Introduction
Hepatic fibrosis, characterized by the excessive
production and deposition of the extracellular matrix (ECM), is a
common pathologic change observed in the progression of various
chronic liver diseases to cirrhosis (1–3).
Transforming growth factor-β1 (TGF-β1), the most potent
profibrogenic cytokine currently known, stimulates the activation
and transformation of hepatic stellate cells (HSCs) into
myofibroblasts producing abnormal and abundant ECM (4,5).
It has been reported that TGF-β1 binds to TGF-β type II receptors
on the cell surface, and then recruits type I receptors; as a
consequence, the type II receptor kinase phosphorylates the type I
receptors and intracellular signal molecules such as Smads.
Phosphorylated (p-)Smad2 and/or p-Smad3 form heteroligomers with
Smad4, which translocate into the nucleus to control the ECM gene
transcription (6–7). On the contrary, blocking TGF-β1
signaling may inhibit collagen production and promote the
degradation of collagen (8,9).
The roles of TGF-β1 in HSCs activation and liver fibrosis have been
intensively studied; however, the transcriptional regulation of
TGF-β1 remains poorly understood.
Cyclic adenosine 3′,5′-monophosphate
(cAMP)-responsive element (CRE) binding protein-1 (CREB-1), a
eukaryotic transcription factor, binds to CRE sites in the
promoters of various cytokines to widely regulate gene
transcription (10,11). The role of CREB-1 in fibrogenesis
remains controversial. Several studies have reported that CREB-1
exerts anti-fibrotic effects, such as in myocardial fibrosis and
pulmonary fibrosis (12,13). On the contrary, CREB-1 facilitated
high glucose induced renal tubulointerstitial fibrosis in diabetic
nephropathy (14). Currently,
there limited research regarding the role of CREB-1 in liver
fibrosis. Our previous study showed that TGF-β3 induced CREB-1
phosphorylation in HSCs, and p-CREB-1 bound to the TGF-β3 promoter
and enhanced its transcriptional activity, suggesting that p-CREB-1
was involved in TGF-β3 auto-regulation (15). However, the effects of p-CREB-1 in
TGF-β1-induced liver fibrosis remain unclear.
Herein, we reported that p-CREB-1 expression was
significantly upregulated in the fibrotic liver tissue of rats as
well as in HSCs treated with exogenous TGF-β1. We also found that
the overexpression of p-CREB-1 increased TGF-β1 expression and
auto-induction, contributing to hepatic fibrogenesis by directly
binding to the CRE site within the TGF-β1 promoter to enhance the
transcriptional activity of TGF-β1.
Materials and methods
Cell culture and treatment
The rat HSC line HSC-T6 was obtained from the Cell
Bank of the Chinese Academy of Sciences (Shanghai, China). The
cells were cultured in Dulbecco's modified Eagle's medium (DMEM)
and supplemented with 10% fetal bovine serum (both from Gibco, New
York, NY, USA) with 5% CO2 at 37°C. Trypsin (2.5%;
Gibco) was used for continuous cell culture, and cells in
logarithmic growth phase were selected for subsequent experiments.
The cells were seeded in 6-well plates for 24 h to ensure 80–90%
confluence, and then treated with or without exogenous TGF-β1
recombinant human protein (10 ng/ml; PeproTech, Rocky Hill, NJ,
USA) for 8 h.
Construction and transfection of
plasmids
The coding sequence of CREB-1 was amplified from rat
liver cDNA by PCR and cloned into XhoI/EcoRI digested
pIRES2-EGFP vector (FulenGen, Co., Ltd, Guangzhou, China) to
generate a CREB-1 overexpression plasmid (pIRES2-CREB-1). The
primers were as follows: 5′-AGACTCGAGATGACCATGGACTCTG-3′
(forward) and 5′-ACGGAATTCTCCCAAATTAATCTGAC-3′
(reverse). The underlined nucleotide sequences represent the
restriction enzyme cutting sites. For sh-CREB-1 plasmid
construction, an siRNA sequence targeting the 877–895 sequence of
rat CREB-1 mRNA (5′-GAAGAAGCAGC ACGAAAGA-3′) was synthesized and
cloned into a pGenesil-1.1 plasmid (Genesil Biotechnology Co.,
Ltd., Wuhan, China) at the BamHI/HindIII sites.
Plasmid transfection was performed using
Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA)
according to the manufacturer's instructions. Briefly, the HSCs
were seeded in 6-well plates and allowed to reach 70–80%
confluence. For each well, transfection was performed using a
mixture containing 250 µl Opti-MEM, with 4 µg plasmid
and 10 µl Lipofectamine 2000. Following transfection for 6
h, the culture medium was replaced with fresh DMEM.
Animals
All experimental protocols and animal maintenance
procedures in this study were approved by the Ethics Review
Committee of the Experimental Animal Center of Tongji Medical
College at the Huazhong University of Science and Technology
(Wuhan, China). Male Sprague-Dawley rats (n=40) weighing 180–200 g
were purchased from the Animal Experiment Center of Wuhan
University and maintained under a 12-h light/dark cycle at a
constant temperature of 20–22°C. The rats were randomly allocated
to one of four treatment groups (n=40 for each group): i) normal
group, ii) model group, iii) Lv-NC group and iv) Lv-CREB-1 group.
