Introduction
Renal interstitial fibrosis (RIF) is a common
pathway through which chronic kidney diseases (CKD) progress to
end-stage renal disease (1,2).
As a dynamic process, RIF is characterized pathologically by the
deposition of extracellular matrix (ECM) components in association
with the loss of tubular epithelial cells, the accumulation of
fibroblasts and the infiltration of inflammatory cells (3). It has been recognized that α-smooth
muscle actin (α-SMA)-positive myofibroblasts play a crucial role in
the progression of RIF following injury, and are responsible for
the synthesis of ECM components such as type I and III collagens
(1). Myofibroblasts are
terminally differentiated cells that drive the development of renal
fibrogenesis. Increasing evidence indicates that a large proportion
of renal myofibroblasts may originate from differentiated tubular
epithelial cells through the epithelial-mesenchymal transition
(EMT) (4,5).
The EMT may be an adaptive response of epithelial
cells following chronic injury and plays an integral role in the
development of RIF (4,5). During the EMT, tubular cells in the
kidneys lose their epithelial phenotypes [reduced levels of
epithelial markers such as E-cadherin and zonula occluden-1
(ZO-1)], and acquire new characteristic features of mesenchymal
cells [the expression of mesenchymal proteins including α-SMA and
vimentin]. As a multifunctional cytokine with profibrogenic
properties, transforming growth factor-β1 (TGF-β1) is regarded as
one of the important factors in the induction of EMT (3). It has been previously demonstrated
that the suppression of the EMT by bone morphogenetic protein-7
(BMP-7), or Smad7, an antagonist of TGF-β1 signaling, markedly
attenuates fibrotic changes following injury (6,7).
Therefore, halting or reversing TGF-β1-mediated EMT and the
acquisition of a myofibroblastic phenotype are promising, novel
strategies for attenuating the progression of RIF.
Sonic hedgehog (SHH) signaling is a stem
cell-related signaling pathway that plays an important role in
embryonic development, tissue regeneration and organogenesis
(8–10). The aberrant activation of the SHH
signaling pathway leads to pathological consequences, including the
development of various types of human tumors (11,12). In addition, previous studies have
also showed that activated SHH signaling is involved in tissue
fibrogenesis (13,14). In sustained and uncontrolled
tissue injury, SHH signaling is activated through the binding of
the ligand SHH, to its membrane receptor patched 1 protein (PTCH1).
The binding of the SHH ligand to PTCH1 relieves the inhibition of a
signal transducer smoothened (SMO), which is repressed by PTCH1,
and then initiates a signaling cascade. As a result, activated SHH
signaling promotes cellular proliferation and inhibits apoptosis
through enhancing the expression of proliferation-associated genes
such as c-Myc and cyclin D (15).
As EMT may involve abnormal proliferation, we hypothesized that
overactive SHH signaling also induces the acquisition of a
myofibroblastic phenotype through the EMT and results in RIF, which
is in agreement with previous studies of fibrogenesis in other
types of tissues (13,14).
To explore the role of the SHH signaling pathway in
RIF as well as the underlying molecular mechanisms responsible for
triggering the fibrogenic response, we examined the expression of
SHH-pathway proteins in rats subjected to either unilateral
ureteral obstruction (UUO) or bilateral ureteral obstruction (BUO).
In addition, the activity of SHH signaling was also evaluated in
rats subjected to BUO followed by recanalization of the ureters
(referred to as the RBUO group) was also evaluated. In
vitro, recombinant protein TGF-β1 was used to examine the
association between SHH signaling and EMT as well as ECM deposition
in renal tubular epithelial cells (RTECs). Moreover, the activity
of SHH signaling was regulated by treatment with exogenous SHH with
or without cyclopamine, an Smo inhibitor, in order to further
explore the detailed role of this pathway in the induction of EMT
and ECM accumulation.
Materials and methods
Establishment of the animal model
Forty-eight male Sprague-Dawley rats that weighed
approximately 180 to 200 g (6 to 8 weeks of age) were purchased
from the Experimental Animal Center of Wenzhou Medical University
(Wenzhou, China). The rats were housed under controlled conditions
[temperature (22–25°C), humidity (40–60%) and light cycle (12 h
dark/light)] and were fed standard rat chow and water, except for
one day of fasting prior to the operation. The weight-matched rats
were randomly assigned to two groups: i) an obstruction group and
ii) a recanalization group. These two groups were further divided
into sub-groups, each containing 12 rats. The obstruction group was
divided into the following sub-groups: sham-operation (for 7 days)
and UUO (for 7 days). The recanalization group was divided into the
following sub-groups: BUO for 1 day and RBUO (BUO for 1 day and
then recanalization of the ureters for 7 days). UUO and BUO surgery
were performed as previously described (16,17), and the longitudinal axis of the
kidneys was examined. In the sham-operation group, the left ureter
was only dissociated rather than ligated. In the rats in the BUO or
UUO groups, the ureter was ligated using a vein clamp at
approximately upper 1/3 near the kidney. In the rats in the RBUO
group, both sides of the ligated ureter were open again. All rats
were sacrificed by cervical dislocation and were anesthetized by
0.2% pentobarbital natrium. The kidney samples were excised on day
2 for the BUO group, and on day 8 for the UUO, sham-operation and
RBUO groups (after BUO operation).
The animal study protocols were approved by the
Institutional Animal Care and Use Committee of Wenzhou Medical
University (Wenzhou, China).
Histopathological examination
The kidney specimens were fixed in formalin,
embedded in paraffin and cut into 4-µm sections, and then
stained with hematoxylin and eosin (H&E) and Masson's trichrome
(both from Yuanye Biotechnology, Shanghai, China). The slides were
examined and images were captured using a DM4000B LED microscope
system and a DFC 420C 5M digital microscope camera (both from Leica
Microsystems GmbH, Wetzlar, Germany). The tubular diameter of
kidney specimens with H&E staining were examined based on the
longest dimension of renal tubule.
Immunohistochemical analysis
Immunohistochemical analysis was performed on
4-µm-thick sections of the kidney samples that had been
dewaxed in xylene and hydrated in graded ethanol (100, 95, 85, and
75%) and distilled water. Endogenous peroxidase was blocked by 3%
hydrogen peroxide. Antigen retrieval was performed by heating in
0.1% sodium citrate buffer (pH 6.0). To examine the SHH signaling
pathway, anti-SHH (bs-1544R, 1:800; Biogot Technology, Shanghai,
China), anti-PTCH1 (sc-9016, 1:1,000), anti-SMO (sc-13943, 1:1,000)
and anti-GLI1 (sc-6153, 1:1,000) (all from Santa Cruz
Biotechnology, Santa Cruz, CA, USA) antibodies were used. To
examine the EMT, anti-α-SMA (sc-32251, 1:1,000; Santa Cruz
Biotechnology), anti-E-caderin (ab53033, 1:1,000; Abcam, Cambridge,
MA, USA), and anti-type III collagen (bs-0549R, 1:800; Biogot
Technology) antibodies were used. To examine proliferation,
anti-proliferating cell nuclear antigen (PCNA) (sc-9857, 1:1,000;
Santa Cruz Biotechnology) antibody was used. Immunohistochemical
studies were semiquantitatively or quantitatively assessed by two
independent investigators in a blinded manner.
Cell culture and drug treatment
The normal rat kidney tubule epithelial (NRK-52E)
cell line was obtained from the Cell Bank of Chinese Academy of
Sciences (Shanghai, China). The NRK-52E cells were maintained in
Dulbecco's modified Eagle's medium supplemented with 5% fetal
bovine serum (FBS), 100 U/ml penicillin and 100 µg/ml
streptomycin (all from Invitrogen, Carlsbad, CA, USA). The NRK-52E
cells were seeded on six-well culture plates to approximately 70%
confluence in the complete medium containing 5% FBS for 24 h, which
was then replaced with serum-free medium for 24 h prior to
treatment with 5 ng/ml TGF-β1, or 10 ng/ml SHH (both from PeproTech
Rocky Hill, NJ, USA) with or without 5 µmol/l cyclopamine
(Merck Chemicals, Darmstadt, Germany).
