Introduction
Serrated polyposis (SPP), which was previously known
as hyperplastic polyposis, is characterized by the presence of
multiple colorectal epithelial polyps with a serrated architecture,
termed serrated (SE) polyps, as well as an increased predisposition
to colorectal cancer (CRC) (1–3).
SE polyps differ from adenomatous (AD) polyps and are comprised of
various lesions, namely: hyperplastic polyps (HPs), non-dysplastic
lesions with normal proliferation and architecture but elongated
crypts with a saw-toothed appearance; sessile serrated adenomas
(SSAs), lesions that present abnormal proliferation and
architecture and may or may not include dysplasia; and traditional
serrated adenomas (TSAs) that are dysplastic polyps with prominent
serration (1,4–6).
The presence of multiple SE polyps has also been associated with
other hereditary conditions, namely serrated pathway syndrome or
Jass syndrome (7–9).
It has been proposed that these SE lesions arise
through the serrated pathway rather than through the
adenoma-carcinoma sequence pathway (7,10–13). It has also been suggested that the
HP is the precursor lesion in this pathway, with SSA as an
intermediate step which then progresses to an adenocarcinoma with
or without microsatellite instability (MSI or MSS, respectively).
At the molecular level, SE lesions associated with those hereditary
serrated syndromes share some genetic alterations, namely the
presence of B-raf proto-oncogene, serine/threonine kinase
(BRAF) mutations and the methylator phenotype, termed CpG
island methylator phenotype (CIMP) (14–16), although these are also common to
the sporadic SE lesions.
However, the analysis of SPP lesions has revealed
specific features which are distinct from the SE lesions occurring
in a sporadic context; accordingly, in SPP, HPs, TSAs and CRC are
preferentially located in the proximal colon, i.e. proximal to the
splenic flexure (17–19). Moreover, patients with SPP have
previously been found to present extensive DNA methylation in the
normal mucosa of the proximal colon (20), suggesting the involvement of
widespread gene promoter methylation (CIMP) (7,14,21). This phenotype appears to be
related to MutL homolog 1 (MLH1) methylation, which has been
associated with MSI status, rather than O-6-methylguanine-DNA
methyltransferase (MGMT) methylation. However, MSI appears
to be less frequent in SPP lesions than in sporadic lesions
(14,22). Similarly, SPP lesions also
exhibited a lower frequency of Kirsten rat sarcoma viral oncogene
homolog (KRAS) mutations when compared with sporadic SE
lesions (21,22). However, it was also found that
most adenomas and CRCs from patients with SPP exhibited a classic
morphology and that few of these had BRAF or KRAS
mutations, although SE lesions presented a high frequency of
mutations in these genes (22).
Previous research has suggested that tumorigenesis associated with
SPP may not necessarily follow the serrated pathway and may follow
an alternate pathway, involving TSAs and tubulovillous adenomas
(TVAs) as intermediate lesions that progress to MSS adenocarcinomas
with KRAS mutations or even a traditional pathway with
adenomatous polyposis coli (APC) mutations as the initiating
events (10).
A review of published case studies of SPP reported
that approximately 10–50% of patients with SPP have been described
as having a family history of CRC (23). In agreement with these findings,
several studies have described an increased risk of CRC in the
first-degree relatives of probands diagnosed with SPP compared with
the general population (2,17,18,24–28).
These studies have contributed to the notion that there are SPP
cases where heredity may play a role in the development of CRC
and/or polyps. Indeed, another review on this subject reinforces
the concept that familial SPP exists and also the importance of
defining the genetic basis of familial SPP and of studying these
families in a systematic manner (29).
Thus, in the present study, we aimed to
characterize, at the clinical and molecular level, SE and AD
lesions from a cohort of patients with SPP who had been stratified
into two groups: patients with or without a family history of SPP
and/or polyps/CRC in first-degree relatives, in order to elucidate
the information available regarding this new SPP entity with an
apparent hereditary component.
Patients and methods
Patients and specimens
Eighteen patients diagnosed with SPP according to
the WHO diagnostic criteria (1)
were included in this study: 12 patients with SPP associated with a
family history of SPP and/or polyps/CRC (multiple or diagnosed at a
young age) in first-degree relatives (designated herein as
SPP-FHP/CRC) (11 index and one affected relative diagnosed
simultaneously with the index patient), and 6 index patients
without a family history of SPP/polyps/CRC (designated herein as
sporadic SPP) from the familial colorectal cancer registry of the
Portuguese Institute of Oncology of Lisbon Francisco Gentil
(Lisbon, Portugal). No evidence of SPP and/or polyps/CRC was found
in the first-degree relatives of the patients with sporadic SPP,
either by regular colonoscopy examination or by the absence of
symptoms. The patients were classified as presenting a family
history of polyps in first-degree relatives (5/11), if at least one
relative had been diagnosed with polyps at or under 52 years of age
or with >10 polyps. We cannot exclude the possibility that some
of these families, namely PH4 or PH6, may have Jass syndrome
instead of SPP, due to the presence of a mixture of AD and SE
lesions (7,9). All patients had developed >10
lesions prior to the date of recruitment.
Sixty-two lesions were included here: 1 hyperplastic
colonic mucosa (HCM), 25 HPs, 8 TSAs, 11 SSAs, 1
adenomatous/serrated (AD/S) carcinoma (Ca), 1 serrated carcinoma
(SCa), 8 tubular adenomas (TAs), 2 TVAs and 5 Ca. Two normal
colonic mucosa (NCM) samples were also included in this study.
Fresh colorectal lesions were obtained from colectomy or
colonoscopy specimens from patients who underwent surgery or
colonoscopy in the Portuguese Institute of Oncology of Lisbon
Francisco Gentil. Sections from corresponding areas of the
specimens submitted for diagnosis were divided into two parts: one
was snap frozen in liquid nitrogen immediately after resection,
while the other was formalin-fixed and paraffin-embedded.
Histological characterization, according to the WHO
guidelines (1), was performed by
experienced pathologists (R.F. and P.C.). The study was conducted
in accordance with local ethical standards and in agreement with
the Helsinki Declaration of 1975, as revised in 1983. Informed
consent for diagnosis and additional investigational studies, which
may result in improving the knowledge about the pathogenesis of the
disease, was obtained from patients included in this study.
Moreover, biological material used for DNA isolation was obtained
from archival sections from colorectal adenomas and carcinomas
specimens, submitted for diagnosis (histological classification)
and derived from patients who underwent surgery or colonoscopy in
the Portuguese Institute of Oncology of Lisbon Francisco Gentil;
only somatic analysis was performed and samples are truly
anonymized.
Methods
DNA isolation
DNA was isolated from fresh-frozen and/or
paraffin-embedded tumor tissue and matched normal tissue. DNA was
isolated from the paraffin-embedded tissues by proteinase K
digestion, which was followed by phenol/chloroform extraction and
ethanol precipitation, as previously described (30). DNA from the fresh frozen tissue
was isolated by proteinase K digestion, followed by precipitation
with a saturated NaCl solution and ethanol as previously described
(31).
Somatic mutation analysis of the APC
gene
APC mutation analysis was performed using the
protein truncation test (PTT) for exon 15, as previously reported
(30). Briefly, APC exon
15 was divided into four overlapping fragments that were amplified
by polymerase chain reaction (PCR) and, subsequently, in
vitro transcription and translation were performed using a TnT
T7-coupled reticulocyte lysate system (Promega, Madison, WI, USA).
In negative cases, the mutational cluster region was subsequently
analyzed by automated sequencing in order to search for missense
mutations. For the samples obtained from paraffin-embedded tissues,
APC mutations were analyzed by single-strand conformational
polymorphism (SSCP) or by automated sequencing (32).
Somatic mutation analysis of catenin beta
1 (CTNNB1)
Genomic DNA from each tumor sample was amplified by
PCR for SSCP analysis of exon 3 of the CTNNB1 gene. The
amplified products were analyzed in a mutation detection
enhancement (MDE) gel and visualized by silver staining (33).
Somatic mutation analysis of the AXIN2
gene
AXIN2 mutation analysis was performed by
amplification of a repetitive sequence containing the
(G)7, (C)6 and (C)5 tracts (where
a considerable mutation frequency was described) (34), followed by electrophoresis in 7%
polyacrilamide gel containing formamide and urea and visualization
by silver staining (33).
Somatic mutation analysis of BRAF, KRAS
and neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS)
genes
BRAF (exon 15: forward primer 5′-TCATAATGCTTG
CTCTGATAGGA-3′; reverse primer 5′-GGCCAAAAATTTAATCAGTGGA-3′,
KRAS (exon 2: forward primer 5′-GTGTGACATGTTCTAATATAGTCA-3′;
reverse primer 5′-GAATGGTCCTGCACCAGTAA) (35) and NRAS (exons 2 and
3-primers were kindly provided by Dr Branca Cavaco), were amplified
by PCR. The DNA samples were amplified in a standard PCR buffer
(Invitrogen, Waltham, MA, USA). Mutations in these genes were
analyzed by automated sequencing.
Sequencing analysis
After amplification, PCR products were purified with
Illustra GFX™ PCR DNA and Gel Band purification kit (GE Healthcare,
Little Chalfont, UK) according to the manufacturer's instructions.
Sequencing reactions were performed using the BigDye Terminator
Cycle Sequencing kit and the respective products were analyzed on
the ABI PRISM™ 310 Genetic Analyzer (both from Applied Biosystems,
Foster City, CA, USA) using Sequencing Analysis software. The
pathogenic relevance of missense variants was evaluated by
comparing aminoacid sequences using PolyPhen software (http://genetics.bwh.harvard.edu/pph/)
and SIFT software (http://sift.jcvi.org/).
