Introduction
Inherited cardiomyopathies are divided into the
following 4 categories according to alterations in ventricular
morphology and function: hypertrophic cardiomyopathy (HCM), dilated
cardiomyopathy (DCM), arrhythmogenic right ventricular
cardiomyopathy (ARVC) and restrictive cardiomyopathy (RCM)
(1,2). It is associated with extensive
genetic heterogeneity; inherited cardiomyopathy-linked mutations
have been found in over 100 disease-causing genes, including
mutations in the genes encoding the following: cardiac sarcomere
proteins, cytoskeletal proteins and nuclear envelope proteins
(3–5), cardiac development and structural
remodeling proteins (6–8), cardiac transcription factors
(9–14), Tax-1-binding protein (15) and the RAS-mitogen-activated
protein kinase pathway (16). As
the leading cause of sudden cardiac death (SCD) in adolescents and
young athletes, HCM is the most common type of inherited
cardiomyopathy with a morbidity rate of approximately 1 in 500
individuals worldwide, which is characterized by unexplained left
ventricular hypertrophy (17,18). DCM is characterized by left
ventricular dilatation and systolic dysfunction [with a left
ventricular ejection fraction (LVEF) of <50%], and affects at
least 1/2,500 individuals in the general population (3); it is primarily caused by pathogenic
gene mutations inherited in a Mendelian autosomal dominant pattern
(19). Molecular genetic testing
is crucial for selecting the correct therapy and management
strategies for the disease, as well as to evaluate the prognosis of
patients with inherited cardiomyopathy and of their family
members.
Currently, conventional capillary-based sequencing
is the gold standard approach for detecting mutations associated
with this disease. However, this method has drawbacks, as not all
types of genetic variation are detectable and it is a
time-consuming and costly technique. In a striking technological
development, the Ion Torrent Personal Genome Machine (PGM) system
was launched in 2011 by Life Technologies, and it has been
demonstrated to be a more rapid, more sensitive and less costly
system, and it also allows the scalable sequencing of samples
(20).
In the present study, we enrolled 16 Chinese
patients diagnosed with inherited cardiomyopathy (either HCM or
DCM) and performed bioinformatics and molecular genetic analyses of
the entire coding sequence and flanking regions of 12 major disease
(cardiomyopathy)-related genes using the Ion Torrent PGM system. As
a result, we identified a novel [myosin, heavy chain 7, cardiac
muscle, β (MYH7), p.Asn885Thr] mutation, a variant of uncertain
significance [troponin T type 2 (cardiac) (TNNT2), p.Arg296His] and
a double heterozygous [protein kinase, AMP-activated, gamma 2
non-catalytic subunit (PRKAG2), p.Gly100Ser plus MYH7, p.Arg719Trp]
mutation. The findings of our study expand the mutational spectrum
of MYH7 and TNNT2 which are associated with HCM, and enhance our
understanding of the molecular mechanisms underlying inherited
cardiomyopathy. Furthermore, we present a useful approach for the
genetic testing of patients with inherited cardiomyopathy.
Subjects and methods
Patients and healthy controls
A total of 16 patients with inherited cardiomyopathy
were recruited, namely 8 patients with DCM and 8 patients with HCM,
who were traditionally diagnosed according to the criteria
specified in the American College of Cardiology Foundation/American
Heart Association (ACCF/AHA) guideline for the diagnosis and
treatment of hypertrophic cardiomyopathy (21) and the European guidelines for the
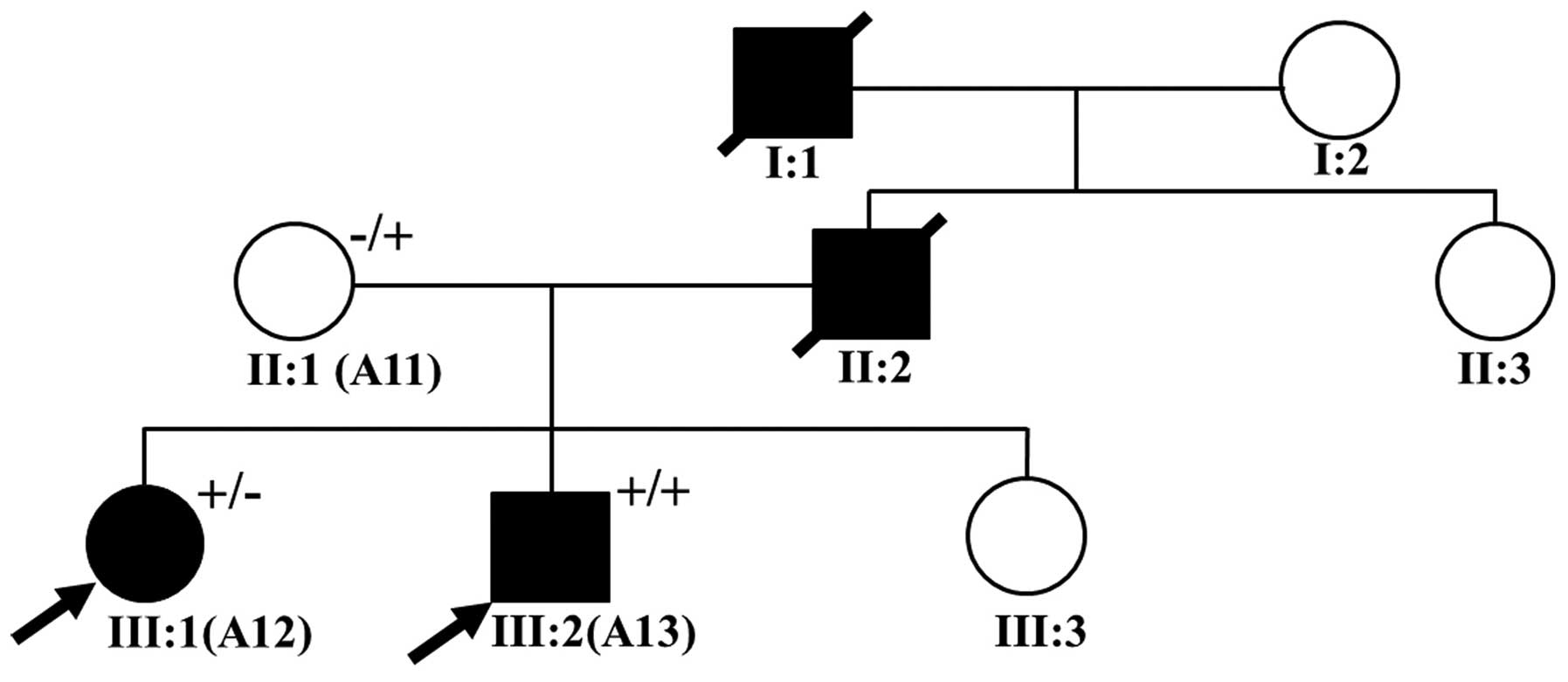
study of familial dilated cardiomyopathies (22). Of the 16 patients, 2 (patients A12
and A13) were considered to have familial HCM (Fig. 1). The demographic and clinical
characteristcs of the patients, including family history, clinical
symptoms, echocardiography results, and 12-lead electrocardiography
(ECG) records, were collected. In addition, 100 healthy individuals
without any symptoms of cardiovascular disease were enrolled into
this study as healthy control subjects. All of the subjects
provided written informed consent prior to participating
voluntarily in this study. The study protocol was approved by the
Ethics Committee of The Affiliated Hospital of Kunming University
of Science and Technology (Kunming, China) and complied with the
principles of the Declaration of Helsinki.
