Introduction
Coronary artery and heart diseases are common
diseases affecting the elderly, and are often associated with a
high morbidity. Myocardial ischemia is the main symptom of coronary
artery and heart diseases, which is characterized by a reduction in
blood perfusion and oxygen supply, causing heart dysfunction and
deficiency (1). Myocardial
ischemia is always followed by myocardial injury and the apoptosis
of the cardiac muscle cells, eventually leading to myocardial
infarction (MI) and finally to the death of patients, commonly
termed sudden cardiac death in this case (2,3).
Hence, research into myocardial ischemic injury may provide an
experimental basis for the exploration of sudden cardiac death. It
has been demonstrated that myocardial apoptosis is the most
destructive event induced by myocardial ischemia and oxygen
reperfusion (4). It is also
widely known that apoptosis is one of the main types of programmed
cell death (5). Consequently,
based on the descriptions given above, we hypothesized that the
inhibition of apoptosis may provide a protective effect against
myocardial ischemia and possible sudden death. Moreover, an
in-depth molecular understanding of the apoptotic pathway would
provide a better understanding of the mechanisms behind myocardial
ischemia and may provide effective treatments for myocardial
ischemic injury, and for the prevention of sudden death.
Hypoxia is closely related to cardiovascular
diseases due to the insufficient oxygen supply; in particular,
hypoxia is one of the harmful consequences of myocardial ischemia,
which triggers a wide range of cellular responses, including gene
regulation and cell apoptosis (6). However, hypoxia-induced cell
apoptosis is rarely reported in human cardiomyocytes. In the
present study, the culture of human cardiomyocytes under hypoxic
conditions was employed to mimic cardiac ischemia.
MicroRNAs (miRNAs or miRs) are a class of 17–25
nucleotide, non-coding small RNA molecules, regulating the
expression of their target genes at the post-transcriptional level
by binding to the 3′-untranslated region (3′-UTR) of the target
mRNA and degenerating the target mRNA (7). A variety of miRNAs have been
reported to regulate myocardial hypoxia-induced apoptosis (8–10).
For example, miR-133a was reported to be downregulated in
hypoxia-treated H9c2 cells, although the overexpression of miR-133a
was shown to inhibit hypoxia-induced apoptosis (11). In addition, in hypoxia-exposed
neonatal rat cardiomyocytes, miR-26b was shown to be upregulated;
however, the downregulation of miR-26b exerted inhibitory effects
on hypoxia-induced apoptosis (12). miR-429 belongs to the miR-200
family, which is dysregulated in various types of cancer. For
example, miR-429 has been reported to play a role in colorectal
cancer (14), hepatocellular
carcinoma (13,15), esophageal carcinoma (16), breast cancer (17) and oral squamous cell carcinoma
(18), and its expression and
functions (inhibitory or promotive effects on cancer development)
vary with and are dependent upon the type of tumor. Moreover, Gao
and Liu reported that in hepatocellular carcinoma cells, miR-429
directly targets Notch1 and reduces both the mRNA and protein
levels of Notch1 (15). However,
the precise functions and effects of miR-429 in myocardial ischemia
and subsequent apoptosis have not been fully investigated to
date.
The Notch pathway participates in various cellular
processes, including cell proliferation, differentiation and
apoptosis during the embryonic and adult stages (19,20). In particular, the Notch pathway
has been reported to be associated with cardiac repair,
regeneration and protection (21). In fact, the Notch pathway controls
mesodermal differentiation during embryonic and adult development,
including cardiac differentiation (22,23). Moreover, the activation of the
Notch pathway has been shown to exert inhibitory effects on the
degree of ischemic injury and to improve heart function following
MI (24). The Notch receptors are
important effectors of the Notch pathway, which regulates
cardiovascular development and homeostasis (25,26). In mammals, 4 Notch receptors
(Notch1-4) and 5 Notch ligands have been identified (27), among which Notch1 is strongly
expressed in cardiomyocytes, smooth muscle cells and endothelial
cells (21). Notch1 has been
reported to play a role in promoting the proliferation of immature
cardiomyocytes in rat models, and the inhibition of the Notch
pathway inhibits proliferation and induces apoptosis (28). Yu and Song demonstrated that
Notch1 reduced the apoptosis of rat cardiomyocytes in ischemic
post-conditioning, and the activation of the Notch1 gene markedly
promoted cell proliferation and inhibited cell apoptosis (29). However, although the results
mentioned above demonstrate the protective and promoting effects of
Notch1 in cardiac development, very little is known regarding the
underlying mechanisms of the function of Notch1 in this area. In
addition, the interactions between miRNAs and Notch1 are not yet
fully understood, particularly in human cardiomyocytes. We
hypothesized that Notch1 may play an important role in alleviating
the hypoxia-induced apoptosis of human cardiomyocytes and,
therefore, we designed the subsequent experiments to verify our
hypothesis.
In this study, the culture of human cardiomyocytes
under hypoxic conditions was employed to mimic the insufficient
oxygen supply induced by myocardial ischemia. The expression of
miR-429 was evaluated, and it was artificially overexpressed and
silenced under hypoxic conditions. Subsequently, the effects of
miR-429 on cell apoptosis were investigated. Notch1 was introduced
into this study as it was identified as one of the target genes of
miR-429 in the homo sapiens species. Moreover, the effects
of Notch1 expression on cell apoptosis were further explored. In
sum, our results provide new insight into cell apoptosis induced by
myocardial ischemia and the cause of sudden death, which may lead
towards the discovery of novel therapeutic methods targeting Notch1
and to the more effective prevention from myocardial ischemia and
ensuing sudden death.
Materials and methods
Cell culture and exposure to hypoxia
Human cardiomyocytes (AC-16), purchased from the
Cell Resource Center of Shanghai Institutes for Biological
Sciences, Chinese Academy of Sciences, were cultured in RPMI-1640
medium supplemented with 10% fetal bovine serum (FBS) (both from
HyClone, Logan, UT, USA) in an atmosphere of 37°C with 5%
CO2. For culture under hypoxic conditions, the cells
were incubated in a hypoxic (1% O2/5% CO2/94%
N2) atmosphere at 37°C for 12 h. The cells cultured
under normoxic conditions (5% CO2/95% air) served as the
controls.
Transfection with miRNA-mimics and
miRNA-inhibitor
To induce miR-429 overexpression or silencing, the
cells were transfected with negative control miRNA (NC miRNA),
miRNA-mimics and miRNA-inhibitor (GenePharma Co., Ltd., Shanghai,
China) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA)
following the manufacturer's insructions.
The ectopic expression of Notch1
Adenoviral vectors were constructed by GenePharma
Co., Ltd. Briefly, the adenoviruses were amplified and titrated in
293 cells. The AC-16 cells were transiently transfected with
adenoviruses carrying the Notch1 gene (Ad-Notch1) without any
obvious signs of cytotoxicity [determined by the fact that the
transfected AC-16 cells survived as usual without sudden death, and
the morphology of transfected AC-16 cells was also unaltered (data
not shown)] using Lipofectamine 2000 (Invitrogen) following the
manufacturer's instructions. Some of the AC-16 cells were
transfected with the empty carrier LacZ (Ad-LacZ) as the negative
control (NC) group. For the co-overexpression of miR-429 and
Notch1, the cells were co-transfected with miRNA-mimics and
Ad-Notch1 harboring no specific binding sequences of miR-429 in the
3′-UTR using Lipofectamine 2000 (Invitrogen) according to the
manufacturer's instructions.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Cell viability was evaluated using the MTT Cell
Proliferation and Cytotoxicity assay kit (Beyotime, Jiangsu,
China). Briefly, the cells were seeded into 96-well dishes at a
density of 1×104 cells/well, and were then subjected to
hypoxic conditions as described above. Subsequently, 20 µl
of MTT solution (5 mg/ml) were added to each well followed by
incubation for a further 4 h. The medium was then discarded and 150
µl of dimethylsulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO,
USA) was added with rotation for 10 min to dissolve the formazan.