MSI/loss of heterozygosity (LOH)
analysis
MSI status was analyzed using the Bethesda panel of
reference markers (36). Each
colonic lesion and paired normal DNA were amplified by PCR for each
of the microsatellite markers and analyzed in the ABI Prism™ 310
Genetic Analyzer using GeneScan software (Applied Biosystems). The
lesions were classified as MSI-high (H) when showing MSI in two or
more of the five markers, MSI-low (L) when MSI was detected in one
of the markers, and MSS when none of the markers revealed
instability (37). In cases
exhibiting MSI-L, BAT-40 and MYCL markers were also analyzed and
the lesions were classified as MSI-H when MSI was detected in
>40% of the 7 markers analyzed; otherwise they were classified
as MSI-L, as previousy described (37).
A total of 4 dinucleotide markers flanking
MGMT (D10S1703, D10S1676, D10S169 and D10S1651) were
analyzed for each colonic lesion and paired normal DNA in order to
evaluate the presence of LOH. Each lesion was subsequently scored
as demonstrating LOH if the ratio between the areas of the normal
and the tumor alleles was >1.5 or <0.67.
Regarding the D5S346 marker, LOH was evaluated (and
confirmed using the D5S1965 marker) as indicative of loss of the
APC gene. LOH of D2S123 and D17S250 was also evaluated as
described above.
Methylation analysis
The analysis of MGMT and mismatch repair
(MMR) gene promoter methylation was performed by
methylation-specific multiplex ligation-dependent probe
amplification (MS-MLPA) (38)
using the SALSA MS-MLPA KIT ME011 MMR, (MRC-Holland, Amsterdam, The
Netherlands). MS-MLPA reactions were performed as described by the
manufacturer. The samples were analyzed using GeneScan software on
the ABI Prism™ 310 Genetic Analyzer (Applied Biosystems). The
results were normalized using MRC Coffalyser MLPA-DAT software
v.9.4 (MRC-Holland). A ratio of 0.15 or higher, corresponding to
15% of methylated DNA, was indicative of promoter methylation as
described elsewhere (32,39).
Statistical analysis
Fisher's exact test (using a two-sided or 2×3 table)
and the χ2 test (http://www.quantitativeskills.com/sisa/index.htm)
were used to compare categorical variables, and the Student's
t-test (http://www.physics.csbsju.edu/stats/t-test.html) was
used to compared continuous variables. A p-value <0.05 was
considered to indicate a statistically significant difference.
Results
Clinical characterization
Table I summarizes
the clinical features of the 18 patients included in this study,
stratified into two groups: SPP-FHP/CRC and sporadic SPP. The
average age at diagnosis (i.e. the age at which they presented
symptoms) among our cohort of SPP patients (n=18) was 55±11 years
(range 25–80); however, it was significantly higher in the
SPP-FHP/CRC group than in the sporadic SPP group [60±10 years
(range 41–80) vs. 46±15 years (range 25–68), p=0.027 (Student's
t-test)] (Table II).
 | Table IClinical features of the patients
evaluated in this study, histological characterization of the
respective lesions and family history of SPP and/or polyps/CRC in
first-degree relatives. |
Table I
Clinical features of the patients
evaluated in this study, histological characterization of the
respective lesions and family history of SPP and/or polyps/CRC in
first-degree relatives.
| Family | Patient ID | Age at diagnosis in
years | Gender | Total number of
lesions | Type of
lesionsa | Preferential
location of lesionsb | CRC | WHO diagnostic
criteriac | No. of affected
individuals in the family | Family
historyd (age at diagnosis in
years) |
|---|
| SPP-FHP/CRC |
| PH1 | A756 | 62 | M | >100 |
HP+TALGD+TSALGD+SSA | Whole colone | No | 3 | 3 | Son, >45 SE
polyps (42); Son, 2 SE polyps
(37) |
| PH3 | A755 | 41 | F | >100 |
HP+TSALGD+TVALGD+SSA+TALGD | Whole colone | No | 3 | 3 | Mother, CRC (67);
maternal aunt, CRC (58) |
| PH4 | CA638 | 66 | M | 19 | TSA
(LGD+HGD)+VA | Whole colon | Yesf | 1 | 5 | Twin brother, 20 AD
and SE polyps (70); son, 2 AD polyps (37); nephew, 18 AD polyps (54) |
| CA636 | 69 | M | 50 |
HP+TALGD+TSALGD+TVALGDg | Proximal | Yesf | 3 | | |
| PH5 | A193 | 69 | F | 40 |
HP+TALGD+TVAHGD | Proximal | Yes | 3 plus 1 | 3 | Sister, 2 AD polyps
(67); son, 1 SE polyp (52) |
| PH6 | A478 | 64 | M | 50–100 |
HP+TSALGD+TALGD+SSA | Distale | No | 3 plus 1 | 5 | Father, CRC
(57); sister, 7 polyps (61);
daughter, 1 SE polyp (39); son,
2 AD polyps (35) |
| PH7 | A759 | 50 | M | 50–100 | HP+TA | Whole colon | Yes | 3 | 3 | Mother, 2 AD polyps
(75); brother, 8 SE and AD polyps (47) |
| PH8 | A760 | 59 | F | 45 |
HP+SSA+TSALGD | Proximal | No | 1 plus 3 | 4 | Maternal
grandfather, CRC; mother, CRC (73); sister, 3 AD polyps (61) |
| PH12 | A758 | 57 | M | 40 | HP | Distal | Yes | 3 | 2 | Brother, 1 AD polyp
(44) |
| PH14 | A686 | 80 | M | 49 |
HP+TSALGD+TALGD | Distal | No | 3 | 4 | Daughter, 2 SE
polyps (54); daughter, 11 SE and
1 AD polyps (42); son, 1 AD
polyp (44) |
| PH19 | A993 | 58 | F | 17 |
HP+TALGD | Distal | No | 1 | 3 | Mother, CRC;
sister, 6 polyps (68) |
| PH33 | A983 | 49 | M | 45 |
HP+TSALGD+TALGD+SSA | Distal | No | 1 plus 3 | 4 | Paternal uncle,
polyps (69); father, 1 AD polyp (77); brother, 1 AD polyp (50) |
| Sporadic SPP | | | | | | | | | |
| PH9 | A500 | 48 | F | 40 | HP+SSA+TSALGD | Whole colon | No | 1 plus 3 | 1 | -h |
| PH10 | A989 | 25 | F | <100 | HP | NA | Yes | 3 plus 1 | 1 | -h |
| PH11 | A757 | 53 | M | 50 | HP+TSA | Distal | Yesf | 3 | 1 | -h |
| PH16 | A990 | 68 | M | 15 | HP+SSA | Whole colon | No | 1 | 1 | -h |
| PH18 | A992 | 46 | F | 10 |
TSALGD+HP | Distal | No | 1 | 1 | -h |
| PH22 | A951 | 33 | F | 31 |
HP+TALGD | Distal | No | 1 plus 3 | 1 | -h |
| Table IIComparison between clinical features
in patients with sporadic SPP and those with SPP-FHP/CRC. |
Table II
Comparison between clinical features
in patients with sporadic SPP and those with SPP-FHP/CRC.
| Clinical
feature | SPP-FHP/CRC | Sporadic SPP | p-value |
|---|
| Age at diagnosis
(years) | 60±10 | 46±15 | 0.027
(Student's t-test) |
| Preferential
location of lesions |
| Whole colon | 4/12 (33%) | 2/5 (40%) | NS |
| Proximal | 3/12 (25%) | 0/5 | NS |
| Distal | 5/12 (42%) | 3/5 (60%) | NS |
| ≥40 lesions | 10/12 (83%) | 3/6 (50%) | NS |
| AD lesions | 10/12
(83%) | 1/6
(17%) | 0.013a |
| ≥3 types of
lesions | 9/12
(75%) | 1/6
(17%) | 0.032a |
The number of lesions was higher in the SPP-FHP/CRC
group than in the sporadic SPP group (threshold, ≥40 lesions):
[10/12 (83%) vs. 3/6 (50%) patients, respectively]. With respect to
histological features, AD lesions were more frequent in the
SPP-FHP/CRC group than in the patients with sporadic SPP [10/12
(83%) vs. 1/6 (17%), p=0.013]. Moreover, the patients with
SPP-FHP/CRC presented a more heterogeneous spectrum of lesions in
comparison with the patients with sporadic SPP. In agreement with
these findings, the presence of three or more types of lesions was
more frequent in the former group [9/12 (75%) vs. 1/6 (17%),
p=0.032].
Regarding the location of the lesions in each
patient, we observed that whereas some of the patients with SPP
presented lesions dispersed uniformly throughout the whole colon,
other patients with SPP presented a predominance of lesions in one
of the two major segments, proximal or distal. Therefore, a
prevalence of proximal or distal location of the lesions was
considered when at least 70% of the lesions (majority SE) were
located in the proximal or distal colon, respectively. In
accordance, a preferential proximal location was observed in 3/12
(25%) of the SPP-FHP/CRC patients and in none of the patients with
sporadic SPP (0/5). A preferential distal location was observed in
3/5 (60%) of the sporadic SPP patients and in 5/12 (42%) of the
patients with SPP-FHP/CRC.