DNA extraction, genomic library
construction and template preparation/amplification
Peripheral whole blood samples (~2 ml) from each
subject were collected in Vacutainer tubes coated with EDTA (BD
Biosciences, Franklin Lakes, NJ, USA) and stored at 4°C until DNA
extraction. Genomic DNA was extracted from anticoagulated whole
blood of each sample using a commercial Blood Genomic DNA Miniprep
kit (Axygen, Union City, CA, USA). The majority of the primers were
designed using the freely available Lasergene PrimerSelect software
(DNASTAR, Madison, WI, USA) and Primer Premier 5.0 (Premier Biosoft
International, Palo Alto, CA, USA), and then 116 pairs of primer
sets were synthesized and we amplified 226 coding exons of the
following genes: MYH7; myosin binding protein C, cardiac
(MYBPC3); TNNT2; troponin I type 3 (cardiac)
(TNNI3); myosin, light chain 2, regulatory, cardiac, slow
(MYL2); lamin A/C (LMNA); myosin, light chain 3,
alkali, ventricular, skeletal, slow (MYL3); PRKAG2;
sodium channel, voltage gated, type V alpha subunit (SCN5A);
myosin, heavy chain 6, cardiac muscle, α (MYH6); actin, α,
cardiac muscle 1 (ACTC1); and tropomyosin 1 (α)
(TPM1); (data available upon request). From this panel,
approximately 15 pairs of primer sets having similar annealing
temperatures and of similar amplicon size were combined in one
reaction pool in order to reduce the reaction times and reagent
costs. Polymerase chain reaction (PCR) amplification was performed
using PrimeSTAR GXL DNA polymerase (Takara, Otsu, Japan) under the
following conditions: DNA denaturation at 98°C for 3 min, followed
by 30 cycles of denaturing at 98°C for 10 sec, annealing at 54 or
57°C for 15 sec and extension at 68°C for 1–3 min, and finalized
with one extension cycle of 68°C for 5 min (data available upon
request). Finally, the multiplex PCR products from each sample were
mixed in microtubes at equal concentrations which were determined
using the SequalPrep Normalization kit (Invitrogen, Carlsbad, CA,
USA). Genomic library construction was performed using the manual
(Publi cation no. 4471989, Revision N) and DNA template preparation
was conducted using an Ion OneTouch 2 instrument and an Ion
OneTouch enrichment system (ES) (both from Life Technologies,
Carlsbad, CA, USA).
Ion Torrent PGM sequencing and
bioinformatics analysis
DNA high-throughput sequencing was performed using
reagents from the Ion PGM Sequencing 400 kit (obtained from Life
Technologies). The prepared samples of Ion Sphere Particles (ISP)
were loaded onto an Ion 314 sequencing chip (Life Technologies),
and DNA sequencing was performed in the Ion PGM instrument using
the Ion PGM 400 sequencing kit set at 640 flows for 160 runs. Raw
data from the PGM runs were processed using the Ion Torrent
platform-specific pipeline software Torrent Suite v4.0.2 (Life
Technologies) to generate sequence reads. The FastQC (v3.4.1.1)
plug-in software was used in order to perform the analysis of the
mean read depth and alignment quality. The short reads alignment
was rapidly and accurately achieved using the Burrows Wheeler
Aligner (BWA) Multi-Vision software package. Coverage Analysis
(v4.0-r77897) plug-in software was used to assess the number of
mapped bases, the percentage of coverage on the target gene.
The sequence variants in the 12 genes in each sample
were identified using the Torrent Suite Variant Caller (TSVC;
v4.0-r76860) plug-in and browser extensible data (BED) files
(chromosome coordinates) that specify the coding regions of the
target genes within the human reference genome (hg19) retrieved
from the NCBI database (build 37) as a reference. The TSVC plug-in
generated files in variant caller format (VCF) were further
annotated using online Ion Reporter software (https://ionreporter.lifetechnologies.com/ir/secure/home.html),
and Integrated Genomic Viewer (IGV) software (23) was then used to complete the
visualization and to eliminate false-positive variants.
Molecular genetic analysis
We analyzed mutations that have been reported to be
associated with the disease phenotype in the PubMed Database
(http://www.ncbi.nlm.nih.gov/pubmed/)
or by the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk). In addition to the above,
the mutations meeting the following criteria were putatively
considered pathogenic (24): i)
if the mutation had a minor allele frequency (MAF) of <10% in
the NCBI dbSNP137 (http://www.ncbi.nlm.nih.gov/projects/SNP/), the 1000
Genomes Project (http://www.1000genomes.org/) and NHLBI Grand
Opportunity Exome Sequencing Project (ESP; https://esp.gs.washington.edu/drupal/) databases; ii)
pathogenicity prediction programs that assess the functional
significance of amino acid substitutions were used, including
PolyPhen-2 (25), SIFT (26) or MutationTaster algorithms
(27), which labeled the mutation
as pathogenic; iii) the novel mutations were absent from the 100
unrelated healthy control subjects in order to rule out the
possibility of the polymorphisms existing in the normal population;
iv) evolutionary conservation analysis of the novel mutation was
performed in a number of vertebrate species (namely, Macaca
mulatta, Felis catus, Mus musculus, Gallus
gallus, Danio rerio and Xenopus tropicalis). The
Clustal W alignment program (http://www.genome.jp/tools/clustalw/) was used to
align orthologs from eukaryotes, and the weblogo (http://weblogo.berkeley.edu/logo.cgi)
format was used for aligning eukaryotic species. All of the
putative pathogenic mutations were verified by PCR and conventional
capillary-based sequencing analysis using an ABI 3730 automatic
sequencer (Applied Biosystems, Foster City, CA, USA).
Results
Demographic and clinical characteristics
of the patients with DCM and HCM
The recorded demographic and clinical
characteristics of all 16 patients (8 patients diagnosed with DCM
and 8 patients diagnosed with HCM) are presented in Table I. The patients with DCM had a
median age at diagnosis of 44 years (ranging from 16 to 70 years
and SD of 17.2 years) and those with HCM had a median age at
diagnosis of 37.1 years (ranging from 10 to 57 years and SD of 19.2
years). Echocardiography revealed a mildly dilated left ventricular
end-systolic diameter (LVESD, an average of 57.8 mm) and left
ventricular end-diastolic diameter (LVED, an average of 68.0 mm)
and LVEF (an average of 32.6%) of <50% in the patients with DCM.
Echocardiography also revealed that the patients with HCM
experienced significant concentric left ventricular hypertrophy
(LVH) with severe alterations in interventricular septum thickness
(IVST, average of 19.2 mm) and left ventricular posterior wall
thickness (LVPWT; average of 11.5 mm).
 | Table IAvailable demographic and clinical
characteristics of patients with inherited cardiomyopathy. |
Table I
Available demographic and clinical
characteristics of patients with inherited cardiomyopathy.
| Subject no. | Gender | Age (years) | Disease | Family History | Echocardiogram
|
|---|
| IVST (mm) | LVED (mm) | LVESD (mm) | LVPWT (mm) | LA (mm) | LVEF (%) |
|---|
| 19 | Male | 70 | DCM | Negative | N/A | N/A | N/A | N/A | N/A | N/A |
| A7 | Male | 16 | DCM | Negative | 8 | 61 | N/A | 8.8 | 35 | 45.0 |
| A63 | Male | 41 | DCM | Positive | 9.3 | 60.6 | 48 | 8.6 | 40 | 38.0 |
| A64 | Female | 56 | DCM | Positive | 9.9 | 67.4 | 59.6 | 8.5 | 58.2 | 24.0 |
| 38 | Male | 40 | DCM | Negative | 9.2 | 90.1 | 81.6 | 8.5 | 61.8 | 19.0 |
| A51 | Male | 59 | DCM | Negative | N/A | N/A | N/A | N/A | N/A | N/A |
| A88 | Male | 30 | DCM | Negative | 7.0 | N/A | N/A | 8.0 | 40 | 22.0 |
| A37 | Female | 40 | DCM | Positive | 7.9 | 61.3 | 42 | 6.8 | 32.6 | 48.0 |
| Mean ± SD | N/A | 44.0±17.2 | N/A | N/A | 8.5±1.0 | 68.0±12.6 | 57.8±17.5 | 8.2±0.7 | 44.6±12.3 | 32.6±12.5 |
| A36 | Female | 49 | HCM | Positive | 22 | 44.2 | 31.7 | 10.9 | 32 | 54.0 |
| A15 | Male | 14 | HCM | Positive | 18.8 | 37.2 | 24.0 | 16.0 | 46.0 | 65.0 |
| 51 | Female | 57 | HCM | Negative | 11.7 | 37.4 | 26.1 | 11.5 | 29.1 | 58.0 |
| A94 | Male | 57 | HCM | Positive | 16.3 | 49.6 | 26.2 | 13.5 | 52.5 | 78.0 |
| A12 | Female | 21 | HCM | Positive | 25.9 | 32.0 | 18.4 | 9.4 | 26.9 | 75.0 |
| A13 | Male | 10 | HCM | Positive | 21.0 | N/A | N/A | 12.0 | 40.0 | 48.0 |
| 64 | Male | 42 | HCM | Negative | 18.1 | 51.7 | 34.4 | 9.8 | 44.0 | 61.0 |
| A86 | Male | 47 | HCM | Positive | 20.0 | 49.2 | 32.3 | 8.8 | 32.0 | 63.0 |
| Mean ± SD | N/A | 37.1±19.2 | N/A | N/A | 19.2±4.2 | 43.0±7.6 | 27.6±5.6 | 11.5±2.4 | 37.8±9.2 | 62.6±10.1 |
Among the patients with familial HCM (Fig. 1), the proband of this family (A13,
III:2) was a young child, who at the age of 10 years was diagnosed
with a malignant HCM phenotype that manifested as a greater LVPWT
with evident septal asymmetry, an enlarged left atrium (LA),
decreased left ventricular systolic and diastolic function (IVST,
21 mm; LVPWT, 12 mm; LA, 40 mm; and LVEF, 48%); at the time of the
initial diagnosis the child was experiencing chest pain and
syncope. The family history revealed that the grandfather of the
proband had died from heart disease at age 42 and that his father
had succumbed to sudden cardiac death at age 40. In the absence of
clinical data, the child's mother (A11, II:1) was assumed to be
free of clinical symptoms. Patient A12 (III:1), who is the
proband's elder sister, was diagnosed at the age of 21 with a
severe HCM phenotype (IVST, 25.9 mm; LVPWT, 9.4 mm; and LA, 26.9
mm).
