Introduction
Non-alcoholic fatty liver disease (NAFLD) and
non-alcoholic steatohepatitis (NASH), the advanced stage of NASH,
are common conditions associated with insulin resistance and
metabolic syndrome (1-3). In addition, NAFLD is associated with
an increased risk of cardiovascular disease (4). Under fasting conditions, the liver
produces glucose via gluconeogenesis in order to maintain blood
glucose at a constant level (5).
After meals, insulin converts glucose into glycogen and suppresses
gluconeogenesis to maintain the level of glycemia (6). However, in patients with NAFLD,
insulin cannot suppress gluconeogenesis and does not convert
glucose to glycogen after meals, a condition known as hepatic
insulin resistance. Previous studies found that the excessive
hepatic accumulation of triglycerides (TGs) and free fatty acids
(FFAs) induced hepatic insulin resistance (7,8).
Peroxisome proliferator-activated receptor γ (PPARγ)
is a member of the nuclear hormone receptor superfamily (9). PPARγ is abundantly expressed in
adipose tissue and plays a key role in lipid metabolism (10). Drugs activating PPARγ such as
thiazolidinediones (TZDs) reverse NAFLD (11,12). However, previous studies have
shown that PPARγ expression is increased in the liver of obese
patients and animals (13–15).
Thus, PPARγ may enhance hepatic steatosis under conditions of
obesity.
Cyclooxygenase (COX), which exists in two isoforms,
COX-1 and COX-2, plays an important role in the production of
eicosanoids (16). COX-1 is
expressed abundantly in many tissues. However, COX-2 is not usually
expressed in the resting state. Following exposure to a stimulus
such as inflammation, the expression of COX-2 increases (17–19). A recent study showed that
development of NAFLD in rats with diet-induced obesity was
suppressed by concomitant treatment with a COX-2 selective
inhibitor (20). However, the
mechanisms by which the COX-2 selective inhibitor suppressed NAFLD
were not fully explained.
In the present study, we aimed to elucidate the
detailed mechanisms through which nimesulide, a COX-2 selective
inhibitor, suppresses the development of NAFLD in a murine model of
high fat diet (HFD)-induced obesity.
Materials and methods
Animals
Male C57BL/6J mice (WT mice, 8 weeks old) were
purchased from CLEA Japan (Osaka, Japan). All animals were housed
in the Center for Laboratory Animal Care of Tottori University
School of Medicine (Yonago, Japan). All experimental procedures
were performed in accordance with Tottori University Animal Care
Guidelines, which conform to the NIH Guide for the Care and Use of
Laboratory Animals (NIH publication no. 85–23, revised in 1985).
Experimental protocols were approved by the Institutional Animal
Care and Use Committee of Faculty of Medicine of Tottori
University.
The mice were randomly assigned to three groups (6
mice/group) and fed either a normal chow diet (NC), a 32% HFD (CLEA
Japan) or a HFD plus nimesulide [HFD-nime; 66 mg/kg/day: nimesulide
was premixed into the 32% HFD (Cayman Chemical Co., Ann Arbor, MI,
USA)] ad libitum for 12 weeks. The concentration of
nimesulide mixed with powdery chow was calculated by measuring the
food consumption, which was monitored daily. Then, after a 12 h
fast, the animals were sacrificed by pentobarbital anesthesia
injection, and blood samples and the livers of these animals were
collected.
Oil Red O staining
The liver was isolated, embedded in Tissue-Tek 4583
Optimal Cutting Temperature compound (Sakura Finetek Japan Co.,
Ltd., Tokyo, Japan) and snap-frozen in liquid nitrogen. Cryostat
sections of mouse liver were washed in water for 5 min and then
stained with Oil Red O solution (Polysciences, Inc., Warrington,
PA, USA) for 30 min. Subsequently, the sections were counterstained
with hematoxylin (Muto Pure Chemicals Co., Ltd., Tokyo, Japan) for
1 min.
Measurement of areas of hepatic fibrosis
using Sirius red stain
Formalin-fixed, paraffin-embedded liver sections
(4-µm thick) were stained with picrosirius red (Sirius red)
and counter-stained with fast green (both from Chroma-Gesellschaft
Schmid GmbH & Co., Münster, Germany). The areas of hepatic
fibrosis were subsequently measured in 10 randomly selected fields
in each specimen (magnification, ×400; Olympus BX51N-34 microscope;
Olympus Corp., Tokyo, Japan) using WinROOF software (version 5.71;
Mitani Corporation, Tokyo, Japan).
Analysis of inflammatory cell
infiltration in liver tissue
The mature mouse cell surface glycoprotein F4/80,
which is expressed at high levels on Kupffer cells, was
immunohistochemically stained using a rat monoclonal anti-F4/80
mouse antibody (cat. no. ab6640; Abcam, Tokyo, Japan) diluted at
1:100 with 0.01 M/l phosphate-buffered saline (PBS), according to
the manufacturer's instructions. Goat anti-rat secondary antibody,
from the Histofine Simple Stain Mouse MAX-PO (Rat) kit (cat. no.
414311F; Nichirei Bioscience Inc., Tokyo, Japan) was used without
dilution. The immunopositive cells were analyzed in 10 intralobular
ocular fields (magnification, ×400; Olympus BX51N-34 microscope) in
each specimen.