Molecular characterization
The molecular alterations found in each SPP-FHP/CRC
lesion, namely mutations in RAS/RAF and Wnt signaling genes, MSI,
MGMT and MMR methylation, LOH of MGMT locus and LOH
at D2S123 and D17S250 markers, are presented in Table III. The spectra of these
molecular alterations led us to observe that the somatic
mutation/promoter hypermethylation spectra differs between those
patients whose lesions were preferentially located in the proximal
colon, or distributed throughout the whole colon, and those whose
lesions were preferentially located in the distal colon, as shown
in Table IV. This led us to
stratify the patients with SPP-FHP/CRC into two groups,
proximal/whole-colon and distal SPP-FHP/CRC.
| Table IIIResults of the molecular analysis of
SE and AD lesions from patients with SPP. |
Table III
Results of the molecular analysis of
SE and AD lesions from patients with SPP.
| A, Results of the
molecular analysis of SE and AD lesions from patients
SPP-FHP/CRC |
|---|
|
|---|
| Family | Patient ID | Lesion ID | Type of lesion | Location of
lesions | MSI | D2S123, D17S250
LOH | LOH of MGMT
locus | Mutations
| Promoter
methylation |
|---|
| APC | CTNNB1 | AXIN2 | BRAF | KRAS | MGMT | MMR genes |
|---|
| Preferential
location of lesions - proximal/whole-colon |
| PH1 | A756 | A756AS | TSALGD | NA | MSS′ | NC, NC | Yes | c.4099C>T
(p.Q1367X) | N | N | – | N | M | MSH6 |
| A756AT1 | TALGD | NA | MSS′ | NC, N | No | c.4123C>T
(p.H1375Y) | N | N | N | N | M | MSH6 |
| A756AT2 | TALGD | NA | MSS′ | NC, N | IC | c.4123C>T
(p.H1375Y) | N | N | N | N | NM | NM |
| A756PH1 | HP | NA | MSS′ | NC, NC | Yes | N | N | – | N | N | M | MSH6,
MSH3 |
| A756PH2 | SSA | NA | MSS′ | NC, N | IC | c.4289delC
(p.T1430TfsX43) | N | N | – | N | M | MSH6 |
| PH3 | A755 | A755AS1 | TSALGD | Distal | MSI-H | NI, D17S250 | IC | NC | N | N | c.1785T>G
(p.F595C) | N | – |
– |
| A755AS2 | TSA | Proximal | MSI-L | NI, D17S250 | IC | c.4235G>A
(p.G1412E) | c.115G>A
(p.A39T) | N | c.1799T>A
(p.V600E) | N | M | MSH6 |
| A755AS3 | TSALGD | Proximal | MSI-H | NI, NI | Yes | NC | N | N | N | N | NM | MSH6 |
| A755PH1 | HP | Proximal | MSI-L | NI, D17S250 | Yes | N | N | N | N | N | M | MSH6 |
| A755PH2 | SSA | Proximal | MSI-L | D2S123,
D17S250 | IC | N | N | N | N | N | NM | NM |
| PH4 | CA638 | CA638AS | TSALGD | Distal | MSS | N, N | No | N | c.122C>T
(p.T41I) | N | N | c.35G>C
(p.G12A) | M | MSH3 |
| CA638C | Ca (AD/S) | Rectum | MSS | N, N | No | LOH | N | N | N | c.35G>A
(p.Gl2D) | M | NM |
| CA636 | CA636AT1 | TALGD | Proximal | MSI-L | NI, D17S250 | Yes | c.4262delG
(p.S1421MfsX52) | N | – | N | N | M | NM |
| CA636C | Ca | Proximal | MSS | D2S123, N | Yes | NC | N | N | N | N | M | MLH3 |
| CA636AT2 | TALGD | Proximal | MSI-L | D2S123, NI | Yes | N | N | N | N | N | NM | NM |
| CA636AT3 | TALGD | Proximal | MSS | D2S123,
D17S250 | Yes | N | N | N | c.1811G>A
(p.W604X) | N | M | NM |
| CA636ATV | TVALGD | Proximal | MSI-H | NI, NI | IC | c.4024T>G
(p.L1342V) | N | N | N | c.38G>A
(p.G13D) | M | NM |
| PH5 | A193 | A193AT | TALGD | Proximal | MSI-L | N, D17S250 | IC | c.4189G>A
(p.E1397K) | N | N | N | N | M | MSH6 |
| A193ATV | TVAHGD | Rectum | MSS | D2S123, N | Yes | c.4123C>T
(p.H1375Y) | N | N | N | c.35G>A
(p.G12D) | M | NM |
| A193C | Ca | Proximal | MSS | D2S123,
D17S250 | Yes | N | N | N | N | N | M | NM |
| A193PH1 | HP | Proximal | MSS | D2S123, N | Yes | LOH, c.4123C>T
(p.H1375Y) | N | N | N | N | M | NM |
| A193PH2 | HP | Proximal | MSS | D2S123,
D17S250 | Yes | NC | N | N | N | N | M | NM |
| PH7 | A759 | A759T | Ca | Proximal | MSI-H | NI, N | IC | N | c.133T>C
(p.S45P) | c.l994delG
(p.G665AfsX24) | N | c.34G>A)
(p.G12S) | NM | NM |
| PH8 | A760 | A643PA | HP | Proximal | MSS | NI, N | No | N | N | N | c.1799T>A
(p.V600E) | N | M | NM |
| A643PB | HP | Distal | MSS | NI, N | No | N | N | N | c.1799T>A
(p.V600E) | N | M | NM |
| A760AS1 | TSALGD | Proximal | MSS′ | NC, NC | Yes | LOH | N | N | c.1799T>A
(p.V600E) | N | – | – |
| A760PH1 | SSA | NA | MSI-L | NI, D17S250 | – | c.3926_3930
delAAAGA (p.E1309DfsX2) | N | N | N | N | NM | NM |
| A760PH2 | SSA | Proximal | MSI-H | NI, NC | Yes | NC | N | N | N | N | – | – |
| A760PH3 | SSA | Proximal | MSI-L | NI, N | Yes | LOH | c.130C>T
(p.P44S) | N | c.1799T>A
(p.V600E) | N | NM | NM |
| A760PH4 | SSA | NA | MSI-L | NI, NC | Yes | LOH | N | N | N | N | NM | NM |
| A760PH5 | SSA | NA | MSI-L | NI, N | No | LOH | N | N | N | N | NM | NM |
| A760PH6 | SSA | NA | MSS | NI, N | No | LOH | N | N | N | N | NM | NM |
| Preferential
location of lesions - distal |
| PH6 | A478 | A478PA2 | HP | NA | MSS | N, N | No | N | N | N | c.1799T>A
(p.V600E) | N | M | NM |
| A478PB | HP | NA | MSS | N, N | – | N | N | N | c.1799T>A
(p.V600E) | N | – | – |
| A478PC | NCM | NA | MSS | N, N | – | N | N | N | N | c.35G>T
(p.G12V) | NM | NM |
| A478PD | NCM | NA | MSS | N, N | – | N | – | – | N | – | – | – |
| A478PE | HCM | NA | MSS | N, N | – | N | N | N | N | c.35G>A
(p.G12D) | NM | NM |
| A478PF | TALGD | NA | MSS | N, N | – | N | N | N | N | N | NM | NM |
| A478PG | HP | NA | MSS | N, N | No | N | N | N | c.1799T>A
(p.V600E) | N | M | NM |
| A478PH | HP | NA | MSS | N, N | No | N | N | N | c.1799T>A
(p.V600E) | N | NM | NM |
| A462PH1 | SSA | NA | MSS | N, N | No | N | N | N | c.1799T>A
(p.V600E) | N | M | NM |
| A462PH2 | SSA | NA | MSS | N, N | IC | N | N | N | c.1799T>A
(p.V600E) | N | M | NM |
| PH12 | A758 | A758C | Ca | Distal | MSI-L | NI, NC | No | LOH | N | N | c.1799T>A
(p.V600E) | N | M | NM |
| A758PH | HP | Rectum | MSI-L | NI, D17S250 | IC | LOH | N | N | c.1799T>A
(p.V600E) | N | M | NM |
| PH14 | A686 | A686P1A | HP | Distal | MSS | N, N | No | N | N | N | N | c.35G>A
(p.G12D) | NM | NM |
| A686P2B | HP | Distal | MSS | N, N | No | N | N | N | N | c.35G>T
(p.G12V) | M | NM |
| A686P3C | HP | Distal | MSS | N, N | No | N | N | N | N | c.35G>A
(p.G12D) | NM | NM |
| A686P4D | HP | Distal | MSS | N, N | No | N | N | N | N | c.35G>A
(p.G12D) | NM | NM |
| A686P5E | HP | Distal | MSS | N, N | – | N | N | N | N | c.35G>T
(p.G12V) | – | – |
| PH19 | A993 | A993PH1 | HP | Rectum |
MSS* | – | Yes | NC | c.115G>A
(p.A39T) | N | c.1799T>A
(p.V600E) | N | – | – |
| PH33 | A983 | A983PA | HP | Proximal |
MSS* | – | No | N | N | N | c.1799T>A
(p.V600E) | N | NM | NM |
| A983PB | HP | Proximal |
MSS* | – | No | N | N | N | c.1799T>A
(p.V600E) | N | NM | NM |
| A983PC | HP | Distal |
MSS* | – | No | N | N | N | c.1799T>A
(p.V600E) | N | NM | NM |
| A983PD | TALGB | Distal |
MSS* | | No | c.4189G>T
(p.E1397X) | N | N | N | N | NM | NM |
| B, Results of the
molecular analysis of SE and AD lesions from patients with SPP
without a family history of SPP and/or polyps/CRC in first-degree
relatives |
|---|
|
|---|
| Family | Patient ID | Lesion ID | Type of lesion | Location of
lesions | MSI | D2S123, D17S250
LOH | Mutations
| Promoter
methylation |
|---|
| APC | CTNNB1 | AXIN2 | BRAF | KRAS | MGMT | MMR genes |
|---|
| Preferential
location of lesions - Proximal/whole-colon |
| PH9 | A500 | A500PA1 | HP | Proximal | MSS | D2S123, NI | N | N | N |
c.l799T>A(p.V600E) | N | NM | MLH1 |
| A500PB2 | HP | Proximal | MSS | N, NI | N | N | N |
c.l799T>A(p.V600E) | N | NM | NM |
| PH16 | A990 | A990ASS | SSA | NA |
MSS* | – | NC | N | N | N | N | M | NM |
| A990PH1 | HP | Rectum |
MSS* | – | NC | N | N | N | N | NM | MSH3 |
| Preferential
location of lesions - Distal |
| PH10 | A989 | A989C | Ca | Distal |
MSS* | – | NC | N | N | N | N | NM | NM |
| PH11 | A757 | A757AS1 | SCa | NA | MSS | N, N | c.4233delT
(p.S1411RfsX4) | N | N | N | N | M | NM |
| PH18 | A992 | A992AS1 | TSALGB | Distal |
MSS* | – | NC | N | N |
c.l799T>A(p.V600E) | N | – | – |
| A992AS2 | TSALGB | Distal |
MSS* | – | NC | N | N | N | N | – | – |
| PH22 | A951 | A951P1 | HP | Rectum |
MSS* | – | N | N | N |
c.l799T>A(p.V600E) | N | NM | NM |
| A951P2 | HP | Distal |
MSS* | – | N | N | N |
c.l799T>A(p.V600E) | N | NM | NM |
| Table IVMolecular characterization of lesions
from patients with SPP-FHP/CRC, stratified by preferential location
of the lesions in each patient. |
Table IV
Molecular characterization of lesions
from patients with SPP-FHP/CRC, stratified by preferential location
of the lesions in each patient.