Analysis of raw data collected by
performing multiplex PCR amplification followed by Ion Torrent PGM
sequencing
To rapidly detect HCM- and DCM-related mutations of
the 12 target genes, 116 primer pairs were designed for multiplex
PCR in order to amplify a total of 154,537 base pairs (bp): the PCR
fragment size ranged from 245 to 2906 bp (data available upon
request). To distinguish the sequence data of each individual
sample, we linked an Ion Xpress Barcode Adapter sequence to each
fragment in the library. Following the optimization of PCR
amplification conditions, we used only 6 microtubes containing
multiplexed PCR pools in order to perform 116 PCR reactions on each
sample (data available upon request).
In general, an Ion 314 semiconductor chip can
generate an amount of 300 kb data on the microporous surface, and
as it only utilizes 50% of the microporous. we assumed that the
average reads length was 200 bp, and each of the nucleotide
sequencing has a depth of 30x (Q30=99.9% certainty that the correct
base was called). Therefore, a pool of library DNA from 5 to 6
patient samples was amplified using an Ion OneTouch 2 system and
then loaded onto an Ion 314 semiconductor chip. Finally, runs of
all 16 samples were performed in 3 independent replicates. The
average 314 semiconductor chip loading obtained was 77.6% (ranging
from 75 to 81%), and an average PGM run generated raw data of
approximately 49.9 Mb on the 314 semiconductor chip (ranging from
49.1 to 50.5 Mb) (data available upon request) and a mean read
length of 160 bp (ranging from 147 to 181 bp) (data available upon
request). The sequencing data quality was assessed using the FastQC
plug-in software, which revealed that the average Q value was
approximately 30 (Q30=99.9% certainty that the correct base was
called) (data available upon request).
Finally, upon completion of the analysis, we
obtained a total of 783,648 raw reads, including 112,378,736 raw
bases, the average for 7,023,671 bases/sample (Table II). This resulted in an average
of 89% of bases per sample sequenced, indicative of a quality of
>Q20 (where Q20=99% certainty that the correct base was called).
Total reads were mapped uniquely to the human reference genome
(hg19) retrieved from the NCBI database (build 37) using the
Burrows Wheeler Aligner (BWA) Multi-Vision software package.
Approximately 96.6% of the bases were matched to hg19 (Table II), and approximately 90% of the
bases were matched to coding regions of the target gene(s) using
the Coverage Analysis plug-in. It was found that the average depth
of coverage of all the exons was at least 28-fold across all 16
samples (Table II). According to
previous research, the majority of the raw sequencing data were
considered as qualified (28,29).
| Table IIIon Torrent PGM run statistics and
potential disease mutations in patients with inherited
cardiomyopathy. |
Table II
Ion Torrent PGM run statistics and
potential disease mutations in patients with inherited
cardiomyopathy.
| Subject no. | Total bases | ≥Q20 bases | Reads | Mapped reads | Mean depth | Variants | Synonymous | Insertions and
deletions | Non-synonymous |
|---|
| 19 | 14,418,988 | 13,103,929 | 107,304 | 103,943 | 50.06 | 90 | 5 | 1 | 1 |
| A15 | 7,791,816 | 7,066,013 | 58,703 | 56,546 | 29.74 | 36 | 7 | 0 | 3 |
| A36 | 5,324,605 | 4,823,023 | 39,257 | 37,978 | 14.74 | 16 | 5 | 0 | 1 |
| A37 | 5,831,466 | 5,307,572 | 40,876 | 39,480 | 21.54 | 37 | 8 | 0 | 2 |
| A7 | 4,812,050 | 4,088,627 | 43,384 | 41,417 | 21.88 | 98 | 4 | 0 | 4 |
| A13 | 6,089,346 | 5,237,958 | 46,431 | 44,261 | 23.60 | 89 | 5 | 0 | 2 |
| A63 | 7,376,806 | 6,320,407 | 53,364 | 51,338 | 32.39 | 84 | 5 | 1 | 0 |
| A64 | 8,285,404 | 7,132,742 | 61,327 | 58,623 | 37.20 | 116 | 6 | 1 | 1 |
| A94 | 4,111,436 | 3,522,751 | 32,388 | 30,602 | 15.55 | 89 | 7 | 1 | 1 |
| A12 | 5,626,106 | 5,101,170 | 39,302 | 38,165 | 26.36 | 91 | 6 | 0 | 3 |
| 38 | 7,807,093 | 7,039,285 | 48,538 | 47,368 | 33.25 | 128 | 7 | 0 | 1 |
| 51 | 7,785,380 | 7,012,358 | 46,493 | 45,430 | 27.94 | 124 | 5 | 0 | 3 |
| 64 | 7,546,099 | 6,803,657 | 46,726 | 45,536 | 27.51 | 127 | 8 | 0 | 2 |
| A51 | 6,792,213 | 6,086,301 | 40,887 | 39,770 | 31.94 | 93 | 5 | 0 | 1 |
| A86 | 5,261,059 | 4,714,393 | 32,785 | 31,808 | 22.73 | 76 | 6 | 0 | 1 |
| A88 | 7,518,870 | 6,751,598 | 45,884 | 44,622 | 31.58 | 105 | 8 | 0 | 1 |
| Mean | 7,023,671 | 6,256,987 | 48,978 | 47,305 | 28 | 87 | – | – | – |
Identification of pathogenic mutations in
order to improve diagnosis
Sequence analysis using the Ion Torrent Variant
Caller (v4.0-r76860) plug-in revealed a total of 1,399 nucleotide
variations in the 16 patient samples. These variations were
positioned in the 5′-UTR, 3′-UTR and in both introns and exons,
with an average of 87 variants per patient (Table II). The identified variations
were annotated using online Ion Reporter software: 1,368 (97.8%) of
them were predicted to be non-coding or synonymous, whereas 31
(2.2%) were non-synonymous and insertion or deletion variants that
lead to alterations in 1 or more amino acids (Table II). Non-synonymous and frame
shift variation sites were described (data available upon
request).