Immunofluorescence staining
The NRK-52E cells were cultured with TGF-β1 or SHH
with or without cyclopamine in the six-well plates containing glass
slides and were then washed with PBS and fixed with 4%
paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) at 4°C for 30
min. Following permeabilization with 0.1% Triton X-100 for 10 min,
the specimens were washed with PBS and then blocked with 10% FBS in
order to eliminate non-specific fluorescence. Immunofluorescence
staining was performed using anti-type III collagen (1:800), α-SMA
(1:1,000), E-cadherin (1:1,000), PTCH1 (1:1,000), SMO (1:1,000),
GLI1 (1:1,000), and PCNA (1:1,000) as the primary antibody, and the
cell preparations were incubated with DyLight 488/594-labeled
secondary antibodies. The immunocytochemical samples were
semiquantitatively or quantitatively assessed by two independent
investigators in a blinded manner.
Reverse transcription-quantitative
polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the rat kidneys using
TRIzol reagent (Invitrogen), reverse transcribed to cDNA templates
using a ReverTra Ace qPCR RT kit (Toyobo Biotechnology, Tokyo,
Japan). RT-qPCR was performed using a SYBR-Green Real-Time PCR
Master Mix-Plus-(Toyobo Biotechnology). The quality was analyzed on
agarose gels, and the quantity was measured using Varioskan Flash
(Thermo Fisher Scientific, Waltham, MA, USA). The sequence-specific
primers of Shh; GLI family zinc finger 1 (Gli1); Ptch1; Smo;
collagen, type I, α 1 (Col1α1); collagen, type III, α 1 (Col3α1);
TGF-β1; TGF-β1R; E-cadherin; vimentin and α-SMA, listed in Table I, were synthesized by Invitrogen,
and β-actin was used as an endogenous reference gene. The samples
were analyzed in triplicate, and the melting curve was examined to
verify that a single product was amplified. The quantitative
analysis of all samples was performed using the ΔΔCT value
method.
 | Table ITwo-step RT-qPCR primers. |
Table I
Two-step RT-qPCR primers.
| Gene | Sequence
(5′→3′) | GenBank accession
no. | Length (bp) |
|---|
| Shh | F:
ACAAGAAACTCCGAACGATT | NM_017221 | 183 |
| R:
ACAAGAAACTCCGAACGATT | | |
| Gli1 | F
CCTCGTGGCTTTCATCAACTCT |
XM_006241443.2 | 185 |
| R:
GAAGCATCATTGAACCCTGAGTAGA | | |
| Ptch1 | F:
TCCAGCCGACCCAGATTG | NM_053566.1 | 252 |
| R:
ACATAGTCGTAGCCCCTGAAGTG | | |
| Smo | F:
TGTGGCTCAGGTAGATGG | NM_012807.1 | 170 |
| R:
GGTGGTTGCTCTTGATGG | | |
| Col1α1 | F:
GATCCTGCCGATGTCGCTAT | NM_053304.1 | 276 |
| R:
GGAGGTCTTGGTGGTTTTGTATTC | | |
| Col3α1 | F:
AAGGCTGAAGGAAATAGC | NM_032085.1 | 147 |
| R:
AATGTCATAGGGTGCGATA | | |
| TGF-β1 | F:
AGGCGGTGCTCGCTTTGT | NM_021578.2 | 137 |
| R:
GATTGCGTTGTTGCGGTCC | | |
| TGF-β1R | F:
TGATCCATCCGTTGAAGAAA |
XM_006238030.2 | 144 |
| R:
CTAGCTGCTCCATTGGCATA | | |
| E-cadherin | F:
GTGCCACCACCAAAGATA | NM_031334.1 | 195 |
| R:
GGCTGAGACAACCCTAAT | | |
| α-SMA | F:
GGCATCCACGAAACCACCT | NM_031004.2 | 212 |
| R:
CCGCCGATCCAGACAGAAT | | |
| Vimentin | F:
TGACCGCTTCGCCAACTAC | NM_031140.1 | 141 |
| R:
CGCAACTCCCTCATCTCCTC | | |
| β-actin | F:
CCCATCTATGAGGGTTACGC | NM_031144.2 | 150 |
| R:
TTTAATGTCACGCACGATTTC | | |
Enzyme-linked immunosorbent assay
(ELISA)
Suspensions were prepared by the homogenization of
the rat kidney tissues and subsequently centrifuged (at 5,000 × g,
at 4°C for 10 min) and the resulting supernatant was collected.
Cells treated with 0–50 ng/ml SHH were cultured for 24 h, and the
culture super natant fluid was collected. The avidin-biotin
complex-ELISA was performed according to the manufacturer's
instructions in order to determine the levels of TGF-β1 and SHH.
The ELISA kits were purchased from XiTang Biotechnology (Shanghai,
China). All experiments were repeated at least three times.
Western blot analysis
Whole proteins from the rat kidneys or the cultured
cells were collected and western blot analysis was performed using
antibodies against PTCH1 (sc-9016, 1:200), SMO (sc-13943, 1:200),
α-SMA (sc-32251, 1:200), Rac1 (sc-95, 1:200; Santa Cruz
Biotechnology) and E-cadherin (1:400). Quantification was performed
by measuring the intensity of the signals using Image-Pro Plus
software (version 6.0; Media Cybernetics, Silver Spring, MD, USA),
and normalized to that of the β-actin antibody (AP0060, 1:3,000;
Biogot Technology).
Statistical analysis
The data are presented as the means ± SEM. All of
the statistical analyses were performed using the Statistical
Package for Social Sciences (version 16.0; SPSS Inc., Chicago, IL,
USA). To analyze the difference between two groups, the two-sided
Student's t-test was used. One-way ANOVA was used when more than
two groups were present. A P-value <0.05 was considered to
indicate a statistically significant difference.
Results
SHH signaling activity increases during
RIF induced by UUO in rats
As expected, the longitudinal axis and the tubular
diameter in the UUO group were significantly increased compared
with the sham-operated group (Fig.
1A). In the obstructed kidneys, H&E staining revealed
marked tubular dilation and atrophy associated with inflammatory
cell infiltration (Fig. 1B, top
panels), and Masson trichrome staining revealed severe
tubulointerstitial fibrosis (Fig.
1B, lower panels). In addition, the results of ELISA showed
that the expression levels of the profibrotic factor TGF-β1 in the
kidneys from the UUO group were significantly elevated (Fig. 1C). The results of
immunohistochemical staining, RT-qPCR and western blot analysis
revealed the high mRNA and protein expression of the mesenchymal
markers, α-SMA, vimentin and Rac1, and the low expression of the
epithelial marker E-cadherin in the UUO group (Fig. 1D–F). Moreover, UUO also increased
Rac1 protein expression (Fig.
1F). Rac1 is regarded as an important indicator of cellular
adhesion and invasion (18) and
the high expression of Rac1 increases the migration ability of
epithelial cells and promotes the transition to myofibroblasts.
Thus, ureteral obstruction induced a myofibroblastic phenotypic
change, resulting in excessive ECM deposition and RIF, as
demonstrated by the upregulated expression of TGF-β1, type I
(Col1α1) and III (Col3α1) collagens in the kidney tissues of the
rats subjected to UUO (Fig. 1D and
E).