| Molecular
characterization | Preferential
location of lesions
| p-value |
|---|
|
Proximal/whole-colon | Distal colon |
|---|
| Total Wnt gene
mutations | 14/26
(54%) | 4/20
(20%) | 0.02b |
| Total RAS/RAF gene
mutations | 12/30
(40%) | 18/20
(90%) |
3.7×10−4b |
| BRAF gene
mutations | 7/30
(23%) | 12/20
(60%) | 0.0089
(χ2 test) |
| KRAS gene
mutations | 5/32 (16%) | 6/20 (30%) | NS |
| MSIa | 15/26
(58%) | 2/15
(13%) |
0.0059b |
| MMR gene
methylation and/or LOH of D2S123 | 17/18
(94%) | 0/11 |
3.0×10−7b |
| MGMT gene
methylation | 19/29 (65%) | 7/17 (41%) | NS |
| LOH of MGMT
locus | 16/23
(70%) | 1/14
(7%) |
2.2×10−4b |
Although the ratio between the different types of
lesions in the two groups, proximal/whole-colon and distal, do not
match, i.e. in the former a higher proportion of AD lesions have
been analyzed [in a recent study, individuals with large and
right-sided SE lesions also had significantly more AD lesions
compared with those without such types of SE lesions (40)], we found differences with respect
to the mutation spectrum, even considering only HPs or SSAs, which
have been analyzed in both groups. Accordingly, HPs and SSAs from
patients with proximal/whole-colon SPP-FHP/CRC presented RAS/RAF
gene mutations less frequently [2/6 (33%) and 1/7 (14%) vs. 14/14
and 2/2, respectively, p=0.003 and p=0.08] whereas MMR gene
methylation or LOH of D2S123 [flanking mutS homolog 6
(MSH6)] occurred more frequently (4/4 and 2/2 vs. 0/7 and
0/2, respectively, p=0.003 and p=0.33), when compared with the same
type of lesions from distal SPP-FHP/CRC patients (Table V). Moreover, SE and AD lesions
presented a similar spectrum of molecular alterations, especially
among each patient. The exception were BRAF mutations that
were significantly more frequent in SE lesions [17/36 (47%) vs.
2/14 (14%), p=0.05] (Table V).
For each patient, either presenting proximal/whole-colon or distal
SPP-FHP/CRC, the spectra of somatic molecular alterations detected
in the lesions were similar regardless of the location of each
specific lesion. For example, in a patient with a prevalence of
proximal lesions, these presented a specific mutation pattern that
was shared by the few distal lesions that were analyzed from the
same patient and different from the proximal lesions from patients
with a predominance of distal lesions and vice versa (Table
IIIA).
| Table VMolecular characterization of
SPP-FHP/CRC lesions stratified by histological type of lesions and
the preferential location of the lesions in each patient
(proximal/whole-colon or distal). |
Table V
Molecular characterization of
SPP-FHP/CRC lesions stratified by histological type of lesions and
the preferential location of the lesions in each patient
(proximal/whole-colon or distal).
| HCM | HP | TSA | SSA | SCa | SE lesions | TA | TVA | Ca | AD lesions | SE+AD lesions |
|---|
| Total Wnt gene
mutations |
|
Proximal/whole-colon | – | 1/4
(25%)a | 4/4a | 6/7(86%)a | 1/1 | 12/16
(75%)b,c | 1/6 (17%) | 0/2 | 1/2 (50%) | 2/10
(20%)b | 14/26 (54%) |
| Distal colon | 0/1 | 2/14 (14%) | – | 0/2 | – | 2/17
(12%)c | 1/2 (50%) | – | 1/1 | 2/3 (67%) | 4/20 (20%) |
| Total RAS/RAF gene
mutations |
|
Proximal/whole-colon | – | 2/6
(33%) | 4/5 (80%) | 1/7 (14%) | 1/1 | 8/19
(42%)d | 1/6 (17%) | 2/2 | 1/3 (33%) | 4/11 (36%) | 12/30 (40%) |
| Distal colon | 1/1 | 14/14 | – | 2/2 | – | 17/17d | 0/2 | – | 1/1 | 1/3 (33%) | 18/20 (90%) |
| BRAF gene
mutations |
|
Proximal/whole-colon | – | 2/6
(33%) | 3/5 (60%) | 1/7 (14%) | 0/1 | 6/19
(32%)e | 1/6 (17%) | 0/2 | 0/3 | 1/11 (9%) | 7/30 (23%) |
| Distal colon | 0/1 | 9/14
(64%) | – | 2/2 | – | 11/17
(65%)e | 0/2 | – | 1/1 | 1/3 (33%) | 12/20 (60%) |
| KRAS gene
mutations |
|
Proximal/whole-colon | – | 0/6 | 1/6 (17%) | 0/8 | 1/1 | 2/21
(10%)f | 0/6 | 2/2 | 1/3 (33%) | 3/11 (27%) | 5/32 (16%) |
| Distal colon | 1/1 |
5/14(36%) | – | 0/2 | – | 6/17
(35%)f | 0/2 | – | 0/1 | 0/3 | 6/20 (30%) |
| MSI |
|
Proximal/whole-colon | – | 1/5
(20%)g | 3/4
(75%)g | 6/7(86%)g | 0/1 | 10/17
(59%)h | 3/4 (75%) | 1/2 (50%) | 1/3 (33%) | 5/9 (56%) | 15/26 (58%) |
| Distal colon | 0/1 | 1/10(10%) | – | 0/2 | – | 1/13
(8%)h | 0/1 | – | 1/1 | 1/2 (50%) | 2/15 (13%) |
| MMR gene
methylation and/or LOH of D2S123 |
|
Proximal/whole-colon | – | 4/4 | 4/4 | 2/2 | 0/1 | 10/11
(91%)i | 4/4 | 1/1 | 2/2 | 7/7 | 17/18 (94%) |
| Distal colon | 0/1 | 0/7 | – | 0/2 | – | 0/10i | 0/1 | – | – | 0/1 | 0/11 |
| MGMT gene
methylation |
|
Proximal/whole-colon | – | 6/6j | 3/4 (75%) | 1/7 (14%) | 1/1 | 11/18(61%) | 4/6 (67%) | 2/2 | 2/3 (67%) | 8/11 (73%) | 19/29 (65%) |
| Distal colon | 0/1 | 4/11
(36%)j | – | 2/2 | – | 6/14 (43%) | 0/2 | – | 1/1 | 1/3 (33%) | 7/17(41%) |
| LOH of MGMT
locus |
|
Proximal/whole-colon | – | 4/6
(67%)k | 3/4 (75%) | 3/5 (60%) | 0/1 | 10/16
(63%)l | 3/4 (75%) | 1/1 | 2/2 | 6/7 (86%) | 16/23 (70%) |
| Distal colon | – | 1/11
(9%)k | – | 0/1 | – | 1/12
(8%)l | 0/1 | – | 0/1 | 0/2 | 1/14 (7%) |
The abovementioned findings led us to analyze the
molecular data from the patients with SPP-FHP/CRC, who were
stratified into two groups: proximal/whole-colon and distal
SPP-FHP/CRC.
Proximal/whole-colon vs. distal
SPP-FHP/CRC
Mutations in the Wnt genes, as well as MSI, were
significantly more frequent in the patients with
proximal/whole-colon SPP-FHP/CRC than in the patients with distal
SPP-FHP/CRC [14/26 (54%) vs. 4/20 (20%), p=0.02; 15/26 (58%) vs.
