogenic factors,
such as nitric oxide and angiopoietins, which function to increase
VEGF production (47). VEGF has
been identified as an angiogenic factor and survival factor that
stimulates angiogenesis and protects cells from stresses (48). In this study, we found that MIF
promoted the expression of VEGF in CSCs and CSC differentiation
into endothelial cells, suggesting that MIF improves cardiac
function by promoting angiogenesis. Our results are consistent with
the pro-angiogenic effects of MIF and PI3K/Akt/mTOR pathway
activation in other organs, including tumors and corneal tissue
(49,50). However, whether MIF regulates
additional angiogenic factors remains unclear.
AMPK orchestrates the regulation of both glycolysis
and glucose uptake and protects the heart against ischemic injury
and apoptosis (51). There is
evidence to suggest that MIF also plays a role in the stimulation
of the AMPK pathway to protect the heart in ischemic heart disease
(18) and promote the survival
and proliferation of neural stem/progenitor cells (22). In this study, we also found that
MIF promotes the phosphorylation of AMPK, and that AMPK inhibition
partly blocked the proliferation of CSC induced by MIF. These
results suggest that MIF promotes the proliferation of CSCs partly
through the activation of AMPK. As MIF can stimulate many signaling
pathways, we cannot rule out other mechanisms contributing to
effects of MIF on resident cardiac stem cells, such as JNK
inhibition.
Taken together, our data suggest that MIF promotes
CSC proliferation and endothelial differentiation, suggesting thatt
MIF not only increased the quantity, but also improved the function
of CSCs. This may be one explanation for why in ischemic heart
failure, the number of multipotent cardiac stem cells in the left
ventricle is higher than that in the atria and MIF can improve
heart function. However, MIF plays a potential role in
inflammation. It has been demonstrated that MIF provokes the
inflammatory response following myocardial infarction to remove
cellular debris and facilitate healing, whereas excessive
inflammation leads to adverse cardiac remodeling (52). It has also been demonstrated that
the role of MIF differs depending on the source of MIF. White et
al found that non-leukocyte MIF enhanced myocardial healing,
whereas leukocyte MIF enhanced damage (34). Koga et al found that the
intracellular overexpression of MIF had oxidoreductase effects,
however, exogenous MIF did not display such an effect (41). These may be one of the reasons why
many studies obtained unexpected results regarding the role of MIF
in post-infarct healing and cardiac remodeling (53). These suggest that further studies
are warranted in order to develop novel therapeutic methods which
preserve the benign effects of MIF to provide cardio-protection in
ischemic heart disease without activating MIF pro-inflammatory
activity. For instance, Luedike et al found that the
S-nitros(yl)ation modification of MIF enhanced the cytoprotective
effects in myocardial reperfusion injury (54). Furthermore, it has been
demonstrated that MIF acts as an oxidoreductase to maintain
intracellular redox homeostasis in cardiomyocytes (41). In the present study, we cannot
completely rule out the possibility that antioxidant or other
effects of MIF compensate for its protective effects on CSCs.
In conclusion, the results presented in this study
demonstrate that CSCs express MIF and its receptor CD74. We also
found that MIF affected cell survival, cell cycle progression,
proliferation and differentiation, by promoting the activation of
the PI3K/Akt/mTOR and AMPK pathways through CD74. Our findings
support the hypothesis that MIF may be a novel potential
therapeutic factor for the treatment for degenerative heart
disorders through CSC activation.
Acknowledgments
We would like to thank Dr Hulun Li and Dr Wei Liu
for their expert assistance with the experimental design and
excellent technical assistance. This study was supported by grants
from the National Natural Science Foundation of China (to B.Y.,
grant nos. 81171430 and 81330033) and the Key Laboratory of
Myocardial Ischemia Mechanism and Treatment (Harbin Medical
University), Ministry of Education (to J.C., grant no.
KF201402).
References
|
1
|
Tang XL, Rokosh G, Sanganalmath SK, Yuan
F, Sato H, Mu J, Dai S, Li C, Chen N, Peng Y, et al: Intracoronary
administration of cardiac progenitor cells alleviates left
ventricular dysfunction in rats with a 30-day-old infarction.
Circulation. 121:293–305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chimenti I, Smith RR, Li TS, Gerstenblith
G, Messina E, Giacomello A and Marbán E: Relative roles of direct
regeneration versus paracrine effects of human cardiosphere-derived
cells transplanted into infarcted mice. Circ Res. 106:971–980.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rufaihah AJ, Haider HK, Heng BC, Ye L, Tan
RS, Toh WS, Tian XF, Sim EK and Cao T: Therapeutic angiogenesis by
transplantation of human embryonic stem cell-derived
CD133+ endothelial progenitor cells for cardiac repair.