Biochemical analysis
Blood samples were collected in microtainer tubes
(BD Biosciences, Tokyo, Japan) and centrifuged (9,000 × g, 10 min)
in order to obtain plasma. Plasma levels of glutamic pyruvic
transaminase (GPT)/alanine transaminase (ALT) and glutamic
oxaloacetic transaminase (GOT)/aspartate transaminase (AST) were
measured using DRI-CHEM 7000 Z (Fujifilm, Tokyo, Japan). The
hepatic TG content was determined using a commercially available TG
quantification kit (BioVision Inc., Mountain View, CA, USA) and a
microtiter plate reader (Sunrise rainbow RC-R; TECAN, Kawasaki,
Japan).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
The hepatic tissue samples were homogenized and
total RNA was extracted using the RNeasy Lipid Tissue Mini kit
(Qiagen, Hilden, Germany). The RNA concentrations were determined
by measuring the absorbance at 260 nm using a NanoDrop 1000
spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), and
the quality of RNA was confirmed by electrophoresis using ethidium
bromide-stained 1% agarose gels. Total RNA (2 µg) was
reverse transcribed in a final volume of 11.5 µl containing
4 µl 5X standard buffer, 2 µl 0.1 M dithiothreitol, 1
µl SuperScript II RNase H-reverse transcriptase (Invitrogen
Life Technologies, Carlsbad, CA, USA), 2 µl 10 M MdNTP
(Promega, Madison, WI, USA), 1 µl 50 pmol/µl Random
Primer (Promega), 0.5 µl 100 pmol/µl Oligo (dt)15
Primer (Promega) and 1 µl 40 U/µl ribonuclease
inhibitor (Wako Pure Chemical Industries, Ltd., Osaka, Japan). The
mixtures were incubated at 37°C for 60 min, 95°C for 5 min and then
cooled to 4°C for 5 min using a MyCycler Thermal Cycler (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Quantitative PCR assays (7900HT Fast Real-Time PCR
system; Applied Biosystems, Carlsbad, CA, USA) were performed in a
final volume of 10 ml containing 250 nM Universal ProbeLibrary
probe (Roche, Basel, Switzerland), 900 nM forward primer, 900 nM
reverse primer, 5 ml EXPRESS qPCR Supermix with Premixed Rox
(Invitrogen) and 2 ml cDNA. The mRNA levels of COX-1 (GenBank:
NM_008969), COX-2 (GenBank: NM_011198), PPARγ (mouse) (GenBank:
NM_011146), PPARγ (human) (GenBank: NM_015869), tissue inhibitor of
metallopro-teinases-1 (TIMP-1; GenBank: NM_011593), procollagen-1
(GenBank: U08020), transforming growth factor-β1 (TGF-β1; GenBank:
NM_011577) and monocyte chemoattractant protein-1 (MCP-1; GenBank:
NM_100127112) were assessed using the 7900HT Fast Real-Time PCR
system with SDS2.3 software (Applied Biosystems). Human β-actin
(GenBank: NM_001101) and mouse β-actin (GenBank: NM_007393) were
used as internal controls for RNA integrity. The PCR products were
eluted into ultra pure water using a QIAquick gel extraction kit
(Qiagen), and a standard curve was prepared using serial dilutions.
The following thermal cycle conditions were used: 95°C for 20 sec
followed by 45 cycles of 1 sec at 95°C and 20 sec at 60°C. The
relative mRNA expression levels were calculated using the
2−ΔΔCT method.
Measurement of hepatic
15-deoxy-Δ12,14-prostaglandin J2
(15d-PGJ2)
The hepatic content of 15d-PGJ2 was
quantified using a commercially available enzyme-linked
immunosorbent assay (Enzo Life Sciences, Tokyo, Japan) in
accordance with the manufacturer's instructions.
Cell culture and 15d-PGJ2
treatment
Human hepatocarcinoma HepG2 cells (a gift from
Professor Shiota, Division of Molecular and Genetic Medicine,
Tottori University, Yonago, Japan) were seeded (30,000
cells/cm2) and maintained for 24 h in DMEM (Wako Pure
Chemical Industries, Ltd.) supplemented with 2 mM glutamine, 1%
penicillin/streptomycin solution and 10% FBS, at 37°C in a 5%