2/15 (13%), p=0.0059] (Table
IV). Interestingly, among the proximal/whole-colon SPP-FHP/CRC
samples, Wnt gene mutations were significantly more frequent in the
SE lesions than in the AD lesions [12/16 (75%) vs. 2/10 (20%),
p=0.0091]. In the SE lesions, Wnt gene mutations, as well as MSI,
were more frequent in TSAs (4/4 and 3/4) and in SSAs (6/7 and 6/7)
and rarely detected in HPs (1/4 and 1/5) (p=0.02 and p=0.017,
respectively) (Table V).
Similarly, LOH of the MGMT locus and
MGMT methylation were also more frequent in lesions from the
patients with proximal/whole-colon SPP-FHP/CRC, in comparison with
lesions from the patients with distal SPP-FHP/CRC [16/23 (70%) vs.
1/14 (7%), p=2.2×10−4; 19/29 (65%) vs. 7/17 (41%),
p=0.1, respectively] (Table IV).
The same difference was observed even considering SE lesions only
(Table V). In particular,
MGMT methylation was detected in all HP lesions (6/6) from
proximal/whole-colon SPP-FHP/CRC, in contrast to the lower
frequency observed in HPs from distal SPP-FHP/CRC (4/11, 36%)
(p=0.017).
Interestingly, among lesions which were deemed
informative for methylation analysis and loss of LOH of D2S123, the
presence of MMR gene methylation or of the D2S123 LOH (flanking
MSH6) was only observed in proximal/whole-colon SPP-FHP/CRC
lesions [17/18 (94%) vs. 0/11, p=3.0×10−7]. These
alterations were found in the majority of early lesions and in all
histological types (Tables IIIA and IV). It is of note that
MLH1 methylation was not detected in any of the SPP-FHP/CRC
lesions, being observed only in one lesion from the sporadic SPP
group (Table IIIB). Moreover, it is also of note that except for
one lesion, MSI, either MSI-L or MSI-H was detected only in
dinucleotide microsatellite markers.
By contrast to the abovementioned molecular
alterations, in the SPP-FHP/CRC samples, BRAF and
KRAS mutations were more frequent in the lesions located in
the distal colon than those located in the proximal/whole-colon
[12/20 (60%) vs. 7/30 (23%), p=0.0089 (χ2 test) and 6/20
(30%) vs. 5/32 (16%), respectively], although the latter was not
statistically significant (Table
IV). Considering only the SE lesions, mutations in the RAS/RAF
genes were detected in all the lesions from the distal SPP-FHP/CRC,
namely in all HPs (14/14), HCM (1/1) and in one NCM (Table IIIA),
but in only 8/19 (42%) lesions from the proximal/whole-colon
SPP-FHP/CRC (p=1.3×10−4).
With respect to KRAS/BRAF mutations, two
groups of patients were observed among those with either
proximal/whole-colon or distal SPP-FHP/CRC: one group whose lesions
presented KRAS mutations (PH4, PH5, PH7 and PH14) and
another group, whose lesions presented BRAF mutations (PH3,
PH8, PH19 and PH33) (Table IIIA). One patient presented either
BRAF- or KRAS-positive lesions (family PH6). Notably,
CRC was more frequent in the patients with proximal/whole-colon
SPP-FHP/CRC associated with KRAS mutations than in the
remaining patients [4/4 vs. 1/8 (12%), p=0.01] (Tables I and
IIIA).
Discussion
SPP-FHP/CRC and sporadic SPP differ at
the clinical level
The patients with SPP-FHP/CRC and sporadic SPP
differed with respect to clinical and histological features. The
older mean age at diagnosis (60 vs. 46 years old) of the former may
underlie a slower process of tumorigenesis. This is in contrast to
that which has been observed in relation to other hereditary CRC
syndromes, namely in familial adenomatous polyposis and in Lynch
syndrome, which are diagnosed at an earlier age (usually ≤50 years
old) compared to the age at diagnosis in patients with sporadic CRC
(>60 years old) (32,41). The presence of a more
heterogeneous histological pattern of lesions in patients with
SPP-FHP/CRC, associated with the concomitant presence of both SE
and AD lesions, compared with the lesions in patients with sporadic
SPP, suggests the involvement of other pathways in the tumorigenic
process associated with SPP-FHP/CRC, in addition to the serrated
pathway of tumorigenesis.
Molecular alterations involved in tumor
initiation distinguish between two forms of SPP-FHP/CRC:
proximal/whole-colon and distal
Two forms of SPP-FHP/CRC appear to exist according
to the preferential location of the lesions in the colon and
rectum, proximal/whole-colon and distal, which differ with respect
to the somatic events involved in tumor initiation. LOH and
methylation of MGMT, MMR gene methylation and/or LOH of
D2S123 and Wnt gene mutations appear to be the major somatic events
that lead to tumor initiation in proximal/whole-colon SPP-FHP/CRC.
By contrast, in distal SPP-FHP/CRC, KRAS or BRAF
mutations were found in the majority of early lesions and thus seem
to play a major role in tumor initiation.
We have previously shown that distinct Wnt gene
mutations are selected in sporadic and hereditary CRC according to
tumor location, i.e. proximal or distal colon (33,42). We and others have also proposed
that this finding is the result of the selection of a specific
level of β-catenin signaling, optimal for tumor formation, which
differs along the colorectum, thus contributing to differences in
lesion distribution in specific types of CRC, such as Lynch
syndrome (42,43). Proving this, variable gradients in
the number of stem cells and physiological Wnt activity have been
demonstrated throughout the length of the intestinal tract
(44). Thus, in a similar
fashion, tumorigenic pathways may also differ between proximal and
distal SPP-FHP/CRC.
MGMT and MMR alterations, followed by Wnt
gene mutations, are involved in the initiation of
proximal/whole-colon SPP-FHP/CRC
The exclusive detection of MMR gene methylation
(mainly of MSH6) and/or LOH of D2S123 (flanking MSH6)
in proximal/whole-colon SPP-FHP/CRC, in the majority of early
lesions and in all histological types, appears to suggest that the
MMR system plays an important role in the initiation of
proximal/whole-colon SPP-FHP/CRC, which is in agreement with the
high frequency of MSI (either MSI-L or MSI-H) in these lesions
(58%). Interestingly, MMR methylation and, consequently, MMR
deficiency were not associated with MLH1 methylation as has
been previously observed in sporadic SE lesions located in the
proximal colon (7), but rather
with MSH6 or mutS homolog 3 (MSH3) methylation.
Accordingly, a high frequency of LOH of the MSH3 locus has
been recently described in sporadic MSI-L CRC, suggesting that the
impairment of other MMR genes such as MSH3 or MSH6,
as observed in the present study, are involved in an MSI-L pathway,
instead of MLH1, which is usually associated with the MSI-H
serrated pathway (45). The
detection of MSI almost exclusively in dinucleotide microsatellite
markers is in agreement with this finding, since this type of MSI
has been described to be a characteristic feature of MSI-L tumors
(46). Interestingly, in the
present study, MSI was detected more frequently at D2S123 followed
by D17S250.
MGMT methylation and LOH of the MGMT
locus were the most frequent alterations in proximal/whole-colon
SPP-FHP/CRC, and MGMT methylation was detected in all HPs,
commonly known as the precursor lesion (12). Therefore, we suggest that,
similarly to MMR alterations, LOH and methylation of MGMT
may also be early events in SPP-FHP/CRC proximal/whole-colon
tumorigenesis. Notably, among the 17 lesions informative for both
MMR and MGMT alterations in this form of SPP-FHP/CRC, in 16
(94%) both events were noted (Table IIIA). It is known that
MGMT deficiency results in the inability to repair
O6-methylguanine in the DNA, caused by genotoxic stress, which,
once accumulated, leads to the translocation of the MutSα complex
(MSH2 and MSH6) into the nucleus, thus increasing GT mismatch
binding activity (47). This may
lead to a selective pressure for molecular changes that impair MSH2
or MSH6 function such as promoter hypermethylation or LOH, thus
explaining the association between the latter and MGMT
methylation in the same early SPP-FHP/CRC lesions. Thus, we suggest
a primary role for MGMT methylation, O6-methylguanine errors
and MMR alterations in tumor initiation of proximal/whole-colon
SPP-FHP/CRC. We further suggest that this molecular signature may
indicate that a germline defect in the mechanisms regulating the
response to genotoxic stress underlies the genetic susceptibility
in this form of SPP-FHP/CRC.
In the present study, the higher frequency of Wnt
gene mutations in proximal SPP-FHP/CRC, particularly in SE lesions,
when compared with the AD lesions, suggests that this pathway also
plays an important role in this form of SPP-FHP/CRC, especially in
the transition to SE adenoma since this frequency was significantly
higher in TSA and SSA than in HP and HCM (p=8.4×10−4).
In agreement, CTNNB1 or AXIN2 mutations, that are
selected almost exclusively in proximal colorectal tumors with MSI
(33), were detected in 4/14
(28%) TSAs and SSAs from proximal/whole-colon SPP-FHP/CRC.
Moreover, among APC nonsense and missense mutations, the
majority (7/8; 88%) were of the transition type which is a
characteristic feature of cells presenting MMR defects (48–50).
The occurrence of LOH of D2S123 and/or D17S250
dinucleotide marker in the present study [17/48 (35%) and 7/51
(14%), respectively], has been previously described in Paneth cell
metaplasia, a condition that is commonly observed in the small
intestine and the proximal colon of elderly individuals (51,52). Therefore, MGMT deficiency
may make these cells more exposed to genotoxic stress, thus leading
to molecular changes in Wnt genes and consequently to commitment to
Paneth cell lineage [to which Wnt gene mutations largely contribute
(53)] and to Paneth cell
metaplasia. Therefore, colonic mucosa with Paneth cell metaplasia
may be one of the pre-neoplastic lesions in the development of
proximal/whole-colon SPP-FHP/CRC.