Regen Med. 5:231–244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leri A, Kajstura J and Anversa P: Role of
cardiac stem cells in cardiac pathophysiology: A paradigm shift in
human myocardial biology. Circ Res. 109:941–961. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Williams AR and Hare JM: Mesenchymal stem
cells: Biology, pathophysiology, translational findings, and
therapeutic implications for cardiac disease. Circ Res.
109:923–940. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Oskouei BN, Lamirault G, Joseph C, Treuer
AV, Landa S, Da Silva J, Hatzistergos K, Dauer M, Balkan W, McNiece
I and Hare JM: Increased potency of cardiac stem cells compared
with bone marrow mesenchymal stem cells in cardiac repair. Stem
Cells Transl Med. 1:116–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ellison GM, Vicinanza C, Smith AJ, Aquila
I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfò M,
et al: Adult c-kit(pos) cardiac stem cells are necessary and
sufficient for functional cardiac regeneration and repair. Cell.
154:827–842. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chimenti C, Kajstura J, Torella D, Urbanek
K, Heleniak H, Colussi C, Di Meglio F, Nadal-Ginard B, Frustaci A,
Leri A, et al: Senescence and death of primitive cells and myocytes
lead to premature cardiac aging and heart failure. Circ Res.
93:604–613. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dimmeler S and Leri A: Aging and disease
as modifiers of efficacy of cell therapy. Circ Res. 102:1319–1330.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Urbanek K, Quaini F, Tasca G, Torella D,
Castaldo C, Nadal-Ginard B, Leri A, Kajstura J, Quaini E and
Anversa P: Intense myocyte formation from cardiac stem cells in
human cardiac hypertrophy. Proc Natl Acad Sci USA. 100:10440–10445.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Urbanek K, Torella D, Sheikh F, De Angelis
A, Nurzynska D, Silvestri F, Beltrami CA, Bussani R, Beltrami AP,
Quaini F, et al: Myocardial regeneration by activation of
multipotent cardiac stem cells in ischemic heart failure. Proc Natl
Acad Sci USA. 102:8692–8697. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Burger-Kentischer A, Goebel H, Seiler R,
Fraedrich G, Schaefer HE, Dimmeler S, Kleemann R, Bernhagen J and
Ihling C: Expression of macrophage migration inhibitory factor in
different stages of human atherosclerosis. Circulation.
105:1561–1566. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calandra T, Echtenacher B, Roy DL, Pugin
J, Metz CN, Hültner L, Heumann D, Männel D, Bucala R and Glauser
MP: Protection from septic shock by neutralization of macrophage
migration inhibitory factor. Nat Med. 6:164–170. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morand EF, Bucala R and Leech M:
Macrophage migration inhibitory factor: An emerging therapeutic
target in rheumatoid arthritis. Arthritis Rheum. 48:291–299. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Calandra T and Roger T: Macrophage
migration inhibitory factor: A regulator of innate immunity. Nat
Rev Immunol. 3:791–800. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morand EF, Leech M and Bernhagen J: MIF: A
new cytokine link between rheumatoid arthritis and atherosclerosis.
Nat Rev Drug Discov. 5:399–410. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu CM, Lai KW, Chen YX, Huang XR and Lan
HY: Expression of macrophage migration inhibitory factor in acute
ischemic myocardial injury. J Histochem Cytochem. 51:625–631. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miller EJ, Li J, Leng L, McDonald C,
Atsumi T, Bucala R and Young LH: Macrophage migration inhibitory
factor stimulates AMP-activated protein kinase in the ischaemic
heart. Nature. 451:578–582. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qi D, Hu X, Wu X, Merk M, Leng L, Bucala R
and Young LH: Cardiac macrophage migration inhibitory factor
inhibits JNK pathway activation and injury during
ischemia/reperfusion. J Clin Invest. 119:3807–3816. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Stumptner-Cuvelette P and Benaroch P:
Multiple roles of the invariant chain in MHC class II function.
Biochim Biophys Acta. 1542:1–13. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leng L, Metz CN, Fang Y, Xu J, Donnelly S,
Baugh J, Delohery T, Chen Y, Mitchell RA and Bucala R: MIF signal
transduction initiated by binding to CD74. J Exp Med.
