Introduction
Endothelial progenitor cells (EPCs) play an
important role in vascular morphogenesis, both during embryonic
development and in postnatal neovascularization, thereby
contributing to tissue maintenance and repair in various
pathological conditions (1,2).
Previous findings have shown that EPCs contribute to blood flow
recovery in ischemic limbs and improve left ventricular function
following myocardial infarction (3,4),
indicating a potential role for EPCs in the cell-based therapeutic
neovascularization of ischemic tissues. However, the numbers and
function of EPCs decline as part of aging-associated senescence;
aged EPCs are less able to contribute to vascular repair and
regeneration and thereby contribute to the increased propensity
toward vascular pathology (5,6).
Therefore, it is critical to further elucidate the mechanism
responsible for the senescence of EPCs in order to provide novel
insights into therapeutic neovascularization.
Oxidized low-density lipoprotein (ox-LDL) is taken
up by EPCs in a receptor-dependent manner wherein it generates
oxidative stress. The level of ox-LDL increases in patients with
coronary artery disease or diabetes and serves as an independent
predictor for future cardiac events in these patients (7,8).
Several studies have shown that ox-LDL functions as an important
factor that adversely affects the growth and bioactivity of EPCs by
triggering apoptosis, inducing cell ossification and inhibiting
endothelialization (9–12). It has also been demonstrated that
ox-LDL also accelerated the senescence of EPCs, leading to cellular
dysfunction (13,14). Lai and Liu showed that
ox-LDL-induced senescence of EPCs was mediated by oxidative stress
and the Akt/hTERT pathway (15).
Visfatin was originally cloned in 1994 as a cytokine
named pre-B cell colony-enhancing factor (PBEF) from a bone marrow
cDNA library based on sequence similarities to cytokines (16). It is now well established that
visfatin is expressed in adipose tissue as well as skeletal muscle,
liver and immune cells. Visfatin has also been found to be produced
within the myocardium localized to cardiomyocytes and fibroblasts,
and within the brain localized to neuronal cells, with particularly
high expression during ischemia. The widespread distribution of
this molecule is indicative of the broad functions of visfatin in
both health and disease. Recently, visfatin has been implicated in
the pathogenesis of various cardiovascular disorders, such as
atherosclerosis and myocardial failure, and in brain ischemia
(16). van der Veer et al
showed that FK866, an antagonist of visfatin, induced premature
senescence in human vascular smooth muscle cells, together with
enhanced resistance to oxidative stress (17). However, the effects of viastatin
on the ox-LDL-induced senescence of EPCs as well as the underlying
mechanism responsible for these effects has not yet been explored,
to the best of our knowledge. In the present study, we report that
visfatin is a senescence-related protein that inhibits the
ox-LDL-induced senescence of EPCs by activating sirtuin 1 (SIRT1)
through the phosphoinositide-3-kinase (PI3K)/Akt/extracellular
signal–regulated kinase (ERK) pathway.
Materials and methods
Cell culture
EPCs were purchased from the American Type Culture
Collection (ATCC; Rockville, MD, USA) and cultured in RPMI-1640
supplemented with 10% fetal bovine serum (FBS) and 100 μg/ml
penicillin/streptomycin (all from Invitrogen, Carlsbad, CA,
USA).
Ox-LDL preparation
The fresh human plasma was collected from patients
at Xiangya Hospital of Central South University (Changsha, China).
Human ox-LDL was isolated from fresh human plasma by sequential
ultracentrifugation, as previously described (18). Briefly, LDL was isolated by
ultracentrifugation at 290,000 × g for 4 h in a sodium bromide
(NaBr) gradient, and the top layer (containing LDL) was collected.
To validate the observations made with copper-oxidized LDL, we
performed additional experiments using L5, an electronegative and
minimally oxidized LDL circulating in patients with
hypercholesterolemia.
Telomeric repeat amplification protocol
(TRAP) assay
For quantitative analysis of telomerase activity,
the TRAP assay, in the which the telomerase reaction product is
amplified by a polymerase chain reaction (PCR), was performed using
the TeloTAGGG PCR ELISAPLUS kit (Roche Molecular
Biochemicals, Mannheim, Germany) according to the manufacturer's
instructions.
Construction of shRNAs
We designed and purchased three different shRNA
duplexes of SIRT1 from Gene Pharma (Shanghai, China). The following
primers were used: shRNA-1 (SIRT1-homo-1075) sense,
5′-CACCGCAACTATACCCAGAACATAGTTCAAGAGACTATGTTCTGGGTATAGTTGCTTTTTTG-3′
and reverse,
5′-GATCCAAAAAAGCAACTATACCCAGAACATAGTCTCTTGAACTATGTTCTGGGTATAGTTGC-3′;
shRNA-2 (SIRT1-homo-1961) sense,
5′-CACCGCTTGATGGTAATCAGTATCTTTCAAGAGAAGATACTGATTACCATCAAGCTTTTTTG-3′
and reverse,
5′-GATCCAAAAAAGCTTGATGGTAATCAGTATCTTCTCTTGAAAGATACTGATTACCATCAAGC-3′;
shRNA-3 (SIRT1-homo- 2214) sense,
5′-CACCGGAGATGATCAAGAGGCAATTTCAAGAGAATTGCCTCTTGATCATCTCCTTTTTTG-3
and reverse,
5′-GATCCAAAAAAGGAGATGATCAAGAGGCAATTCTCTTGAAATTGCCTCTTGATCATCTCC-3.
Transfection
Twenty-four hours prior to transfection, the cells
were seeded in a 6-well culture plate. SIRT1 shRNAs were
transfected using Lipofectamine 2000 (Invitrogen) according to the
manufacturer's instructions. Twenty-four or forty-eight hours
later, the EPCs were collected and subjected to further analysis.