Finally, the optical density (OD) value of each well at 490 nm was
measured and recorded using a microplate spectrophotometer
(NanoDrop Technology, Wilmington, DE, USA); this was set as the
Y-axis for the viability curve. Each well was measured in
triplicate.
5-Bromo-2-deoxyuridine (BrdU) assay
Cell proliferation was assessed by BrdU assay using
the BrdU Labeling and Detection kit II (Roche, Basel, Switzerland)
following the manufacturer's instructions. Briefly, the cells were
seeded in 96-well dishes at a density of 1×104
cells/well, and were then subjected to indicated
treatments/conditions. Subsequently, BrdU labeling solution was
added to each well at a final concentration of 10 µM
followed by a further 4 h of incubation at 37°C. The cells were
then fixed with 4% cold paraformaldehyde for 20 min and washed
thrice with phosphate-buffered saline (PBS), followed by incubation
in 1.5 M HCl for 30 min for denaturalization. The cells were then
incubated with peroxidase-labeled anti-BrdU antibody (1:100,
provided with the kit) at room temperature. The cells were
counterstained with hematoxylin, and the BrdU-positive cells and
total cells were counted in 5 randomly selected fields at ×400
magnification. Cell proliferation was demonstrated as the labeling
index (LI) of BrdU-positive cells vs. total cells.
Terminal deoxynucleotide
transferase-mediated nick end-labeling (TUNEL) assay
The cells were seeded in 6-well dishes at a density
of 1×105 cells/well, followed by exposure to hypoxic
conditions and transfection as described above. TUNEL assay was
used to detect the apoptotic cardiomyocytes following myocardial
ischemia. Briefly, the cells were fixed with 4% paraformaldehyde
for 30 min, washed and incubated on ice with 0.1% Triton X-100
(Sigma-Aldrich) for 2 min. TUNEL staining was performed using the
TUNEL detection kit (Roche) according to the manufacturer's
instructions. Finally, the cells were counterstained with
hematoxylin after TUNEL staining. Five microscopic fields in each
well were randomly selected to count the TUNEL-positive cells and
the total cells at ×400 magnification. The ratio of the
TUNEL-positive cells vs. the total number of cells was calculated
as the apoptotic rate.
Hoechst staining
The cells were digested and subcultured into 96-well
plates at a density of 5×103 cells/well. Subsequently,
the cells were subjected to transfection as described above. Cell
apoptosis was detected using the Hoechst staining kit (Beyotime) by
counting the number of stained cell nuclei according to the
manufacturer's instructions. The positively stained cells were
visualized under a fluorescence microscope (Olympus, Tokyo, Japan)
and counted in randomly selected fields for calculating the
apoptotic rate.
Detection of caspase-3 activity
The supernatant of the treated cells was collected
and examined using the colorimetric caspase-3 assay kit (Beyotime)
to detect the caspase-3 activity according to the manufacturer's
instructions. Briefly, the AC-16 cells were lysed on ice, and 5 mg
of the caspase-3 substrate were then added to the lysates, and the
samples were incubated for 4 h at 37°C. The absorbance at 405 nm
was measured using a microplate spectrophotometer (NanoDrop
Technology).
Target prediction
The target genes of miR-429 were predicted from 3
established miRNA-target prediction programs: TargetScan
(http://www.targetscan.org/), Pictar
(http://pictar.mdc-berlin.de/cgi-bin/new_PicTar_mouse.cgi)
and microRNA.org (http://www.microrna.org/microrna/home.do) at the same
time.
Dual-luciferase reporter assay
The mutant type (Mut) 3′-UTR was constructed by
replacing certain miR-429 binding sites in the 3′-UTR of the Notch1
with other bases. The Notch1 wild-type (WT) 3′-UTR and Mut 3′-UTR
were amplified, and subsequently transfected into a luciferase
reporter vector (pmiR-REPORT; Ambion, Rockville, MD, USA) to
generate the recombinant plasmids of the pmiR-Notch1-WT and
pmiR-Notch1-Mut. The NC miRNA and miRNA-mimics were co-transfected
with the pmiR-Notch1-WT and pmiR-Notch1-Mut, mutually, into the 293
cells using Lipofectamine 2000 reagent (Invitrogen) according to
the manufacturer's instructions. The luciferase activity was
examined 24 h later using the Dual-Luciferase Reporter assay system
(Promega, Madison, WI, USA).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the AC-16 cells
usingTRIzol® reagent (Invitrogen) and then
reverse-transcribed into cDNA using the RevertAid First Strand cDNA
Synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA). qPCR
was performed using SYBR® Fast qPCR Mix (Takara
Biotechnology, Dalian, China) on the CFX96 Real-Time PCR Detection
System (Bio-Rad, Hercules, CA, USA). qPCR was then conducted under
the following conditions: 95°C for 10 min, 40 cycles at 95°C for 2
min, 60°C for 12 sec, and 72°C for 30 sec. U6 served as the
internal reference gene for miRNAs and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) for the other genes. The relative expression
level was normalized to that of the internal reference gene using
the 2−ΔΔCt cycle threshold method.
Western blot analysis
Total protein was extracted using RIPA buffer
(Sigma-Aldrich) as follows: the medium was discarded and RIPA
buffer was added, with rotation for 10 min on ice for protein
lysis. The protein lysates were collected using a scraper and
separated by 10% sodium dodecyl sulfonate-polyacrylamide gel
electrophoresis (SDS-PAGE) and then electroblotted onto the PVDF
membranes (Millipore, Billerica, MA, USA), which were subsequently
incubated with the primary antibodies, including rabbit
anti-caspase-3 (ab408; 1:2,000), goat anti-Bcl-2 (ab37899;
1:2,000), rabbit anti-Bax (ab104156; 1:1,500) and rabbit anti-GAPDH
(ab57062; 1:2,000) overnight at 4°C, followed by incubation with
secondary antibodies (ab97196 and ab6880; 1:4,000) (all from Abcam,
Cambridge, UK) at 37°C for 4 h. An enhanced chemiluminescence
detection system was used for visualizing the bound proteins.
Densitometric analysis was performed using Image-Pro Plus (Roper
Industries, New York, NY, USA), and the densitometry of the
immunoblots was normalized to GAPDH. The densitometry of the most
abundant band in each experiment was set as 100%, and other bands
were normalized to 100%.