The entire potential non-synonymous and frameshift
variation sites were filtered (24). Among these variants, 5 known
heterozygous mutations (MYH7, p.Arg719Trp, p.Ala26Val and
p.Arg663Cys; PRKAG2, p.Gly100Ser and MYBPC3, p.Ser236Gly) are
already known to be associated with inherited cardiomyopathy, and
one variant was of uncertain significance (TNNT2, p.Arg296His). Of
note, we identified a novel A>C mutation located at nucleotide
position c.2654 (according to the cDNA reference sequence, GenBank
accession number NM_000257.3) within exon 22 of MYH7, which
resulted in the replacement of asparagine at the 885th amino acid
with threonine (p.Asn885Thr), as shown in Table IV. These HCM-and DCM-related
pathogenic mutations were validated by conventional capillary-based
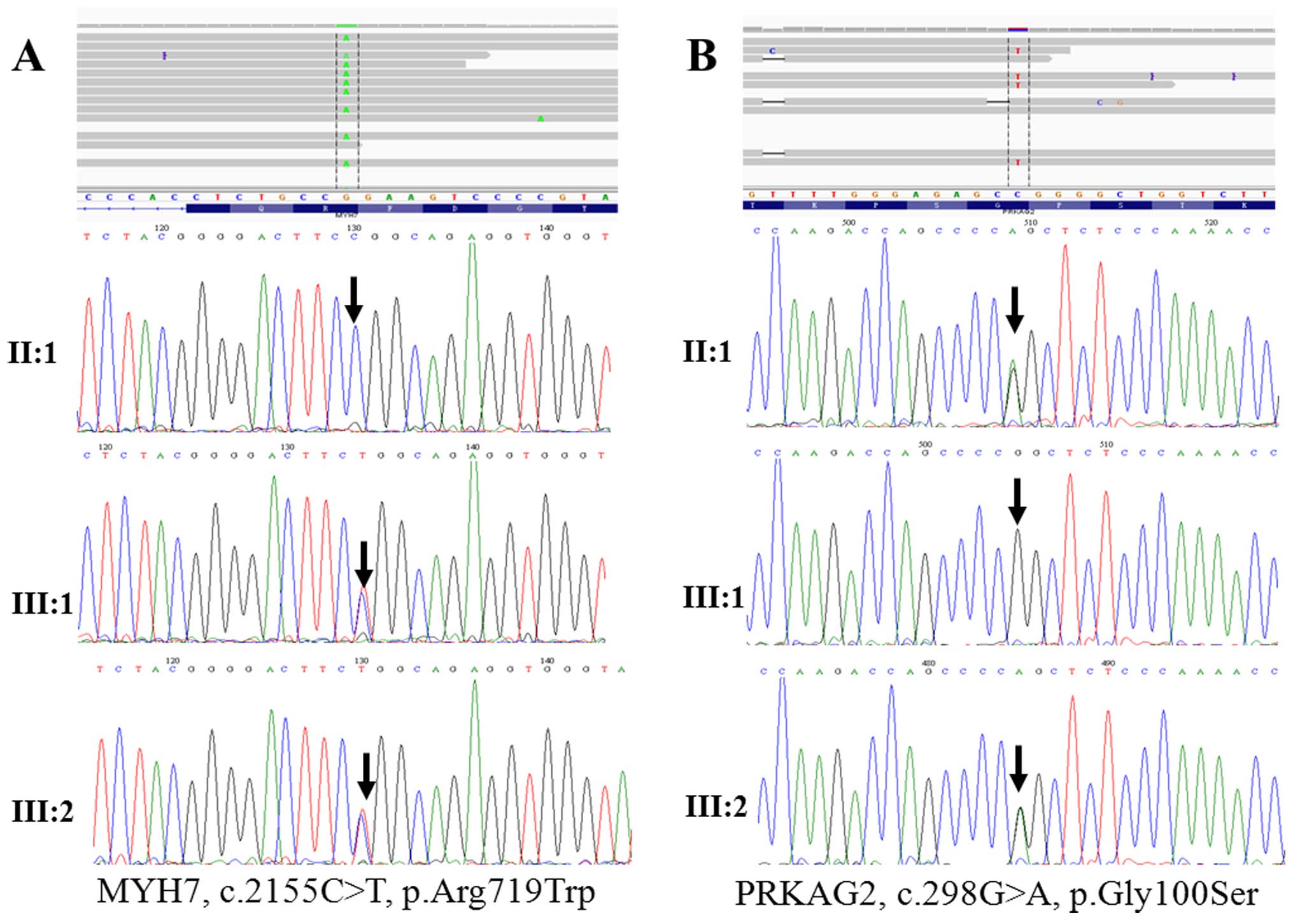
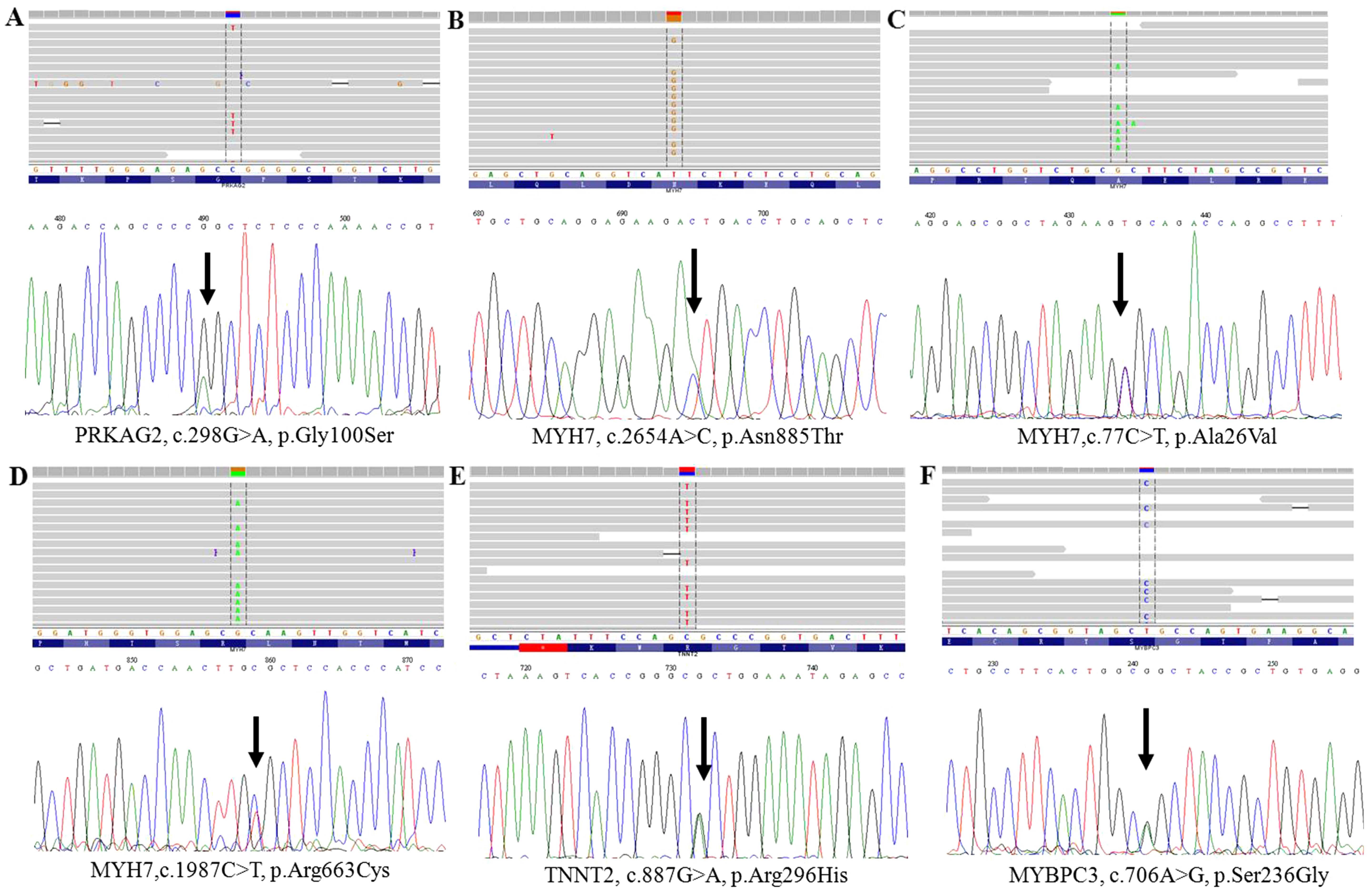
sequencing (Figs. 2A and B and
3), and the PCR primers for the
first generation sequencing are listed in Table III. All of the putative
mutations were verified by Sanger sequencing (Figs. 1Figure 2–3), demonstrating that Ion Torrent PGM
sequencing achieves a high degree of accuracy with regard to
detecting rare mutations.
| Table IVPathogenic mutations detected in
subjects. |
Table IV
Pathogenic mutations detected in
subjects.