Total RNA or protein was extracted from the indicated cells for
analysis. The experiment was peformed in triplicate. More than nine
wells were treated with the same type of shRNA.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from the cells using TRIzol
reagent (Invitrogen) according to the manufacturer's instructions.
qPCR was performed using the StepOne Plus sequence detection system
(Applied Biosystems, Foster City, CA, USA). To ensure the
reproducibility of the results, all the genes were tested in
triplicate. The following primers were used: SIRT1 sense
(5′-TAGCCTTGTCAGATAAGGAAGGA-3′) and antisense
(5′-ACAGCTTCACAGTCAACTTTGT-3′); p53 sense
(5′-ACTTGTCGCTCTTGAAGCTAC-3′) and antisense
(5′-GATGCGGAGAATCTTTGGAACA-3′); AKT1 sense
(5′-AGCGACGTGGCTATTGTGAAG-3′) and antisense
(5′-GCCATCATTCTTGAGGAGGAAGT-3′); PI3K/p85 sense
(5′-ACACCACGGTTTGGACTATGG-3′) and antisense
(5′-GGCTACAGTAGTGGGCTTGG-3′); ERK sense
(5′-ACACCACGGTTTGGACTATGG-3′) and antisense
(5′-GGCTACAGTAGTGGGCTTGG-3′); and β-actin sense,
(5′-CATTAAGGAGAAGCTGTGCT-3′) and antisense
(5′-GTTGAAGGTAGTTTCGTGGA-3′). The cycling conditions were an
initial denaturation for 2 min at 94°C; followed by 40 cycles of 10
sec at 94°C, 30 sec at 55°C, and 30 sec at 72°C.
Western blot analysis
Whole cell extracts were prepared with a cell lysis
reagent (Sigma-Aldrich, St. Louis, MO, USA) according to the
manufacturer's instructions, and then, the protein was quantified
using a BCA assay (Pierce, Rockford, IL, USA). The protein samples
were then separated by SDS-PAGE (10%) and detected by western blot
analysis using polyclonal rabbit anti-PI3K p85 antibody (#4257;
Cell Signaling Technology, Danvers, MA, USA,), rabbit anti-p53
antibody (sc-6243; Santa Cruz Biotechnology, Santa Cruz, CA, USA),
mouse anti-β-galactosidase (Gal) antibody (Cell Signaling
Technology, #2372), rabbit anti-Akt antibody (#9272; Cell Signaling
Technology), rabbit anti-SIRT1 antibody (ab110304; Abcam,
Cambridge, UK), rabbit anti-phosphorylated (p-)Akt(Ser473) antibody
(ab81283; Abcam), rabbit anti-ERK antibody (#4695; Cell Signaling
Technology), rabbit anti-p-ERK antibody (#9101; Cell Signaling
Technology) and monoclonal mouse anti-GAPDH (SC-365062; Santa Cruz
Biotechnology) as a control. Goat anti-rabbit IgG (Pierce)
secondary antibody conjugated to horseradish peroxidase and ECL
detection systems (SuperSignal West Femto; Pierce) were used for
detection.
Statistical analysis
Each experiment was repeated at least three times.
Data are shown as the means ± SD and were analyzed using SPSS 18.0.
Statistical comparisons between groups were analyzed using the
Student's t-test and a two-tailed p<0.05 was considered to
indicate a statistically significant difference.
Results
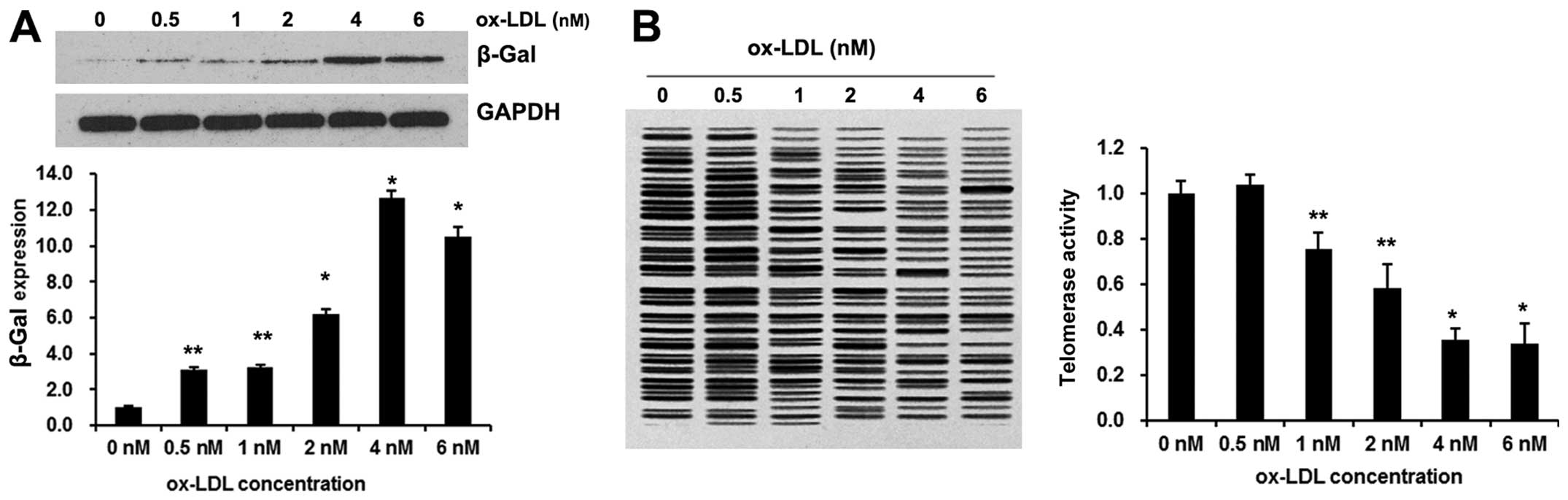
Ox-LDL induces EPC senescence
To assess the onset of senescence, β-Gal expression
and telomerase activity were detected as biochemical markers of
cellular senescence. As shown in Fig.
1A, ox-LDL exposure evidently increased β-Gal expression. The
acceleration of EPC senescence occurred dose dependently, with a
maximal effect achieved at a concentration of 4.0 nM ox-LDL.
Cellular senescence is critically influenced by
telomerase, which elongates telomeres, thereby counteracting the
telomere length reduction induced by each cell division.
Breitschopf et al have demonstrated that pro-atherosclerotic
factors impair telomerase activity in mature endothelial cells
(19). Therefore, we measured
telomerase activity using the TeloTAGGG PCR
ELISAPLUS kit. As demonstrated in Fig. 1B, 4.0 nM ox-LDL significantly
diminished telomerase activity to approximately 60%. These results
clearly demonstrated that 4.0 nM ox-LDL significantly accelerates
the senescence of EPCs.