197:1467–1476. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ohta S, Misawa A, Fukaya R, Inoue S,
Kanemura Y, Okano H, Kawakami Y and Toda M: Macrophage migration
inhibitory factor (MIF) promotes cell survival and proliferation of
neural stem/progenitor cells. J Cell Sci. 125:3210–3220. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schrans-Stassen B, Lue H, Sonnemans D,
Bernhagen J and Post M: Stimulation of vascular smooth muscle cell
migration by macrophage migration inhibitory factor. Antioxid Redox
Signal. 7:1211–1216. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xiong CJ, Huang B, Zhou Y, Cun YP, Liu LT,
Wang J, Li CQ, Pan Y and Wang H: Macrophage migration inhibitory
factor inhibits the migration of cartilage end plate-derived stem
cells by reacting with CD74. PLoS One. 7:e439842012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xia W, Zhang F, Xie C, Jiang M and Hou M:
Macrophage migration inhibitory factor confers resistance to
senescence through CD74-dependent AMPK-FOXO3a signaling in
mesenchymal stem cells. Stem Cell Res Ther. 6(82)2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Palumbo S, Tsai TL and Li WJ: Macrophage
migration inhibitory factor regulates AKT signaling in hypoxic
culture to modulate senescence of human mesenchymal stem cells.
Stem Cells Dev. 23:852–865. 2014. View Article : Google Scholar
|
|
27
|
Ciuffreda L, Di Sanza C, Incani UC and
Milella M: The mTOR pathway: A new target in cancer therapy. Curr
Cancer Drug Targets. 10:484–495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao N, Zhang Z, Jiang BH and Shi X: Role
of PI3K/AKT/mTOR signaling in the cell cycle progression of human
prostate cancer. Biochem Biophys Res Commun. 310:1124–1132. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Morgensztern D and McLeod HL:
PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer
Drugs. 16:797–803. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smith AJ, Lewis FC, Aquila I, Waring CD,
Nocera A, Agosti V, Nadal-Ginard B, Torella D and Ellison GM:
Isolation and characterization of resident endogenous
c-Kit+ cardiac stem cells from the adult mouse and rat
heart. Nat Protoc. 9:1662–1681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu J, Wang Y, Du W, Liu W, Liu F, Zhang
L, Zhang M, Hou M, Liu K, Zhang S and Yu B: Wnt1 inhibits hydrogen
peroxide-induced apoptosis in mouse cardiac stem cells. PLoS One.
8:e588832013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hou M, Cui J, Liu J, Liu F, Jiang R, Liu
K, Wang Y, Yin L, Liu W and Yu B: Angiopoietin-like 4 confers
resistance to hypoxia/serum deprivation-induced apoptosis through
PI3K/Akt and ERK1/2 signaling pathways in mesenchymal stem cells.
PLoS One. 9:e858082014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen J, Baydoun AR, Xu R, Deng L, Liu X,
Zhu W, Shi L, Cong X, Hu S and Chen X: Lysophosphatidic acid
protects mesenchymal stem cells against hypoxia and serum
deprivation-induced apoptosis. Stem Cells. 26:135–145. 2008.
View Article : Google Scholar
|
|
34
|
White DA, Su Y, Kanellakis P, Kiriazis H,
Morand EF, Bucala R, Dart AM, Gao XM and Du XJ: Differential roles
of cardiac and leukocyte derived macrophage migration inhibitory
factor in inflammatory responses and cardiac remodelling post
myocardial infarction. J Mol Cell Cardiol. 69:32–42. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Beltrami AP, Urbanek K, Kajstura J, Yan
SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A,
Beltrami CA and Anversa P: Evidence that human cardiac myocytes
divide after myocardial infarction. N Engl J Med. 344:1750–1757.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Usui T, Yamagami S, Kishimoto S, Seiich Y,
Nakayama T and Amano S: Role of macrophage migration inhibitory
factor in corneal neovascularization. Invest Ophthalmol Vis Sci.
48:3545–3550. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X, Chen T, Leng L, Fan J, Cao K, Duan
Z, Zhang X, Shao C, Wu M, Tadmori I, et al: MIF produced by bone
marrow-derived macrophages contributes to teratoma progression
after embryonic stem cell transplantation. Cancer Res.
72:2867–2878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chu L, Hao H, Luo M, Huang Y, Chen Z, Lu
T, Zhao X, Verfaillie CM, Zweier JL and Liu Z: Ox-LDL modifies the
behaviour of bone marrow stem cells and impairs their endothelial
differentiation via inhibition of Akt phosphorylation. J Cell Mol
Med. 15:423–432. 2011. View Article : Google Scholar
|
|
39
|
Zhou Y, Pan P, Yao L, Su M, He P, Niu N,
McNutt MA and Gu J: CD117-positive cells of the heart: progenitor
cells or mast cells? J Histochem Cytochem. 58:309–316. 2010.