CO2 humidified atmosphere. The compound
15d-PGJ2 was then added at a concentration of 5 or 10
μM and the cells were incubated for a further 48 h. The
solvent DMSO was used as a vehicle control. RNA was extracted using
an RNeasy mini kit, and the expression of β-actin and PPARγ was
quantified using the real-time RT-PCR system.
Intraperitoneal glucose tolerance test
(ipGTT) and plasma insulin measurement
To assess glucose tolerance after 12 weeks, the mice
were injected with glucose (1 mg/g body weight) intraperitoneally
after a 12-h fast. Blood samples were drawn from the tail vein at
baseline and at 15, 30, 60, 90 and 120 min after the injection. The
blood glucose level was measured using an automatic blood glucose
meter (Glucose Pilot; Iwai Chemicals Ltd., Tokyo, Japan). Plasma
insulin levels was measured using a Mercodia mouse insulin EIA kit
(Mercodia Inc., Winston-Salem, NC, USA).
Statistical analysis
Data were processed using StatView J-4.5 software
(SAS Institute Inc., Cary, NC, USA). Values are expressed as the
means ± standard error of mean (SEM). Values of groups were
analyzed by one-way ANOVA with post-hoc tests for multiple
comparisons. Perfusion recovery among groups was assessed using
ANOVA with repeated measures. The Student's t-test was used for
comparisons between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Animal model of obesity and NAFLD
As shown in Fig.
1, the body weights of the mice in the HFD group significantly
increased compared with those in the NC group. Histopathologically,
Oil Red O-positive areas, which indicated TG accumulation in the
liver, were clearly larger in the HFD group (Fig. 1B). COX-2 mRNA expression in the
liver was significantly increased in the HFD group compared with
the NC group, although no difference was observed in COX-1 mRNA
expression between the two groups (Fig. 1C and D).
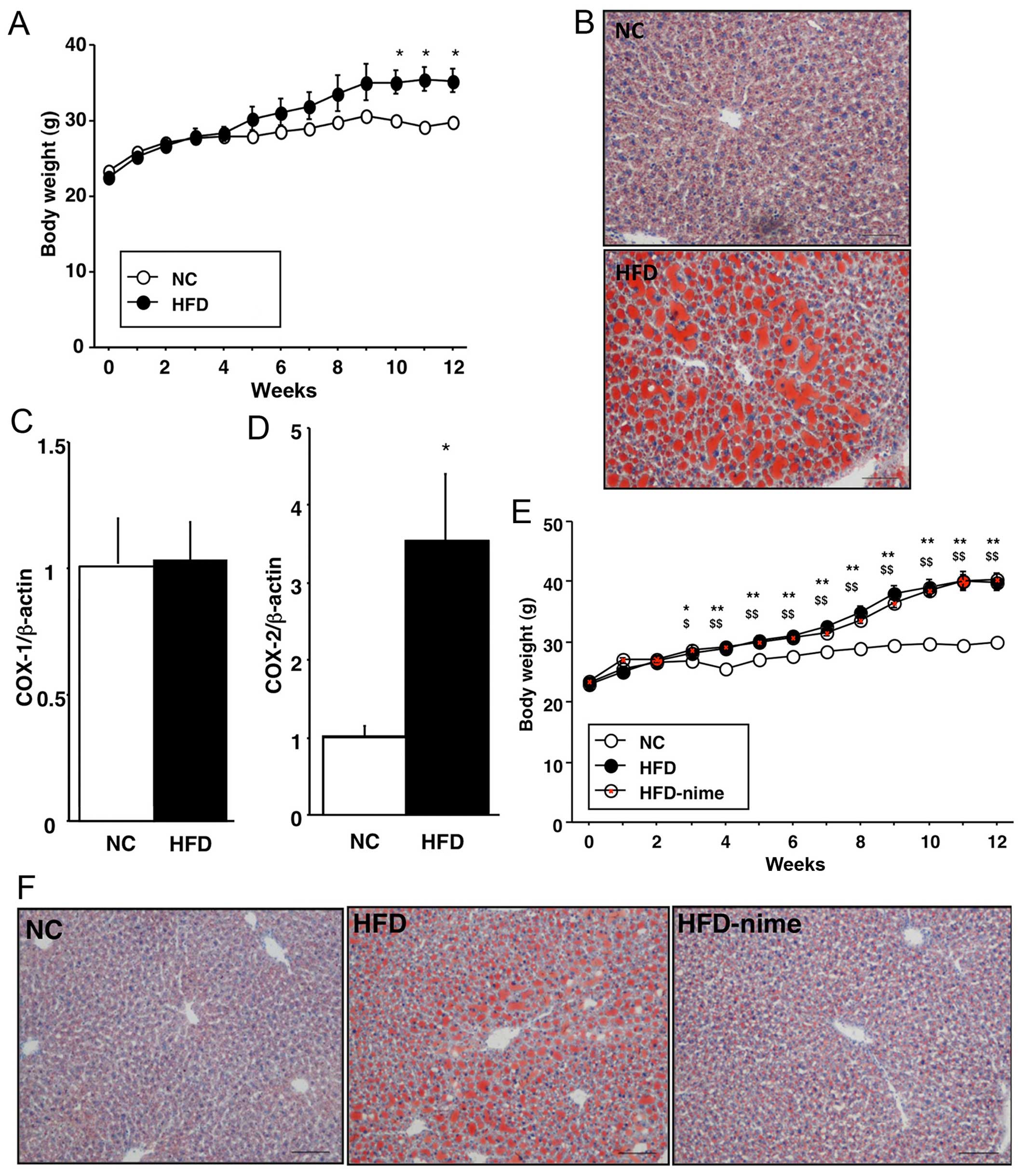 | Figure 1(A) Weekly body weight measurements
of the mice in the NC and HFD groups (n=3 per group). (B) Oil Red O
staining of the liver in the NC and HFD groups (original
magnification, ×100). (C and D) Cyclooxygenase (COX)-1 and -2 mRNA
expression in the livers of mice in the NC and HFD groups (n=9 per
group. *P<0.05 vs. NC. (E) Weekly body weight
measurements of the mice in the NC, HFD and HFD-nime groups (n=6
per group). *P<0.05 HFD vs. NC, **P<0.01 HFD vs.
NC, $P<0.05 HFD-nime vs. NC, $$P<0.01
HFD-nime vs. NC. (F) Oil Red O staining of the liver in the NC, HFD
and HFD-nime groups (original magnification, ×100). NC, normal chow
diet; HFD, high fat diet; HFD-nime, high fat plus nimesulide. |
Effect of the COX-2 selective inhibitor
on body weight and NAFLD
The body weights of mice in the HFD-nime group
increased and were comparable with those in the HFD group (Fig. 1E). Histopathological findings
indicated that TG accumulation was suppressed in the HFD-nime group
compared with that in the HFD group (Fig. 1F). Similarly, the increased
hepatic TG content observed in the HFD group was also significantly
suppressed by nimesulide (Fig.
2A). Plasma GPT levels in the HFD group were significantly
increased compared with those in the NC group. Nimesulide
significantly suppressed the increase of plasma GPT in the HFD
group (Fig. 2B). Plasma GOT
levels exhibited a similar tendency although they did not reach a
statistically significant level (Fig.
2C).
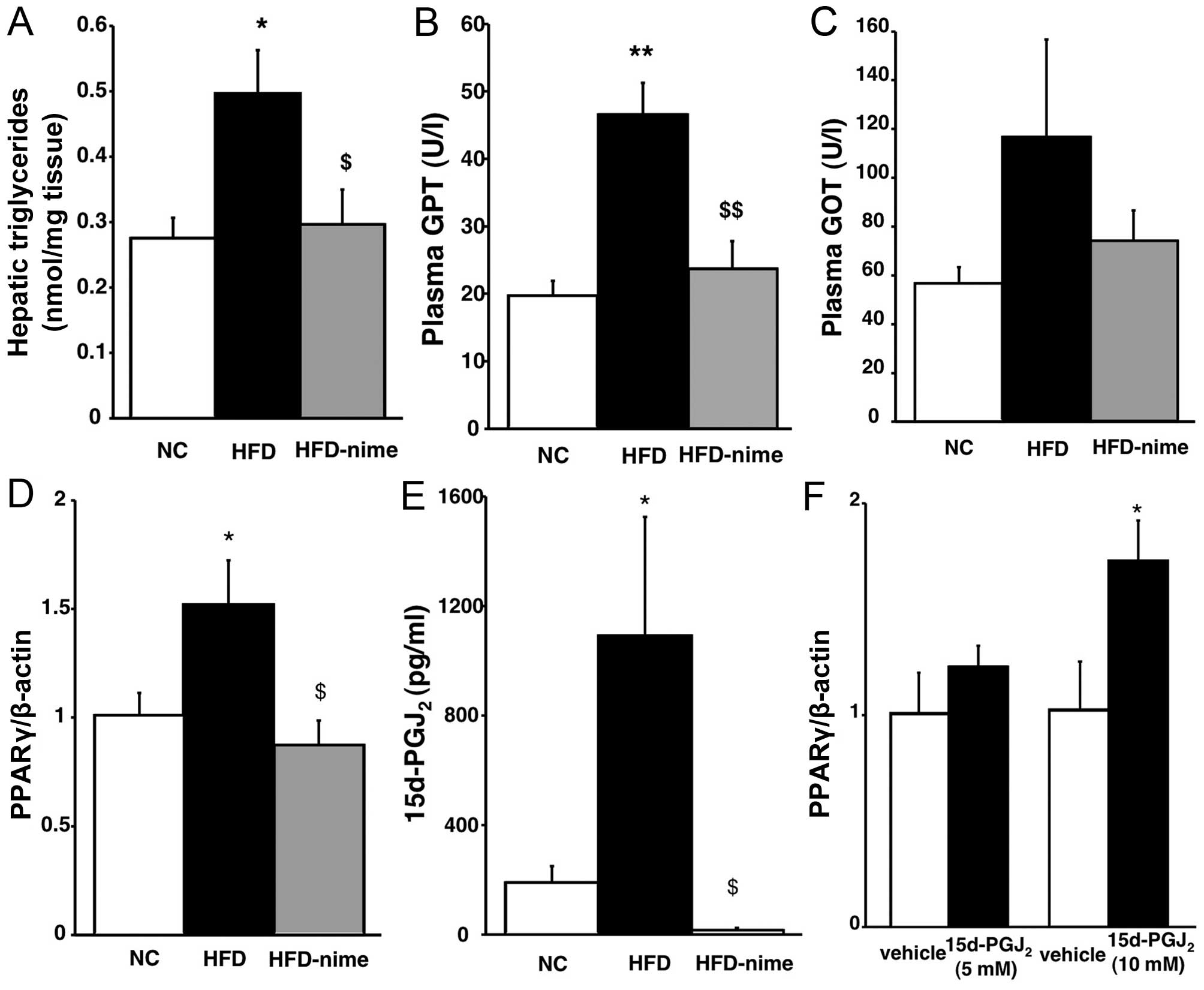 | Figure 2(A) Triglyceride content in the
livers of mice in the three groups (n=5 per group). (B and C)
Plasma levels of GPT and GOT in the three groups (n=6 per group).