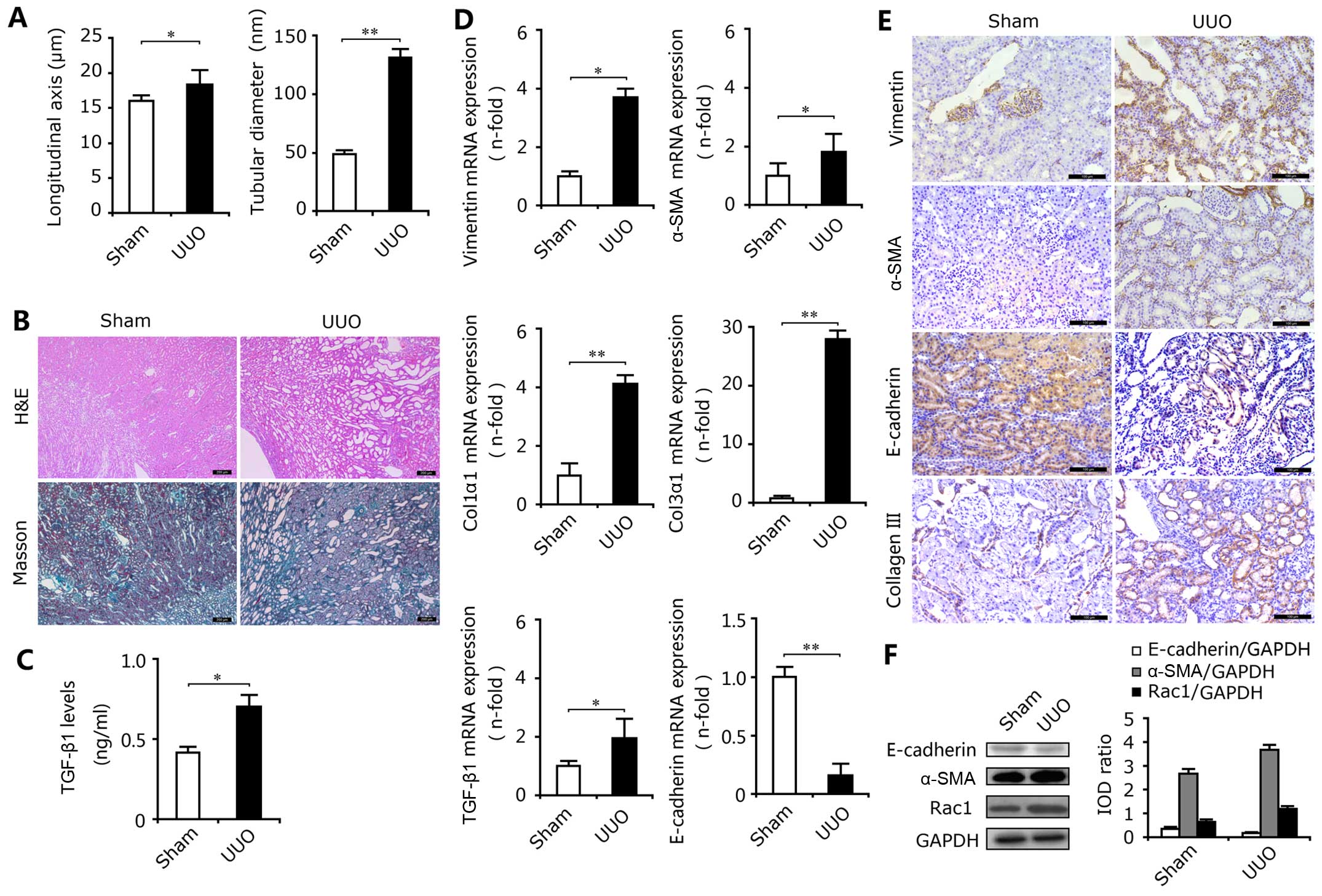 | Figure 1Epithelial-mesenchymal transition
(EMT) and renal interstitial fibrosis (RIF) in rats with unilateral
ureteral obstruction (UUO). (A) Ureteral obstruction induced marked
increases in the longitudinal axis and tubular diameter in the rat
kidneys. *P<0.05, **P<0.01 vs. the sham
group. (B) H&E staining showed obvious kidney injury in the
kidneys from the UUO group and Masson's trichrome staining revealed
excessive deposition of total collagen. (C) The levels of TGF-β1,
determined by ELISA, in the rats with UUO were enhanced. Results
are expressed as the means ± SEM (n=3), *P<0.05 vs.
the sham group. (D) RT-qPCR showed that mRNA expression levels of
vimentin, α-smooth muscle actin (α-SMA), collagen, type I, α 1
(Col1α1), collagen, type III, α 1 (Col3α1) and TGF-β1 were
increased in the UUO group, and the mRNA expression level of
E-cadherin was decreased. Results are expressed as the means ± SEM
(n=6), *P<0.05, **P<0.01 vs. the sham
group. (E) Upregulated expression of vimentin, α-SMA and type III
collagen and downregulated expression of E-cadherin in kidney
tissues of rats subjected to UUO compared with the sham groups,
were determined by immunohistochemical staining. Bar, 100
µm. (F) Western blot analysis revealed the enhanced protein
expression of α-SMA and Rac1, and the reduced expression of
E-cadherin in the kidneys of rats with UUO. |
In the obstructed kidneys, the enhanced expression
of PCNA in the renal cortex, particularly around the renal tubules,
revealed that the proliferation of RTECs may involve tubular EMT
and RIF (Fig. 2A and B). Thus, we
examined the activation of the proliferation-associated SHH
signaling pathway in the kidneys from the UUO group. Our results
revealed that obstruction not only enhanced the Shh levels
(Fig. 2C), but also upregulated
the protein expression of SMO, SHH and GLI1, and downregulated the
expression of PTCH1 (Fig. 2D). In
addition, the mRNA expression levels as indicated by RT-qPCR also
confirmed the results of immunohistochemical staining described
above (Fig. 2E). Thus, these
results showed that SHH signaling was activated following
obstruction. Furthermore, PTCH1 is mainly expressed in the
epithelial cells around the renal tubules, which suggests that the
activation of SHH signaling may be principally responsible for the
induction of the proliferation of RTECs and transdifferentiation
into myofibroblasts. The activation of SHH signaling may be closely
associated with the induction of EMT.
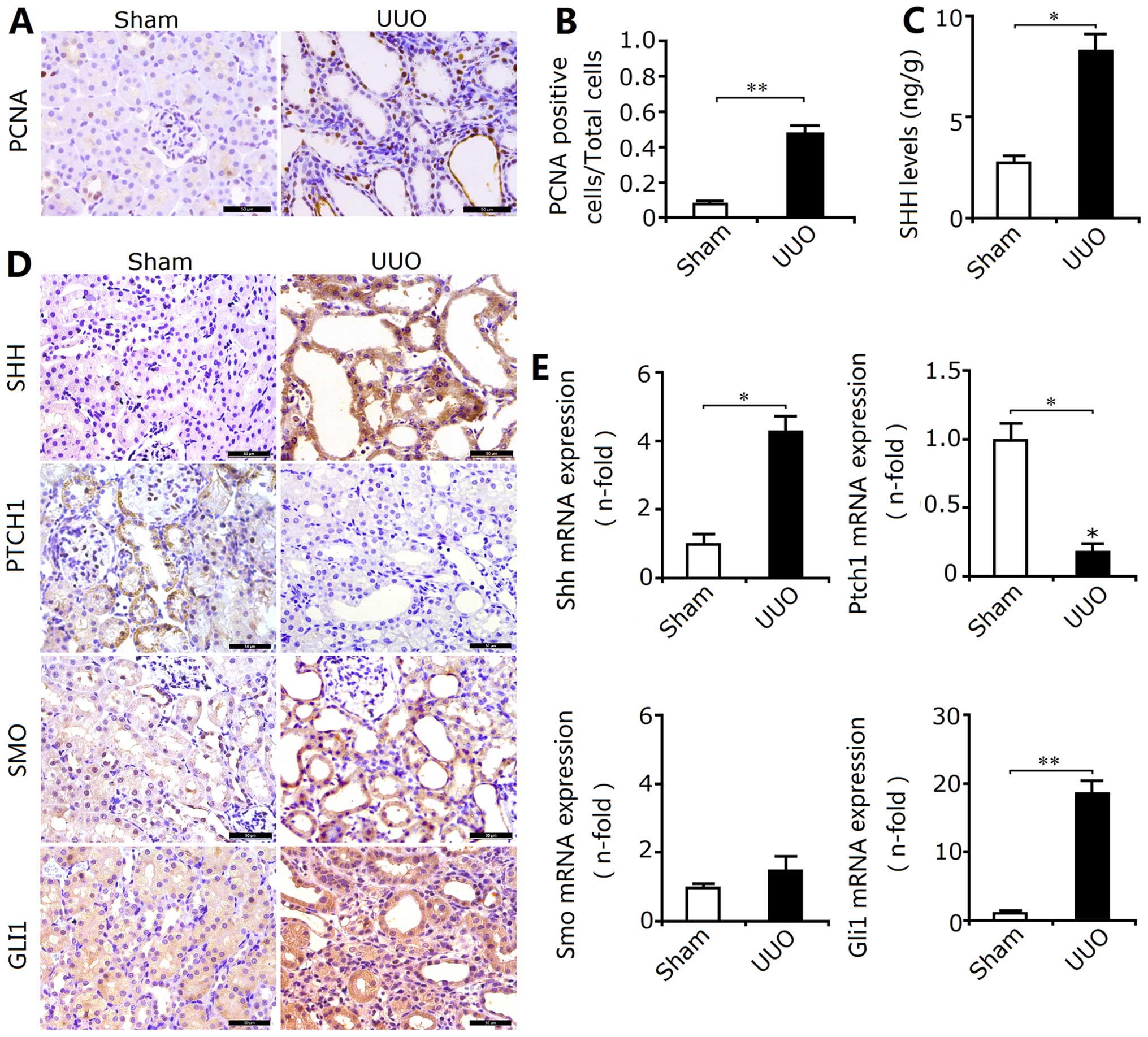 | Figure 2Activation of sonic hedgehog (SHH)
signaling during renal interstitial fibrosis (RIF) in rats with
unilateral ureteral obstruction (UUO). (A) The expression of
proliferating cell nuclear antigen (PCNA) was determined by
immunohistochemical staining. Bar, 50 µm. (B) The ratio of
PCNA-positive cells/total cells (determined according to Fig. 2A in the rats with UUO) was
significantly increased. Results are expressed as the means ± SEM
(n=10), **P<0.01 vs. the sham group. (C) The levels
of sonic hedgehog (SHH) determined by ELISA in the rats with UUO
were enhanced. Results are expressed as the means ± SEM (n=3),
*P<0.05 vs. the sham group. (D) The location and
expression of SHH, patched 1 protein (PTCH1), smoothened (SMO), and
GLI family zinc finger 1 (GLI1) were determined by
immunohistochemical staining. Bar, 100 µm. The results
showed that the protein expression of SHH, SMO and GLI1 in the rats
with UUO was increased, and the expression of Ptch1 was decreased.