At the beginning of the 2nd week, the rats in the Lv-NC and
Lv-CREB-1 groups were tail-vein injected with 5×108 TU
Lv-NC or Lv-CREB-1 (Biowit Technologies, Shenzhen, China), and the
rats in the normal and model groups were injected with an
equivalent dose of sterilized saline water. From the 3rd to the
10th week, a subcutaneous injection of 40% tetrachoromethane
(CCl4; Alpha Biotechnology Co., Ltd., Wuhan, China)
olive oil solution twice a week was administered to establish a rat
model of hepatic fibrosis (initial dose 5 ml/kg body weight,
thereafter 3 ml/kg), and equivalent olive oil was injected into the
rats of normal group as control. After 8 weeks of CCl4
injections, the rats were intraperitoneally anesthetized with
pentobarbital sodium (30 mg/kg body weight). Blood samples were
collected from the heart, and serum was then separated by
centrifugation. The rats were then sacrificed by exsanguination,
and the liver tissues were removed and fixed with 4%
phosphate-buffered paraformaldehyde for Masson's staining (Goodbio
Technology Co., Ltd., Wuhan, China), or frozen for the extraction
of RNA and protein. Serum TGF-β1 levels were detected using
enzyme-linked immunosorbent assay (ELISA) kits (Dakewe, Beijing,
China) according to the manufacturer's instructions.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen) according to the manufacturer's instructions. Total
cDNA was prepared by reverse transcription using PrimeScript™ RT
Master mix (Takara, Otsu, Japan). RT-qPCR was then performed using
SYBR-Green PCR Master mix (Takara) in a 20 µl volume
containing 10 µl SYBR-Green I, 2 µl cDNA, 0.8
µl forward primer, 0.8 µl reverse primer and 6.4
µl ddH2O. All PCRs were performed on an ABI
StepOne™ (Applied Biosystems, Foster City, CA, USA) under the
following conditions: 95°C for 10 min, followed by 40 cycles of
95°C for 5 sec and 60°C for 1 min. The reactions were performed in
triplicate with GAPDH as the internal control and the relative
expression levels were calculated using the 2−ΔΔCt
method. The specific primers were as follows: TGF-β1 forward,
5′-CACCGCGTACCAAATGAAGA-3′ and TGF-β1 reverse,
5′-TGGTGCCCTCTGAAATGAAAG-3′; collagen I forward,
5′-GGCGAGTGCTGTCCTTTCTG-3′ and collagen I reverse,
5′-GGGTCCCTCGACTCCTATGAC-3′; CREB-1 forward,
5′-CCAAACTAGCAGTGGGCAGTATATT-3′ and CREB-1 reverse,
5′-GGTACCATTGTTAGCCAGCTGTATT-3′; and GAPDH forward,
5′-GTATGACTCTACCCACGGCAAGT-3′ and GAPDH reverse, 5′-TTCCCGTT
GATGACCAGCTT-3′.
Western blot analysis
Proteins were extracted using RIPA lysis buffer
(Beyotime Institute of Biotechnology, Jiangsu, China), and then
quantified using a bicinchoninic acid kit (Applygen Technologies,
Inc., Beijing, China). After separation by 10% sodium dodecyl
sulfate (also known as sodium lauryl sulfate)-polyacrylamide gel
electrophoresis, the proteins were transferred onto a PVDF membrane
(Millipore, Billerica, MA, USA). The membrane was blocked with 8%
skim milk powder in TBST for 1 h at room temperature, and then
incubated with rabbit anti-rat primary antibodies CREB-1 (1:1,000,
cat. no. 9197) and phospho-CREB-1 Ser133 (1:1,000, cat. no. 9198)
(both from Cell Signaling Technology, Danvers, MA, USA); TGF-β1
(1:1,000, ab92486) and collagen I (1:5,000, ab34710) (both from
Abcam, Cambridge, UK); and GAPDH (1:5,000, cat. no. 5174; Cell
Signaling Technology) at 4°C overnight. After washing thrice with
1% TBST for 10 min, the horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody (1:5,000, cat. no. 7074; Cell
Signaling Technology) was added and the membrane was incubated for
1 h at room temperature. Finally, the proteins were detected using
an enhanced chemiluminescence detection system (Thermo Fisher
Scientific, Waltham, MA, USA). The expression of the target
proteins was quantified using AlphaEaseFC software, and the ratios
of target proteins against GAPDH were calculated.
Chromatin immunoprecipitation (ChIP)
assay
The HSCs were seeded in 10 cm cell culture dishes
and allowed to reach 60–70% confluence, and were transfected with
either a pIRES2-CREB-1 or pIRES2-EGFP vector. Forty-eight hours
after transfection, a ChIP assay was performed using an EpiQuik™
ChIP kit (Epigentek, Farmingdale, NY, USA) according to the
manufacturer's instructions. The antibodies used for ChIP were
anti-p-CREB-1 (Cell Signaling Technology) and normal rabbit IgG.
Briefly, the cells were trypsinized and cross-linked in 1%
formaldehyde at room temperature for 10 min. After washing with
phosphate-buffered saline, the cells were lysed and the chromatin
was fragmented to approximately 200–1,000 bp by sonication. The
chromatin was then incubated in the antibody-coating microwells.
After washing, the immunoprecipitated DNA was retrieved from the
microwell and the purified DNA was analyzed by qPCR. The amplified
region was 183 bp near the transcription start site of the TGF-β1
gene, and a distant region (164 bp) was used as a negative control.
The primers for ChIP were as follows: CRE site,
5′-CGGACCTGCTGGCAATAG-3′ (forward) and 5′-CCCAAGGAAAGGTAGGTGATAG-3′
(reverse); distant region, 5′-GGGAGGACACAAATACGACC-3′ (forward) and
5′-ACGGGACTAAAACAGGGAGT-3′ (reverse).
Reporter plasmid construction
To construct a TGF-β1 promoter reporter plasmid, a
2209 bp fragment of the TGF-β1 gene promoter (−2168/+41 relative to
the transcription start site) was amplified by PCR and cloned into
the pGL3-basic plasmid (Promega, Madison, WI, USA) between the
KpnI and HindIII sites, generating pGL3-2209. The
primers were as follows: 5′-GAAGGTACCACTGGGGAAGAAAGGAAAC-3′
(forward) and 5′-GCAAAGCTTCCAAGGAAAGGTAGGTGAT-3′
(reverse). The pGL3-2209 plasmid was used as template for
generating several deleted TGF-β1 promoter reporter plasmids using
the same reverse primer. The forward primers for the deleted
reporter plasmids were as follows: pGL3-1710,
5′-ACTGGTACCTAGCAGCCCAGGCACTC-3′; pGL3-1078,
5′-CTAGGTACCGACCCTGTTTTCTCACGA-3′; pGL3-448,
5′-AGTGGTACCTGCAAGTCAGAGACGGG-3′; and pGL3-66,
5′-AGAGGTACCCCGCGACTCCTGCTG-3′.
To generate a pGL3-mut plasmid, the predicted CREB-1
binding site (CGTCA) located in the TGF-β1 promoter was converted
to 'AATTC' by PCR using pGL3-2209 as a template. The primers were
as follows: 5′-ACTCTGGTGTCAGAGAATTCCCGCGACTCCTGCTGC-3′ (forward)
and 5′-CAGCAGGAGTCGCGGGAATTCTCTGACACCAGAGTG-3′ (reverse).