*P<0.05 vs. NC, **P<0.01 vs. NC,
$P<0.05 vs. HFD, $$P<0.01 vs. HFD. (D)
PPARγ mRNA expression in the livers of mice in the NC, HFD and
HFD-nime groups (n=6 per group). (E) 15d-PGJ2 content in
the liver of mice in the three groups (n=6 per group).
*P<0.05 vs. NC, $P<0.01 vs. HFD. (F)
PPARγ mRNA expression at 48 h after treatment with
15d-PGJ2 (n=6 per group). *P<0.05 vs.
vehicle. GPT, Plasma glutamic pyruvic transaminase; GOT, glutamic
oxaloacetic transaminase; NC, normal chow diet; HFD, high fat diet;
HFD-nime, high fat plus nimesulide; PPARγ, peroxisome
proliferator-activated receptor γ; 15d-PGJ2,
15-deoxy-Δ12,14-prostaglandin J2. |
Hepatic PPARγ mRNA expression and
5d-PGJ2 content
PPARγ mRNA expression in the liver was significantly
increased in the HFD group whereas nimesulide significantly
suppressed PPARγ mRNA expression in the HFD-nime group (Fig. 2D). The findings of previous
studies suggested that the mRNA expression and activation of PPARγ
were regulated by 15d-PGJ2, which is one of the
metabolites of COX-2 and a PPARγ natural agonist, in various types
of cell (21,22). Therefore, we examined whether
15d-PGJ2 was increased in the livers of obese mice, and
whether it was suppressed by nimesulide, using an enzyme-linked
immunosorbent assay. As shown in Fig.
2E, the hepatic 15d-PGJ2 content was higher in the
HFD group than in the NC group, and nimesulide completely
suppressed 15d-PGJ2 content in the liver. In order to
determine whether 15d-PGJ2 enhanced the expression of
PPARγ at the mRNA and protein level, we performed in vitro
experiments using human hepatocarcinoma HepG2 cells. As shown in
Fig. 2F, 15d-PGJ2
enhanced the mRNA expression of PPARγ in HepG2 cells in a
dose-dependent manner and 10 µM 15d-PGJ2
significantly increased the mRNA expression of PPARγ.
Effect of the COX-2 selective inhibitor
on glucose metabolism in obese mice
Previous studies have suggested that NAFLD is a risk
factor for disordered glucose metabolism (1–3).
Therefore, in order to determine whether nimesulide is capable of
reversing disordered glucose metabolism, we performed an ipGTT. The