(E) RT-qPCR revealed that changes in the mRNA expression of Shh,
Ptch1, Smo and Gli1 were in accordance with the changes in protein
expression. Results are expressed as the means ± SEM (n=6),
*P<0.05, **P<0.01 vs. the sham
group. |
Relief of ureteral obstruction reduces
SHH signaling activity, as well as attenuating the EMT and RIF
In this experiment, the rats with BUO were selected
as the controls for the rats subjected to recanalization. As
observed in the rats subjected to UUO, BUO induced marked
tubulointerstitial injury in the kidney tissues. Although the
deposition of total collagen determined by Masson's trichrome
staining showed that the extent of RIF in the rat kidneys from the
BUO group was less severe due to the shorter injury time (Fig. 3A), the upregulated expression of
α-SMA and type III collagen and the downregulated expression of
E-cadherin confirmed the induction of EMT and the excessive
deposition of ECM components (Fig.
3B). Recanalization attenuated the extent of tubulointerstitial
injury and reduced the deposition of total collagen. In addition,
recanalization also decreased the TGF-β1 levels, inhibited EMT
induction and reduced ECM accumulation (Fig. 3B–D). Thus, recanalization
attenuates obstruction-induced EMT and RIF.
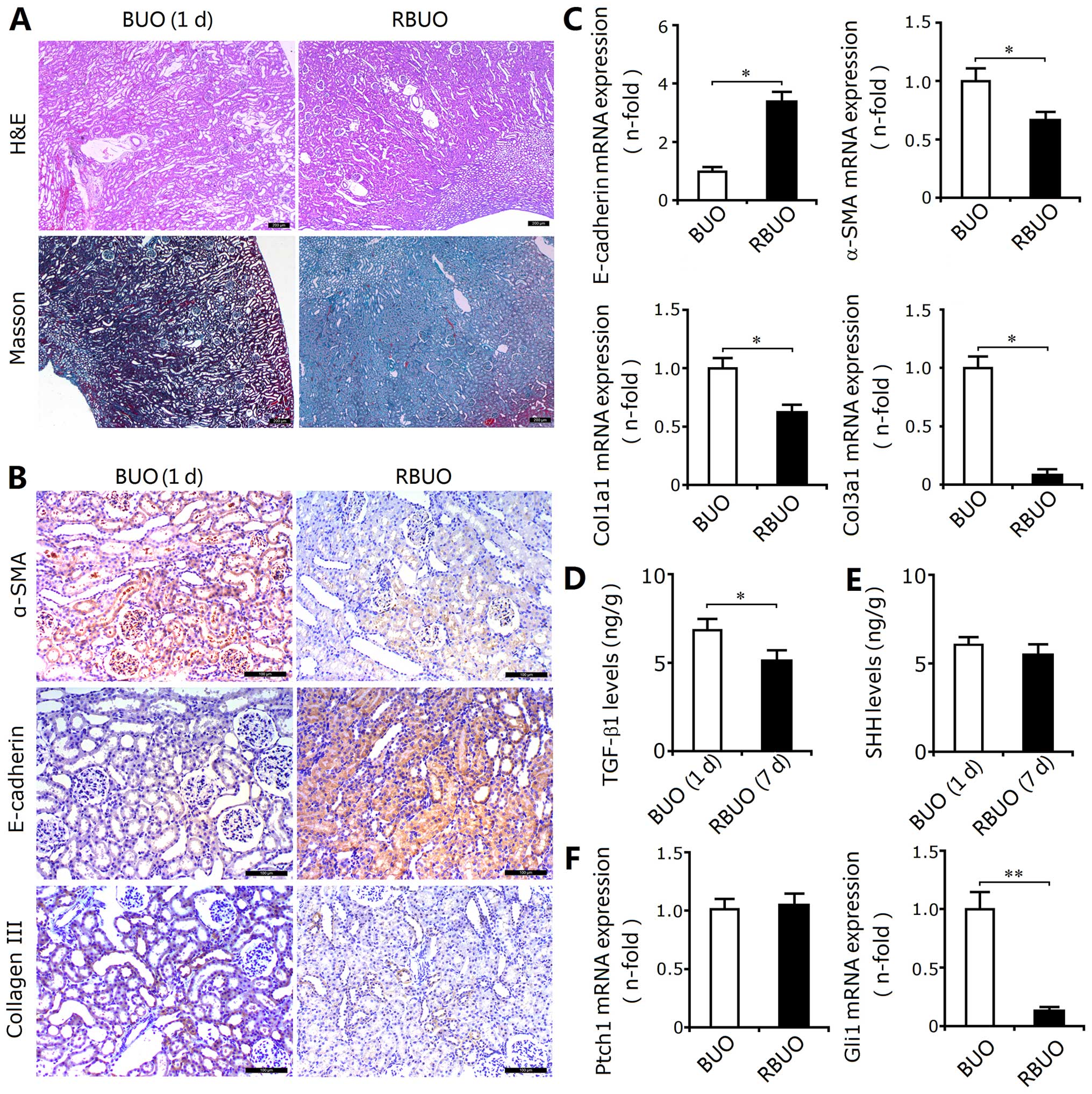 | Figure 3Sonic hedgehog (SHH) signaling
activity, epithelial-mesenchymal transition (EMT) and renal
interstitial fibrosis (RIF) in the rats subjected to bilateral
ureteral obstruction (BUO) followed by recanalization (RBUO group).
(A) Compared with the rats subjected to BUO, H&E and Masson's
trichrome staining revealed that recanalization attenuated kidney
injury and reduced collagen deposition in the RBUO group,
respectively. Bar, 200 µm. (B) The recanalization operation
decreased the protein expression of α-smooth muscle actin (α-SMA)
and type III collagen (determined by immunohistochemical staining)
in the kidneys from the RBUO group, and increased the expression of
E-cadherin. Bar, 100 µm. (C) Recanalization increased the
mRNA expression of E-cadherin (determined by RT-qPCR) in the rat
kidneys and decreased the expression of α-SMA, Col1α1 and Col3α1.
Results are expressed as the means ± SEM (n =6),
*P<0.05 vs. the BUO group. (D) Downregulated levels
of transforming growth factor-β1 (TGF-β1) in the RBUO group
compared with those in the BUO group. Results are expressed as the
means ± SEM (n=3), *P<0.05 vs. the BUO group. (E)
There were no significant differences between the levels of sonic
hedgehog (SHH) in the RBUO and BUO groups. (F) Recanalization
downregulated the mRNA expression of GLI family zinc finger 1
(Gli1), but not of patched 1 protein (Ptch1). Results are expressed
as the means ± SEM (n=6), **P<0.01 vs. the BUO
group. |
In the rats subjected to UUO, SHH signaling was
activated as mentioned above. In the rats subjected to BUO followed
by recanalization, we also identified the decreased mRNA expression
of Gli1, a target gene of SHH signaling; however, the protein
levels of SHH and mRNA expression of Ptch1 did not show any
significant differences (Fig. 3E and
F). Thus, we speculated that recanalization-induced reduction
of renal EMT and RIF may be associated with downregulated SHH
signaling activity in a noncanonical manner. The inhibition of SHH
signaling activation may be an important strategy for the
attenuation of RIF.