Statistical analysis
All data are presented as the means ± standard
deviation (SD). Statistical analysis was performed using SPSS 16.0
software (SPSS, Inc., Chicago, IL, USA). The Student's t-test was
used for comparisons between 2 groups. For multiple-group
comparisons, one-way analysis of variance (ANOVA) was performed. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
The expression of miR-429 is upregulated
under hypoxic conditions
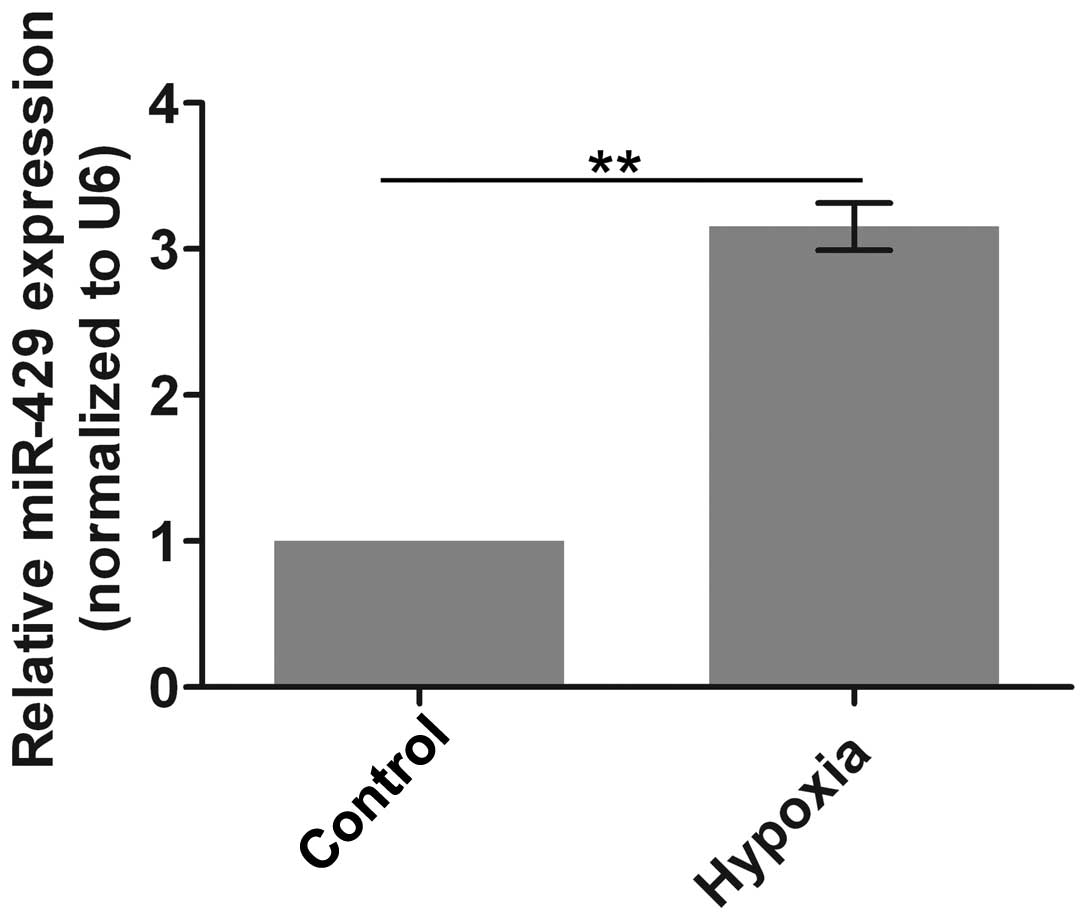
Firstly, the expression of miR-429 was evaluated.
miR-429 expression was significantly increased after the cells were
subjected to culture under hypoxic conditions compared with the
control group (Fig. 1). This
result suggests that the expression of miR-429 is upregulated under
conditions mimicking myocardial ischemia.
miR-429 is effectively verexpressed and
silenced
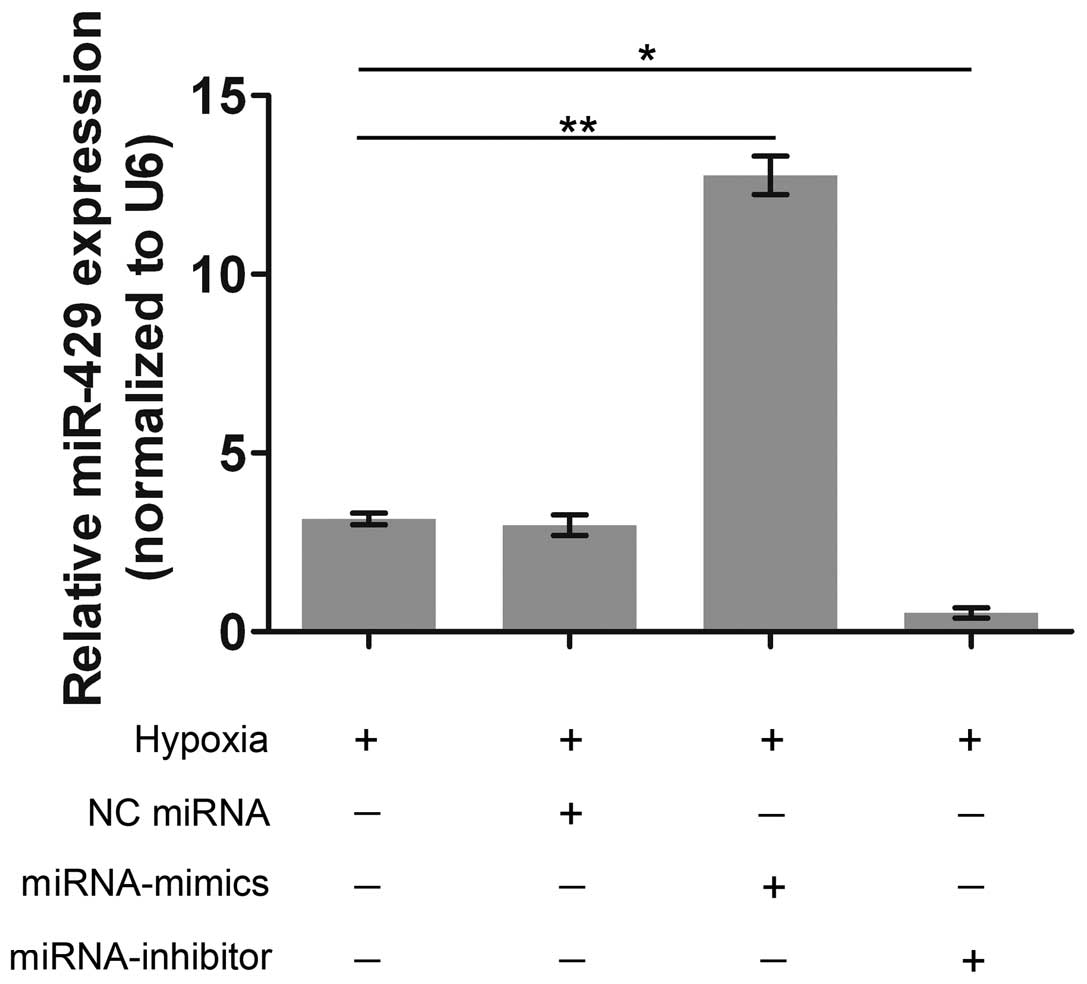
To examine the role of miR-429 in hypoxia-induced
apoptosis, miR-429 was artificially overexpressed and silenced by
transfection with miRNA-mimics and miRNA-inhibitor, respectively.
The expression of miR-429 was significantly higher in the
miRNA-mimics group than that in the hypoxia group (Fig. 2). On the contrary, miR-429
expression was barely observed in the miRNA-inhibitor group. These
results demonstrated that the transfection with miRNA-mimics and
miRNA-inhibitor was effective in altering miR-429 expression in the
AC-16 cells.