| Subject no. | Gene symbol | Ref Chr. | Transcript | Nucleotide
changes | Amino acid
changes | SIFT | Poly Phen-2 | Mutation
Taster | MAF in 1000G | MAF in ExAC
(EA) | Novel/known
(Refs.) |
|---|
| A12 | MYH7 | chr14:23895180 | NM_000257.3 | c.2155C>T | p.Arg719Trp | N/A | N/A | N/A | 0.000 | 0.000 | Known (46) |
| A13 | MYH7 | chr14:23895180 | NM_000257.3 | c.2155C>T | p.Arg719Trp | N/A | N/A | N/A | 0.000 | 0.000 | Known (46) |
| A11 | PRKAG2 | chr7:151478406 | NM_016203.3 | c.298G>A | p.Gly100Ser | N/A | N/A | N/A | 0.071 | 0.035 | Known (47) |
| A13 | PRKAG2 | chr7:151478406 | NM_016203.3 | c.298G>A | p.Gly100Ser | N/A | N/A | N/A | 0.071 | 0.035 | Known (47) |
| A15 | PRKAG2 | chr7:151478406 | NM_016203.3 | c.298G>A | p.Gly100Ser | N/A | N/A | N/A | 0.071 | 0.035 | Known (47) |
| A37 | MYH7 | chr14:23902865 | NM_000257.3 | c.77C>T | p.Ala26Val | N/A | N/A | N/A | 0.008 | 0.006 | Known (35) |
| A94 | MYBPC3 | chr11:47370041 | NM_000256.3 | c.706A>G | p.Ser236Gly | N/A | N/A | N/A | 0.031 | 0.030 | Known (52) |
| A36 | MYH7 | chr14:23894003 | NM_000257.3 | c.2654A>C | p.Asn885Thr | NT | PD | DC | 0.000 | 0.000 | Novel |
| 64 | MYH7 | chr14:23896043 | NM_000257.3 | c.1987C>T | p.Arg663Cys | N/A | N/A | N/A | 0.000 | 0.000 | Known (53) |
| A86 | TNNT2 | chr1:201328348 | NM_001276345.1 | c.887G>A | p.Arg296His | NT | PD | DC | 0.000 | 0.000 | VSU |
| Table IIIPCR primers used for the validation
of the gene sequence variants. |
Table III
PCR primers used for the validation
of the gene sequence variants.
| Gene symbol | Nucleotide
changes | Primers
(5′→3′) | Fragment size
(bp) |
|---|
| MYH7 | c.2155C>T | Sense:
GCTAATCAGTGACAAAGCCAGGATC
Antisense: AGGGTGGAAGAGCCAACAGTAGC | 1,434 |
| PRKAG2 | c.298G>A | Sense:
CAGTCCTGTGTGGTCAGAACTTGG
Antisense: GGACCAGAAGGATTACGCTTTGAT | 907 |
| MYH7 | c.77C>T | Sense:
AGCCAGCTTCTGCTCACTCCAG
Antisense: GCCACTTGTAAGGGTTGACGGT | 1,013 |
| MYBPC3 | c.706A>G | Sense:
CACCATACTTGGCTAATTTTCGT
Antisense: GGATGACTGTTGACGGGACATAATGT | 1,542 |
| MYH7 | c.2654A>C | Sense:
GCTAATCAGTGACAAAGCCAGGATC
Antisense: AGGGTGGAAGAGCCAACAGTAGC | 1,434 |
| MYH6 | c.5410C>A | Sense:
TGATGGAGGAGGGAAAGGTGATT
Antisense: GGGTGCCAGGTGAACGGTTAA | 2,286 |
| MYH7 | c.1987C>T | Sense:
GCAGAATCCATGTCCACCTGT
Antisense: TGTCCTAGGAGGTCCTGTTCC | 1,248 |
| TNNT2 | c.887G>A | Sense:
AGGGTGATTGTGAGGGTTACAG
Antisense: GAGGGTCAAGAGAATGTGTCGT | 2,007 |
Notably, the novel MYH7, p.Asn885Thr mutation was
consistently predicted to be deleterious by the PolyPhen-2
(25), SIFT (26), and MutationTaster (27) algorithms which showed that the
mutation is probably damaging with scores of 0.997 and 0.996 on the
HumDiv and HumVar models, respectively (Fig. 4C). Secondly, this mutation was not
found in reference alleles from the 100 healthy controls, and was
also absent from the HGMD database (www.hgmd.cf.ac.uk), NCBI dbSNP137 (http://www.ncbi.nlm.nih.gov/projects/SNP/) and 1000
Genomes project (http://www.1000genomes.org/). Finally, the mutation,
p.Asn885Thr, occurs within a 3 helix bundle with the second helix
interrupted and it was highly conserved across a number of species
(Fig. 4A). As with the mutation
of p.Asn885Thr in MYH7, notably, a variant of uncertain
significance (TNNT2, p.Arg296His) was predicted to be probably
damaging to amino acids using in silico programs, which is
relevant for the pathogenicity of HCM (Fig. 4B and D).
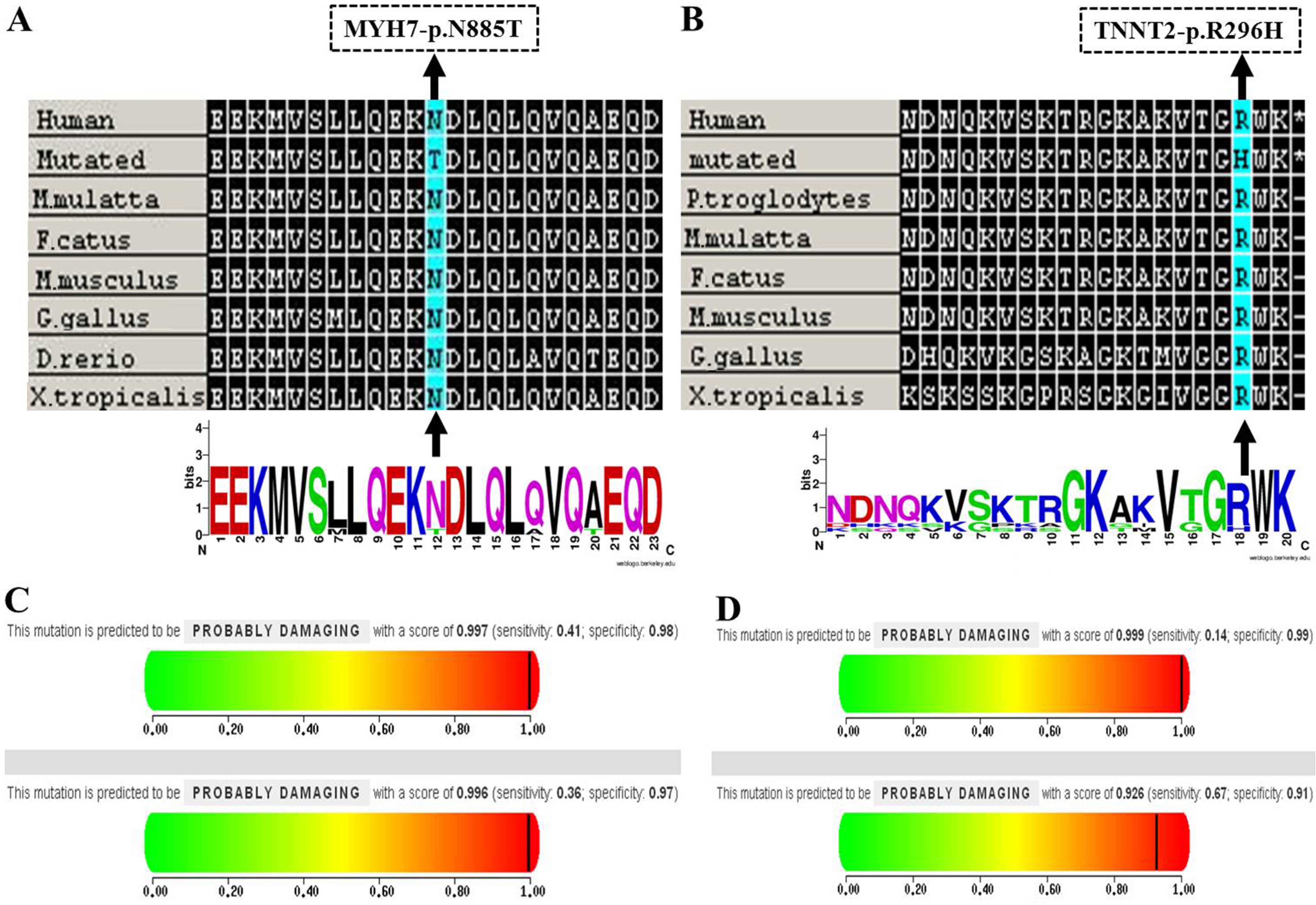 | Figure 4(A and B) Evolutionary conservation
analyses of the MYH7, p.Asn885Thr and TNNT2, p.Arg296His mutations
were performed, respectively. Clustal W was used to align sequences
from Homo sapiens, Macaca mulatta, Felis
catus, Mus musculus, Gallus gallus, Danio
rerio and Xenopus tropicalis and WebLogo was used to
generate sequence logos. The MYH7, p.Asn885Thr and TNNT2,
p.Arg296His mutations are marked by a black arrow; (C and D)
Results of the PolyPhen-2 analysis predicting the pathogenicity of
the mutations of MYH7, p.Asn885Thr and TNNT2, p.Arg296His,
respectively. |
Discussion
The identification of pathogenic mutations is
critical for understanding the molecular pathogenesis of inherited
cardio-myopathy, as this in turn will aid the clinical diagnosis of
this disease. In the present study, we established a method for the
rapid detection of potentially pathogenic mutations in a panel of
12 major genes closely associated with the occurrence of HCM or DCM
using the Ion Torrent PGM system. A novel heterozygous mutation
(MYH7, p.Asn885Thr) and a variant of uncertain significance (TNNT2,
p.Arg296His) were identified.