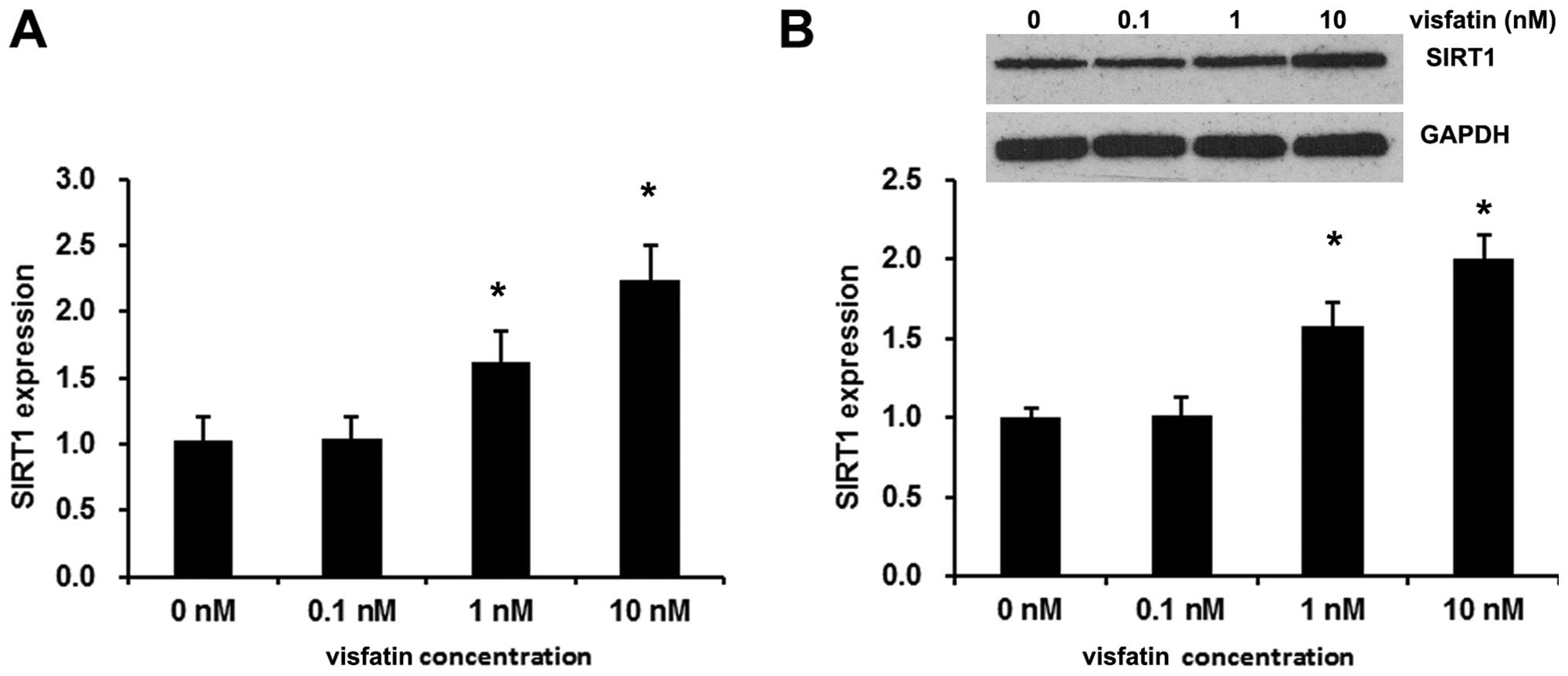
Visfatin induces SIRT1 expression in
EPCs
SIRT1 is an anti-aging molecule and SIRT1 expression
and activity reduction are involved in aging-associated diseases
(20,21). In the present study, exogenetic
visfatin evidently increased SIRT1 expression. The acceleration of
the mRNA and protein expression of SIRT1 occurred dose dependently,
with a maximal effect achieved at a concentration of 10 nM visfatin
(Fig. 2).
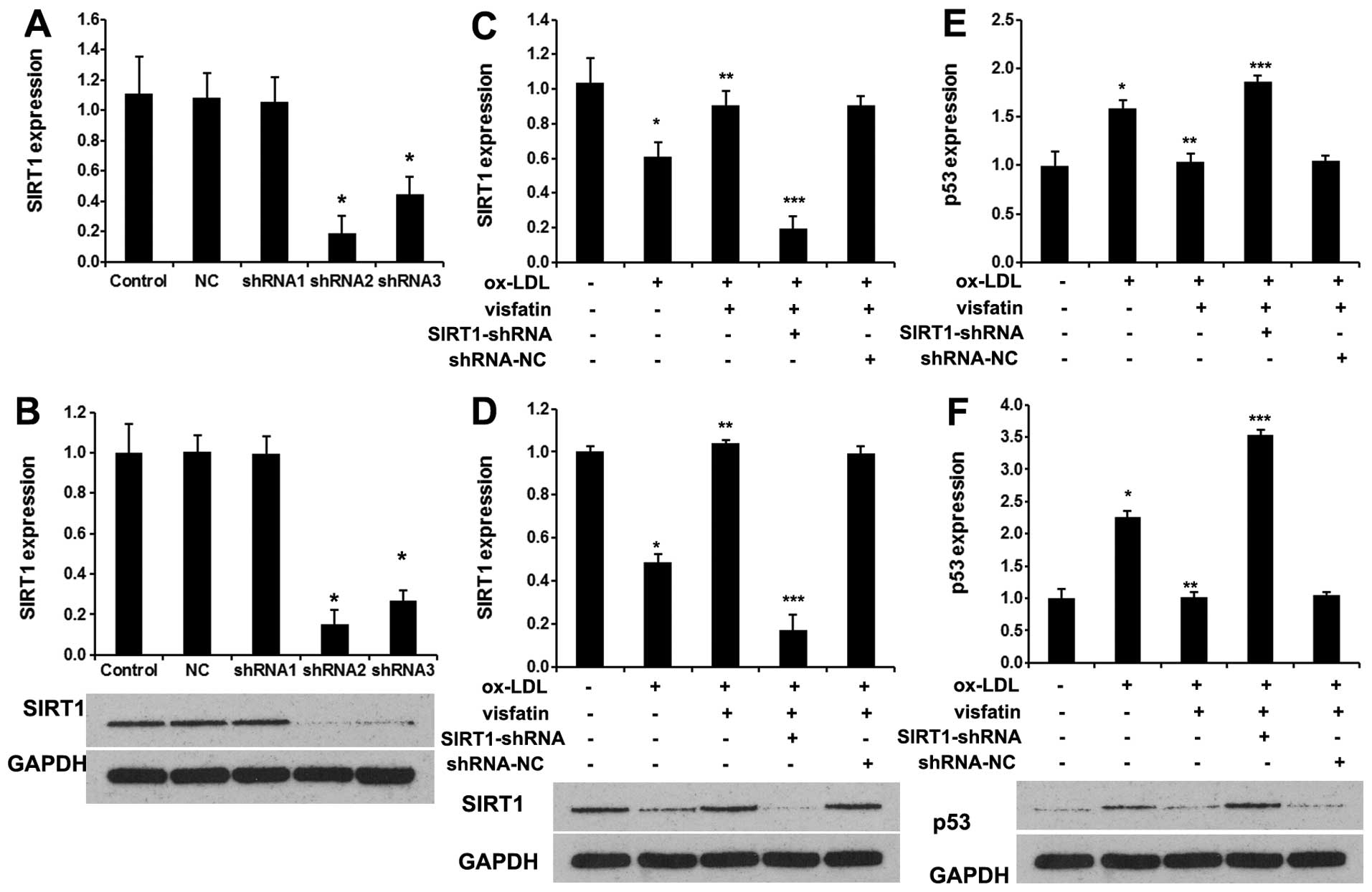
Visfatin abolishes ox-LDL-induced
senescence in EPCs by upregulating SIRT1
Herein, we analyzed the effects of visfatin on SIRT1
and p53 in ox-LDL-stimulated EPCs. Firstly, 3 SIRT1 shRNAs were
used to silence SIRT1 expression. As shown in Fig. 3A, shRNA2 effectively blocked the
expression of SIRT1 mRNA. Western blot analysis revealed marked
reductions of 82.2% with shRNA2, 72.6% with shRNA3, and 3.2% with
shRNA1 in the protein levels of SIRT1 following the transfection of
EPCs with SIRT1 shRNAs, compared with the control (untransfected)
group and NC (negtive control) group (Fig. 3B) (p<0.01). Thus, EPCs
transfected with SIRT1 shRNA2 were used for subsequent experiments.
Next, 10 nM visfatin evidently increased the mRNA (Fig. 3C) and protein (Fig. 3D) expression levels of SIRT1 in