Cell viability, proliferation and
apoptosis are affected by miR-429 expression
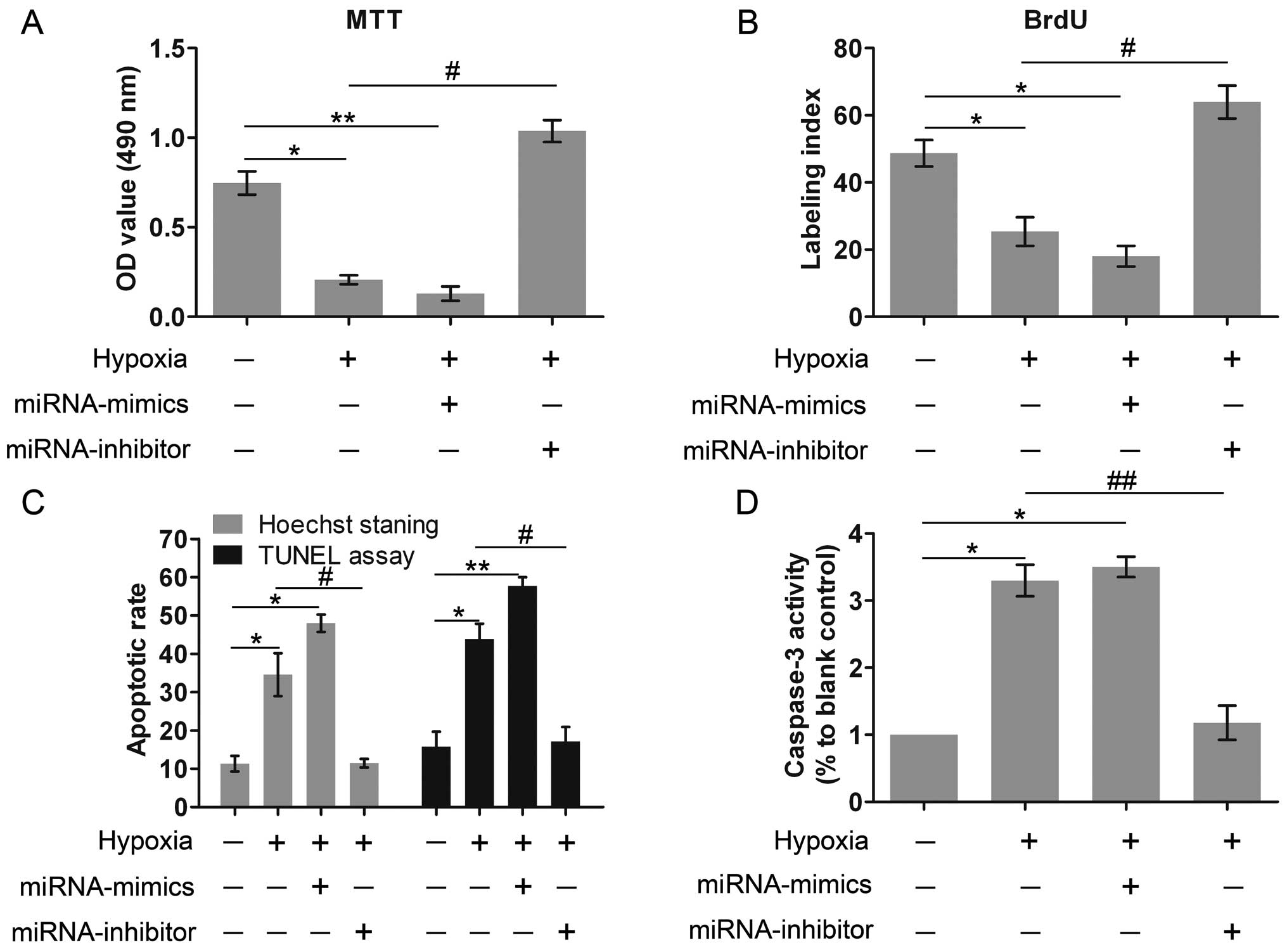
We then examined the effects of miR-429 on cell
viability, proliferation and apoptosis. Compared with the control
group, the culture under hypoxic conditions and miR-429
overexpression markedly inhibited cell viability, whereas the
knockdown of miR-429 expression significantly restored cell
viability under hypoxic conditions, which was even higher than
those of the control group (Fig.
3A). We hypothesized that miR-429 was associated with key genes
involved in modulating cell viability, but the detailed mechanism
remains to be investigated. Similar results were also recorded with
the BrdU assay to evaluate cell proliferation (Fig. 3B). In addition, the results of
Hoechst staining and TUNEL assay revealed that hypoxia markedly
promoted cell apoptosis, and miR-429 overexpression also increased
the apoptotic rate (Fig. 3C). By
contrast, the silencing of miR-429 exerted potent protective
effects against apoptosis; the apoptotic rate was markedly reduced
following the silencing of miR-429. Caspase-3 is an indicator of
cell apoptosis. Exposure to hypoxia and the high expression of
miR-429 markedly enhanced caspase-3 activity; however, the
silencing of miR-429 exerted inhibitory effects on caspase-3
activity (Fig. 3D). On the whole,
all these results indicated that hypoxia and the overexpression of
miR-429 both impaired cell survival, whereas the knockdown of
miR-429 expression greatly contributed to cell survival.
The expression of apoptosis-related genes
is altered by the miR-429 expression
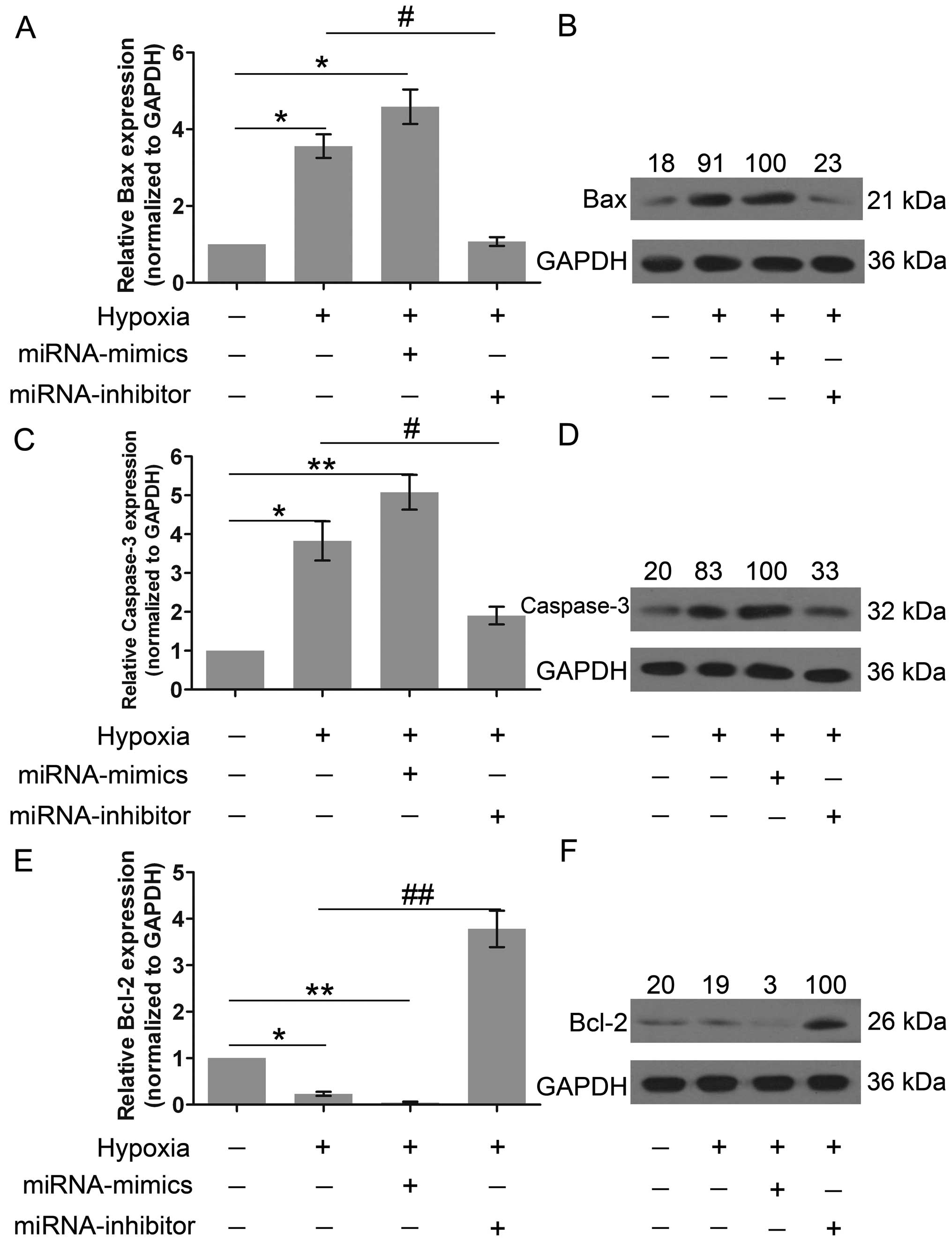
Subsequently, to further investigate the role of
miR-429 in cell apoptosis, the expression of Bax, caspase-3 and
Bcl-2 was explored. Compared with the control group, the level of
expression of Bax and caspase-3 was markedly increased following
culture under hypoxic conditions and by miR-429 overexpression
(Fig. 4A–D). However, Bax and
caspase-3 were minimally expressed in the cells lacking miR-429
expression. Fig. 4E and F
demonstrate that Bcl-2 was highly expressed in the miRNA-inhibitor
group, indicating that miR-429 is a negative regulator of Bcl-2
expression. In short, these results suggest that miR-429 regulates
cell apoptosis and survival.