Luciferase reporter assay
The 293T cells (purchased from the Cell Bank of the
Chinese Academy of Sciences) were seeded in 96-well plates and
allowed to reach 70–80% confluence. Then, 150 ng of either
pIRES2-CREB-1 plasmid or pIRES2-EGFP plasmid, 50 ng of constructed
reporter plasmid and 5 ng of pRL-TK Renilla plasmid were
cotransfected into the 293T cells using Lipofectamine 2000 reagent
(Invitrogen) according to the manufacturer's instructions.
Forty-eight hours after transfection, the luciferase activity was
measured using the Dual Luciferase Reporter assay kit
(Promega).
Statistics analysis
Data were analyzed by the independent-samples t-test
or one-way analysis of variance using SPSS 17.0 software (IBM
Corp., Armonk, NY, USA). Correlation analysis was performed using
Pearson's correlation test. A p-value <0.05 indicated a
statistically significant difference.
Results
p-CREB-1 is significantly upregulated in
rat fibrotic liver tissues
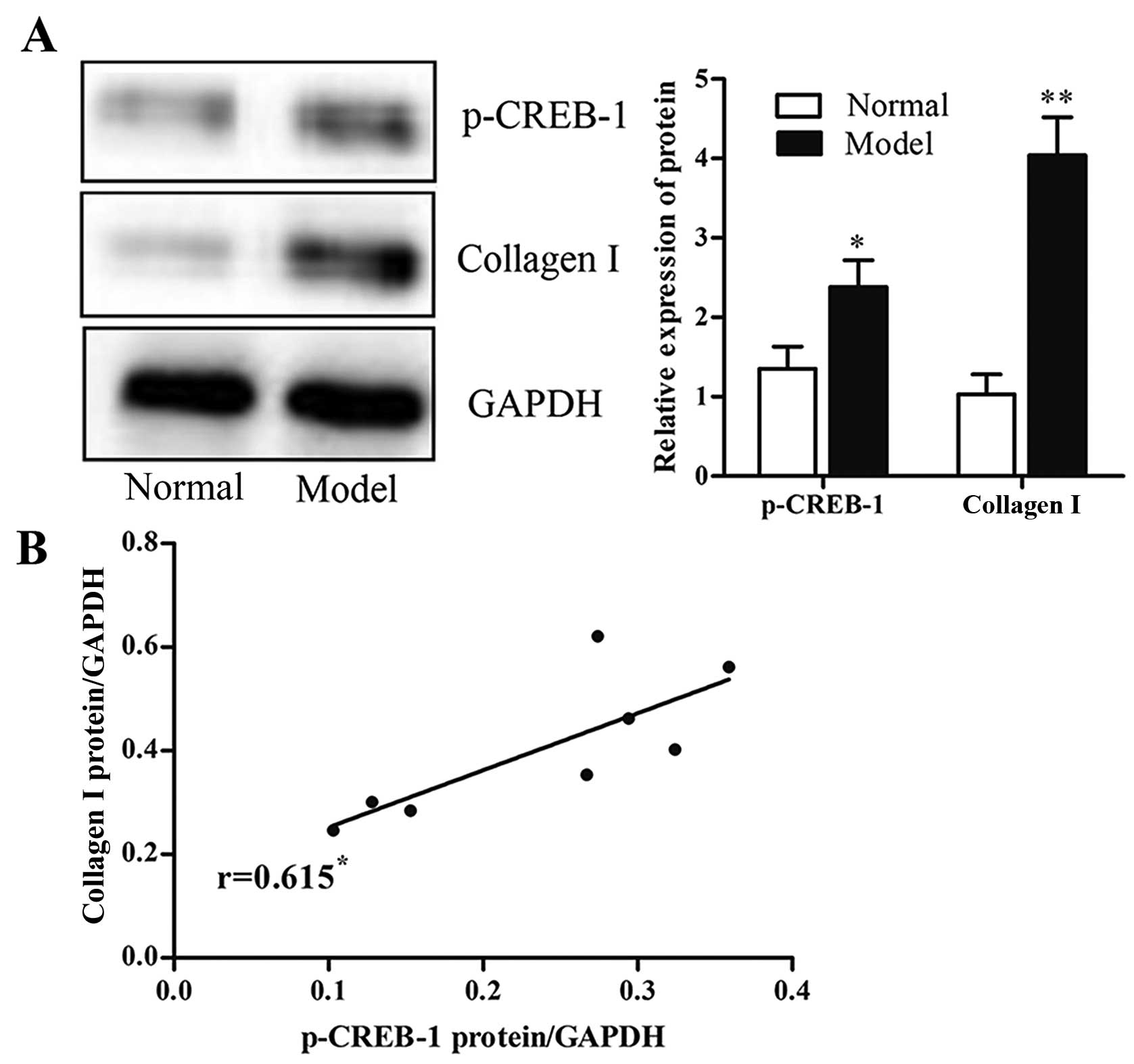
To examine the potential role of p-CREB-1 in liver
fibrogenesis, we detected the expression of p-CREB-1 in rat liver
tissues with CCl4-induced fibrosis, and the results
showed that rats with liver fibrosis had higher p-CREB-1 expression
than those without liver fibrosis (1.8-fold, p<0.05) (Fig. 1A). Additionally, there was a
positive correlation between p-CREB-1 expression and collagen
contents in a rat model of liver fibrosis (Fig. 1B). These results suggested that
p-CREB-1 may play a role in liver fibrosis.