ox-LDL-stimulated EPCs, which were reversed by SIRT1 shRNA.
Moreover, 10 nM visfatin evidently decreased the mRNA (Fig. 3E) and protein (Fig. 3F) expression levels of p53 in
ox-LDL-stimulated EPCs, which were also reversed by SIRT1
shRNA.
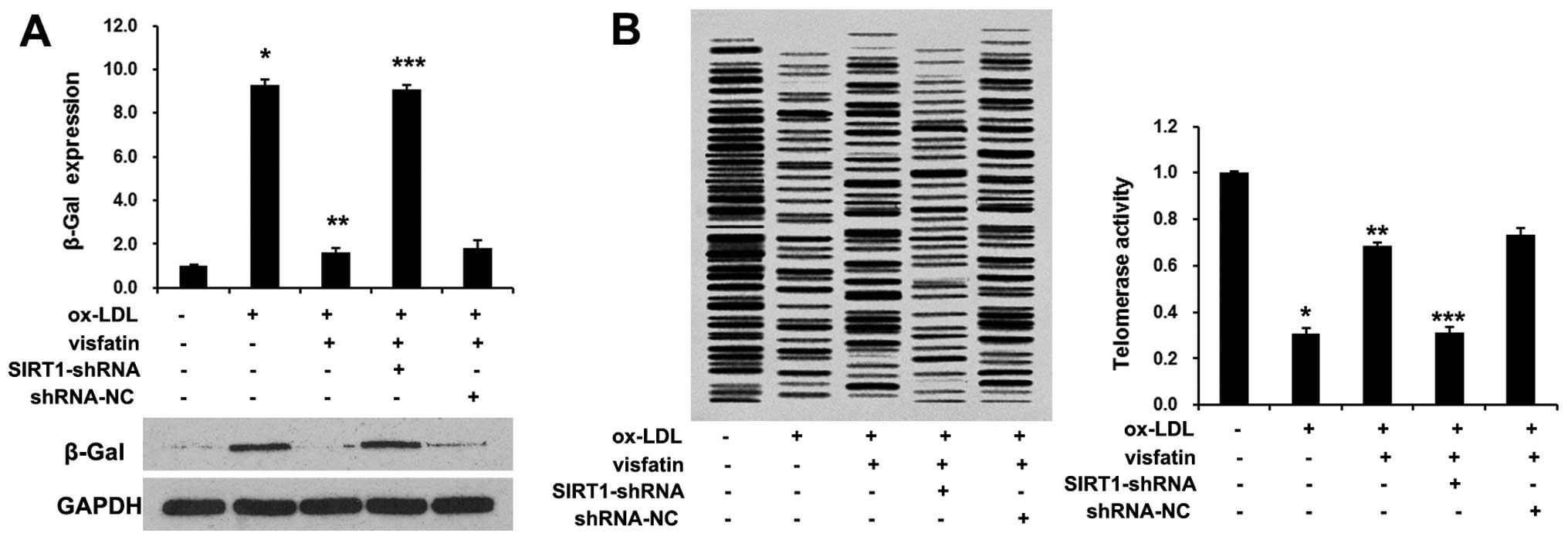
We then analyzed the role of visfatin in the
ox-LDL-induced senescence of EPCs. The treatment of EPCs with
visfatin decreased senescence-associated-β-Gal expression (Fig. 4A). Moreover, visfatin evidently
reversed the ox-LDL-induced decrease in telomerase activity
(Fig. 4B). These results raised
the possibility that visfatin may abolish the ox-LDL-induced
senescence of EPCs. We then analyzed the role of SIRT1 in the
ox-LDL-induced senescence of EPCs. We found that SIRT1-shRNA
abolished the effects of visfatin on the ox-LDL-induced senescence
of EPCs. In cultured mesangial cells, it has been demonstrated that
SIRT1 inhibits oxidative stress-related apoptosis through p53
deacetylation (22). Herein, we
found that visfatin evidently decreased p53 expression in EPCs,
which was reversed by SIRT1-shRNA. In conclusion, these results
indicated that visfatin abolished the ox-LDL-induced senescence of
EPCs by upregulating SIRT1 expression.