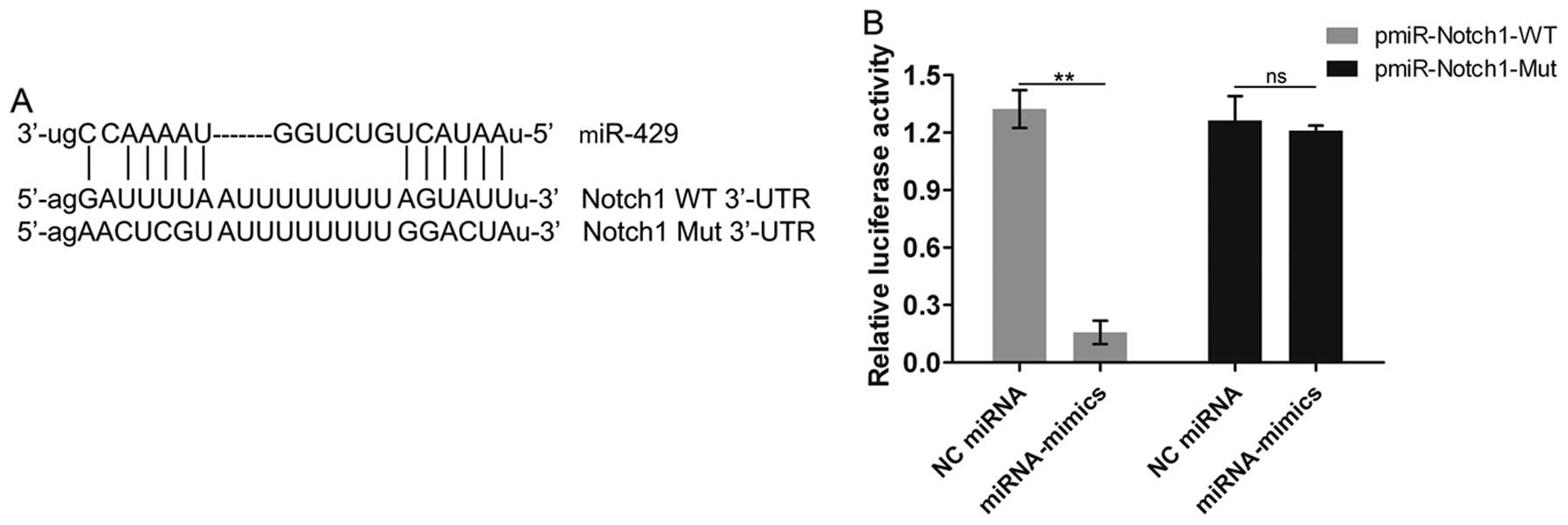
Notch1 is identified as the target gene
of miR-429
We then aimed to explore the mechanisms underlying
the regulatory effects of miR-429 on cell apoptosis. We predicted
that Notch1 was a potential target gene of miR-429 through our
analysis using the TargetScan, Pictar and microRNA. org databases.
We then aimed to confirmed this prediction through Dual-Luciferase
Reporter assay. miR-429 was shown to have some binding sites in the
3′-UTR of the WT Notch1 mRNA (Fig.
5A). Moreover, miR-429 overexpression markedly inhibited
luciferase activity in the pmiR-Notch1-WT, not in the
pmiR-Notch1-Mut (Fig. 5B),
demonstrating that miR-429 interacts with the Notch1 gene and
possibly regulates the expression of Notch1; namely, Notch1 is the
target gene of miR-429.
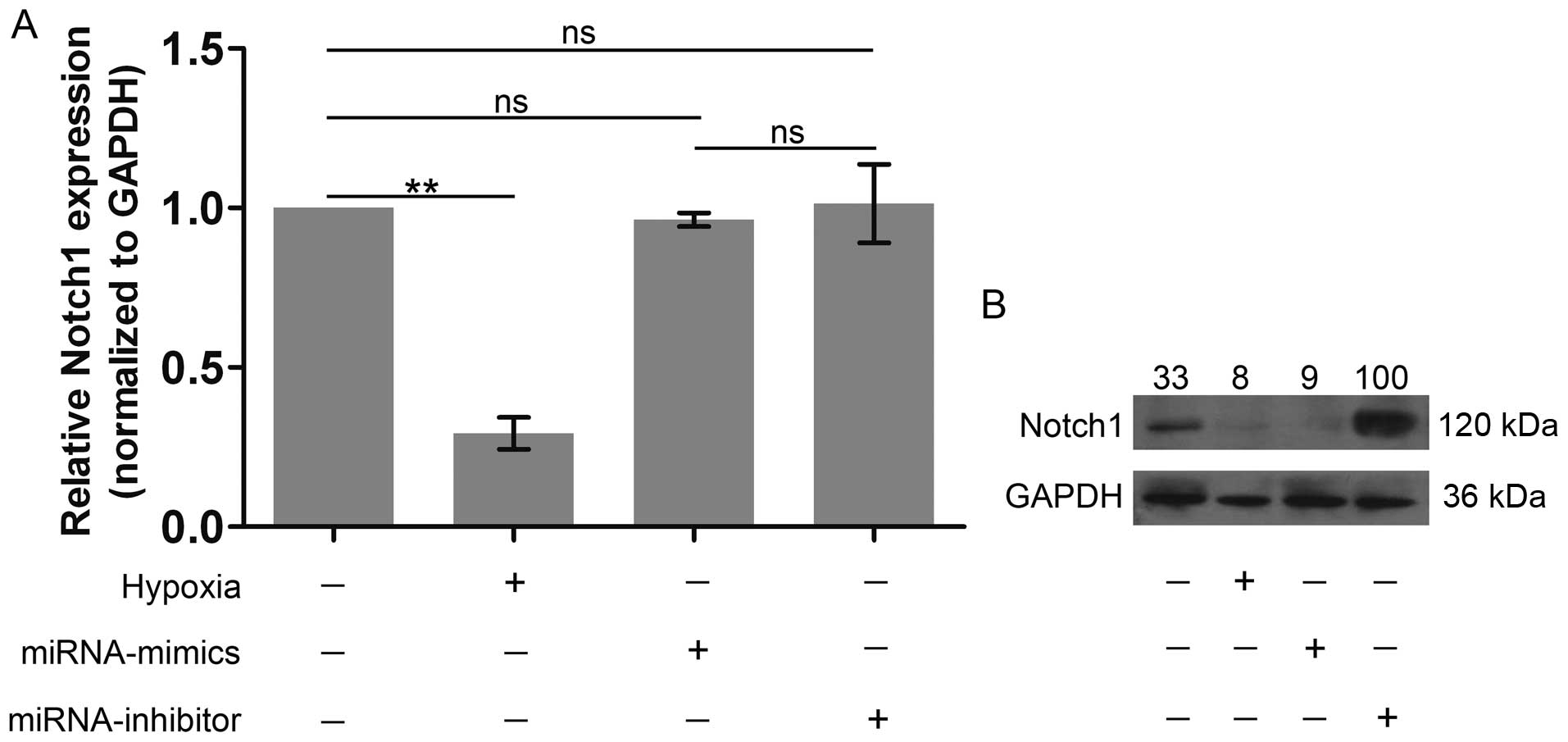
Notch1 expression is inhibited by miR-429
overexpression, but is promoted by the silencing of miR-429 at the
protein level
As we already confirmed that Notch1 was one of the
target genes of miR-429, we further examined the role of miR-429 in
regulating the expression of Notch1. Notch1 expression was notably
suppressed following culture under hypoxic conditions (Fig. 6). It was noteworthy that the
expression of Notch1 at the mRNA level was not significantly
altered by miR-429 overexpression or miR-429 silencing (Fig. 6A). In particular, in the protein
level, Notch1 expression after the knockdown of miR-429 expression
significantly exceeded its own expression in the hypoxia group,
whereas miR-429 overexpression markedly inhibited Notch1 expression
(Fig. 6B), indicating that the
silencing of miR-429 promoted Notch1 expression. On the whole,
these results further confirm that Notch1 is the target gene of
miR-429.