Activation of SHH signaling occurs during
the EMT and ECM deposition in TGF-β1-treated RTECs
In vivo, the above-mentioned findings
demonstrated that the activation of the SHH signaling pathway was
involved in the induction of EMT and RIF. However, confirmation
in vitro is required; thus, we investigated the role of SHH
signaling in TGF-β1-induced EMT in cultured RTECs (NRK-52E). We
found that TGF-β1 significantly induced the overexpression of α-SMA
and type III collagen and reduced the expression of E-cadherin as
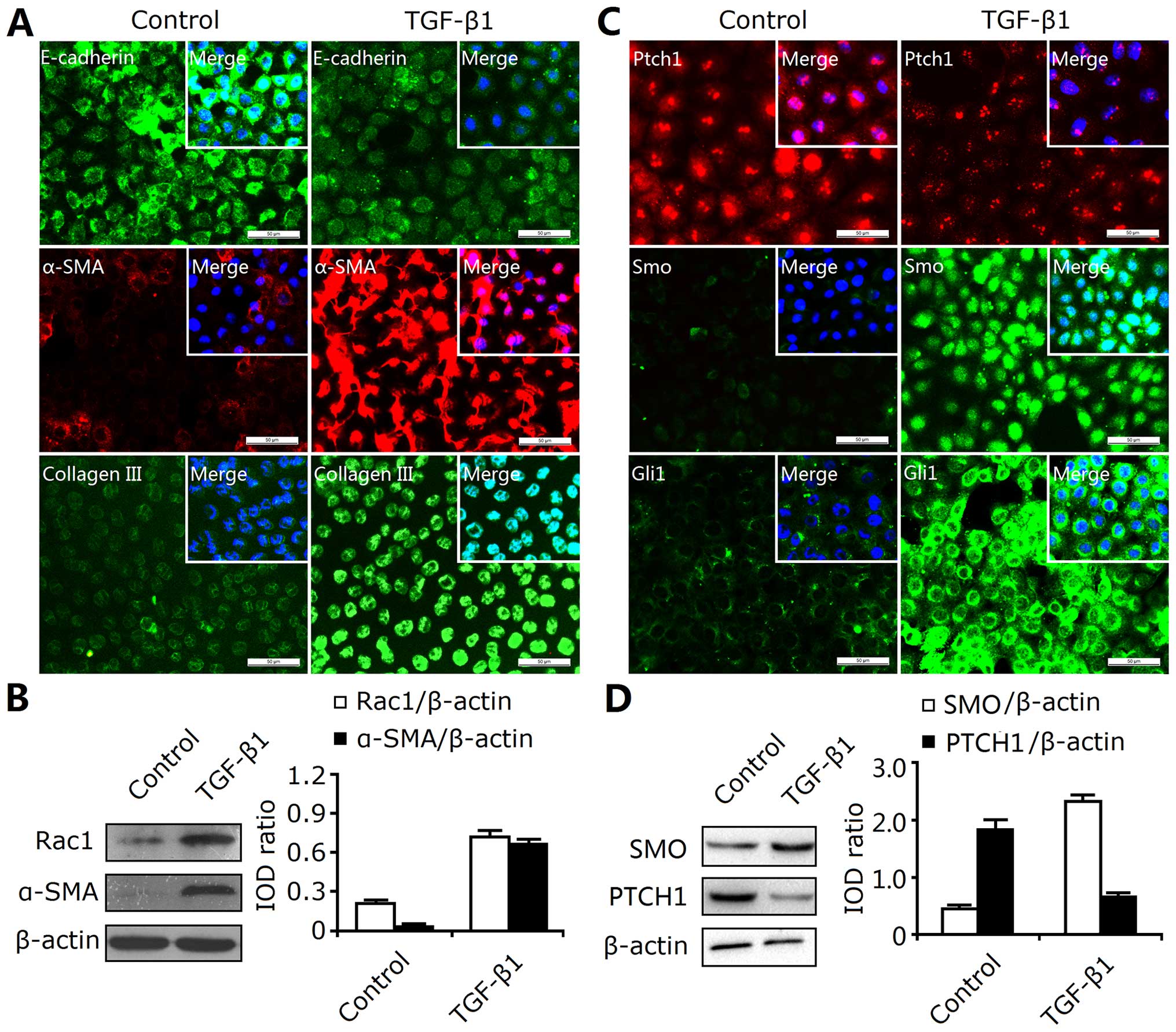
indicated by immunofluorescence staining (Fig. 4A). Western blot analysis revealed
that TGF-β1 not only enhanced α-SMA expression, but also enhanced
Rac1 expression (Fig. 4B).
Therefore, the EMT response in the cultured NRK-52E cells was
induced by TGF-β1.
In addition, TGF-β1 also induced the activation of
the SHH signaling pathway. As shown in Fig. 4C, the protein expression of SMO
and GLI1 were significantly increased, and the expression of PTCH1
was decreased in the TGF-β1-treated NRK-52E cells. Moreover,
western blot analysis revealed the enhanced expression of SMO and
the decreased expression of PTCH1 (Fig. 4D). Thus, these findings indicated
that the SHH signaling pathway was activated during the EMT induced
by TGF-β1 treatment.
Exogenous SHH promotes TGF-β1 expression
and the EMT in RTECs
Given that TGF-β1 induced EMT and ECM deposition,
and was accompanied by the activation of SHH signaling, we then
aimed to determine whether activated SHH signaling directly
promotes the EMT and ECM deposition. In this experiment using
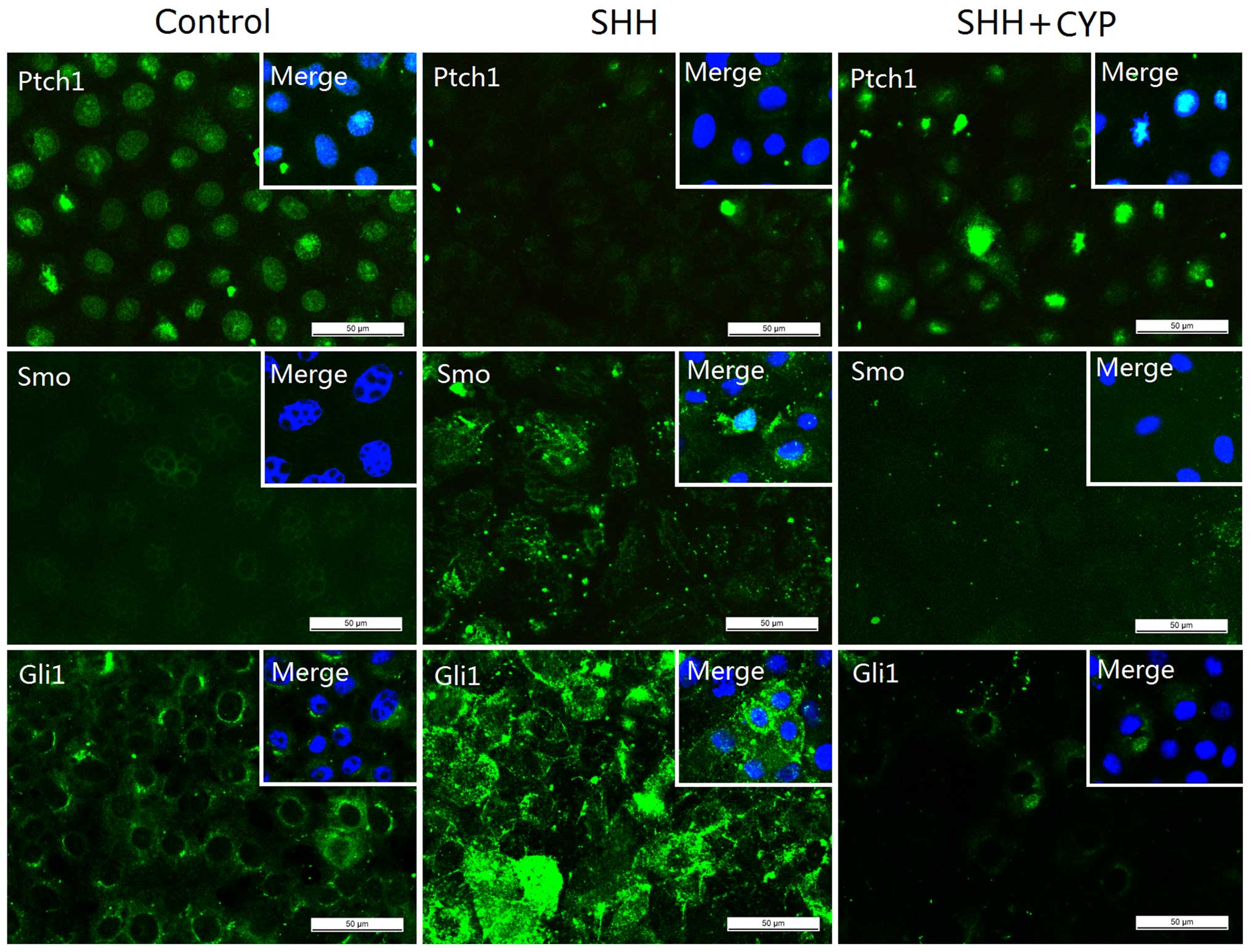
cultured NRK-52E cells, exogenous recombinant protein SHH was used
as an activator of the SHH pathway. As expected, SHH enhanced the
protein expression of SMO and GLI1, and reduced the expression of
PTCH1, and thus, activated the SHH signaling pathway (Fig. 5). The activation of SHH signaling
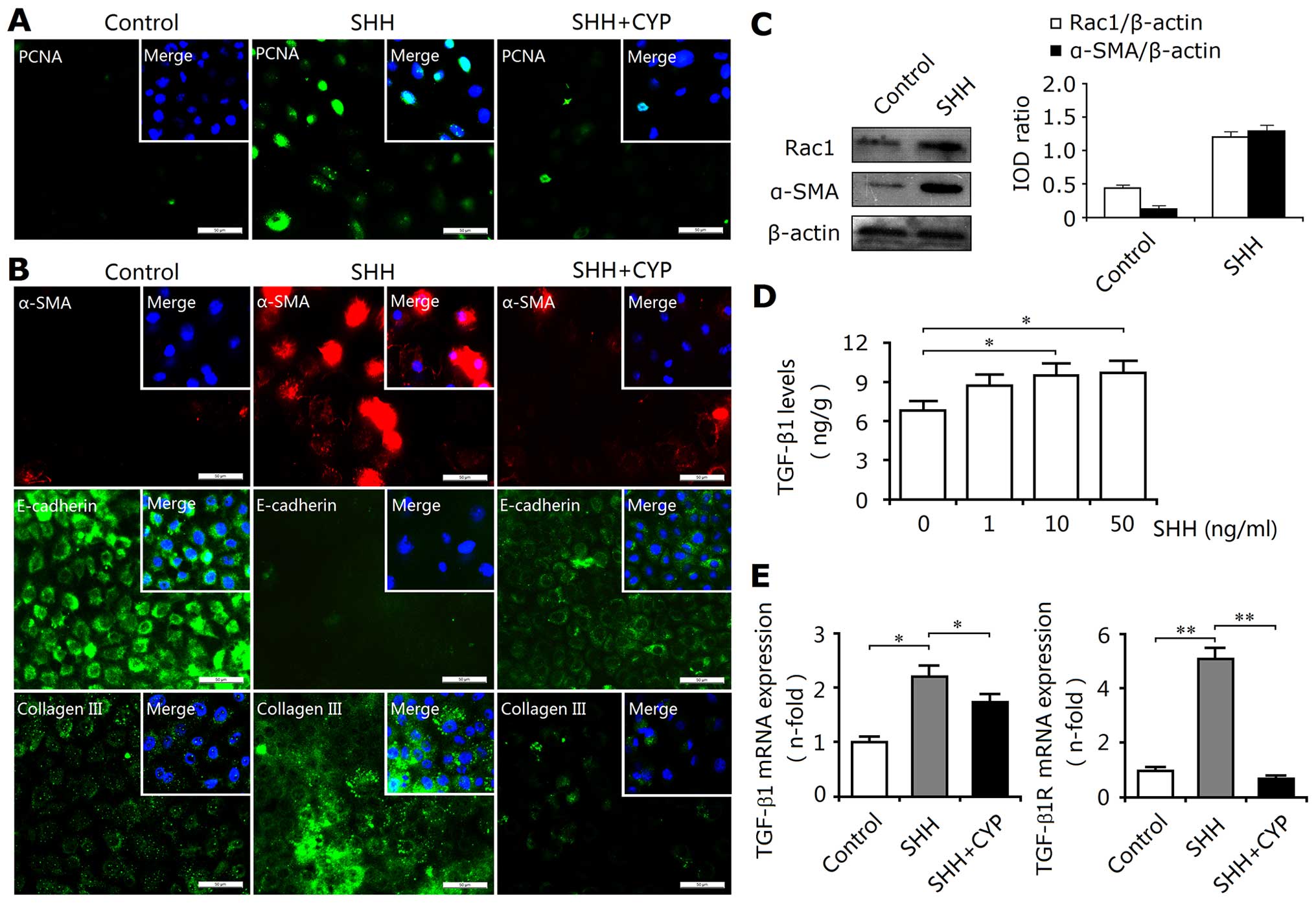
resulted in the upregulated expression of PCNA and cellular
proliferation (Fig. 6A). This
proliferative activity did not induce an increase in cell numbers;
however, phenotypic changes were induced. Thus, activated SHH
signaling may play an important role in promoting the deposition of
ECM-producing myofibroblasts. Our findings also confirmed this
hypothesis. As shown in Fig. 6B,
Shh upregulated the protein expression levels of α-SMA and type III
collagen, and downregulated the expression of E-cadherin.
Additionally, Shh also enhanced Rac1 expression (Fig. 6C). Furthermore, this Shh-mediated