mice in the HFD group showed impaired glucose tolerance, but this
was significantly ameliorated by the administration of nimesulide
(Fig. 3A). In addition, the mice
in the HFD group developed hyperinsulinemia, which was also
suppressed by nimesulide (Fig.
3B).
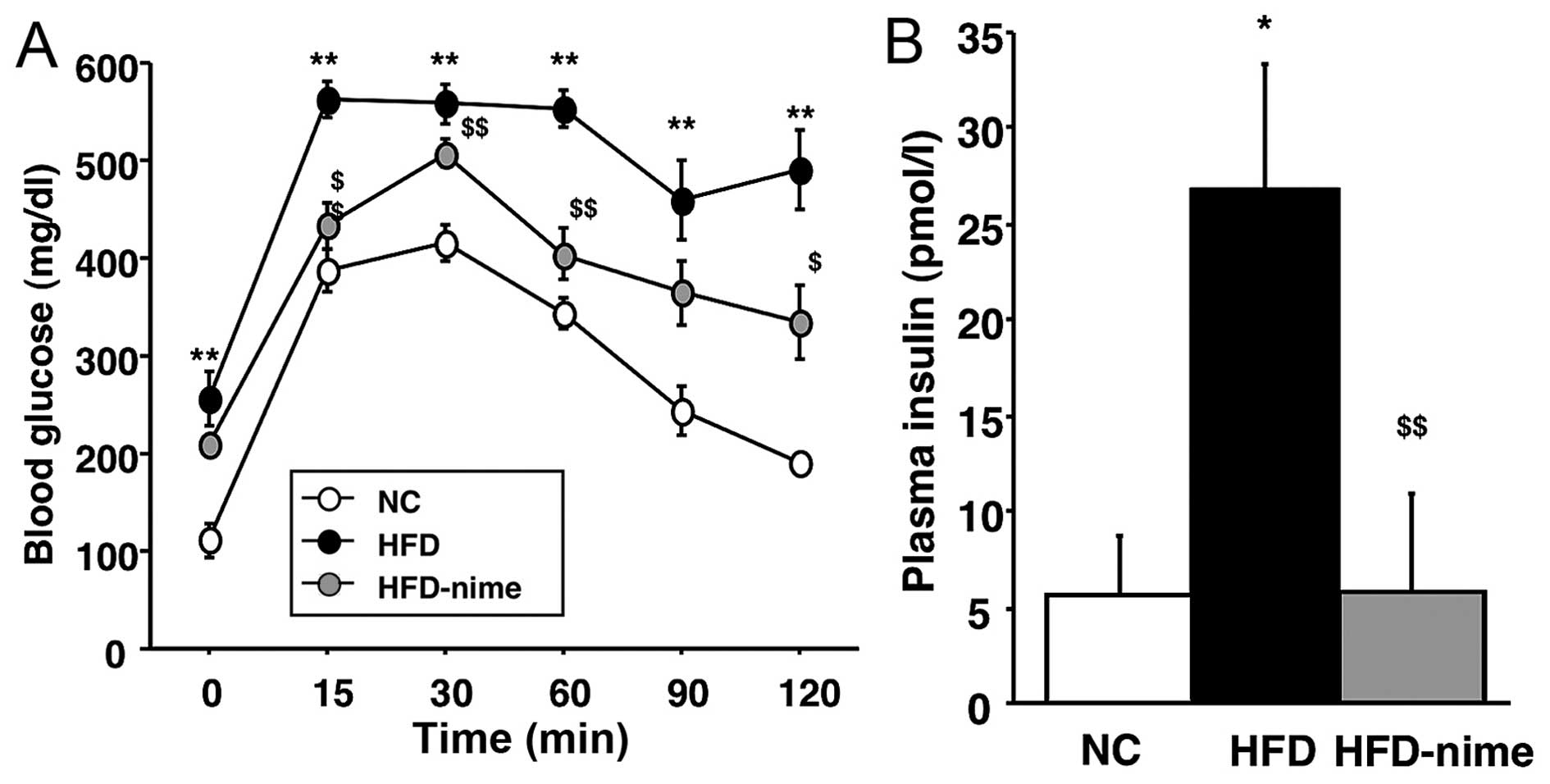 | Figure 3(A) Measurement of blood glucose by
the ipGTT. Blood glucose was measured prior to glucose injection
and at 15, 30, 60, 90, 120 min thereafter. (n=6 per group). (B)
Fasting blood insulin concentration in the NC, HFD and HFD-nime
groups (n=5, 6, 6, respectively). *P<0.05 vs. NC,
**P<0.01 vs. NC, $P<0.05 vs. HFD,
$$P<0.01 vs. HFD. ipGTT, intraperitoneal glucose
tolerance test; NC, normal chow diet; HFD, high fat diet; HFD-nime,
high fat plus nimesulide. |
Effects of COX-2 on hepatic fibrosis
There was no significant difference in the extent of
the area immunostained with Sirius red between the NC group and the
HFD group. Furthermore, the administration of nimesulide did not
significantly affect the area of Sirius red immunostaining
(Fig. 4A and C). The mRNA
expression of TIMP-1 and procollagen-1 in the liver were
significantly increased in the HFD group compared with the NC group
(Fig. 5A and B). Nimesulide
significantly suppressed the mRNA expression of TIMP-1 and
procollagen-1 in the HFD-nime group (Fig. 5A and B). However, nimesulide did
not alter the mRNA expression of TGF-β1 (Fig. 5C).
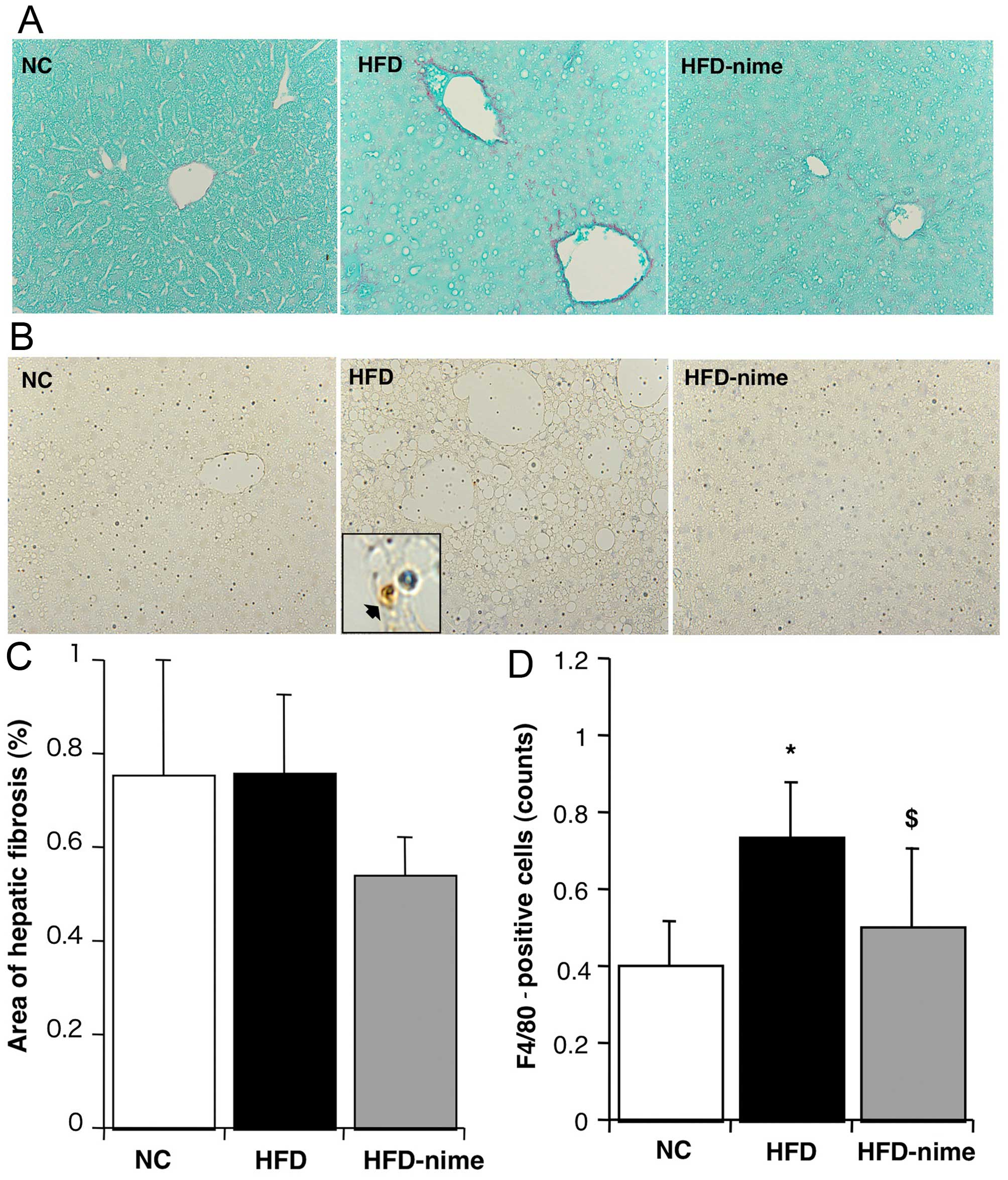 | Figure 4(A and C) Areas of hepatic fibrosis
determined by Sirius red immunostaining. Areas of Sirius red
immunostaining (original magnification, ×400) measured using image
analysis (n=6, 6, 8, respectively). Areas of hepatic fibrosis did
not change among the groups. (B) F4/80 immunostaining for Kupffer
cells (original magnification, ×400, and within the box, high
maginification ×1,000, immunopositive-F4/80 cell denoted by black
arrow). (D) Numbers of immunopositive-F4/80 cells in each group
(n=6, 6, 8, respectively). *P<0.05 vs. NC,
$P<0.05, vs. HFD. NC, normal chow diet; HFD, high fat
diet; HFD-nime, high fat plus nimesulide. |
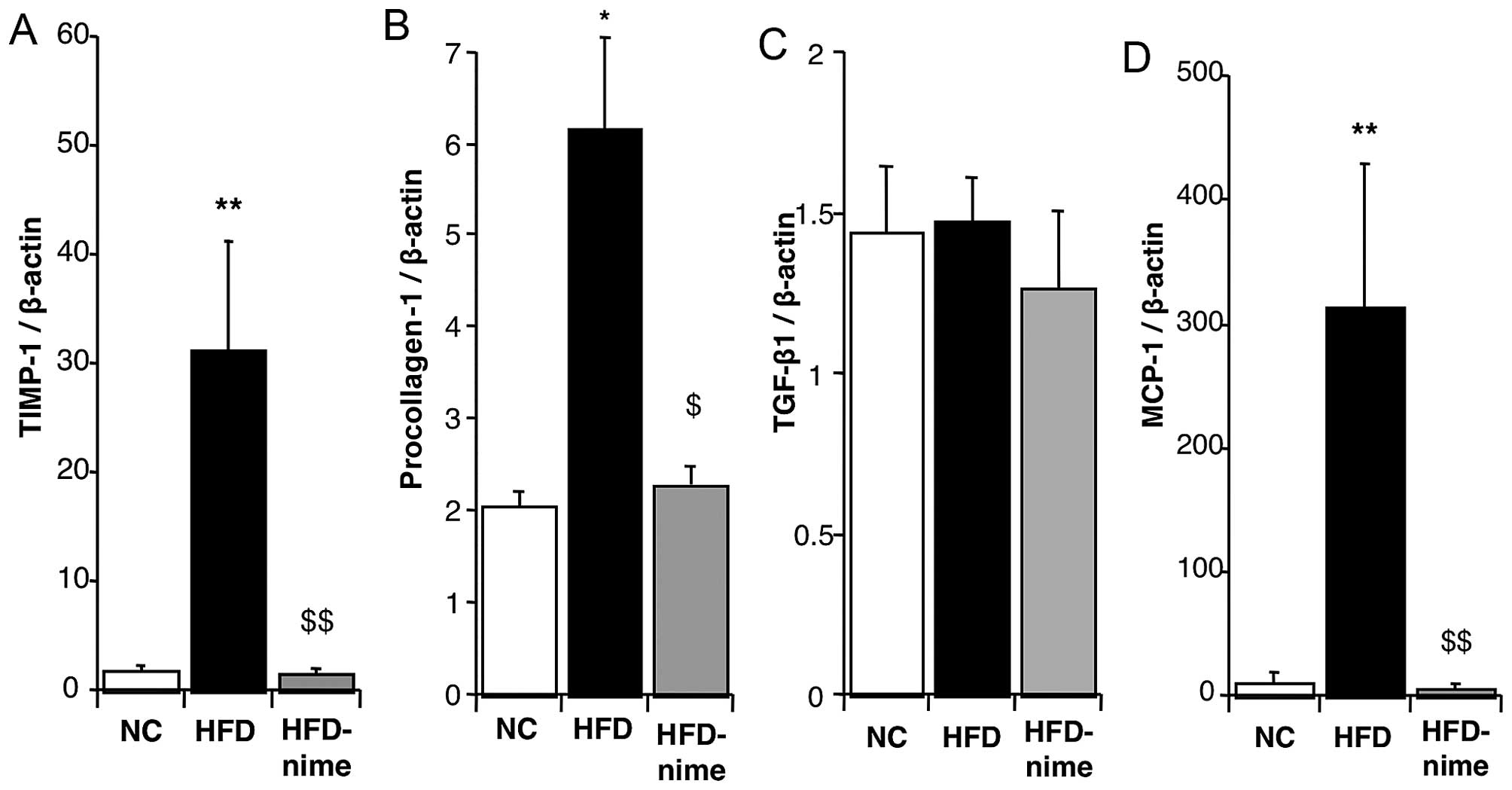 | Figure 5(A) TIMP-1 mRNA expression in the