BRAF and KRAS mutations play different
roles in proximal and distal SPP-FHP/CRC
Our finding that patients with proximal/whole-colon
or distal SPP-FHP/CRC may carry, preferentially, either KRAS
or BRAF mutations, supports previous observations suggesting
that different forms of SPP exist, depending on whether lesions
follow a KRAS or a BRAF pathway (22).
In addition, in the present study, we noted distinct
roles for these mutations between proximal and distal SPP-FHP/CRC.
BRAF or KRAS mutations were detected in the majority
of distal SPP-FHP/CRC lesions, mostly SE early lesions, thus
underlining their importance in early stages, whereas in
proximal/whole-colon SPP-FHP/CRC these mutations (mainly of
KRAS) appear to be more important in tumor progression
(mostly detected in TSAs, TVAs and Cas). Accordingly, a KRAS or
alternate pathway has been proposed to be involved in the
transition from TSA or TVA to CRC in SPP (10). Indeed, in patients with
proximal/whole-colon SPP-FHP/CRC, KRAS mutations were found
only in TSAs, TVAs or Cas (PH4, PH5 and PH7) which is in accordance
with their association with the development of a villous
architecture and hence with malignant transformation (10,54–56). Therefore, as TSAs and TVAs shared
early somatic events with HPs and TAs, respectively, from the same
patients, and based on the model of SPP tumorigenesis previously
presented by Leggett and Whitehall (10), we propose that
proximal/whole-colon SPP-FHP/CRC tumorigenesis may follow an
alternate or KRAS pathway, where TVA and TSA may develop from TA
and HP, respectively, finally leading to CRC, that may or may not
present with MSI (Fig. 1A, upper
panel). Alternatively, a serrated or BRAF pathway (families PH3 and
PH8) where SSA or TSA will probably lead to MSI-H cancer carrying
BRAF mutations may also occur (Fig. 1B, upper panel). In this model, a
deficient DNA repair pathway characterized by MGMT and MMR
gene methylation and/or LOH followed by Wnt gene mutations, appears
to be predominant in proximal SPP-FHP/CRC (Fig. 1, upper panel).
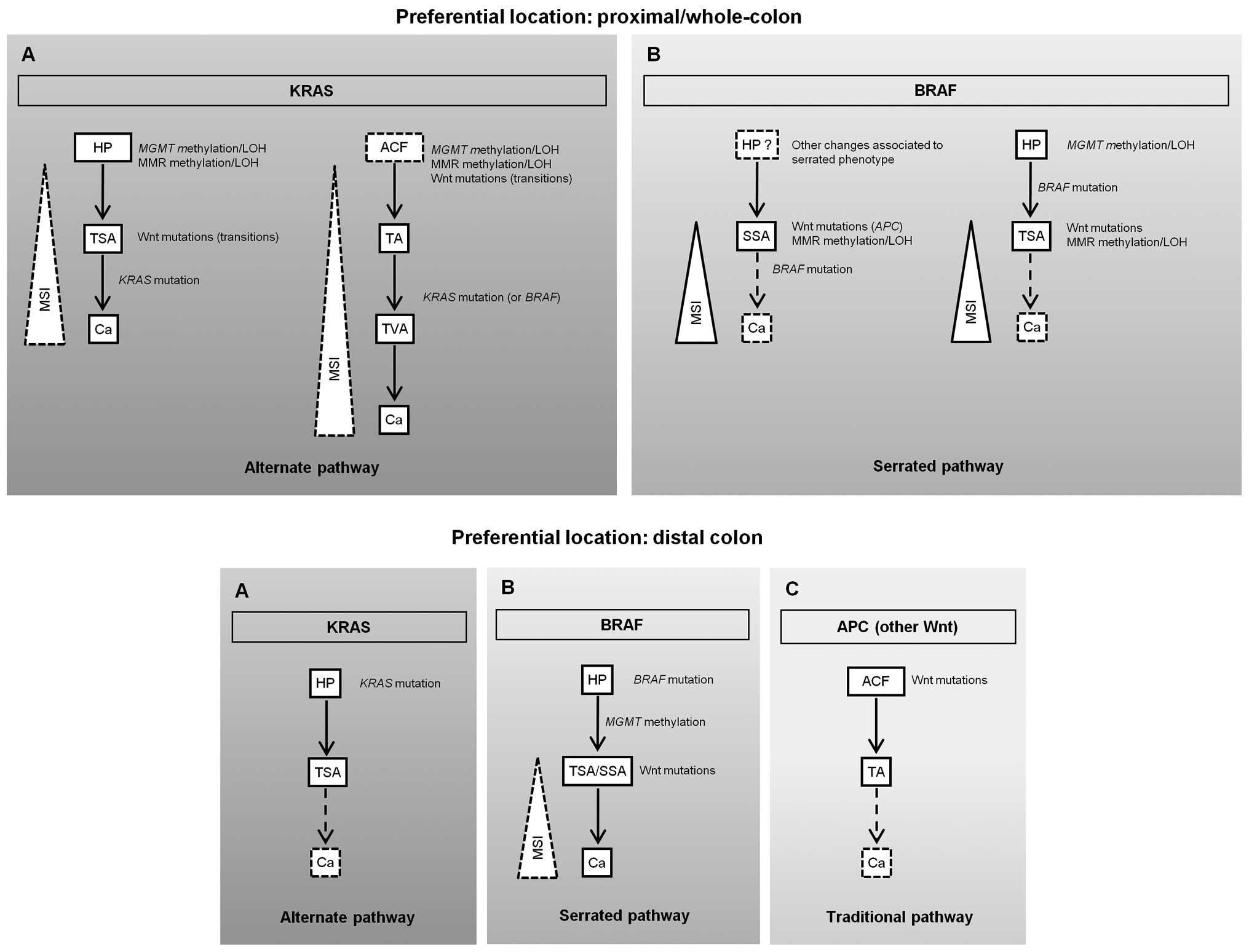 | Figure 1Proposed pathways for colorectal
tumorigenesis in proximal/whole-colon (upper panel) and distal
serrated polyposis (SPP) associated with a family history of SPP
and/or polyps/colorectal cancer (SPP-FHP/CRC) (bottom panel). Both
of these forms may follow a KRAS (alternate) or a BRAF (serrated)
pathway, (A and B), respectively, in the upper and lower panels. In
addition, in distal SPP, an adenomatous polyposis coli (APC)
(traditional) pathway may also occur [(C) in bottom panel]. Each
lesion or molecular alteration in these proposed pathways is
hypothesized based on the results obtained in the present study and
we do not exclude that, in some cases, some of these molecular
alterations may not occur, or even that other molecular alterations
may also be found. The steps involving lesions that were not
analyzed in this study and about which we have previously published
information are represented by broken arrows. In some pathways a
broken line was used to represent the increase in microsatellite
instability (MSI) with tumor progression, to suggest a lower
frequency in those cases. Ca, carcinoma; H P, hyperplastic polyp;
MMR, mismatch repair; SSA, sessile serrated adenoma; TA, tubular
adenoma; TSA, traditional serrated adenoma; TVA, tubulovillous
adenoma. |
In distal SPP-FHP/CRC, either KRAS (PH6 and
PH14) or BRAF (PH6, PH12, PH19 and PH33) mutations play a
major role in tumor initiation, either through an alternate or a
serrated pathway (Fig. 1A and B,
bottom panel, respectively). Wnt gene mutations and MMR defects
were detected in the only carcinoma presented by these patients and
thus are likely involved in tumor progression. As PH6 and PH33 also
presented TAs and did not present TVAs, we hypothesize that, in
distal SPP-FHP/CRC, some AD lesions may also develop through a
traditional pathway initiated by APC mutations (Fig. 1C, bottom panel).
CRC is more frequent in patients with
proximal/whole-colon SPP-FHP/CRC with TSAs, TVAs and KRAS
mutations
The association of KRAS mutations in TSA or
TVA with the development of CRC in proximal/whole-colon SPP-FHP/CRC
suggests a higher contribution of the alternate pathway in the
development of CRC in patients with SPP-FHP/CRC. Supporting our
hypothesis, KRAS mutations have been previously described as
more prevalent than BRAF mutations in a series of SE
carcinomas occurring in the sporadic context, mainly in those
presenting adjacent serrated adenomas (51%) (57). Moreover, in the same study, SE
carcinomas were frequently MSS and presented a higher frequency of
MGMT loss compared with traditional carcinomas, which is in
agreement with our findings demonstrating that MGMT
deficiency plays a prominent role in SPP-FHP/CRC, mainly in the
proximal colon.
The findings that KRAS and BRAF
mutations promote serrated and hyperplastic features, despite being
incapable of initiating colonic adenoma development by themselves
(58,59), may contribute to the apparent
lower incidence of CRC in distal SPP-FHP/CRC, as according to the
results of our present study, KRAS or BRAF mutations
appear to be the initial molecular events in this form. However,
additional studies involving more families are warranted.
In conclusion, SPP-FHP/CRC appears to be a distinct
clinical and histological entity differing from sporadic SPP.