EMT response may be associated with increased levels of TGF-β1 and
its receptor TGF-β1R (Fig. 6D and
E). Thus, these results suggested that activated SHH signaling
induced the expression of TGF-β1 as well as EMT and ECM
accumulation.
Blockade of SHH signaling inhibits the
Shh-mediated EMT and ECM deposition
As stated above, the activated SHH signaling pathway
was involved in the induction of EMT and ECM deposition. Herein, we
examined whether the downregulated activity of the SHH signaling
pathway exerts a protective effect on these fibrotic-like changes.
A small molecule antagonist of SHH signaling, cyclopamine was added
to the NRK-52E cell culture following SHH treatment. As shown in
Fig. 5, cyclopamine significantly
inhibited the Shh-mediated downregulation of PTCH1 protein
expression as well as the SHH-mediated upregulation of SMO and GLI1
expression. Thus, cyclopamine inhibited the SHH-induced activation
of SHH signaling. This inhibition of SHH signaling significantly
decreased the proliferation of the NRK-52E cells (Fig. 6A), and resulted in the inhibition
of the EMT process and the reduced synthesis of ECM components, as
indicated by the downregulated expression of α-SMA and type III
collagen, and the upregulated expression of E-cadherin (Fig. 6B). Moreover, cyclopamine also
reduced the expression levels of TGF-β1 and TGF-β1R (Fig. 6E). Thus, the in vitro
blockade of the SHH signaling pathway efffectively exerts
inhibitory effects on the EMT and ECM deposition.
Discussion
The present study provides evidence that ureteral
obstruction enhances the expression levels of SHH-pathway proteins,
mesenchymal markers, and decreases the expression of epithelial
markers in the kidney tissues of rats, suggesting that RIF is
associated with tubular EMT and the overactivity of the SHH
signaling pathway. The EMT as well as fibrotic changes, both of
which are associated with SHH signaling, were inhibited by
performing recanalization of the ureter. We have also shown that
SHH signaling is activated during the EMT and ECM accumulation in
the TGF-β1-treated RTECs. Exogenous Shh recombinant protein
activated SHH signaling, resulting in the upregulated expression of
mesenchymal genes, the profibrogenic cytokine TGF-β1 and the
downregulated expression of epithelial markers. Notably, we have
found that the blockade of SHH signaling with cyclopamine abolished
SHH-mediated EMT, the acquisition of a myofibroblastic phenotype,
and decreased TGF-β1 expression and ECM production. Thus, the SHH
signaling pathway plays an important role in tubular EMT and
RIF.