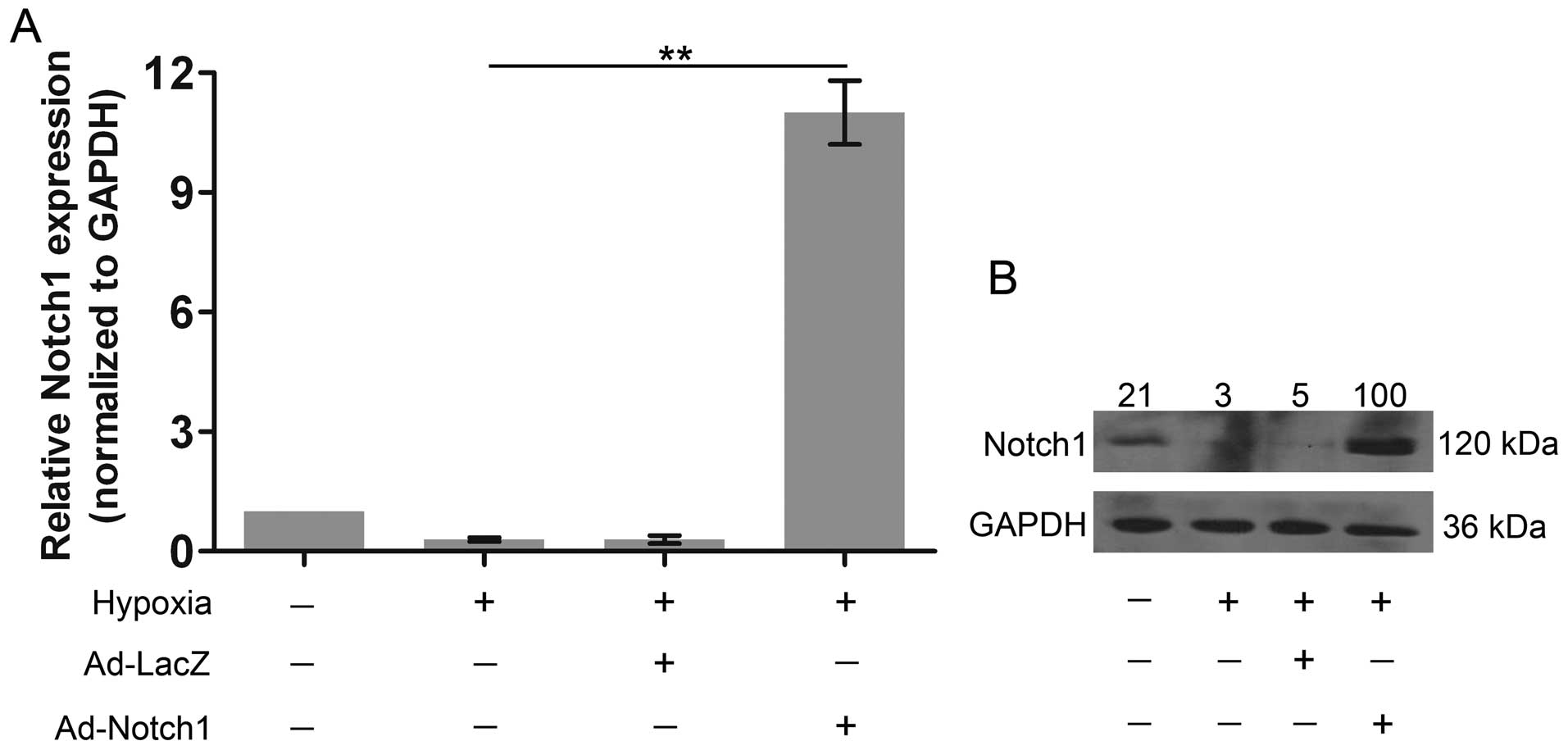
Notch1 is overexpressed in AC-16
cells
Finally, to investigate whether miR-429 regulates
hypoxia-induced apoptosis by regulating Notch1 expression, we
overexpressed Notch1 to explore its role in cell apoptosis. Notch1
was markedly over-expressed at the mRNA level (Fig. 7A); moreover, its protein
expression was also markedly increased compared with both the
control group and the hypoxia group (Fig. 7B).