The Ion Torrent PGM technique is based on the PCR
amplification of DNA obtained from subjects which is followed by
pooling and barcode labeling of the fragments in each sample and
high-throughput sequencing (30,31). The multiplex amplification primers
were designed to amplify the principal HCM and DCM disease-related
genes in a condensed format that required only 6 microtubes. This
process avoids the need for multiple amplifications and reduces the
reagent cost per patient. Approximately 90% of the obtained
sequences were matched to the coding regions of the target genes
using the Coverage Analysis plug-in; however, 10% of the coding
region was not covered. This may be due to several factors.
Firstly, as the primer pairs were mixed in one reaction, multiplex
PCR has a low specific amplification. In addition, the sequence
stretches of low complexity and as well as GC-rich regions are
difficult to capture (30). Such
uncovered regions must be completed using conventional
capillary-based sequencing, although there is a risk of missing
some mutations using this technique. Despite the drawback regarding
lower sequence coverage, the Ion Torrent PGM-based approach using
multiplex PCR remains a high-throughput, low-cost method for the
detection of mutations.
Herein, we constructed a library to sequence the
genomic DNA isolated from patients (19, A15, A36, A37 and A7).
Similarly, the second (A13, A63, A64, A94, A12 and 38) and third
(51, 64, A51, A86 and A88) pool were also used to construct
libraries, respectively, and a total of 8 patients were identified
as carriers of pathogenic mutations (Table IV). We detected mutations in 12
disease genes in 7 (7/8) patients with HCM and in 2 (2/8) of the 8
patients with DCM. Of these 7 mutations, 5 are known (MYH7,
p.Arg719Trp, p.Ala26Val and p.Arg663Cys; PRKAG2, p.Gly100Ser and
MYBPC3, p.Ser236Gly), one is a variant of uncertain significance
(TNNT2, p.Arg296His) and one is a novel mutation (MYH7,
p.Asn885Thr). Our DCM mutation detection rates are consistent with
those of a previous study (32),
but the HCM detection rate was higher than that in earlier studies
(33,34). As shown in Table IV, 9 subjects [a total of 7
subjects with HCM, one subject (A37) with DCM, and one (A11)
subject who was clinically asymptomatic] carried mutations; subject
A13 carried a double heterozygous mutation (PRKAG2, p.Gly100Ser
plus MYH7, p.Arg719Trp). Mutations were distributed mostly in
MYH7 (50%, 5/10) and PRKAG2 (30%, 3/10), followed by
MYBPC3 (10%, 1/10) and TNNT2 (10%, 1/10). Mutations
were not found in the LMNA, MYH6, MYL2,
TPM1, MYL3, SCN5A, ACTC1 and
TNNI3 genes. No pathogenic mutations were detected in 87.5%
(7/8) of the patients with DCM. This may be due to the fact that
there are fewer known DCM-related genes compared with the number of
HCM-related genes (3,35); thus, the likelihood of detecting a
mutation using our limited gene panel is low. Alternatively,
lifestyle and enviornmental factors may play a more important role
in the etiology of DCM which is not taken into account when
performing genetic screening alone (32).
To the best of our knowledge, this is the first
study to describe the novel MYH7 mutation (p.Asn885Thr) in patients
with HCM. Sequence conservation analysis revealed that this residue
is highly conserved across a number of species (Fig. 4A), thereby impairing its
contractile function. Our results suggest that this novel mutation
may be functionally deleterious and thus, play an important role in
the pathogenesis of HCM, and it expands the mutational spectrum of
the MYH7 gene. Moreover, to the best of our knowledge we are
the first to report a variant of uncertain significance,
p.Arg296His in TNNT2 which is associated with HCM; this illustrated
that the status of the variant TNNT2, p. Arg296His may be upgraded
to pathogenic. Determining the pathogenicity of a mutation remains
a major clinical challenge (36);
further independent studies with in vitro or animal models
(37,38) are essential in order to validate
our results.
His558Arg polymorphism of the SCN5A gene is
associated with dilated cardiomyopathy (39), atrial fibrillation (40), idiopathic cardiac conduction
disorders (41) and Brugada
syndrome (42). His558Arg is a
common polymorphism that interacts with the other mutation,
eventually modifying or modulating the disease phenotypes (39,42). We identified the common
polymorphism of His558Arg in the SCN5A gene in 4 patients with DCM
(subject nos. 19, A51, A7 and A37) and validated these findings
using Sanger sequencing (data available upon request). Of note, the
mutation MYH7, p.Ala26Val plus the common polymorphism of SCN5A,
His558Arg were detected in one patient (A37) with DCM. Potentially,
a polymorphism of His558Arg modifies or modulates the variant MYH7,
p.Ala26Val which causes DCM, and may affect the age of onset, the
severity and rate of progression of DCM.
Some patients with inherited cardiomyopathy may have
more than one disease-causing mutation, which can occur in either
the same gene (compound heterozygotes) or in different genes
(double heterozygotes) (43,44). As a consequence of these complex
mutations, the individual usually has a malignant phenotype of
inherited cardiomyopathy (45).
Notably, we have identified, for the first time to the best of our
knowledge, a double heterozygous (MYH7, p.Arg719Trp plus PRKAG2,
p.Gly100Ser) mutation in a proband (III:2) with familial HCM. He
exhibited a malignant phenotype of HCM that manifested with
increased interventricular septum thickness (Table I and Fig. 2A and B). His elder sister (III:1)
was also diagnosed with HCM and carried MYH7, p.Arg719Trp, but not
PRKAG2, p.Gly100Ser. Family screening revealed that the proband's
45-year-old mother (II:1), who was asymptomatic, was also affected,
and carried the PRKAG2, p.Gly100Ser mutation (Fig. 2A and B). These results suggest
that the pathogenic MYH7, Arg719Trp mutation probably originated in
the proband's father and grandfather. The MYH7, p.Arg719Trp
(46) and PRKAG2, p.Gly100Ser
(47) mutations have previously
been shown to be associated with malignant familial HCM and
sporadic HCM, respectively. We suggest that PRKAG2, p.Gly100Ser
exacerbates the clinical severity of HCM in individuals with the
MYH7 p.Arg719Trp mutation and thus, have a 'double dose' gene
mutation effect (48,49). This is associated with a poor
prognosis, and explains why the proband exhibited an early onset
malignant phenotype of HCM (50).
As a mutation carrier, the proband's mother (A11) is a family
member at risk who was clinically asymptomatic (51); long-term follow-up is therefore
essential in this subject. However, her children are at high risk
of developing HCM, and thus genetic testing may be particularly
helpful in this group. Indeed, we have recommended genetic testing
for all first-degree relatives of the proband, since it may
facilitate clinical decisions that impact diagnosis and treatment
strategies.