Visfatin regulates SIRT1 expression
through the PI3K/Akt/ERK pathway
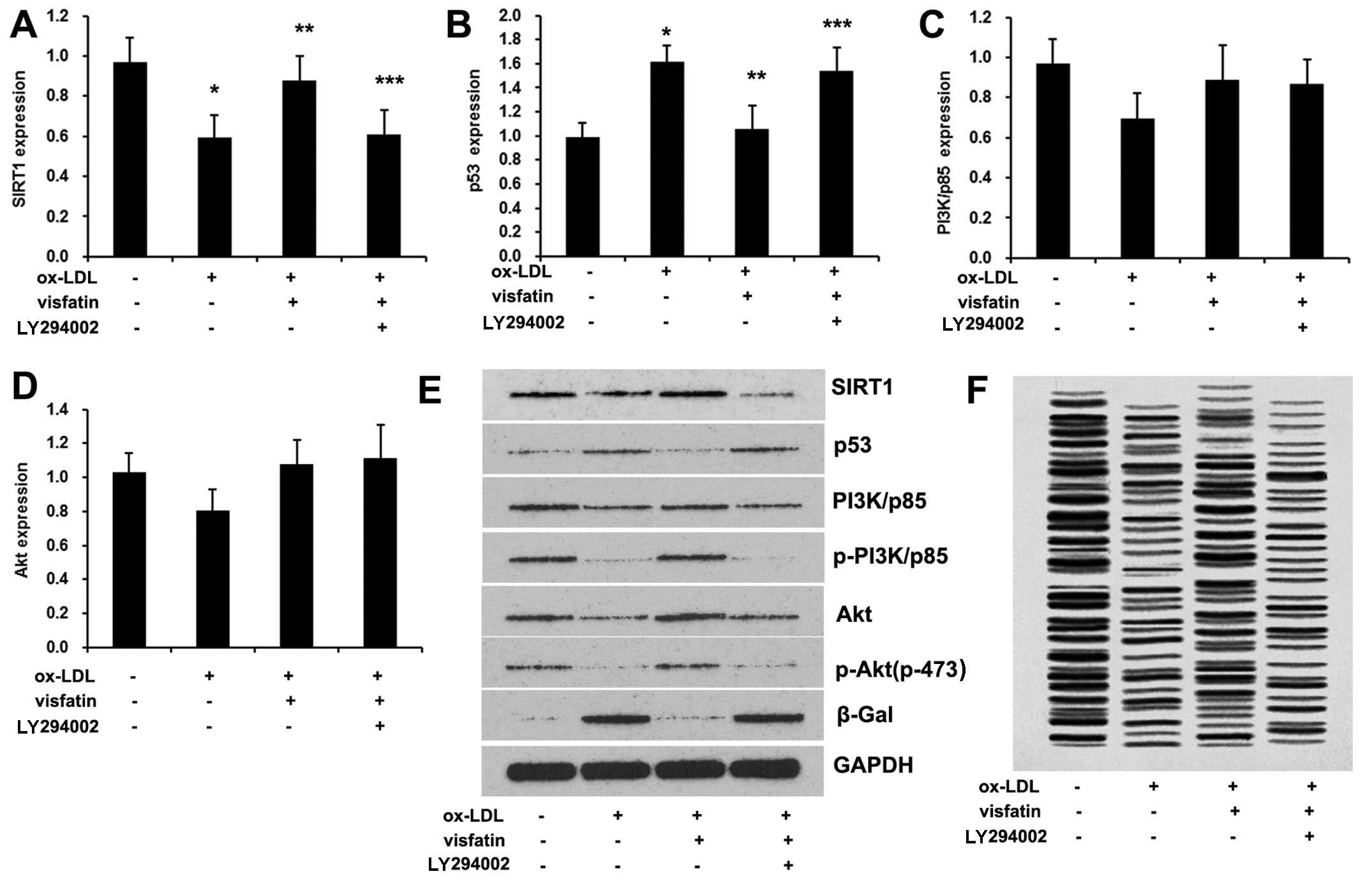
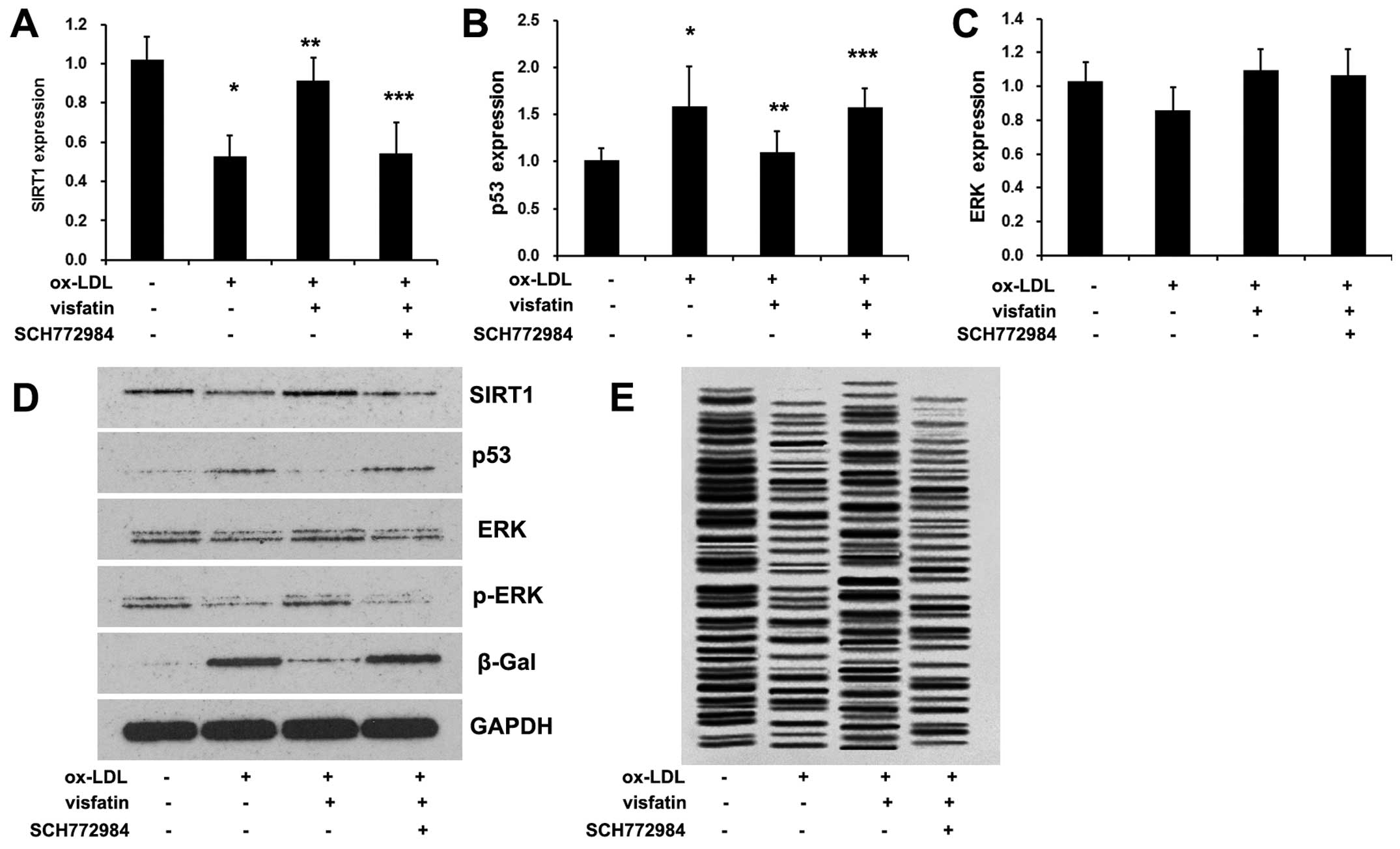
To examine the role of the PI3K/Akt/ERK pathway in
the visfatin-mediated senescence of EPCs, the cells were treated
with ox-LDL and then visfatin, finally the inhibitors, LY294002,
SCH772984. SCH772984 (Active Biochem, Maplewood, NJ, USA) was used
to inhibit ERK and LY294002 (Selleck Chemicals, Houston, TX, USA)
was used to inhibit PI3K. Consistent with previous findings
(18), ox-LDL disrupts the
PI3K/Akt signaling pathway by reducing the p-Akt/Akt and
p-PI3K/PI3K (p85) ratio in EPCs (Fig.
5), which was reversed by visfatin treatment. Furthermore,
LY294002 abolished visfatin-induced PI3K phosphorylation and Akt
phosphorylation (Fig. 5E). In
addition, LY294002 abrogated the visfatin-induced increase in the
expression levels of SIRT1, as well as the visfatin-induced
decrease in the expression of p53 (Fig. 5A, B and E), and diminished
telomerase activity (Fig. 5F).
These results suggested that the PI3K/Akt pathway was regulated by
visfatin in order to mediate SIRT1 expression and ox-LDL-induced
senescence of EPCs. Moreover, ox-LDL also reduced the p-ERK/ERK
ratio in EPCs (Fig. 6D), which
was reversed by visfatin treatment. Pre-treatment with SCH772984
abolished the visfatin-induced increase in ERK phosphorylation
(Fig. 6D). It should be noted
that no significant effects were observed on ERK mRNA expression
(Fig. 6C). In addition, SCH772984
abolished the visfatin-induced increase in the expression levels of
SIRT1, as well the visfatin-induced decrease in p53 expression
(Fig. 6A, B and D), and
diminished telomerase activity (Fig.
5F), suggesting that visfatin-regulated SIRT1 expression and
ox-LDL-induced senescence of EPCs involved ERK phosphorylation. In
conclusion, visfatin regulates SIRT1 expression in ox-LDL-induced
EPC senescence through the PI3K/Akt/ERK pathway.
Discussion
Previous findings suggest that circulating EPCs
serve as a biological marker for vascular function (23) and that the number of EPCs is a
significant predictor of future cardiovascular events (24,25). EPCs also play an imporant role in
post-ischemic vascular repair (26,27). Ox-LDL, as one of the most
important risk factors for cardiovascular disease, has been
revealed to exert a detrimental effect on the growth and
bioactivity of EPCs by triggering apoptosis, inducing cell