liver of mice in the NC, HFD and HFD-nime groups (n=3, 6, 8,
respectively). (B) Procollagen-1 mRNA expression in the liver of
mice in the NC, HFD and HFD-nime groups (n=3, 6 8, respectively).
(C) TGF-β1 mRNA expression in the liver of mice in the NC, HFD and
HFD-nime groups (n=3, 6, 8, respectively). (D) MCP-1 mRNA
expression in the liver of mice in the NC, HFD and HFD-nime groups
(n=3, 6, 8, respectively). *P<0.05 vs. NC,
**P<0.01 vs. NC, $P<0.05 vs. HFD,
$$P<0.01 vs. HFD. TIMP-1, tissue inhibitor of
metalloproteinases-1; NC, normal chow diet; HFD, high fat diet;
HFD-nime, high fat plus nimesulide, TGF-β1, transforming growth
factor-β1; MCP-1, monocyte chemoattractant protein-1. |
Effect of COX-2 on inflammatory reactions
in the liver
The number of F4/80-positive Kupffer cells was
significantly increased in the HFD group (Fig. 4B and D). Nimesulide significantly
suppressed the number of F4/80-positive cells in the HFD-nime group
(Fig. 4B and D). Similarly, the
mRNA expression of MCP-1 in the liver was significantly increased
in the HFD group and significantly suppressed by nimesulide in the
HFD-nime group (Fig. 5D).
Discussion
A previous study showed that COX-2 selective
inhibitors suppressed hepatic steatosis in rats with HFD-induced
obesity (20). However, the
detailed mechanism responsible for this effect was not elucidated.
PPARγ, which belongs to the PPAR family of nuclear receptors, is
mainly expressed in adipose tissue and it plays an important role
in lipid metabolism (10). In
previous studies, PPARγ expression was found to be increased in the
liver of obese rats and to be involved in the development of NAFLD
(13–15,23,24). In the present study, we confirmed
that the mRNA expression of PPARγ was increased in the livers of
mice with HFD-induced obesity, and that it was suppressed by
nimesulide (Fig. 2D). The
histopathological examination of mouse liver tissues showed that
nimesulide suppressed hepatic steatosis (Fig. 1F). These results suggest that the
COX-2 selective inhibitor suppressed hepatic steatosis by
suppressing the expression and activation of PPARγ.