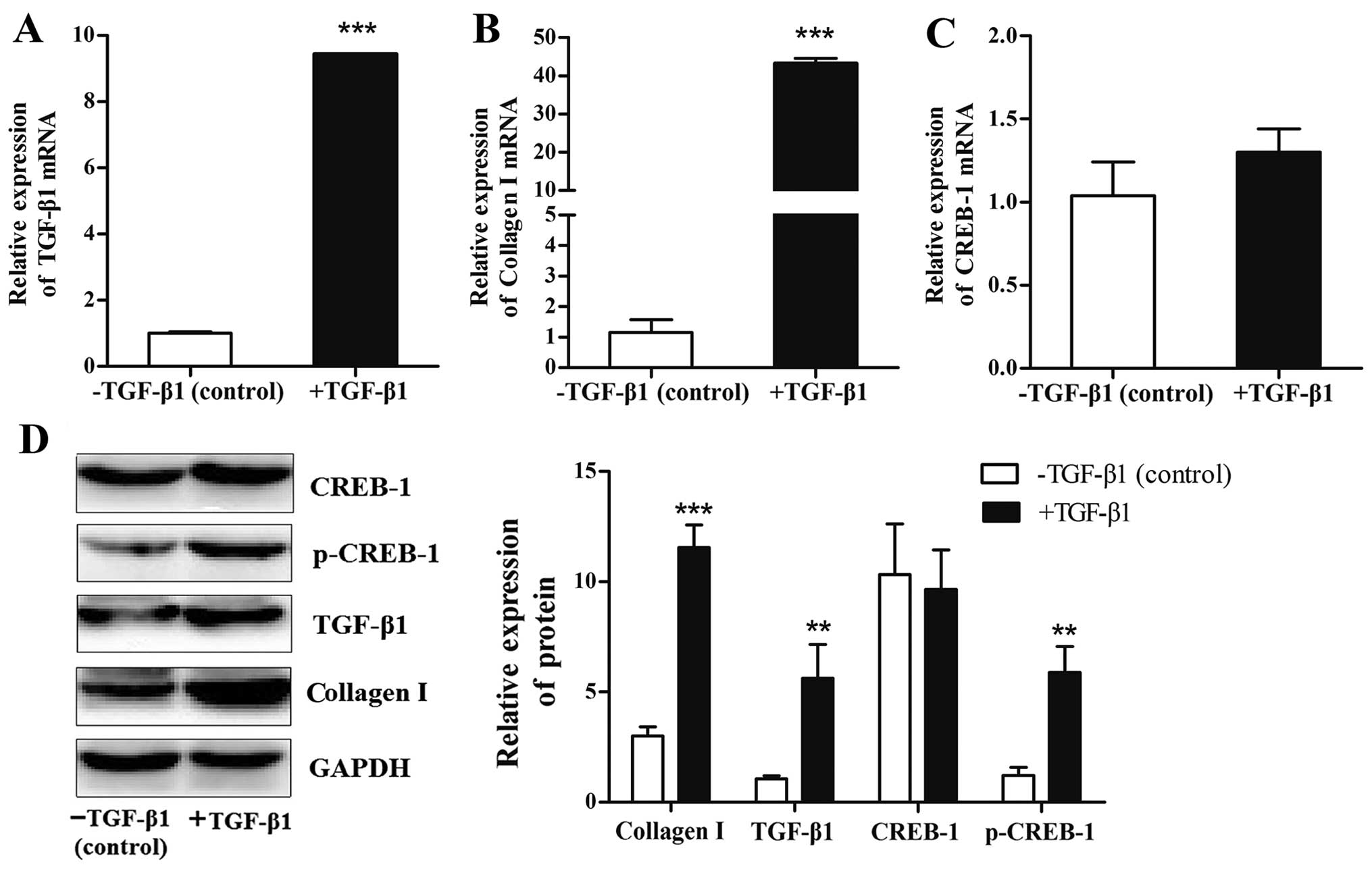
Expression of p-CREB-1 is induced by
exogenous TGF-β1 in HSCs
We then examined the regulatory mechanism
responsible for the effects of p-CREB-1 in liver fibrosis. It is
well known that TGF-β1 may be induced by CCl4 and plays
a crucial role in rat liver fibrogenesis (16), thus it is of interest to explore
the association between p-CREB-1 and TGF-β1. Herein, we used
recombinant human TGF-β1 protein (10 ng/ml) to activate the HSCs,
and found that the protein expression levels of p-CREB-1 were
significantly increased in the TGF-β1-treated HSCs compared with
the control (p<0.01) (Fig.
2D), and this was accompanied by increases in endogenous TGF-β1
mRNA and protein (9.4-fold, p<0.001; and 4.0-fold, p<0.01;
respectively) (Fig. 2A and D).
Additionally, the TGF-β1-treated HSCs also showed notable increases
in the mRNA and protein expression of collagen I (43.3-fold,
p<0.001; and 3.5-fold, p<0.001; respectively), compared with
the HSCs treated with TGF-β1 diluents (Fig. 2B and D). However, the mRNA and
protein expression of CREB-1 was unaffected by TGF-β1 (Fig. 2C and D).
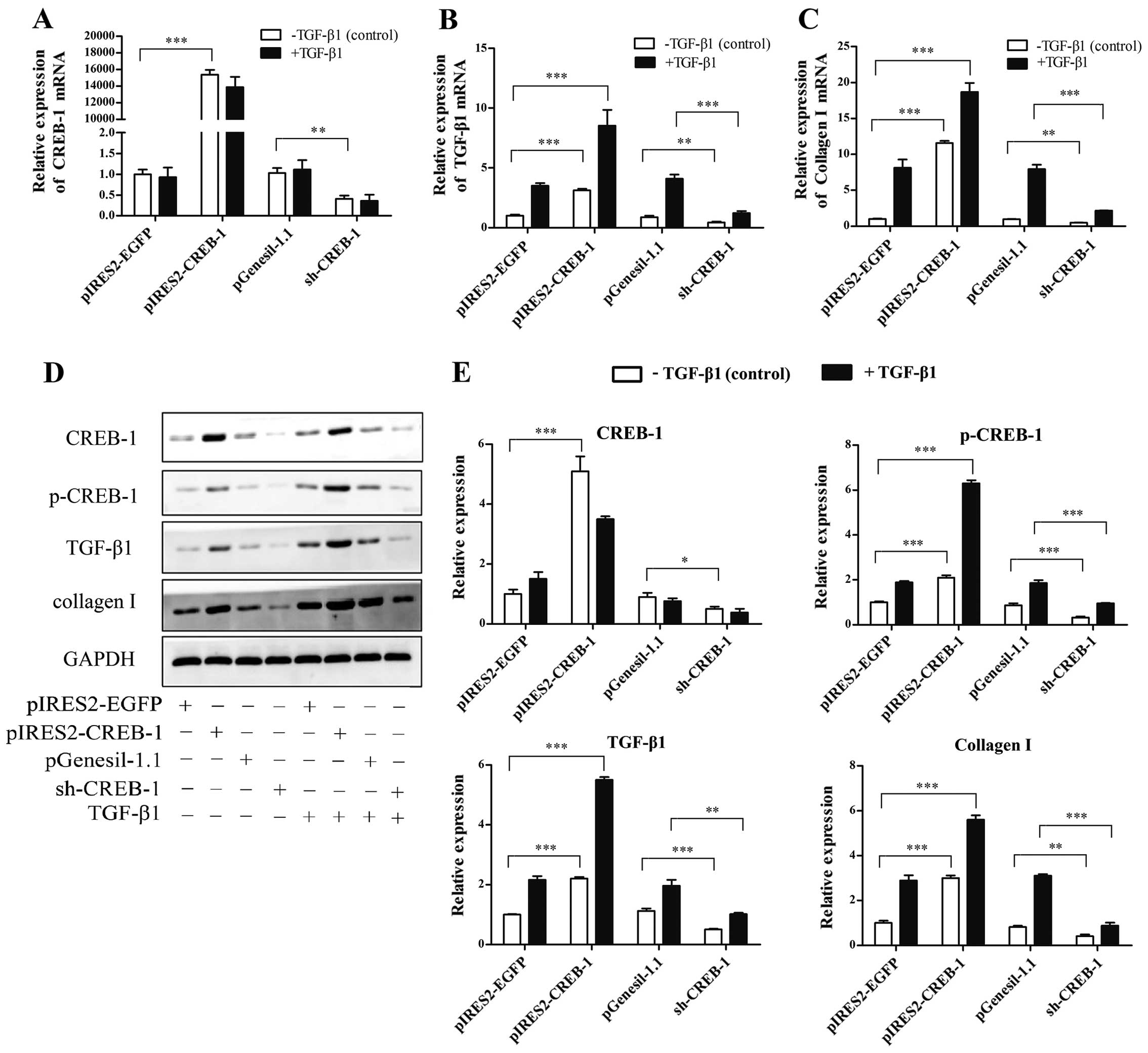
p-CREB-1 increases TGF-β1 expression and
is required for TGF-β1 auto-induction in HSCs
The above result indicates that the TGF-β1 induction
of p-CREB-1 is accompanied by an induction of endogenous TGF-β1. It
is of interest to determine whether p-CREB-1 promotes TGF-β1
expression and is required for TGF-β1 auto-induction. Herein, we
performed loss- and gain-of-function studies. The HSCs were
transfected with the pIRES2-CREB-1 vector in order to upregulate
CREB-1, with or without exogenous TGF-β1 treatment. As shown in
Fig. 3, there were significant
increases in the mRNA and protein expression of TGF-β1 and collagen
I in the pIRES2-CREB-1-transfected HSCs (TGF-β1 mRNA, 3.1-fold,