ossification and inhibiting endothelialization (9–12).
For example, Ji et al showed that ox-LDL significantly
decreased the proliferation, migration and adhesion capacity of
EPCs, whereas ROS production and NADPH oxidase expression was
significantly increased (28).
Ox-LDL stimulates p53-dependent activation of pro-apoptotic Bax
leading to apoptosis of differentiated EPCs (29). Ox-LDL induced dose- and
time-dependent activation of p38 MAPK, which plays a critical role
in regulating the numbers and functions of EPCs (30). Previous findings have demonstrated
that ox-LDL accelerates the onset of EPC senescence, which may be
related to telomerase inactivation, leading to the impairment of
proliferative capacity and network formation (13). Consistent with this study
(13), the present study found
that ox-LDL induces EPC senescence by evidently increasing β-Gal
expression and diminishing telomerase activity.
Visfatin has been implicated in the pathogenesis of
various cardiovascular disorders including atherosclerosis and
myocardial failure, and in brain ischemia (16,31). Previously, van der Veer et
al showed that FK866, an antagonist of visfatin, induced
premature senescence in human vascular smooth muscle cells,
indicating visfatin is a potential anti-senescence molecule
(17). In this study, we found
that 10 nM visfatin evidently abolished ox-LDL-induced EPC
senescence, thereby providing a novel therapeutic target for the
reversal of EPC dysfunction in neovascularization.
The family of sirtuin proteins are a class of
NAD+-dependent protein deacetylases which are proteins
regulating key cellular processes including cell cycle regulation,
apoptosis, metabolic regulation and inflammation (20). SIRT1 is reported to be a regulator
of ageing and its increased expression or activation prolonged
lifespan in lower forms of animals (32). For example, Braidy et al
demonstrated that SIRT1 expression increases with age, concurrently
with increased acetylated p53 levels in all brain regions
investigated (21). Kume et
al showed that SIRT1 attenuates oxidative stress-induced
mesangial cell apoptosis through p53 deacetylation (22). A previous study showed that SIRT1
is a critical modulator of EPC dysfunction during alterations of
glucose metabolism (33),
indicating that the modulation of SIRT1 activity and/or expression
may provide an opportunity for counteracting the impairment of EPC
levels and functionality associated with the ageing of EPCs.