In conclusion, Ion Torrent PGM targeted sequencing
is a rapid and cost-effective method for the clinical genetic
screening of patients with inherited cardiomyopathy. Correct
recognition of the responsible gene mutations is essential for
providing optimal presymptomatic intervention and genetic
counseling for probands and their family members. The gene panel
testing of 12 major disease-related genes in patients with
inherited cardiomyopathy reported in this study has the potential
to assist in revealing the etiology of genetically heterogeneous
HCM, and it is a highly reliable and effective method for the
screening of pathogenic mutations in candidate genes. However, the
coverage of these targeted regions must be further improved.
Furthermore, we detected a novel (MYH7, p.Asn885Thr) mutation and a
variant of uncertain significance (TNNT2, p.Arg296His) that we
suggest be upgraded to pathogenic status; these add new data to the
spectrum of mutations in HCM. Moreover, a double heterozygous
(PRKAG2, p.Gly100Ser plus MYH7, p.Arg719Trp) mutation was found in
a proband with familial HCM. This data has the potential to allow
us to better facilitate risk stratification and guide familial
management of the disease. Finally, we note that genes beyond this
initial 12-gene panel should be included in future tests as their
relevance to DCM becomes clear.
Acknowledgments
We would like to thank all the participants of this
study from the inherited cardiomyopathy registry, and we would like
to thank the staff of the First Hospital of Yunnan Province for
their support. The present study was supported by the Major Program
of Applied Basic Research of Yunnan Province, China (no.
2013FC007).
References
|
1
|
Callis TE, Jensen BC, Weck KE and Willis
MS: Evolving molecular diagnostics for familial cardiomyopathies:
at the heart of it all. Expert Rev Mol Diagn. 10:329–351. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hughes SE and McKenna WJ: New insights
into the pathology of inherited cardiomyopathy. Heart. 91:257–264.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hershberger RE, Hedges DJ and Morales A:
Dilated cardiomyopathy: the complexity of a diverse genetic
architecture. Nat Rev Cardiol. 10:531–547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hinson JT, Chopra A, Nafissi N, Polacheck
WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, et
al: HEART DISEASE. Titin mutations in iPS cells define sarcomere
insufficiency as a cause of dilated cardiomyopathy. Science.
349:982–986. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Spaendonck-Zwarts KY, Posafalvi A, van
den Berg MP, Hilfiker-Kleiner D, Bollen IA, Sliwa K, Alders M,
Almomani R, van Langen IM, van der Meer P, et al: Titin gene
mutations are common in families with both peripartum
cardiomyopathy and dilated cardiomyopathy. Eur Heart J.
35:2165–2173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou YM, Dai XY, Qiu XB, Yuan F, Li RG, Xu
YJ, Qu XK, Huang RT, Xue S and Yang YQ: HAND1 loss-of-function
mutation associated with familial dilated cardiomyopathy. Clin Chem
Lab Med. Nov 18–2015.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao CM, Bing-Sun, Song HM, Wang J, Xu WJ,
Jiang JF, Qiu XB, Yuan F, Xu JH and Yang YQ: TBX20 loss-of-function
mutation associated with familial dilated cardiomyopathy. Clin Chem
Lab Med. 54:325–332. 2016. View Article : Google Scholar
|
|
8
|
Qu XK, Yuan F, Li RG, Xu L, Jing WF, Liu
H, Xu YJ, Zhang M, Liu X, Fang WY, et al: Prevalence and spectrum
of LRRC10 mutations associated with idiopathic dilated
cardiomyopathy. Mol Med Rep. 12:3718–3724. 2015.PubMed/NCBI
|
|
9
|
Zhang XL, Qiu XB, Yuan F, Wang J, Zhao CM,
Li RG, Xu L, Xu YJ, Shi HY, Hou XM, et al: TBX5 loss-of-function
mutation contributes to familial dilated cardiomyopathy. Biochem
Biophys Res Commun. 459:166–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou W, Zhao L, Jiang JQ, Jiang WF, Yang
YQ and Qiu XB: A novel TBX5 loss-of-function mutation associated
with sporadic dilated cardiomyopathy. Int J Mol Med. 36:282–288.
2015.PubMed/NCBI
|
|
11
|
Zhang XL, Dai N, Tang K, Chen YQ, Chen W,
Wang J, Zhao CM, Yuan F, Qiu XB, Qu XK, et al: GATA5
loss-of-function mutation in familial dilated cardiomyopathy. Int J
Mol Med. 35:763–770. 2015.
|
|
12
|
Xu L, Zhao L, Yuan F, Jiang WF, Liu H, Li
RG, Xu YJ, Zhang M, Fang WY, Qu XK, et al: GATA6 loss-of-function
mutations contribute to familial dilated cardiomyopathy. Int J Mol
Med. 34:1315–1322. 2014.PubMed/NCBI
|
|
13
|
Zhao L, Xu JH, Xu WJ, Yu H, Wang Q, Zheng
HZ, Jiang WF, Jiang JF and Yang YQ: A novel GATA4 loss-of-function
mutation responsible for familial dilated cardiomyopathy. Int J Mol
Med. 33:654–660. 2014.
|
|
14
|
Li J, Liu WD, Yang ZL, Yuan F, Xu L, Li RG
and Yang YQ: Prevalence and spectrum of GATA4 mutations associated
with sporadic dilated cardiomyopathy. Gene. 548:174–181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reinstein E, Orvin K, Tayeb-Fligelman E,
Stiebel-Kalish H, Tzur S, Pimienta AL, Bazak L, Bengal T, Cohen L,
Gaton DD, et al: Mutations in TAX1BP3 cause dilated cardiomyopathy
with septo-optic dysplasia. Hum Mutat. 36:439–442. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dhandapany PS, Razzaque MA, Muthusami U,
Kunnoth S, Edwards JJ, Mulero-Navarro S, Riess I, Pardo S, Sheng J,
Rani DS, et al: RAF1 mutations in childhood-onset dilated
cardiomyopathy. Nat Genet. 46:635–639. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H,
Lu S, Wu P, Zhang Y, Shen L, et al: Prevalence of idiopathic
hypertrophic cardiomyopathy in China: A population-based
echocardiographic analysis of 8080 adults. Am J Med. 116:14–18.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maron BJ, Gardin JM, Flack JM, Gidding SS,
Kurosaki TT and Bild DE: Prevalence of hypertrophic cardiomyopathy
in a general population of young adults. Echocardiographic analysis
of 4111 subjects in the CARDIA Study. Coronary Artery Risk
Development in (Young) Adults. Circulation. 92:785–789. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan F, Qiu XB, Li RG, Qu XK, Wang J, Xu
YJ, Liu X, Fang WY, Yang YQ and Liao DN: A novel NKX2-5
loss-of-function mutation predisposes to familial dilated
cardiomyopathy and arrhythmias. Int J Mol Med. 35:478–486.
2015.
|
|
20
|
Rothberg JM, Hinz W, Rearick TM, Schultz
J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M,
et al: An integrated semiconductor device enabling non-optical
genome sequencing. Nature. 475:348–352. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gersh BJ, Maron BJ, Bonow RO, Dearani JA,
Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR and Rakowski H;
American College of Cardiology Foundation/American Heart
Association Task Force on Practice Guidelines: 2011 ACCF/AHA
Guideline for the Diagnosis and Treatment of Hypertrophic
Cardiomyopathy a report of the American College of Cardiology
Foundation/American Heart Association Task Force on Practice
Guidelines. Developed in collaboration with the American
Association for Thoracic Surgery, American Society of
Echocardiography, American Society of Nuclear Cardiology, Heart
Failure Society of America, Heart Rhythm Society, Society for
Cardiovascular Angiography and Interventions, and Society of
Thoracic Surgeons. J Am Coll Cardiol. 58:e212–260. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mestroni L, Maisch B, McKenna WJ, Schwartz
K, Charron P, Rocco C, Tesson F, Richter A, Wilke A and Komajda M:
Collaborative Research Group of the European Human and Capital
Mobility Project on Familial Dilated Cardiomyopathy: Guidelines for
the study of familial dilated cardiomyopathies. Eur Heart J.