p<0.001; TGF-β1 protein, 2.2-fold, p<0.001; collagen I mRNA,
11.5-fold, p<0.001; collagen I protein, 3.0-fold, p<0.01), as
well as in the expression of p-CREB-1 which was also upregulated
compared with the control; and these increases were more
significant when the HSCs were additionally treated with exogenous
TGF-β1 (TGF-β1 mRNA, 8.5-fold, p<0.001; TGF-β1 protein,
3.5-fold, p<0.001; collagen I mRNA, 18.2-fold, p<0.001;
collagen I protein: 5.6-fold, p<0.001), suggesting that the
increased TGF-β1 expression induced by exogenous TGF-β1 was further
enhanced through the upregulation of p-CREB-1 in HSCs.
Moreover, we used a sh-CREB-1 plasmid to
downregulate CREB-1 expression in the HSCs, and examined whether a
reduction of p-CREB-1 suppressed TGF-β1 expression and
auto-induction. As shown in Fig.
3, sh-CREB-1 significantly repressed the expression of TGF-β1
and collagen I (TGF-β1 mRNA, 0.5-fold, p<0.01; TGF-β1 protein,
0.4-fold, p<0.001; collagen I mRNA, 0.4-fold, p<0.01;
collagen I protein, 0.3-fold, p<0.01), compared with
pGenesil-1.1. Furthermore, a reduction of p-CREB-1 decreased the
exogenous TGF-β1 induction of endogenous TGF-β1 and collagen I
expression. The above findings indicated that p-CREB-1 plays a
central role in TGF-β1 expression and auto-regulation.
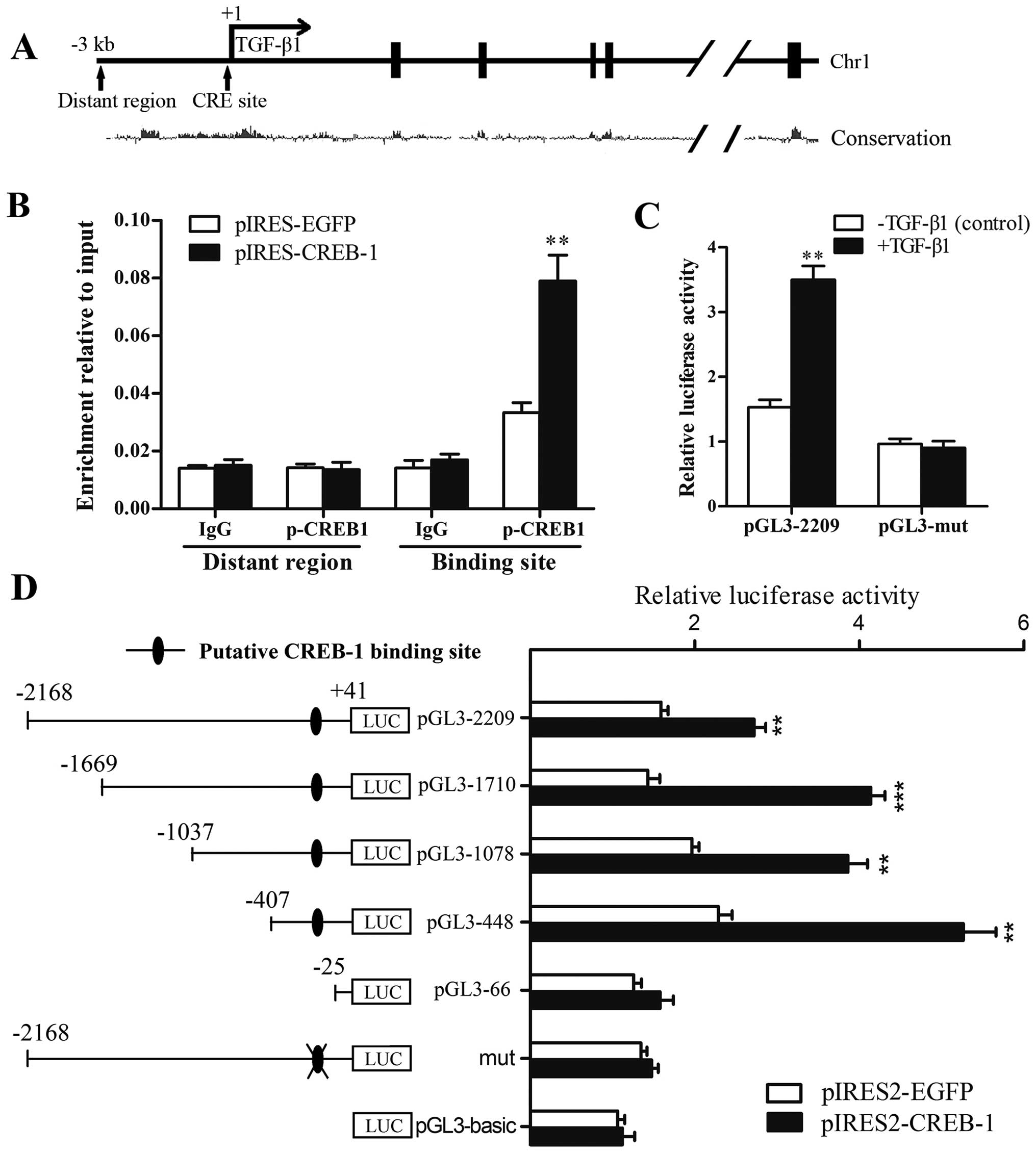
p-CREB-1 transactivates the TGF-β1
promoter through directly binding to the CRE site within the
promoter
To confirm whether the transcription factor p-CREB-1
binds to the TGF-β1 promoter, we performed ChIP assays. As shown in
Fig. 4B, we found a recruitment
of p-CREB-1 to the CRE site in the TGF-β1 promoter. By contrast,
when the HSCs were transfected with pIRES2-CREB-1 vector, the
recruitment was significantly enhanced (2.3-fold) compared with the
control, suggesting that p-CREB-1 directly bind to the TGF-β1
promoter in HSCs.