Moreover, dimethylarginine dimethylaminohydrolase 2
(DDAH)/asymmetric dimethylarginine (ADMA) is involved in the
accelerated senescence of EPCs in diabetes, which is associated
with the activation of SIRT1 (34). In this study, we found that
down-regulated SIRT1 expression is associated with ox-LDL-induced
EPC senescence. We further analyzed the association between
visfatin and SIRT1. We found that visfatin significantly
upregulated SIRT1 expression and silenced SIRT1 evidently abolished
the inhibition of senescence in EPCs and the suppression of p53
expression induced by visfatin. In conclusion, these results
indicated that visfatin abolished the ox-LDL-induced senescence of
EPCs by upregulating SIRT1 expression.
The PI3K/Akt signaling pathway is critical for the
function and survival of EPCs (35), based largely on the downstream
activation of endothelial nitric oxide synthase (eNOS) and
subsequent production of NO (9,36).
Imanishi et al (11)
reported that ox-LDL impairs EPC function through effects on Akt.
Imanishi et al showed that the ox-LDL effects on Akt within
EPCs are mediated indirectly, by an upstream disruption of PI3K,
specifically via nitrosylation of the p85 subunit and subsequent
dissociation of the p85 and p110 subunits (13). In the present study, LY294002, a
PI3K inhibitor, significantly reversed the inhibition of
ox-LDL-induced senescence in EPCs as well as SIRT1 expression
induced by visfatin, indicating that the PI3K/Akt signaling pathway
was regulated by visfatin in order to mediate SIRT1 expression and
ox-LDL-induced senescence of EPCs. The ERK pathway is often
associated with SIRT1 expression in various types of cell (37). In the present study, we further
analyzed whether the ERK signaling pathway is also important in
ox-LDL-induced EPC senescence. We found that ox-LDL induced a
decrease in the p-ERK/ERK ratio, which was reversed by visfatin.
SCH772984, a selective inhibitor of ERK, significantly reduced the
inhibition of ox-LDL-induced EPC senescence and the increase in
SIRT1 expression induced by visfatin.
In conclusion, for the first time to the best of our
knowledge, we demonstrate the anti-senescence effects of visfatin
in ox-LDL-stimulated EPCs, and show that visfatin markedly
attenuates ox-LDL-induced EPC senescence by upregulating SIRT1
expression through the PI3K/Akt/ERK pathway, providing a novel
therapeutic strategies for the reversal of EPC dysfunction.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (no. 81300204, 81302850,
81301106), the Hunan Provincial Natural Science Foundation of China
(2015JJ4056) and the Changsha Science and Technology Key Project
(K1406004-31,K1308032-31).
References
|
1
|
Khoo CP, Pozzilli P and Alison MR:
Endothelial progenitor cells and their potential therapeutic
applications. Regen Med. 3:863–876. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fadini GP, Losordo D and Dimmeler S:
Critical reevaluation of endothelial progenitor cell phenotypes for
therapeutic and diagnostic use. Circ Res. 110:624–637. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang C, Zhang ZH, Li ZJ, Yang RC, Qian GQ
and Han ZC: Enhancement of neovascularization with cord blood
CD133+ cell-derived endothelial progenitor cell
transplantation. Thromb Haemost. 91:1202–1212. 2004.PubMed/NCBI
|
|
4
|
Ott I, Keller U, Knoedler M, Götze KS,
Doss K, Fischer P, Urlbauer K, Debus G, von Bubnoff N, Rudelius M,
et al: Endothelial-like cells expanded from CD34 blood cells
improve left ventricular function after experimental myocardial
infarction. FASEB J. 19:992–994. 2005.PubMed/NCBI
|
|
5
|
Toda N: Age-related changes in endothelial
function and blood flow regulation. Pharmacol Ther. 133:159–176.
2012. View Article : Google Scholar
|
|
6
|
Loomans CJ, de Koning EJ, Staal FJ,
Rookmaaker MB, Verseyden C, de Boer HC, Verhaar MC, Braam B,
Rabelink TJ and van Zonneveld AJ: Endothelial progenitor cell
dysfunction: a novel concept in the pathogenesis of vascular
complications of type 1 diabetes. Diabetes. 53:195–199. 2004.
View Article : Google Scholar
|
|
7
|
Shimada K, Mokuno H, Matsunaga E, Miyazaki
T, Sumiyoshi K, Miyauchi K and Daida H: Circulating oxidized
low-density lipoprotein is an independent predictor for cardiac
event in patients with coronary artery disease. Atherosclerosis.
174:343–347. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shimada K, Mokuno H, Matsunaga E, Miyazaki
T, Sumiyoshi K, Kume A, Miyauchi K and Daida H: Predictive value of
circulating oxidized LDL for cardiac events in type 2 diabetic
patients with coronary artery disease. Diabetes Care. 27:843–844.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma FX, Zhou B, Chen Z, Ren Q, Lu SH,
Sawamura T and Han ZC: Oxidized low density lipoprotein impairs
endothelial progenitor cells by regulation of endothelial nitric
oxide synthase. J Lipid Res. 47:1227–1237. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rosso A, Balsamo A, Gambino R, Dentelli P,
Falcioni R, Cassader M, Pegoraro L, Pagano G and Brizzi MF: p53
mediates the accelerated onset of senescence of endothelial
progenitor cells in diabetes. J Biol Chem. 281:4339–4347. 2006.
View Article : Google Scholar
|
|
11
|
Imanishi T, Hano T, Matsuo Y and Nishio I:
Oxidized low-density lipoprotein inhibits vascular endothelial
growth factor-induced endothelial progenitor cell differentiation.