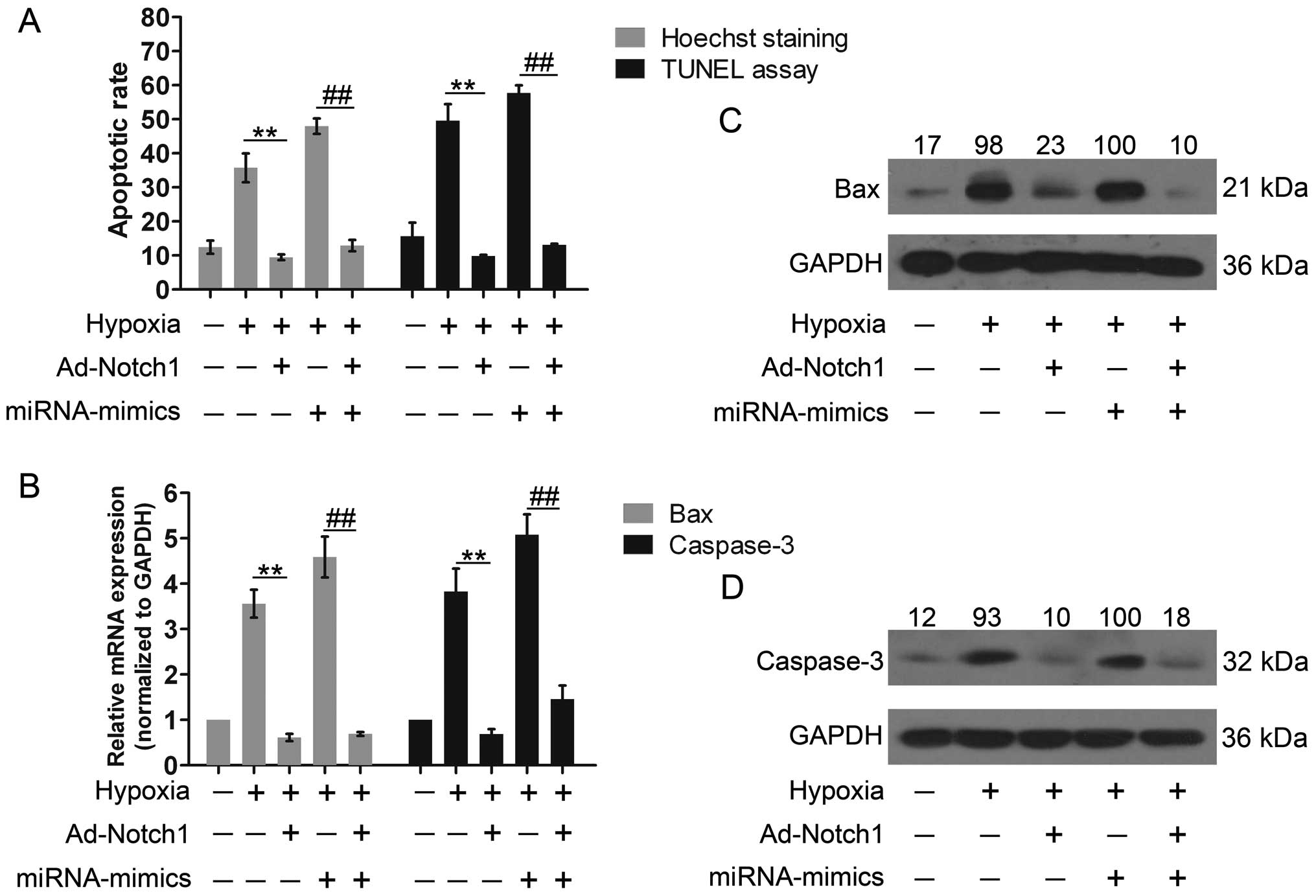
Notch1 overexpression reverses the
effects of the miR-429 overexpression on cell apoptosis
Cell apoptosis was evaluated again following the
co-overexpression of Notch1 and miR-429. The cells were
co-transfected with miRNA-mimics and Ad-Notch1, harboring no
specific binding sequences of the miR-429 in the 3′-UTR. Cell
apoptosis was notably decreased by Notch1 overexpression compared
with the hypoxia group (Fig. 8A).
Moreover, Notch1 overexpression inhibited cell apoptosis induced by
miR-429 overexpression. In addition, Bax expression was markedly
inhibited and caspase-3 expression was significantly reduced by
Notch1 overexpression (Fig. 8B and
C). Similarly, the increased expression of Bax and caspase-3
induced by miR-429 overexpression was reversed by the ectopic
expression of Notch1. These results indicated that the high
expression of Notch1 led to reduced apoptosis and provided
cytoprotective effects against apoptosis, which also suggests that
miR-429 regulates apoptosis by controlling Notch1 expression.
Discussion
In the present study, under hypoxic conditions
mimicking myocardial ischemia, the expression of miR-429 was
significantly upregulated, indicating that miR-429 is a
characteristic molecule in myocardial ischemia. We then further
explored the role of miR-429 in hypoxia-induced cell apoptosis. The
results revealed that the miR-429-deprived cells were more
resistant to hypoxia, implying that the downregulation of miR-429
provides cytoprotective effects against myocardial ischemia and
hypoxia. In addition, Notch1 was also proven to be one of the
target genes of miR-429 and to be involved in the anti-apoptotic
effects. These findings may lead to the development of novel drugs
to induce the downregulation of miR-429 and the upregulation of
Notch1, which could possibly reduce myocardial ischemia disease and
consequent MI and sudden death.
Cardiovascular disease is the leading cause of
mortaliy worldwide (30). In
particular, MI caused by coronary artery and myocardial ischemia
remains one of the most challenging clinical concerns; myocardial
ischemia plays a key role in the progression of cell apoptosis,
cardiac damage and finally, heart failure (31). Heart-derived sudden death is also
an issue which needs to be overcome. Traditionally, the rapid
reperfusion of blood and oxygen is employed in the treatment of
myocardial ischemia. However, rapid reperfusion often results in
poor prognosis, characterized by cardiac injury and the apoptosis
of cardiomyocytes. Other methods, including coronary artery bypass
grafting and percutaneous coronary intervention, are effective in
the majority of patients (32);
however, the associated costs and secondary injuries are
exorbitant. Hence, there is an urgent need for identifying novel
therapeutic targets. Yang et al reported that
radio-protective 105 kDa protein (RP105) suppressed
ischemia/reperfusion-induced myocardial injury by attenuating
myocardial apoptosis (33). This
finding inspired us to embark on a search to find an miRNA molecule
that would regulate cell survival under hypoxic conditions. Some
research into this matter has already been undertaken. Chen et
al reported that in the H2O2-induced
apoptosis of rat cardiomyocytes, the downregulation of miR-100
exerted cytoprotective effects (34). Another study by Tong et al
indicated that the overexpression of miR-21 protected rat
cardiomyocytes against apoptosis (35). These above-mentioned results
demonstrate that the effects of miRNAs in cell apoptosis vary
greatly. Compared with the results from the studies by Chen et
al (34) and Tong et
al (35), our results
indicated for the first time, to the best of our knowledge, that
the downregulation of miR-429 protects human cardio-myocytes under
hypoxic conditions. miR-429 was naturally upregulated following
culture under hypoxic conditions and was artificially silenced by
transfection with miRNA-mimics. The silencing of miR-429 enhanced
cell viability and proliferation, along with the decreased
apoptotic rate and caspase-3 activity, implying that cell apoptosis
was inhibited. Moreover, the decreased expression of Bax and
caspase-3, and the increased expression of Bcl-2 confirmed the
effects of miR-429 silencing. Besides, Notch1, as one of the target
genes of miR-429, has been reported to contribute to the inhibition
of the apoptosis of cardiomyocytes (29,36). In this study, the overexpression
of miR-429 significantly inhibited Notch1 expression. We
consequently investigated the role of Notch1 in human
cardiomyocytes. Its overexpression reversed the effects of miR-429
overexpression and reduced apoptosis, indicating that miR-429
regulates cell apoptosis possibly by regulating Notch1
expression.
In conclusion, our results demonstrate for the first
time that the downregulation of miR-429 contributes to the survival
of human cardiomyocytes, and inhibits apoptosis under hypoxic
conditions. Notably, the overexpression of Notch1 significantly
suppressed the apoptosis of human cardiomyocytes compared with the
hypoxia group, thus providing a novel therapeutic target for the
treatment of cardiac ischemia and the prevention of possible sudden
death.
References
|
1
|
Jianqiang P, Ping Z, Xinmin F, Zhenhua Y,
Ming Z and Ying G: Expression of hypoxia-inducible factor 1 alpha
ameliorate myocardial ischemia in rat. Biochem Biophys Res Commun.