To determine whether the upregulation of p-CREB-1
was responsible for the increased transcriptional activity of the
TGF-β1 promoter, a series of 5′-deletion mutants were constructed
and transfected respectively into 293T cells, which were
cotransfected with the pIRES2-CREB-1 plasmid or pIRES2-EGFP
plasmid, and pRL-TK Renilla plasmid. Reporter gene assays
were performed and showed that the luciferase activities of
pGL3-2209, pGL3-1710, pGL3-1078 and pGL3-448 reporter plasmids were
significantly augmented in cells transfected with the pIRES2-CREB-1
plasmid, whereas that of the pGL3-66 plasmid was not markedly
increased compared with the control. These results suggested that
TGF-β1 transcription was activated by p-CREB-1, and the region
between −407 and −25 was most important for the transactivation by
p-CREB-1 (Fig. 4D). Furthermore,
5′-CTGCA-3′ sequences in this region were identified as the
putative CREB-1 binding site, and converted to 5′-AATTC-3′ in the
pGL3-mut plasmid. Reporter gene assays showed that the luciferase
activity of the pGL3-mut plasmid was not markedly increased by
CREB-1 overexpression, indicating that mutation of the site
completely abrogated the transcriptional activation of the TGF-β1
reporter by p-CREB-1 (Fig. 4D).
Moreover, as shown in Fig. 4C,
exogenous TGF-β1 increased the luciferase activity of pGL3-2209
(2.3-fold higher than control), whereas pGL3-mut was unaffected.
These results suggest that the CRE site is essential for the
transactivation effects of p-CREB-1 on the TGF-β1 promoter.
p-CREB-1 upregulates TGF-β1 expression
and promotes liver fibrogenesis in rats
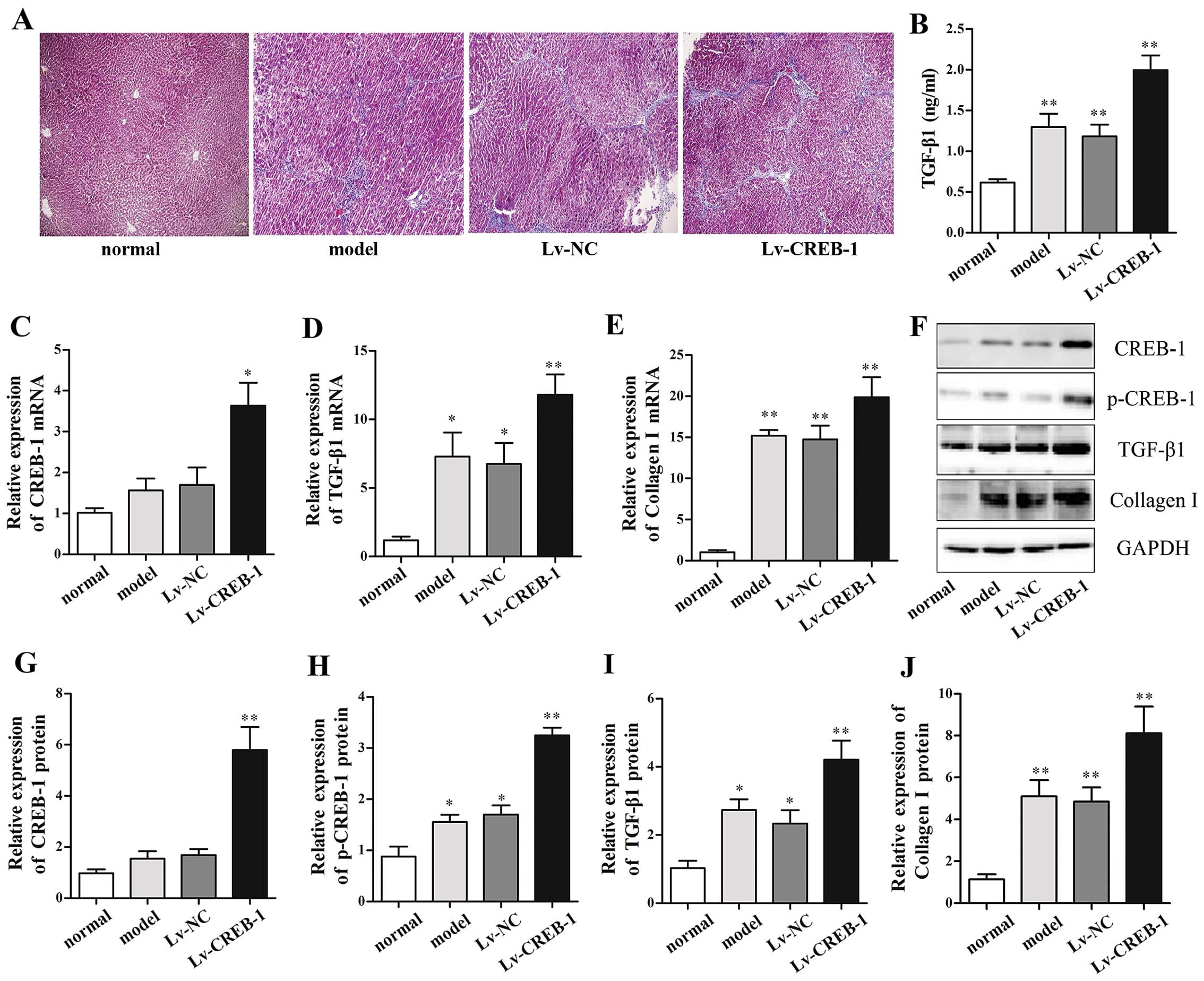
All rats survived in the normal control group
whereas 8, 9 and 7 rats survived in the model group, Lv-NC group
and Lv-CREB-1 group, respectively, until the rats were sacrificed.