15d-PGJ2 is a prostanoid derived from
prostaglandin D2 and it is a natural PPARγ agonist
(25). In the current study, we
showed that the 15d-PGJ2 concentration was increased in
the livers of mice with HFD-induced obesity. The results of our
in vitro experiment as well as those of a previous study
revealed that 15d-PGJ2 increased not only PPARγ activity
but also its expression in hepatocytes (26). PPARγ increases lipogenic gene
expression, such as fatty acid synthase and sterol regulatory
element-binding protein-1, as evidenced by increased levels of a
lipogenic gene, and induces lipid accumulation (23,27,28). Moreover, hepatocyte-specific
PPARγ-deficient mice showed decreased lipogenic gene expression,
and did not accumulate fat in the liver despite consuming an HFD
(29). As a result, it was
hypothesized that 15d-PGJ2 stimulates the expression and
activation of PPARγ in the livers of mice with HFD-induced obesity,
and that NAFLD development is mediated by the increased expression
of this lipogenic gene. However, in clinical cases, TZDs, which are
well-known PPARγ activators, are often used to treat NAFLD and
diabetes mellitus (11,12). It remains unclear whether TZDs
ameliorate hepatic steatosis as a consequence of their primary
insulin-sensitizing effects on adipose tissue (12). However, the following findings of
previous studies suggest that the beneficial effects of TZDs on
NAFLD and insulin resistance were induced by the activation of
PPARγ in the adipose tissue, rather than in the liver or striated
muscle. Transgenic mice characterized by adipocyte-specific PPARγ
activation showed reduced insulin resistance, similar to that
observed in a model of mice with HFD-induced obesity treated with
TZDs. Muscle-specific PPARγ-deficient mice showed improved free
fatty acid metabolism and insulin resistance under treatment with
TDZs (30–32). In addition, in liver-specific
PPARγ-deficient mice, the development of HFD-induced NAFLD and
insulin resistance was suppressed (29). Therefore, we suggest that the
presence of PPARγ in adipose tissue is important in the treatment
of NAFLD and insulin resistance, and that PPARγ in the liver plays
a crucial role in the development of NAFLD in mice with HFD-induced
obesity.
It is well-known that insulin regulates
gluconeogenesis and glycogen synthesis in the liver to maintain the
blood glucose levels (6,33). Previous studies suggested that the
excessive accumulation of TGs or FFAs in the liver suppressed the
metabolic pathway of glucose by activating protein kinase Cε (PKCε)
(34,35), thereby leading to hepatic insulin
resistance and disorders of glucose metabolism. In addition,
ezetimibe, which is known to prevent TG accumulation, suppresses
the development of NAFLD in the livers of obese Zucker rats
(36,37). Thus, the inhibition of fat
accumulation in the liver reduces hepatic insulin resistance. In
this study, the HFD group showed impaired glucose metabolism and
this was ameliorated in the HFD-nime group (Fig. 3). Consequently, nimesulide may
reverse hepatic insulin resistance by suppressing the development
of NAFLD. In addition, a study showed that prostaglandin
E2 (PGE2), which is one of the main products
of COX-2, also induced hepatic insulin resistance via its receptor
EP3β (38). Thus, the increased
expression of COX-2 in the livers of obese mice may be one of the
main contributors to excessive NAFLD and hepatic insulin
resistance.
In the present study, it was found that nimesulide
attenuated hepatic inflammation as another factor involved in the
second hit of the progress of NAFLD. Nimesulide reduced the number
of Kupffer cells infiltrating the liver and decreased the gene
expression of MCP-1, which directs trafficking of immune cell to
sites of tissue damage. The products of lipid peroxidation can
mediate inflammatory recruitment by activating NF-κB and COX-2
(39). The activation of NF-κB
was accompanied by the increased hepatic expression of several
inflammatory cytokines [interleukin (IL)-1β, TNF-α and IL-6] and by
an increase in plasma MCP-1 levels, all known to be NF-κB-dependent
inflammatory cytokines (40).
Kupffer cells are crucial for the early development of NASH by
promoting blood monocyte infiltration through the production of
interferon γ-induced protein-10 and MCP-1 (41). MCP-1 gene expression and the
number of Kupffer cells infiltrating the liver were reduced by
nimesulide and thereby regulated inflammation.
In the present study, we examined whether nimesulide
attenuated the progression of liver fibrosis. As a HFD did not
induce immunohistological liver fibrosis, the effect of nimesulide
on liver fibrosis could not be examined. However, the increased
hepatic gene expression of TIMP-1 and procol-lagen-1 in the HFD
group were suppressed by nimesulide. These findings suggest that
nimesulide may attenuate hepatic fibrogenesis in NAFLD.
Notably, the body weights of the mice in the
HFD-nime group were comparable with those in the HFD group whereas
hepatic TG contents were decreased by nimesulide administration
almost to the control level. It has been previously reported that
in rats fed a HFD in combination with celecoxib (COX-2 selective
inhibitor), TG accumulation in the liver was attenuated, without
affecting body weight (20).
Thus, these findings taken together with the results of the present
study, suggest that the reduction of hepatic TG content by
nimesulide does not affect body weight. However, further
experiments are necessary to examine this possibility in more
detail.
This study has some limitations. Firstly, a clear,
casual association between the decrease in hepatic
15d-PGJ2 content and histological amelioration of NAFLD
by pre-treatment with nimesulide in vivo was not shown,
although it was demonstrated that 15d-PGJ2 induced PPARγ
expression in HepG2 cells. This suggests that other factors may
contribute to the histological amelioration obtained with
nimesulide. Secondly, it has been shown that the upregulation of
COX-2 in adipocytes increases the production of inflammatory
PGE2, leading to insulin resistance (20). However, the effects of nimesulide
on adipose tissue were not examined in the present study. Examining
the effects of nimesulide on 15d-PGJ2 in adipose tissue
would further clarify the amelioration observed in this murine
model of NAFLD. Thirdly, the reduction in insulin resistance
induced by nimesulide may also be attributed to the ability of
nimesulide to restore hepatic TG accumulation. Further experiments
concerning hepatic glucose output or hepatic insulin receptor
substrate phosphorylation are necessary for confirmation. Fourthly,
our study provided no information that would confirm whether
hepatocytic COX-2 and 15d-PGJ2 are upregulated in
steatohepatitis and successfully blocked by nimesulide, which is a
clear limitation. Finally, information to validate the functions of
PPARγ on hepatocytes is lacking in the present study as the
expression of lipogenesis/lipid oxidation genes was not evaluated
in vivo or in vitro and the production of reactive
oxygen species was not examined in hepatocytes.
In conclusion, we suggest that COX-2 expression in
the livers of mice with HFD-induced obesity is involved in the
development of NAFLD through the 15d-PGJ2-PPARγ pathway.
The inhibition of COX-2 by nimesulide suppressed hepatic
inflammation and fibrogenesis, and reduced insulin resistance in
NAFLD. Nimesulide may contribute to the treatment of NAFLD and
metabolic syndrome. Thus, COX-2 may emerge as a molecular target
for preventing the development of NAFLD and insulin resistance in
diet-related obesity.