As a key pathway associated with animal development,
the SHH signaling pathway has been reported to be involved in the
pathogenesis of chronic tissue destruction and impaired renal
function (19,20). As SHH signaling plays an important
role in the formation of nephrons and kidney development (21,22), some researchers have hypothesized
that the aberrant activation of this signaling most likely leads to
RIF (23,24). Using a panel of hedgehog-report
mice, Fabian et al showed that the ligand Shh is expressed
in RTECs, whereas the effector Gli1 is expressed in perivascular
fibroblasts and pericytes, suggesting that the SHH signaling
pathway is activated in a paracrine manner during renal
fibrogenesis (24). Our results
supported the finding that the SHH-signaling ligand Shh is mainly
expressed in RTECs. However, evidence from immunohistochemical
staining in the present study indicated that the effector Gli1 is
also expressed in tubular epithelial cells as well as in
interstitial cells in the kidney tissues of rats subjected to UUO,
suggesting that obstruction also induces the activation of renal
SHH signaling in an autocrine and a paracrine manner. These results
regarding RIF are consistent with a study of liver fibrosis by Yang
et al (29), and they
identified that the SHH signaling pathway acts in an autocrine
manner in the pathogenesis of cirrhosis. In the obstructed kidney,
we hypothesized that the GLI1 protein may be a common downstream
effector not only of the SHH pathway, but also of other signaling
pathways, such as the WNT and BMP pathways (25–27). These activated signaling pathways
may regulate Gli1 expression directly or indirectly in tubular
epithelial cells, interstitial pericytes and fibroblasts.
Another study, by Ding et al (23), also demonstrated that ureteral
obstruction induces SHH expression, predominantly in the renal
tubular epithelium of fibrotic kidneys. In addition, they
identified interstitial fibroblasts as SHH-responding cells using
Gli1lacZ knock-in mice, and in vitro activated
SHH signaling promoted myofibroblast activation and matrix
production. They hypothesized that SHH signaling-mediated EMT may
be the principal cause of myofibroblast accumulation. This
deduction was also confirmed by our study; activated SHH signaling
is associated with the induction of EMT and the excessive
accumulation of ECM components in the kidneys of mice subjected to
UUO. Recanalization abolished these above-mentioned changes. In
addition, activated SHH signaling, the acquisition of a
myofibroblast phenotype and ECM deposition occurred in the
TGF-β1-treated RTECs. These findings indicated that SHH signaling
may be involved in the induction of EMT and RIF.
Although the study of renal fibroblasts by Ding
et al showed that activated SHH signaling induces a
myofibroblastic phenotype derived from the epithelium (23), further studies from the
perspective of epithelial cells are warranted. Thus, in cultured
RTECs exogenous SHH recombinant protein was used to stimulate the
activation of SHH signaling. As a result, the expression of PCNA
was enhanced. Additionally, the upregulated expression of the
mesenchymal marker α-SMA, Rac1 and type III collagen, and the
downregulated expression of the epithelial marker E-cadherin were
observed in the RTECs. Thus, activated SHH signaling promotes
cellular proliferation, and it is accompanied by the transition of
RTECs to myofibroblasts. We hypothesized that the proliferation of
RTECs, which may be associated with a feedback mechanism, is
necessary for the adaptation to injury within the micro-environment
and may be one outcome of cell cycle arrest (28,29). However, the abnormal proliferation
of RTECs may result in the acquisition of a myofibroblastic
phenotype through the EMT (1,30).
As an adaptive response of epithelial cells following injury, EMT
is increasingly recognized not only as a part of the repair process
(unless it is uncontrolled), but also as an integral part of renal
fibrogenesis. Compared to epithelial cells, myofibroblasts possess
a greater ability to adapt to injury or an abnormal
micro-environment. Our in vitro findings confirmed again
that activated SHH signaling plays an important role in EMT
induction and RIF.
In addition, our results notably showed that TGF-β1
may induce the activation of the SHH signaling pathway in RTECs.
Similarly, activated SHH signaling may enhance the expression of
TGF-β1 and its receptor TGF-β1R, suggesting that there is a
feedback loop between the SHH and TGF-β1 signaling pathways, and
their crosstalk induces EMT and ECM accumulation. However, in order
to clarify and confirm this finding, further investigation is also
warranted.
Given the importance of SHH signaling in EMT
induction and RIF, the blockade of the SHH signaling may exert
anti-fibrotic effects. IPI-926, an Smo antagonist, has been
reported to possess several advantages including a long half-life,
increased potency and oral bioavailability in various animal models
(31). However, a study by Fabian
et al demonstrated that IPI-926 may completely abolish Gli1
induction whereas it did not affect Gli2 or reduce RIF induced by
UUO (24). Thus, IPI-926 may be
an ineffective drug for the treatment of RIF. Cyclopamine, another
well-characterized Smo inhibitor, is considered to be limited in
vivo by its short half-life and off-target effects at higher
doses (32,33). However, a study by Ding et
al showed that cyclopamine not only prevents fibroblast
activation and matrix production in vitro, but also
suppresses the expression of Gli1 and matrix genes, and reduces RIF
in vivo (23). Thus,
cyclopamine efficiently exerts an anti-fibrotic effect. In this
study, we found that cyclopamine inhibited the induction of the EMT
and reduced the synthesis of ECM components in the cultured RTECs.
In addition, cyclopamine also decreased the expression of TGF-β1
and its receptor TGF-β1R. These findings suggested that cyclopamine
inhibited renal EMT as well as exerting an anti-fibrotic effect
in vitro. Unlike its analogue IPI-926; however, the
anti-fibrotic effects of cyclopamine and the underlying molecular
mechanisms responsible for these effects warrant further study.
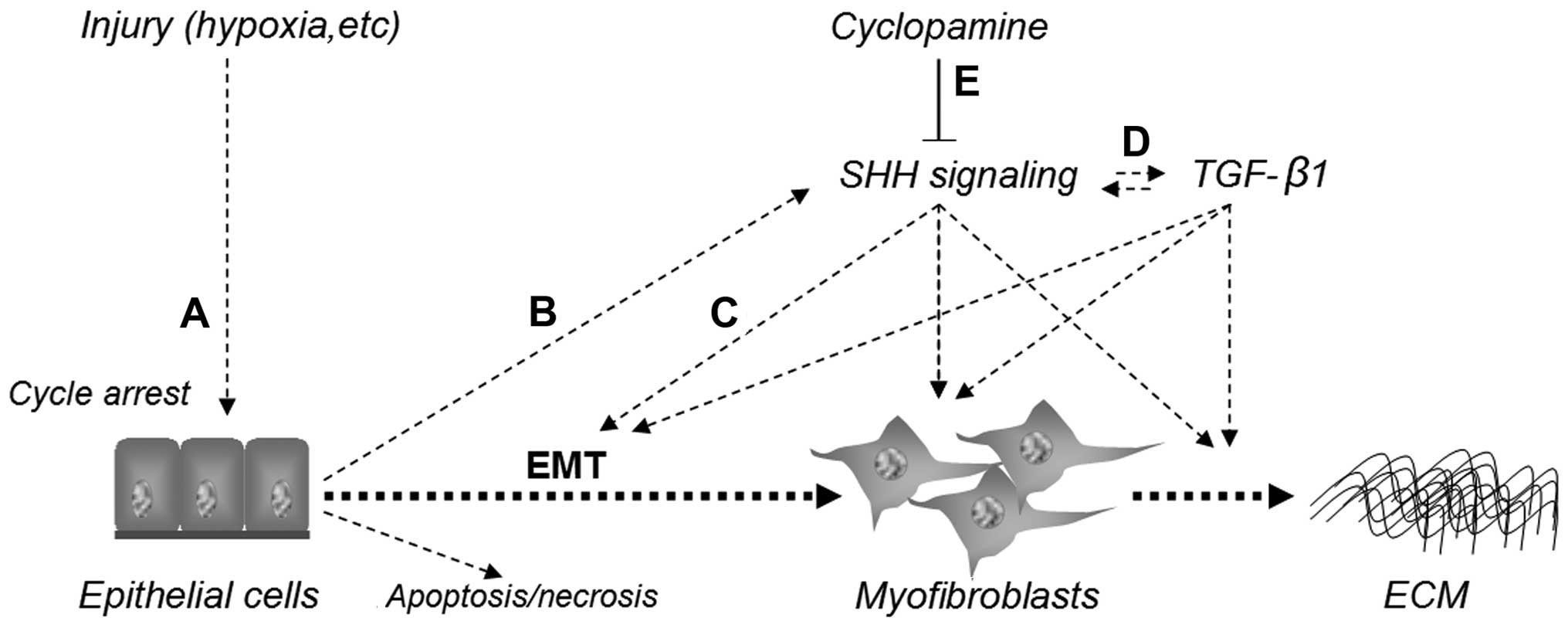
Taken together, as shown in Fig. 7, these findings demonstrate that
injury induces epithelial G2/M cell cycle arrest, and then
stimulates the activation of the SHH signaling pathway. The
activated SHH signaling pathway, which interacts with the TGF-β1
pathway, induces the EMT response, promotes the acquisition of a
myofibroblastic phenotype and ECM deposition and results in RIF.