Masson's staining revealed that the content of collagen in liver
samples from the model group is higher than that in the normal
control group and lower than that in Lv-CREB-1 group (Fig. 5A), suggesting that overexpression
of CREB-1 promoted CCl4-induced hepatic fibrogenesis in
rats.
Furthermore, we determined the hepatic expression of
CREB-1, p-CREB-1, TGF-β1 and collagen I by RT-qPCR and/or western
blot analysis, and the results indicated that CREB-1 expression was
markedly elevated in the Lv-CREB-1 group (Fig. 5C, F and G). p-CREB-1 expression
was 3.7-fold and 1.8-fold upregulated in the Lv-CREB-1 group and
the model group, compared with the normal control group (p<0.01
and p<0.05, respectively) (Fig. 5F
and H). Moreover, the mRNA and protein expression of TGF-β1 in
the model group were also significantly increased compared with the
normal control, and the Lv-CREB-1 groups exhibited a more
significant increase (Fig. 5D, F and
I). Serum TGF-β1 levels detected by ELISA displayed similar
changes among these groups (Fig.
5B). Consistent with these findings, the mRNA and protein
expression of collagen I were also markedly increased in the
Lv-CREB-1 group (Fig. 5E, F and
J). These results suggest that p-CREB-1 also increases the
expression of TGF-β1 and collagen I in vivo.
Discussion
CREB-1 is an important transcription factor in gene
regulation which responds to multiple extracellular signals
(17), and its transcriptional
activity is positively regulated by the phosphorylation of a Ser
residue, Ser133 (10).
Non-phosphorylated CREB-1 is not capable of activating gene
transcription. Once phosphorylated, CREB-1 translocates into the
nucleus and binds to target DNA sequences, and then p-CREB-1
interacts with CREB-binding protein (CBP) to initiate the
transcription of CREB-1-responsive genes (18). In the present study, we examined
the expression level of activated CREB-1 and showed that p-CREB-1
expression was markedly upregulated in a rat model of
CCl4-induced liver fibrosis, indicating that p-CREB-1
may play an important role in liver fibrogenesis. Our study is
consistent with a recent study, which reported a similar finding;
namely that CREB phosphorylation was significantly increased in
alcoholic liver fibrosis (19).
Furthermore, we examined the underlying regulatory mechanism as
well as the role of upregulated p-CREB-1 in liver fibrosis. It is
well known that TGF-β1 is a potent cytokine which promotes the
activation of HSCs and liver fibrogenesis, and is highly expressed
in activated HSCs and CCl4-induced hepatic fibrosis
(16,20,21). Herein, we explored the association
between p-CREB-1 and TGF-β1 in liver fibrosis. Firstly, we
detemined whether p-CREB-1 was induced by TGF-β1. We used TGF-β1
recombinant protein to activate the HSCs, and demonstrated that
TGF-β1 induced the protein expression of p-CREB-1 in HSCs.
Similarly, another study by Chin et al reported that TGF-β1
promoted CREB-1 phosphorylation through the activation of
mitogen-activated protein kinase (MAPK) in neurons, resulting in
the enhanced excitability of neurons (22). Jang et al demonstrated that
TGF-β1 induced CREB-1 phosphorylation in macrophages (23). It appears that p-CREB-1 induced by
TGF-β1 represents the common mechanism responsible for CREB-1
activation. Moreover, we found that the TGF-β1 induction of
p-CREB-1 was accompanied by an auto-induction of TGF-β1. This led
us to explore the potential functional role of p-CREB-1 in liver
fibrosis; whether p-CREB-1 is capable of promoting TGF-β1
expression.
It has been previously established that TGF-β1
functions as a potent pro-fibrotic factor. However, the regulatory
mechanism responsible for TGF-β1 expression in liver fibrosis
remains poorly understood. Herein, the loss- and gain-of-function
studies showed that p-CREB-1 upregulated TGF-β1 expression and was
required for TGF-β1 auto-induction in HSCs. Previous studies
demonstrated that TGF-β1 expression may be regulated by several
transcription factors such as AP-1, SP-1, nuclear factor (NF)-κB,
Kruppel-like factor 4 (gut) (KLF4) and signal transducer and
activator of transcription 3 (STAT3) in various experimental model
systems including liver fibrosis (24-27). In the present study, we reported
that another transcription factor, p-CREB-1, was capable of
transactivating TGF-β1 expression. Moreover, we identified the
region between −407 and −25 was crucial for the transactivation of
TGF-β1, which was consistent with the aforementioned studies.
Additionally, we also found that the overexpression of p-CREB-1
promoted hepatic fibrogenesis through the transactivation of TGF-β1
expression in rats. Our study is consistent with the findings of a
recent study, which reported that the expression of p-CREB-1 was
downregulated by caffeine through its blockade of adenosine A2A
receptor and the cAMP/PKA/CREB signaling pathway, thereby
inhibiting the activation of HSCs and liver fibrogenesis (19). Taken together, our findings
provide evidence for the pro-fibrotic role of p-CREB-1 in liver
fibrogenesis.
In the present study, we found that p-CREB-1
expression was significantly upregulated in fibrotic liver tissues
in rats, as well as in HSCs treated with exogenous TGF-β1. We also
found that p-CREB-1 increased TGF-β1 expression and auto-induction,
contributing to hepatic fibrogenesis by directly binding to the CRE
site within the TGF-β1 promoter to enhance the transcriptional
activity of TGF-β1. Our study characterizes p-CREB-1 as a
profibrogenic factor in hepatic fibrosis, and provides a novel
insight into the regulatory mechanism responsible for TGF-β1
expression.