465:691–695. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elsässer A, Suzuki K, Lorenz-Meyer S, Bode
C and Schaper J: The role of apoptosis in myocardial ischemia: A
critical appraisal. Basic Res Cardiol. 96:219–226. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Freude B, Masters TN, Robicsek F, Fokin A,
Kostin S, Zimmermann R, Ullmann C, Lorenz-Meyer S and Schaper J:
Apoptosis is initiated by myocardial ischemia and executed during
reperfusion. J Mol Cell Cardiol. 32:197–208. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kloner RA: Does reperfusion injury exist
in humans? J Am Coll Cardiol. 21:537–545. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bracken CP, Whitelaw ML and Peet DJ: The
hypoxia-inducible factors: Key transcriptional regulators of
hypoxic responses. Cell Mol Life Sci. 60:1376–1393. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choudhuri S: Small noncoding RNAs:
Biogenesis, function, and emerging significance in toxicology. J
Biochem Mol Toxicol. 24:195–216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fang J, Song XW, Tian J, Chen HY, Li DF,
Wang JF, Ren AJ, Yuan WJ and Lin L: Overexpression of microRNA-378
attenuates ischemia-induced apoptosis by inhibiting caspase-3
expression in cardiac myocytes. Apoptosis. 17:410–423. 2012.
View Article : Google Scholar
|
|
9
|
Hu S, Huang M, Li Z, Jia F, Ghosh Z,
Lijkwan MA, Fasanaro P, Sun N, Wang X, Martelli F, et al:
MicroRNA-210 as a novel therapy for treatment of ischemic heart
disease. Circulation. 122(Suppl 11): S124–S131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qian L, Van Laake LW, Huang Y, Liu S,
Wendland MF and Srivastava D: miR-24 inhibits apoptosis and
represses Bim in mouse cardiomyocytes. J Exp Med. 208:549–560.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li AY, Yang Q and Yang K: miR-133a
mediates the hypoxia-induced apoptosis by inhibiting TAGLN2
expression in cardiac myocytes. Mol Cell Biochem. 400:173–181.
2015. View Article : Google Scholar
|
|
12
|
Wang X, Li C and Dai Q: Downregulation of
microRNA-26b rescued hypoxia-induced apoptosis in cultured neonatal
rat cardiac myocytes by regulating PTEN. Int J Clin Exp Med.
8:4073–4079. 2015.
|
|
13
|
Tang J, Li L, Huang W, Sui C, Yang Y, Lin
X, Hou G, Chen X, Fu J, Yuan S, et al: MiR-429 increases the
metastatic capability of HCC via regulating classic Wnt pathway
rather than epithelial-mesenchymal transition. Cancer Lett.
364:33–43. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li J, Du L, Yang Y, Wang C, Liu H, Wang L,
Zhang X, Li W, Zheng G and Dong Z: MiR-429 is an independent
prognostic factor in colorectal cancer and exerts its
anti-apoptotic function by targeting SOX2. Cancer Lett. 329:84–90.
2013. View Article : Google Scholar
|
|
15
|
Gao H and Liu C: miR-429 represses cell
proliferation and induces apoptosis in HBV-related HCC. Biomed
Pharmacother. 68:943–949. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Y, Li M, Zang W, Ma Y, Wang N, Li P,
Wang T and Zhao G: MiR-429 upregulation induces apoptosis and
suppresses invasion by targeting Bcl-2 and SP-1 in esophageal
carcinoma. Cell Oncol (Dordr). 36:385–394. 2013. View Article : Google Scholar
|
|
17
|
Ye ZB, Ma G, Zhao YH, Xiao Y, Zhan Y, Jing
C, Gao K, Liu ZH and Yu SJ: miR-429 inhibits migration and invasion
of breast cancer cells in vitro. Int J Oncol. 46:531–538. 2015.
|
|
18
|
Lei W, Liu YE, Zheng Y and Qu L: MiR-429
inhibits oral squamous cell carcinoma growth by targeting ZEB1. Med
Sci Monit. 21:383–389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
von Boehmer H: Coming to grips with Notch.
J Exp Med. 194:F43–F46. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miele L and Osborne B: Arbiter of
differentiation and death: Notch signaling meets apoptosis. J Cell
Physiol. 181:393–409. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Hiroi Y and Liao JK: Notch signaling
as an important mediator of cardiac repair and regeneration after
myocardial infarction. Trends Cardiovasc Med. 20:228–231. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Del Monte G, Grego-Bessa J, González-Rajal
A, Bolós V and De La Pompa JL: Monitoring Notch1 activity in
development: Evidence for a feedback regulatory loop. Dev Dyn.
236:2594–2614. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grego-Bessa J, Luna-Zurita L, del Monte G,
Bolós V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama YS,
Chen H, et al: Notch signaling is essential for ventricular chamber
development. Dev Cell. 12:415–429. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gude NA, Emmanuel G, Wu W, Cottage CT,
Fischer K, Quijada P, Muraski JA, Alvarez R, Rubio M, Schaefer E
and Sussman MA: Activation of Notch-mediated protective signaling
in the myocardium. Circ Res. 102:1025–1035. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chiba S: Notch signaling in stem cell
systems. Stem Cells. 24:2437–2447. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
del Monte G, Casanova JC, Guadix JA,
MacGrogan D, Burch JB, Pérez-Pomares JM and de la Pompa JL:
Differential Notch signaling in the epicardium is required for
cardiac inflow development and coronary vessel morphogenesis. Circ
Res. 108:824–836. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bolós V, Grego-Bessa J and de la Pompa JL:
Notch signaling in development and cancer. Endocr Rev. 28:339–363.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Collesi C, Zentilin L, Sinagra G and
Giacca M: Notch1 signaling stimulates proliferation of immature
cardiomyocytes. J Cell Biol. 183:117–128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu B and Song B: Notch 1 signalling
inhibits cardiomyocyte apoptosis in ischaemic postconditioning.
Heart Lung Circ. 23:152–158. 2014. View Article : Google Scholar
|
|
30
|
Gersh BJ, Sliwa K, Mayosi BM and Yusuf S:
Novel therapeutic concepts: the epidemic of cardiovascular disease
in the developing world: global implications. Eur Heart J.
31:642–648. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Eefting F, Rensing B, Wigman J, Pannekoek
WJ, Liu WM, Cramer MJ, Lips DJ and Doevendans PA: Role of apoptosis
in reperfusion injury. Cardiovasc Res. 61:414–426. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Velazquez EJ and Bonow RO:
Revascularization in severe left ventricular dysfunction. J Am Coll
Cardiol. 65:615–624. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang J, Guo X, Yang J, Ding JW, Li S, Yang
R, Fan ZX and Yang CJ: RP105 protects against apoptosis in
ischemia/reperfusion-induced myocardial damage in rats by
suppressing TLR4-mediated signaling pathways. Cell Physiol Biochem.
36:2137–2148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen A, Li G, Chen L, Guo J and Liu Y:
Downregulation of microRNA-100 protects
H2O2-induced apoptosis in neonatal
cardiomyocytes. Int J Clin Exp Pathol. 8:5491–5496. 2015.
|
|
35
|
Tong Z, Jiang B, Wu Y, Liu Y, Li Y, Gao M,
Jiang Y, Lv Q and Xiao X: MiR-21 protected cardiomyocytes against
doxorubicin-induced apoptosis by targeting BTG2. Int J Mol Sci.
16:14511–14525. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Boccalini G, Sassoli C, Formigli L, Bani D
and Nistri S: Relaxin protects cardiac muscle cells from
hypoxia/reoxygenation injury: Involvement of the Notch-1 pathway.
FASEB J. 29:239–249. 2015. View Article : Google Scholar
|