However, we suggest that two forms of SPP-FHP/CRC appear to exist,
proximal/whole-colon and distal, which differ mainly in the
molecular alterations detected in early lesions. We further propose
that a germline defect in the mechanisms regulating the response to
genotoxic damage may underlie the genetic susceptibility in the
former. In addition, our results suggest that CRC appears to
develop more frequently in proximal/whole-colon SPP-FHP/CRC
following an alternate KRAS pathway, thus underlining the
importance of a complete clinical, histological and molecular
characterization for CRC risk evaluation in further studies
involving families with SPP. The results of these studies may be
used to design appropriate guidelines for the clinical management
of proximal and distal colonic presentations of SPP that assumes
major relevance considering the increased risk of CRC and/or polyps
in first-degree relatives.
Abbreviations:
|
AD
|
adenomatous
|
|
APC
|
adenomatous polyposis coli
|
|
AD/S
|
adenomatous/serrated
|
|
BRAF
|
B-raf proto-oncogene,
serine/threonine kinase
|
|
Ca
|
carcinoma
|
|
CIMP
|
CpG island methylator phenotype
|
|
CRC
|
colorectal cancer
|
|
CTNNB1
|
catenin beta 1
|
|
KRAS
|
Kirsten rat sarcoma viral oncogene
homolog
|
|
HCM
|
hyperplastic colonic mucosa
|
|
HP
|
hyperplastic polyp
|
|
LOH
|
loss of heterozygosity
|
|
MDE
|
mutation detection enhancement
|
|
MGMT
|
O-6-methylguanine-DNA
methyltransferase
|
|
MLH1
|
mutL homolog 1
|
|
MMR
|
mismatch repair
|
|
MSI
|
microsatellite instability
|
|
MSI-H
|
microsatellite instability-high
|
|
MSI-L
|
microsatellite instability-low
|
|
MSS
|
microsatellite stable
|
|
MSH3
|
mutS homolog 3
|
|
MSH6
|
mutS homolog 6
|
|
NCM
|
normal colonic mucosa
|
|
NRAS
|
neuroblastoma RAS viral (v-ras)
oncogene homolog
|
|
PCR
|
polymerase chain reaction
|
|
PTT
|
protein truncation test
|
|
SCa
|
serrated carcinoma
|
|
SE
|
serrated
|
|
SPP
|
serrated polyposis
|
|
SPP-FHP/CRC
|
SPP associated with a family history
of SPP and/or polyps/CRC (multiple or diagnosed at a young age) in
first-degree relatives
|
|
SSA
|
sessile serrated adenoma
|
|
SSCP
|
single-strand conformational
polymorphism
|
|
TA
|
tubular adenoma
|
|
TSA
|
traditional serrated adenoma
|
|
TVA
|
tubulovillous adenoma
|
Acknowledgments
The authors thank all the patients who participated
in this study, Dr Inês Francisco for the DNA extraction and Dr
Branca Cavaco for the primers for the NRAS gene. This study
was sponsored and financed by a grant from Núcleo Regional do Sul,
Liga Portuguesa Contra o Cancro, Terry Fox (NRS/LPCC-Terry Fox),
and Fundação para a Ciência e Tecnologia (FCT),
POCI/SAU-OBS/56921/2004.
References
|
1
|
Snover DC, Ahnen DJ, Burt RW and Odze RD:
Serrated polyps of the colon and rectum and serrated polyposis. WHO
Classification of Tumours of the Digestive System. Bosman FT,
Carneiro F, Hruban RH and Theise ND: IARC; Lyon: pp. 160–165.
2010
|
|
2
|
Kalady MF, Jarrar A, Leach B, LaGuardia L,
O'Malley M, Eng C and Church JM: Defining phenotypes and cancer
risk in hyper-plastic polyposis syndrome. Dis Colon Rectum.
54:164–170. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosty C, Parry S and Young JP: Serrated
polyposis: an enigmatic model of colorectal cancer predisposition.
Pathol Res Int. 2011:1570732011. View Article : Google Scholar
|
|
4
|
Snover DC, Jass JR, Fenoglio-Preiser C and
Batts KP: Serrated polyps of the large intestine: a morphologic and
molecular review of an evolving concept. Am J Clin Pathol.
124:380–391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aust DE and Baretton GB; Members of the
Working Group GI-Pathology of the German Society of Pathology:
Serrated polyps of the colon and rectum (hyperplastic polyps,
sessile serrated adenomas, traditional serrated adenomas, and mixed
polyps)-proposal for diagnostic criteria. Virchows Arch.
457:291–297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rosty C, Hewett DG, Brown IS, Leggett BA
and Whitehall VL: Serrated polyps of the large intestine: current
understanding of diagnosis, pathogenesis, and clinical management.
J Gastroenterol. 48:287–302. 2013. View Article : Google Scholar :
|
|
7
|
Young J and Jass JR: The case for a
genetic predisposition to serrated neoplasia in the colorectum:
hypothesis and review of the literature. Cancer Epidemiol
Biomarkers Prev. 15:1778–1784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lindor NM: Hereditary colorectal cancer:
MYH-associated polyposis and other newly identified disorders. Best
Pract Res Clin Gastroenterol. 23:75–87. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roberts A, Nancarrow D, Clendenning M,
Buchanan DD, Jenkins MA, Duggan D, Taverna D, McKeone D, Walters R,
Walsh MD, et al: Linkage to chromosome 2q32.2-q33.3 in familial
serrated neoplasia (Jass syndrome). Fam Cancer. 10:245–254. 2011.
View Article : Google Scholar :
|
|
10
|
Leggett B and Whitehall V: Role of the
serrated pathway in colorectal cancer pathogenesis.
Gastroenterology. 138:2088–2100. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jass JR, Iino H, Ruszkiewicz A, Painter D,
Solomon MJ, Koorey DJ, Cohn D, Furlong KL, Walsh MD, Palazzo J, et
al: Neoplastic progression occurs through mutator pathways in
hyperplastic polyposis of the colorectum. Gut. 47:43–49. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jass JR, Young J and Leggett BA:
Hyperplastic polyps and DNA microsatellite unstable cancers of the
colorectum. Histopathology. 37:295–301. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Snover DC: Update on the serrated pathway
to colorectal carcinoma. Hum Pathol. 42:1–10. 2011. View Article : Google Scholar
|
|
14
|
Chan AO, Issa JP, Morris JS, Hamilton SR
and Rashid A: Concordant CpG island methylation in hyperplastic
polyposis. Am J Pathol. 160:529–536. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kambara T, Simms LA, Whitehall VL, Spring
KJ, Wynter CV, Walsh MD, Barker MA, Arnold S, McGivern A, Matsubara
N, et al: BRAF mutation is associated with DNA methylation in
serrated polyps and cancers of the colorectum. Gut. 53:1137–1144.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weisenberger DJ, Siegmund KD, Campan M,
Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D,
Buchanan D, et al: CpG island methylator phenotype underlies
sporadic microsatellite instability and is tightly associated with
BRAF mutation in colorectal cancer. Nat Genet. 38:787–793. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chow E, Lipton L, Lynch E, D'Souza R,
Aragona C, Hodgkin L, Brown G, Winship I, Barker M, Buchanan D, et
al: Hyperplastic polyposis syndrome: phenotypic presentations and
the role of MBD4 and MYH. Gastroenterology. 131:30–39. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yeoman A, Young J, Arnold J, Jass J and
Parry S: Hyperplastic polyposis in the New Zealand population: a
condition associated with increased colorectal cancer risk and
European ancestry. N Z Med J. 120:U28272007.
|
|
19
|
Boparai KS, Mathus-Vliegen EM, Koornstra
JJ, Nagengast FM, van Leerdam M, van Noesel CJ, Houben M, Cats A,
van Hest LP, Fockens P and Dekker E: Increased colorectal cancer
risk during follow-up in patients with hyperplastic polyposis
syndrome: a multicentre cohort study. Gut. 59:1094–1100. 2010.
View Article : Google Scholar
|
|
20
|
Minoo P, Baker K, Goswami R, Chong G,
Foulkes WD, Ruszkiewicz AR, Barker M, Buchanan D, Young J and Jass
JR: Extensive DNA methylation in normal colorectal mucosa in
hyperplastic polyposis. Gut. 55:1467–1474. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wynter CV, Walsh MD, Higuchi T, Leggett
BA, Young J and Jass JR: Methylation patterns define two types of
hyperplastic polyp associated with colorectal cancer. Gut.