Acknowledgments
The authors gratefully acknowledge the financial
support of the National Natural Science Foundation of China (grant
nos. 30871153 and 81370525).
Abbreviations:
|
p-CREB-1
|
phosphorylated cyclic adenosine
3′,5′-monophosphate-responsive element binding protein-1
|
|
TGF-β1
|
transforming growth factor-β1
|
|
HSCs
|
hepatic stellate cells
|
|
CCl4
|
tetrachloromethane
|
|
RT-qPCR
|
reverse transcription
quantitative-polymerase chain reaction
|
|
ChIP
|
chromatin immunoprecipitation
|
References
|
1
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee UE and Friedman SL: Mechanisms of
hepatic fibrogenesis. Best Pract Res Clin Gastroenterol.
25:195–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Trautwein C, Friedman SL, Schuppan D and
Pinzani M: Hepatic fibrosis: concept to treatment. J Hepatol.
62(Suppl 1): S15–S24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gressner OA, Rizk MS, Kovalenko E,
Weiskirchen R and Gressner AM: Changing the pathogenetic roadmap of
liver fibrosis? Where did it start; where will it go? J
Gastroenterol Hepatol. 23:1024–1035. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bissell DM, Roulot D and George J:
Transforming growth factor beta and the liver. Hepatology.
34:859–867. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inagaki Y and Okazaki I: Emerging insights
into transforming growth factor beta Smad signal in hepatic
fibrogenesis. Gut. 56:284–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-beta signalling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Friedman SL: Mechanisms of disease:
mechanisms of hepatic fibrosis and therapeutic implications. Nat
Clin Pract Gastroenterol Hepatol. 1:98–105. 2004. View Article : Google Scholar
|
|
9
|
Dooley S, Hamzavi J, Breitkopf K,
Wiercinska E, Said HM, Lorenzen J, Ten Dijke P and Gressner AM:
Smad7 prevents activation of hepatic stellate cells and liver
fibrosis in rats. Gastroenterology. 125:178–191. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang X, Odom DT, Koo SH, Conkright MD,
Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen
E, et al: Genome-wide analysis of cAMP-response element binding
protein occupancy, phosphorylation, and target gene activation in
human tissues. Proc Natl Acad Sci USA. 102:4459–4464. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Altarejos JY and Montminy M: CREB and the
CRTC co-activators: sensors for hormonal and metabolic signals. Nat
Rev Mol Cell Biol. 12:141–151. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chan EC, Dusting GJ, Guo N, Peshavariya
HM, Taylor CJ, Dilley R, Narumiya S and Jiang F: Prostacyclin
receptor suppresses cardiac fibrosis: role of CREB phosphorylation.
J Mol Cell Cardiol. 49:176–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baarsma HA, Engelbertink LH, van Hees LJ,
Menzen MH, Meurs H, Timens W, Postma DS, Kerstjens HA and Gosens R:
Glycogen synthase kinase-3 (GSK-3) regulates TGF-β1-induced
differentiation of pulmonary fibroblasts. Br J Pharmacol.
169:590–603. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Visavadiya NP, Li Y and Wang S: High
glucose upregulates upstream stimulatory factor 2 in human renal
proximal tubular cells through angiotensin II-dependent activation
of CREB. Nephron Exp Nephrol. 117:e62–e70. 2011. View Article : Google Scholar
|
|
15
|
Deng L, Li Y, Huang JM, Zhou G, Qian W and
Xu K: Effects of p-CREB-1 on transforming growth factor-β3
auto-regulation in hepatic stellate cells. J Cell Biochem.
112:1046–1054. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Doh KO, Jung HK, Moon IJ, Kang HG, Park JH
and Park JG: Prevention of CCl4-induced liver cirrhosis by ribbon
antisense to transforming growth factor-β1. Int J Mol Med.
21:33–39. 2008.
|
|
17
|
Wen AY, Sakamoto KM and Miller LS: The
role of the transcription factor CREB in immune function. J
Immunol. 185:6413–6419. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sakamoto KM and Frank DA: CREB in the
pathophysiology of cancer: implications for targeting transcription
factors for cancer therapy. Clin Cancer Res. 15:2583–2587. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Q, Dai X, Yang W, Wang H, Zhao H,
Yang F, Yang Y, Li J and Lv X: Caffeine protects against
alcohol-induced liver fibrosis by dampening the cAMP/PKA/CREB
pathway in rat hepatic stellate cells. Int Immunopharmacol.
25:340–352. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang S, Sun WY, Wu JJ and Wei W: TGF-β
signaling pathway as a pharmacological target in liver diseases.
Pharmacol Res. 85:15–22. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Purps O, Lahme B, Gressner AM,
Meindl-Beinker NM and Dooley S: Loss of TGF-beta dependent growth
control during HSC transdifferentiation. Biochem Biophys Res
Commun. 353:841–847. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chin J, Liu RY, Cleary LJ, Eskin A and
Byrne JH: TGF-beta1-induced long-term changes in neuronal
excitability in aplysia sensory neurons depend on MAPK. J
Neurophysiol. 95:3286–3290. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jang YS, Kim JH, Seo GY and Kim PH: TGF-β1
stimulates mouse macrophages to express APRIL through Smad and
p38MAPK/CREB pathways. Mol Cells. 32:251–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y, Wang Y, Liu Y, Wang N, Qi Y and
Du J: Krüppel-like factor 4 transcriptionally regulates TGF-β1 and
contributes to cardiac myofibroblast differentiation. PLoS One.
8:e634242013. View Article : Google Scholar
|
|
25
|
Lin W, Tsai WL, Shao RX, Wu G, Peng LF,
Barlow LL, Chung WJ, Zhang L, Zhao H, Jang JY and Chung RT:
Hepatitis C virus regulates transforming growth factor beta1
production through the generation of reactive oxygen species in a
nuclear factor kappaB-dependent manner. Gastroenterology.
138:2509–2518. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Presser LD, McRae S and Waris G:
Activation of TGF-β1 promoter by hepatitis C virus-induced AP-1 and
Sp1: role of TGF-β1 in hepatic stellate cell activation and
invasion. PLoS One. 8:e563672013. View Article : Google Scholar
|
|
27
|
Hosui A, Kimura A, Yamaji D, Zhu BM, Na R
and Hennighausen L: Loss of STAT5 causes liver fibrosis and cancer
development through increased TGF-beta and STAT3 activation. J Exp
Med. 206:819–831. 2009. View Article : Google Scholar : PubMed/NCBI
|