View Article : Google Scholar :
|
|
40
|
Moran AE, Forouzanfar MH, Roth GA, Mensah
GA, Ezzati M, Flaxman A, Murray CJ and Naghavi M: The global burden
of ischemic heart disease in 1990 and 2010: the Global Burden of
Disease 2010 study. Circulation. 129:1493–1501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Koga K, Kenessey A, Powell SR, Sison CP,
Miller EJ and Ojamaa K: Macrophage migration inhibitory factor
provides cardioprotection during ischemia/reperfusion by reducing
oxidative stress. Antioxid Redox Signal. 14:1191–1202. 2011.
View Article : Google Scholar
|
|
42
|
Ma H, Wang J, Thomas DP, Tong C, Leng L,
Wang W, Merk M, Zierow S, Bernhagen J, Ren J, et al: Impaired
macrophage migration inhibitory factor-AMP-activated protein kinase
activation and ischemic recovery in the senescent heart.
Circulation. 122:282–292. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Losordo DW and Dimmeler S: Therapeutic
angiogenesis and vasculogenesis for ischemic disease: part II:
cell-based therapies. Circulation. 109:2692–2697. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tongers J, Losordo DW and Landmesser U:
Stem and progenitor cell-based therapy in ischaemic heart disease:
Promise, uncertainties, and challenges. Eur Heart J. 32:1197–1206.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hilfiker-Kleiner D, Hilfiker A, Fuchs M,
Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z,
Podewski E, et al: Signal transducer and activator of transcription
3 is required for myocardial capillary growth, control of
interstitial matrix deposition, and heart protection from ischemic
injury. Circ Res. 95:187–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Salh B, Wagey R, Marotta A, Tao JS and
Pelech S: Activation of phosphatidylinositol 3-kinase, protein
kinase B, and p70 S6 kinases in lipopolysaccharide-stimulated Raw
264.7 cells: Differential effects of rapamycin, Ly294002, and
wortmannin on nitric oxide production. J Immunol. 161:6947–6954.
1998.PubMed/NCBI
|
|
47
|
Karar J and Maity A: PI3K/AKT/mTOR pathway
in angiogenesis. Front Mol Neurosci. 4(51)2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Byrne AM, Bouchier-Hayes DJ and Harmey JH:
Angiogenic and cell survival functions of vascular endothelial
growth factor (VEGF). J Cell Mol Med. 9:777–794. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Choudhary S, Hegde P, Pruitt JR, Sielecki
TM, Choudhary D, Scarpato K, Degraff DJ, Pilbeam CC and Taylor JA
III: Macrophage migratory inhibitory factor promotes bladder cancer
progression via increasing proliferation and angiogenesis.
Carcinogenesis. 34:2891–2899. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Amin MA, Volpert OV, Woods JM, Kumar P,
Harlow LA and Koch AE: Migration inhibitory factor mediates
angiogenesis via mitogen-activated protein kinase and
phosphatidylinositol kinase. Circ Res. 93:321–329. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Russell RR III, Li J, Coven DL, Pypaert M,
Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ and Young LH:
AMP-activated protein kinase mediates ischemic glucose uptake and
prevents postischemic cardiac dysfunction, apoptosis, and injury. J
Clin Invest. 114:495–503. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dayawansa NH, Gao XM, White DA, Dart AM
and Du XJ: Role of MIF in myocardial ischaemia and infarction:
insight from recent clinical and experimental findings. Clin Sci
(Lond). 127:149–161. 2014. View Article : Google Scholar
|
|
53
|
Gao XM, Liu Y, White D, Su Y, Drew BG,
Bruce CR, Kiriazis H, Xu Q, Jennings N, Bobik A, et al: Deletion of
macrophage migration inhibitory factor protects the heart from
severe ischemia-reperfusion injury: A predominant role of
anti-inflammation. J Mol Cell Cardiol. 50:991–999. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Luedike P, Hendgen-Cotta UB, Sobierajski
J, Totzeck M, Reeh M, Dewor M, Lue H, Krisp C, Wolters D, Kelm M,
et al: Cardioprotection through S-nitros(yl)ation of macrophage
migration inhibitory factor. Circulation. 125:1880–1889. 2012.
View Article : Google Scholar : PubMed/NCBI
|