The blockade of SHH signaling with cyclopamine abolishes
Shh-mediated EMT, the acquisition of a myofibroblastic phenotype
and decreases TGF-β1 expression and ECM production. Thus, SHH
signaling may play a critical role in EMT induction and the
development of RIF, and the pharmacological inhibition of SHH
signaling may play a therapeutic role in the management of fibrotic
kidney diseases.
Acknowledgments
The present study was supported by the Natural
Science Foundation of Zhejiang province, China (LQ12H05001 and
LY12H05004) and the Wenzhou Municipal Science and Technology Plan
Project (Y20110028). The project was also sponsored by the National
Natural Science Foundation of China (81572087).
References
|
1
|
Meran S and Steadman R: Fibroblasts and
myofibroblasts in renal fibrosis. Int J Exp Pathol. 92:158–167.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vilayur E and Harris DC: Emerging
therapies for chronic kidney disease: what is their role? Nat Rev
Nephrol. 5:375–383. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu Y: Cellular and molecular mechanisms
of renal fibrosis. Nat Rev Nephrol. 7:684–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu Y: Epithelial to mesenchymal
transition in renal fibrogenesis: pathologic significance,
molecular mechanism, and therapeutic intervention. J Am Soc
Nephrol. 15:1–12. 2004. View Article : Google Scholar
|
|
5
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010. View Article : Google Scholar
|
|
6
|
Saika S, Ikeda K, Yamanaka O, Flanders KC,
Ohnishi Y, Nakajima Y, Muragaki Y and Ooshima A: Adenoviral gene
transfer of BMP-7, Id2, or Id3 suppresses injury-induced
epithelial-to-mesenchymal transition of lens epithelium in mice. Am
J Physiol Cell Physiol. 290:C282–C289. 2006. View Article : Google Scholar
|
|
7
|
Zeisberg M, Hanai J, Sugimoto H, Mammoto
T, Charytan D, Strutz F and Kalluri R: BMP-7 counteracts
TGF-beta1-induced epithelial-to-mesenchymal transition and reverses
chronic renal injury. Nat Med. 9:964–968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bhardwaj G, Murdoch B, Wu D, Baker DP,
Williams KP, Chadwick K, Ling LE, Karanu FN and Bhatia M: Sonic
hedgehog induces the proliferation of primitive human hematopoietic
cells via BMP regulation. Nat Immunol. 2:172–180. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ingham PW and McMahon AP: Hedgehog
signaling in animal development: paradigms and principles. Genes
Dev. 15:3059–3087. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pasca di Magliano M and Hebrok M: Hedgehog
signalling in cancer formation and maintenance. Nat Rev Cancer.
3:903–911. 2003. View
Article : Google Scholar
|
|
11
|
Berman DM, Karhadkar SS, Maitra A, Montes
De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman
JR, Watkins DN and Beachy PA: Widespread requirement for Hedgehog
ligand stimulation in growth of digestive tract tumours. Nature.
425:846–851. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thayer SP, di Magliano MP, Heiser PW,
Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del
Castillo C, Yajnik V, et al: Hedgehog is an early and late mediator
of pancreatic cancer tumorigenesis. Nature. 425:851–856. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Omenetti A, Porrello A, Jung Y, Yang L,
Popov Y, Choi SS, Witek RP, Alpini G, Venter J, Vandongen HM, et
al: Hedgehog signaling regulates epithelial-mesenchymal transition
during biliary fibrosis in rodents and humans. J Clin Invest.
118:3331–3342. 2008.PubMed/NCBI
|
|
14
|
Syn WK, Jung Y, Omenetti A, Abdelmalek M,
Guy CD, Yang L, Wang J, Witek RP, Fearing CM, Pereira TA, et al:
Hedgehog-mediated epithelial-to-mesenchymal transition and
fibrogenic repair in nonalcoholic fatty liver disease.
Gastroenterology. 137:1478–1488.e8. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hooper JE and Scott MP: Communicating with
hedgehogs. Nat Rev Mol Cell Biol. 6:306–317. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bai Y, Lu H, Zhang G, Wu C, Lin C, Liang Y
and Chen B: Sedum sarmentosum Bunge extract exerts renal
anti-fibrotic effects in vivo and in vitro. Life Sci. 105:22–30.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pat B, Yang T, Kong C, Watters D, Johnson
DW and Gobe G: Activation of ERK in renal fibrosis after unilateral
ureteral obstruction: modulation by antioxidants. Kidney Int.
67:931–943. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Katoh H, Hiramoto K and Negishi M:
Activation of Rac1 by RhoG regulates cell migration. J Cell Sci.
119:56–65. 2006. View Article : Google Scholar
|
|
19
|
Gill PS and Rosenblum ND: Control of
murine kidney development by sonic hedgehog and its GLI effectors.
Cell Cycle. 5:1426–1430. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu MC, Mo R, Bhella S, Wilson CW, Chuang
PT, Hui CC and Rosenblum ND: GLI3-dependent transcriptional
repression of Gli1, Gli2 and kidney patterning genes disrupts renal
morphogenesis. Development. 133:569–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cain JE and Rosenblum ND: Control of
mammalian kidney development by the Hedgehog signaling pathway.
Pediatr Nephrol. 26:1365–1371. 2011. View Article : Google Scholar
|
|
22
|
Yu J, Carroll TJ and McMahon AP: Sonic
hedgehog regulates proliferation and differentiation of mesenchymal
cells in the mouse metanephric kidney. Development. 129:5301–5312.
2002.PubMed/NCBI
|
|
23
|
Ding H, Zhou D, Hao S, Zhou L, He W, Nie
J, Hou FF and Liu Y: Sonic hedgehog signaling mediates
epithelial-mesenchymal communication and promotes renal fibrosis. J
Am Soc Nephrol. 23:801–813. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fabian SL, Penchev RR, St-Jacques B, Rao
AN, Sipilä P, West KA, McMahon AP and Humphreys BD: Hedgehog-Gli
pathway activation during kidney fibrosis. Am J Pathol.
180:1441–1453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Borello U, Berarducci B, Murphy P, Bajard
L, Buffa V, Piccolo S, Buckingham M and Cossu G: The
Wnt/beta-catenin pathway regulates Gli-mediated Myf5 expression
during somitogenesis. Development. 133:3723–3732. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Daoud G, Kempf H, Kumar D, Kozhemyakina E,
Holowacz T, Kim DW, Ionescu A and Lassar AB: BMP-mediated induction
of GATA4/5/6 blocks somitic responsiveness to SHH. Development.
141:3978–3987. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakamura I, Fernandez-Barrena MG,
Ortiz-Ruiz MC, Almada LL, Hu C, Elsawa SF, Mills LD, Romecin PA,
Gulaid KH, Moser CD, et al: Activation of the transcription factor
GLI1 by WNT signaling underlies the role of SULFATASE 2 as a
regulator of tissue regeneration. J Biol Chem. 288:21389–21398.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wynn TA: Fibrosis under arrest. Nat Med.
16:523–525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang L, Besschetnova TY, Brooks CR, Shah
JV and Bonventre JV: Epithelial cell cycle arrest in G2/M mediates
kidney fibrosis after injury. Nat Med. 16:535–543, 1p following
143. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bai Y, Lu H, Hu L, Hong D, Ding L and Chen
B: Effect of Sedum sarmentosum Bunge extract on aristolochic
acid-induced renal tubular epithelial cell injury. J Pharmacol Sci.
124:445–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tremblay MR, Lescarbeau A, Grogan MJ, Tan
E, Lin G, Austad BC, Yu LC, Behnke ML, Nair SJ, Hagel M, et al:
Discovery of a potent and orally active hedgehog pathway antagonist
(IPI-926). J Med Chem. 52:4400–4418. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang X, Harrington N, Moraes RC, Wu MF,
Hilsenbeck SG and Lewis MT: Cyclopamine inhibition of human breast
cancer cell growth independent of Smoothened (Smo). Breast Cancer
Res Treat. 115:505–521. 2009. View Article : Google Scholar
|
|
33
|
Zhao C, Chen A, Jamieson CH, Fereshteh M,
Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D, et al:
Hedgehog signalling is essential for maintenance of cancer stem
cells in myeloid leukaemia. Nature. 458:776–779. 2009. View Article : Google Scholar : PubMed/NCBI
|