53:573–580. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Carvajal-Carmona LG, Howarth KM, Lockett
M, Polanco-Echeverry GM, Volikos E, Gorman M, Barclay E, Martin L,
Jones AM, Saunders B, et al: Molecular classification and genetic
pathways in hyperplastic polyposis syndrome. J Pathol. 212:378–385.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guarinos C, Sánchez-Fortún C,
Rodríguez-Soler M, Alenda C, Payá A and Jover R: Serrated polyposis
syndrome: molecular, pathological and clinical aspects. World J
Gastroenterol. 18:2452–2461. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boparai KS, Reitsma JB, Lemmens V, van Os
TA, Mathus-Vliegen EM, Koornstra JJ, Nagengast FM, van Hest LP,
Keller JJ and Dekker E: Increased colorectal cancer risk in
first-degree relatives of patients with hyperplastic polyposis
syndrome. Gut. 59:1222–1225. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Win AK, Walters RJ, Buchanan DD, Jenkins
MA, Sweet K, Frankel WL, de la Chapelle A, McKeone DM, Walsh MD,
Clendenning M, et al: Cancer risks for relatives of patients with
serrated polyposis. Am J Gastroenterol. 107:770–778. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Caetano AC, Ferreira H, Soares J, Ferreira
A, Gonçalves R and Rolanda C: Phenotypic characterization and
familial risk in hyperplastic polyposis syndrome. Scand J
Gastroenterol. 48:1166–1172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hazewinkel Y, Koornstra JJ, Boparai KS,
van Os TA, Tytgat KM, van Eeden S, Fockens P and Dekker E: Yield of
screening colonoscopy in first-degree relatives of patients with
serrated polyposis syndrome. J Clin Gastroenterol. 2014.PubMed/NCBI
|
|
28
|
Jasperson KW, Kanth P, Kirchhoff AC,
Huismann D, Gammon A, Kohlmann W, Burt RW and Samadder NJ: Serrated
polyposis: colonic phenotype, extracolonic features, and familial
risk in a large cohort. Dis Colon Rectum. 56:1211–1216. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lanspa SJ, Ahnen DJ and Lynch HT: Serrated
polyposis: the last (or only the latest?) frontier of familial
polyposis? Am J Gastroenterol. 107:779–781. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Albuquerque C, Breukel C, van der Luijt R,
Fidalgo P, Lage P, Slors FJ, Leitão CN, Fodde R and Smits R: The
'just-right' signaling model: APC somatic mutations are selected
based on a specific level of activation of the beta-catenin
signaling cascade. Hum Mol Genet. 11:1549–1560. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Miller SA, Dykes DD and Polesky HF: A
simple salting out procedure for extracting DNA from human
nucleated cells. Nucleic Acids Res. 16:12151988. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Francisco I, Albuquerque C, Lage P, Belo
H, Vitoriano I, Filipe B, Claro I, Ferreira S, Rodrigues P, Chaves
P, et al: Familial colorectal cancer type X syndrome: two distinct
molecular entities? Fam Cancer. 10:623–631. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Albuquerque C, Baltazar C, Filipe B, Penha
F, Pereira T, Smits R, Cravo M, Lage P, Fidalgo P, Claro I, et al:
Colorectal cancers show distinct mutation spectra in members of the
canonical WNT signaling pathway according to their anatomical
location and type of genetic instability. Genes Chromosomes Cancer.
49:746–759. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu W, Dong X, Mai M, Seelan RS, Taniguchi
K, Krishnadath KK, Halling KC, Cunningham JM, Boardman LA, Qian C,
et al: Mutations in AXIN2 cause colorectal cancer with defective
mismatch repair by activating beta-catenin/TCF signalling. Nat
Genet. 26:146–147. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Boland CR, Thibodeau SN, Hamilton SR,
Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA,
Fodde R, Ranzani GN and Srivastava S: A National Cancer Institute:
Workshop on Microsatellite Instability for cancer detection and
familial predisposition: development of international criteria for
the determination of microsatellite instability in colorectal
cancer. Cancer Res. 58:5248–5257. 1998.PubMed/NCBI
|
|
37
|
Umar A: Lynch syndrome (HNPCC) and
microsatellite instability. Dis Markers. 20:179–180. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nygren AO, Ameziane N, Duarte HM,
Vijzelaar RN, Waisfisz Q, Hess CJ, Schouten JP and Errami A:
Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG
methylation and copy number changes of up to 40 sequences. Nucleic
Acids Res. 33:e1282005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gylling A, Ridanpää M, Vierimaa O,
Aittomäki K, Avela K, Kääriäinen H, Laivuori H, Pöyhönen M,
Sallinen SL, Wallgren-Pettersson C, et al: Large genomic
rearrangements and germline epimutations in Lynch syndrome. Int J
Cancer. 124:2333–2340. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yamada A, Minamiguchi S, Sakai Y,
Horimatsu T, Muto M, Chiba T, Boland CR and Goel A: Colorectal
advanced neoplasms occur through dual carcinogenesis pathways in
individuals with coexisting serrated polyps. PLoS One.
9:e980592014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Filipe B, Baltazar C, Albuquerque C,
Fragoso S, Lage P, Vitoriano I, Mão de Ferro S, Claro I, Rodrigues
P, Fidalgo P, et al: APC or MUTYH mutations account for the
majority of clinically well-characterized families with FAP and
AFAP phenotype and patients with more than 30 adenomas. Clin Genet.
76:242–255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Albuquerque C, Bakker ER, van Veelen W and
Smits R: Colorectal cancers choosing sides. Biochim Biophys Acta.
1816:219–231. 2011.PubMed/NCBI
|
|
43
|
Christie M, Jorissen RN, Mouradov D,
Sakthianandeswaren A, Li S, Day F, Tsui C, Lipton L, Desai J, Jones
IT, et al: Different APC genotypes in proximal and distal sporadic
colorectal cancers suggest distinct WNT/β-catenin signalling
thresholds for tumourigenesis. Oncogene. 32:4675–4682. 2013.
View Article : Google Scholar
|
|
44
|
Leedham SJ, Rodenas-Cuadrado P, Howarth K,
Lewis A, Mallappa S, Segditsas S, Davis H, Jeffery R,
Rodriguez-Justo M, Keshav S, et al: A basal gradient of Wnt and
stem-cell number influences regional tumour distribution in human
and mouse intestinal tracts. Gut. 62:83–93. 2013. View Article : Google Scholar :
|
|
45
|
Plaschke J, Preussler M, Ziegler A and
Schackert HK: Aberrant protein expression and frequent allelic loss
of MSH3 in colorectal cancer with low-level microsatellite
instability. Int J Colorectal Dis. 27:911–919. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hatch SB, Lightfoot HM Jr, Garwacki CP,
Moore DT, Calvo BF, Woosley JT, Sciarrotta J, Funkhouser WK and
Farber RA: Microsatellite instability testing in colorectal
carcinoma: choice of markers affects sensitivity of detection of
mismatch repair-deficient tumors. Clin Cancer Res. 11:2180–2187.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Christmann M and Kaina B: Nuclear
translocation of mismatch repair proteins MSH2 and MSH6 as a
response of cells to alkylating agents. J Biol Chem.
275:36256–36262. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Sohn KJ, Choi M, Song J, Chan S, Medline
A, Gallinger S and Kim YI: Msh2 deficiency enhances somatic Apc and
p53 mutations in Apc+/−Msh2−/− mice.
Carcinogenesis. 24:217–224. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Oliveira C, Westra JL, Arango D,
Ollikainen M, Domingo E, Ferreira A, Velho S, Niessen R, Lagerstedt
K, Alhopuro P, et al: Distinct patterns of KRAS mutations in
colorectal carcinomas according to germline mismatch repair defects
and hMLH1 methylation status. Hum Mol Genet. 13:2303–2311. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mark SC, Sandercock LE, Luchman HA, Baross
A, Edelmann W and Jirik FR: Elevated mutant frequencies and
predominance of G:C to A:T transition mutations in Msh6(−/−) small
intestinal epithelium. Oncogene. 21:7126–7130. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wada R, Yamaguchi T and Tadokoro K:
Colonic Paneth cell metaplasia is pre-neoplastic condition of
colonic cancer or not? J Carcinog. 4:52005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wada R: Proposal of a new hypothesis on
the development of colorectal epithelial neoplasia:nonspecific
inflammation - colorectal Paneth cell metaplasia - colorectal
epithelial neoplasia. Digestion. 79(Suppl 1): 9–12. 2009.
View Article : Google Scholar
|
|
53
|
Andreu P, Peignon G, Slomianny C, Taketo
MM, Colnot S, Robine S, Lamarque D, Laurent-Puig P, Perret C and
Romagnolo B: A genetic study of the role of the Wnt/beta-catenin
signalling in Paneth cell differentiation. Dev Biol. 324:288–296.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sada M, Mitomi H, Igarashi M, Katsumata T,
Saigenji K and Okayasu I: Cell kinetics, p53 and bcl-2 expression,
and c-Ki-ras mutations in flat-elevated tubulovillous adenomas and
adenocarcinomas of the colorectum: comparison with polypoid
lesions. Scand J Gastroenterol. 34:798–807. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Maltzman T, Knoll K, Martinez ME, Byers T,
Stevens BR, Marshall JR, Reid ME, Einspahr J, Hart N, Bhattacharyya
AK, et al: Ki-ras proto-oncogene mutations in sporadic colorectal
adenomas: relationship to histologic and clinical characteristics.
Gastroenterology. 121:302–309. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jass JR, Baker K, Zlobec I, Higuchi T,
Barker M, Buchanan D and Young J: Advanced colorectal polyps with
the molecular and morphological features of serrated polyps and
adenomas: concept of a 'fusion' pathway to colorectal cancer.
Histopathology. 49:121–131. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
García-Solano J, Conesa-Zamora P,
Carbonell P, Trujillo-Santos J, Torres-Moreno DD, Pagán-Gómez I,
Rodríguez-Braun E and Pérez-Guillermo M: Colorectal serrated
adenocarcinoma shows a different profile of oncogene mutations, MSI
status and DNA repair protein expression compared to conventional
and sporadic MSI-H carcinomas. Int J Cancer. 131:1790–1799. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Feng Y, Bommer GT, Zhao J, Green M, Sands
E, Zhai Y, Brown K, Burberry A, Cho KR and Fearon ER: Mutant KRAS
promotes hyperplasia and alters differentiation in the colon
epithelium but does not expand the presumptive stem cell pool.
Gastroenterology. 141:1003–1013. e1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Carragher LA, Snell KR, Giblett SM,
Aldridge VS, Patel B, Cook SJ, Winton DJ, Marais R and Pritchard
CA: V600EBraf induces gastrointestinal crypt senescence and
promotes tumour progression through enhanced CpG methylation of
p16INK4a. EMBO Mol Med. 2:458–471. 2010. View Article : Google Scholar : PubMed/NCBI
|