Clin Exp Pharmacol Physiol. 30:665–670. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Liu ZZ, Chen H, Zhang GJ, Kong YH
and Kang XX: Oxidized low-density lipoprotein and
β-glycerophosphate synergistically induce endothelial progenitor
cell ossification. Acta Pharmacol Sin. 32:1491–1497. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Imanishi T, Hano T, Sawamura T and Nishio
I: Oxidized low-density lipoprotein induces endothelial progenitor
cell senescence, leading to cellular dysfunction. Clin Exp
Pharmacol Physiol. 31:407–413. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hill JM, Zalos G, Halcox JP, Schenke WH,
Waclawiw MA, Quyyumi AA and Finkel T: Circulating endothelial
progenitor cells, vascular function, and cardiovascular risk. N
Engl J Med. 348:593–600. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lai P and Liu Y: Angelica sinensis
polysaccharides inhibit endothelial progenitor cell senescence
through the reduction of oxidative stress and activation of the
Akt/hTERT pathway. Pharm Biol. 53:1842–1849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dahl TB, Holm S, Aukrust P and Halvorsen
B: Visfatin/NAMPT: a multifaceted molecule with diverse roles in
physiology and pathophysiology. Annu Rev Nutr. 32:229–243. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van der Veer E, Ho C, O'Neil C, Barbosa N,
Scott R, Cregan SP and Pickering JG: Extension of human cell
lifespan by nicotinamide phosphoribosyltransferase. J Biol Chem.
282:10841–10845. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tie G, Yan J, Yang Y, Park BD, Messina JA,
Raffai RL, Nowicki PT and Messina LM: Oxidized low-density
lipoprotein induces apoptosis in endothelial progenitor cells by
inactivating the phosphoinositide 3-kinase/Akt pathway. J Vasc Res.
47:519–530. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Breitschopf K, Zeiher AM and Dimmeler S:
Pro-atherogenic factors induce telomerase inactivation in
endothelial cells through an Akt-dependent mechanism. FEBS Lett.
493:21–25. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kume S, Kitada M, Kanasaki K, Maegawa H
and Koya D: Anti-aging molecule, Sirt1: a novel therapeutic target
for diabetic nephropathy. Arch Pharm Res. 36:230–236. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Braidy N, Poljak A, Grant R, Jayasena T,
Mansour H, Chan-Ling T, Smythe G, Sachdev P and Guillemin GJ:
Differential expression of sirtuins in the aging rat brain. Front
Cell Neurosci. 9:1672015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kume S, Haneda M, Kanasaki K, Sugimoto T,
Araki S, Isono M, Isshiki K, Uzu T, Kashiwagi A and Koya D: Silent
information regulator 2 (SIRT1) attenuates oxidative stress-induced
mesangial cell apoptosis via p53 deacetylation. Free Radic Biol
Med. 40:2175–2182. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bakogiannis C, Tousoulis D, Androulakis E,
Briasoulis A, Papageorgiou N, Vogiatzi G, Kampoli AM, Charakida M,
Siasos G, Latsios G, et al: Circulating endothelial progenitor
cells as biomarkers for prediction of cardiovascular outcomes. Curr
Med Chem. 19:2597–2604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Reynolds JA, Robertson AC, Bruce IN and
Alexander MY: Improving cardiovascular outcomes in rheumatic
diseases: therapeutic potential of circulating endothelial
progenitor cells. Pharmacol Ther. 142:231–243. 2014. View Article : Google Scholar
|
|
25
|
Werner N, Kosiol S, Schiegl T, Ahlers P,
Walenta K, Link A, Böhm M and Nickenig G: Circulating endothelial
progenitor cells and cardiovascular outcomes. N Engl J Med.
353:999–1007. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takahashi T, Kalka C, Masuda H, Chen D,
Silver M, Kearney M, Magner M, Isner JM and Asahara T: Ischemia-
and cytokine-induced mobilization of bone marrow-derived
endothelial progenitor cells for neovascularization. Nat Med.
5:434–438. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Aicher A, Rentsch M, Sasaki K, Ellwart JW,
Fändrich F, Siebert R, Cooke JP, Dimmeler S and Heeschen C: Nonbone
marrow-derived circulating progenitor cells contribute to postnatal
neovascularization following tissue ischemia. Circ Res.
100:581–589. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ji KT, Qian L, Nan JL, Xue YJ, Zhang SQ,
Wang GQ, Yin RP, Zhu YJ, Wang LP, Ma J, et al: Ox-LDL induces
dysfunction of endothelial progenitor cells via activation of
NF-κB. BioMed Res Int. 2015:1752912015. View Article : Google Scholar
|
|
29
|
Cheng J, Cui R, Chen CH and Du J: Oxidized
low-density lipoprotein stimulates p53-dependent activation of
proapoptotic Bax leading to apoptosis of differentiated endothelial
progenitor cells. Endocrinology. 148:2085–2094. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu Y, Wang Q, Cheng L, Wang J and Lu G:
Effect of oxidized low-density lipoprotein on survival and function
of endothelial progenitor cell mediated by p38 signal pathway. J
Cardiovasc Pharmacol. 53:151–156. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Revollo JR, Körner A, Mills KF, Satoh A,
Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR,
et al: Nampt/PBEF/visfatin regulates insulin secretion in beta
cells as a systemic NAD biosynthetic enzyme. Cell Metab. 6:363–375.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Poulose N and Raju R: Sirtuin regulation
in aging and injury. Biochim Biophys Acta. 1852:2442–2455. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Balestrieri ML, Rienzo M, Felice F,
Rossiello R, Grimaldi V, Milone L, Casamassimi A, Servillo L,
Farzati B, Giovane A and Napoli C: High glucose downregulates
endothelial progenitor cell number via SIRT1. Biochim Biophys Acta.
1784:936–945. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yuan Q, Hu CP, Gong ZC, Bai YP, Liu SY, Li
YJ and Jiang JL: Accelerated onset of senescence of endothelial
progenitor cells in patients with type 2 diabetes mellitus: role of
dimethylarginine dimethylaminohydrolase 2 and asymmetric
dimethylarginine. Biochem Biophys Res Commun. 458:869–876. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ackah E, Yu J, Zoellner S, Iwakiri Y,
Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, et
al: Akt1/protein kinase Balpha is critical for ischemic and
VEGF-mediated angiogenesis. J Clin Invest. 115:2119–2127. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lefevre J, Michaud SE, Haddad P, Dussault
S, Ménard C, Groleau J, Turgeon J and Rivard A: Moderate
consumption of red wine (cabernet sauvignon) improves
ischemia-induced neovascularization in ApoE-deficient mice: effect
on endothelial progenitor cells and nitric oxide. FASEB J.
21:3845–3852. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chou WW, Chen KC, Wang YS, Wang JY, Liang
CL and Juo SH: The role of SIRT1/AKT/ERK pathway in ultraviolet B
induced damage on human retinal pigment epithelial cells. Toxicol
In Vitro. 27:1728–1736. 2013. View Article : Google Scholar : PubMed/NCBI
|