20:93–102. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Robinson JT, Thorvaldsdóttir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Richards CS, Bale S, Bellissimo DB, Das S,
Grody WW, Hegde MR, Lyon E and Ward BE; Collaborative Research
Group of the European Human and Capital Mobility Project on
Familial Dilated Cardiomyopathy: ACMG recommendations for standards
for interpretation and reporting of sequence variations: Revisions
2007. Genet Med. 10:294–300. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sikkema-Raddatz B, Johansson LF, de Boer
EN, Almomani R, Boven LG, van den Berg MP, van Spaendonck-Zwarts
KY, van Tintelen JP, Sijmons RH, Jongbloed JD and Sinke RJ:
Targeted next-generation sequencing can replace Sanger sequencing
in clinical diagnostics. Hum Mutat. 34:1035–1042. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tarabeux J, Zeitouni B, Moncoutier V,
Tenreiro H, Abidallah K, Lair S, Legoix-Né P, Leroy Q, Rouleau E,
Golmard L, et al: Streamlined ion torrent PGM-based diagnostics:
BRCA1 and BRCA2 genes as a model. Eur J Hum Genet. 22:535–541.
2014. View Article : Google Scholar :
|
|
30
|
Costa JL, Sousa S, Justino A, Kay T,
Fernandes S, Cirnes L, Schmitt F and Machado JC: Nonoptical massive
parallel DNA sequencing of BRCA1 and BRCA2 genes in a diagnostic
setting. Hum Mutat. 34:629–635. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li X, Buckton AJ, Wilkinson SL, John S,
Walsh R, Novotny T, Valaskova I, Gupta M, Game L, Barton PJ, et al:
Towards clinical molecular diagnosis of inherited cardiac
conditions: a comparison of bench-top genome DNA sequencers. PLoS
One. 8:e677442013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lakdawala NK, Funke BH, Baxter S, Cirino
AL, Roberts AE, Judge DP, Johnson N, Mendelsohn NJ, Morel C, Care
M, et al: Genetic testing for dilated cardiomyopathy in clinical
practice. J Card Fail. 18:296–303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brito D, Miltenberger-Miltenyi G, Vale
Pereira S, Silva D, Diogo AN and Madeira H: Sarcomeric hypertrophic
cardio-myopathy: genetic profile in a Portuguese population. Rev
Port Cardiol. 31:577–587. 2012.PubMed/NCBI
|
|
34
|
Zou Y, Wang J, Liu X, Wang Y, Chen Y, Sun
K, Gao S, Zhang C, Wang Z, Zhang Y, et al: Multiple gene mutations,
not the type of mutation, are the modifier of left ventricle
hypertrophy in patients with hypertrophic cardiomyopathy. Mol Biol
Rep. 40:3969–3976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao Y, Feng Y, Zhang YM, Ding XX, Song
YZ, Zhang AM, Liu L, Zhang H, Ding JH and Xia XS: Targeted
next-generation sequencing of candidate genes reveals novel
mutations in patients with dilated cardiomyopathy. Int J Mol Med.
36:1479–1486. 2015.PubMed/NCBI
|
|
36
|
Das KJ, Ingles J, Bagnall RD and Semsarian
C: Determining pathogenicity of genetic variants in hypertrophic
cardiomyopathy: importance of periodic reassessment. Genet Med.
16:286–293. 2014. View Article : Google Scholar
|
|
37
|
Hassel D, Dahme T, Erdmann J, Meder B,
Huge A, Stoll M, Just S, Hess A, Ehlermann P, Weichenhan D, et al:
Nexilin mutations destabilize cardiac Z-disks and lead to dilated
cardiomyopathy. Nat Med. 15:1281–1288. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Geisterfer-Lowrance AA, Christe M, Conner
DA, Ingwall JS, Schoen FJ, Seidman CE and Seidman JG: A mouse model
of familial hypertrophic cardiomyopathy. Science. 272:731–734.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cheng J, Morales A, Siegfried JD, Li D,
Norton N, Song J, Gonzalez-Quintana J, Makielski JC and Hershberger
RE: SCN5A rare variants in familial dilated cardiomyopathy decrease
peak sodium current depending on the common polymorphism H558R and
common splice variant Q1077del. Clin Transl Sci. 3:287–294. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen L, Zhang W, Fang C, Jiang S, Shu C,
Cheng H, Li F and Li H: Polymorphism H558R in the human cardiac
sodium channel SCN5A gene is associated with atrial fibrillation. J
Int Med Res. 39:1908–1916. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nikulina SY, Chernova AA, Shulman VA,
Maksimov VN, Gavrilyuk OA, Tretyakova SS and Marilovceva OV: An
investigation of the association of the H558R polymorphism of the
SCN5A gene with idiopathic cardiac conduction disorders. Genet Test
Mol Biomarkers. 19:288–294. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Marangoni S, Di Resta C, Rocchetti M,
Barile L, Rizzetto R, Summa A, Severi S, Sommariva E, Pappone C,
Ferrari M, et al: A Brugada syndrome mutation (p.S216L) and its
modulation by p.H558R polymorphism: standard and dynamic
characterization. Cardiovasc Res. 91:606–616. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Roncarati R, Viviani Anselmi C, Krawitz P,
Lattanzi G, von Kodolitsch Y, Perrot A, di Pasquale E, Papa L,
Portararo P, Columbaro M, et al: Doubly heterozygous LMNA and TTN
mutations revealed by exome sequencing in a severe form of dilated
cardiomyopathy. Eur J Hum Genet. 21:1105–1111. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lekanne Deprez RH, Muurling-Vlietman JJ,
Hruda J, Baars MJ, Wijnaendts LC, Stolte-Dijkstra I, Alders M and
van Hagen JM: Two cases of severe neonatal hypertrophic
cardiomyopathy caused by compound heterozygous mutations in the
MYBPC3 gene. J Med Genet. 43:829–832. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ho CY: Genetics and clinical destiny:
improving care in hyper-trophic cardiomyopathy. Circulation.
122:2430–2440. 2010. View Article : Google Scholar
|
|
46
|
Anan R, Greve G, Thierfelder L, Watkins H,
McKenna WJ, Solomon S, Vecchio C, Shono H, Nakao S and Tanaka H:
Prognostic implications of novel beta cardiac myosin heavy chain
gene mutations that cause familial hypertrophic cardiomyopathy. J
Clin Invest. 93:280–285. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang BL, Xu RL, Zhang J, Zhao XX, Wu H,
Ma LP, Hu JQ, Zhang JL, Ye Z, Zheng X and Qin YW: Identification
and functional analysis of a novel PRKAG2 mutation responsible for
Chinese PRKAG2 cardiac syndrome reveal an important role of non-CBS
domains in regulating the AMPK pathway. J Cardiol. 62:241–248.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Van Driest SL, Vasile VC, Ommen SR, Will
ML, Tajik AJ, Gersh BJ and Ackerman MJ: Myosin binding protein C
mutations and compound heterozygosity in hypertrophic
cardiomyopathy. J Am Coll Cardiol. 44:1903–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhao Y, Feng Y, Zhang YM, Ding XX, Song
YZ, Zhang AM, Liu L, Zhang H, Ding JH and Xia XS: Targeted
next-generation sequencing reveals hot spots and doubly
heterozygous mutations in Chinese patients with familial
cardiomyopathy. BioMed Res Int. 2015:5618192015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brisca G, Fiorillo C, Nesti C, Trucco F,
Derchi M, Andaloro A, Assereto S, Morcaldi G, Pedemonte M, Minetti
C, et al: Early onset cardiomyopathy associated with the
mitochondrial tRNALeu((UUR)) 3271T>C MELAS mutation. Biochem
Biophys Res Commun. 458:601–604. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang AL, Kong DH, Chen DX, Wan J and Yu
YX: Mutation of V896M in cardiac myosin binding protein-c gene in
two Chinese families with hypertrophic cardiomyopathy. Mol Med Rep.
3:759–763. 2010.
|
|
52
|
Millat G, Chanavat V and Rousson R:
Evaluation of a new NGS method based on a custom AmpliSeq library
and Ion Torrent PGM sequencing for the fast detection of genetic
variations in cardiomyopathies. Clin Chim Acta. 433:266–271. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Glotov AS, Kazakov SV, Zhukova EA,
Alexandrov AV, Glotov OS, Pakin VS, Danilova MM, Poliakova IV,
Niyazova SS, Chakova NN, et al: Targeted next-generation sequencing
(NGS) of nine candidate genes with custom AmpliSeq in patients and
a cardiomyopathy risk group. Clin Chim Acta. 446:132–140. 2015.
View Article : Google Scholar : PubMed/NCBI
|