References
|
1
|
Angulo P: Nonalcoholic fatty liver
disease. N Engl J Med. 346:1221–1231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pagano G, Pacini G, Musso G, Gambino R,
Mecca F, Depetris N, Cassader M, David E, Cavallo-Perin P and
Rizzetto M: Nonalcoholic steatohepatitis, insulin resistance, and
metabolic syndrome: further evidence for an etiologic association.
Hepatology. 35:367–372. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Petersen KF, Dufour S, Befroy D, Lehrke M,
Hendler RE and Shulman GI: Reversal of nonalcoholic hepatic
steatosis, hepatic insulin resistance, and hyperglycemia by
moderate weight reduction in patients with type 2 diabetes.
Diabetes. 54:603–608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scorletti E, Calder PC and Byrne CD:
Non-alcoholic fatty liver disease and cardiovascular risk:
metabolic aspects and novel treatments. Endocrine. 40:332–343.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Angrand PO, Coffinier C and Weiss MC:
Response of the phosphoenolpyruvate carboxykinase gene to
glucocorticoids depends on the integrity of the cAMP pathway. Cell
Growth Differ. 5:957–966. 1994.PubMed/NCBI
|
|
6
|
Cross DA, Alessi DR, Cohen P, Andjelkovich
M and Hemmings BA: Inhibition of glycogen synthase kinase-3 by
insulin mediated by protein kinase B. Nature. 378:785–789. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Samuel VT, Petersen KF and Shulman GI:
Lipid-induced insulin resistance: unravelling the mechanism.
Lancet. 375:2267–2277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Greenberg AS, Coleman RA, Kraemer FB,
McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H and Mashek DG: The
role of lipid droplets in metabolic disease in rodents and humans.
J Clin Invest. 121:2102–2110. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Evans RM, Barish GD and Wang YX: PPARs and
the complex journey to obesity. Nat Med. 10:355–361. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rosen ED, Hsu CH, Wang X, Sakai S, Freeman
MW, Gonzalez FJ and Spiegelman BM: C/EBPalpha induces adipogenesis
through PPARgamma: a unified pathway. Genes Dev. 16:22–26. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ratziu V, Giral P, Jacqueminet S,
Charlotte F, Hartemann-Heurtier A, Serfaty L, Podevin P, Lacorte
JM, Bernhardt C, Bruckert E, et al LIDO Study Group: Rosiglitazone
for nonalcoholic steatohepatitis: one-year results of the
randomized placebo-controlled Fatty Liver Improvement with
Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 135:100–110.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ratziu V, Charlotte F, Bernhardt C, Giral
P, Halbron M, Lenaour G, Hartmann-Heurtier A and Bruckert E:
Long-term efficacy of rosiglitazone in nonalcoholic
steato-hepatitis: results of the fatty liver improvement by
rosiglitazone therapy (FLIRT 2) extension trial. Hepatology.
51:445–453. 2010. View Article : Google Scholar
|
|
13
|
Pettinelli P and Videla LA: Up-regulation
of PPAR-gamma mRNA expression in the liver of obese patients: an
additional reinforcing lipogenic mechanism to SREBP-1c induction. J
Clin Endocrinol Metab. 96:1424–1430. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Edvardsson U, Ljungberg A and Oscarsson J:
Insulin and oleic acid increase PPARgamma2 expression in cultured
mouse hepatocytes. Biochem Biophys Res Commun. 340:111–117. 2006.
View Article : Google Scholar
|
|
15
|
Matsusue K, Kusakabe T, Noguchi T,
Takiguchi S, Suzuki T, Yamano S and Gonzalez FJ: Hepatic steatosis
in leptin-deficient mice is promoted by the PPARgamma target gene
Fsp27. Cell Metab. 7:302–311. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dubois RN, Abramson SB, Crofford L, Gupta
RA, Simon LS, Van De Putte LB and Lipsky PE: Cyclooxygenase in
biology and disease. FASEB J. 12:1063–1073. 1998.PubMed/NCBI
|
|
17
|
Murakami M, Matsumoto R, Austen KF and Arm
JP: Prostaglandin endoperoxide synthase-1 and -2 couple to
different transmembrane stimuli to generate prostaglandin D2 in
mouse bone marrow-derived mast cells. J Biol Chem. 269:22269–22275.
1994.PubMed/NCBI
|
|
18
|
Anderson GD, Hauser SD, McGarity KL,
Bremer ME, Isakson PC and Gregory SA: Selective inhibition of
cyclooxygenase (COX)-2 reverses inflammation and expression of
COX-2 and interleukin 6 in rat adjuvant arthritis. J Clin Invest.
97:2672–2679. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ricciotti E and FitzGerald GA:
Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol.
31:986–1000. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsieh PS, Jin JS, Chiang CF, Chan PC, Chen
CH and Shih KC: COX-2-mediated inflammation in fat is crucial for
obesity-linked insulin resistance and fatty liver. Obesity (Silver
Spring). 17:1150–1157. 2009.
|
|
21
|
Muzio G, Trombetta A, Maggiora M,
Martinasso G, Vasiliou V, Lassen N and Canuto RA: Arachidonic acid
suppresses growth of human lung tumor A549 cells through
down-regulation of ALDH3A1 expression. Free Radic Biol Med.
40:1929–1938. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Avis I, Martínez A, Tauler J, Zudaire E,
Mayburd A, Abu-Ghazaleh R, Ondrey F and Mulshine JL: Inhibitors of
the arachidonic acid pathway and peroxisome proliferator-activated
receptor ligands have superadditive effects on lung cancer growth
inhibition. Cancer Res. 65:4181–4190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schadinger SE, Bucher NL, Schreiber BM and
Farmer SR: PPARgamma2 regulates lipogenesis and lipid accumulation
in steatotic hepatocytes. Am J Physiol Endocrinol Metab.
288:E1195–E1205. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gavrilova O, Haluzik M, Matsusue K, Cutson
JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ and
Reitman ML: Liver peroxisome proliferator-activated receptor gamma
contributes to hepatic steatosis, triglyceride clearance, and
regulation of body fat mass. J Biol Chem. 278:34268–34276. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Forman BM, Tontonoz P, Chen J, Brun RP,
Spiegelman BM and Evans RM: 15-Deoxy-delta 12, 14-prostaglandin J2
is a ligand for the adipocyte determination factor PPAR gamma.
Cell. 83:803–812. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maggiora M, Oraldi M, Muzio G and Canuto
RA: Involvement of PPARα and PPARγ in apoptosis and proliferation
of human hepatocarcinoma HepG2 cells. Cell Biochem Funct.
28:571–577. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lessard SJ, Rivas DA, Chen ZP, Bonen A,
Febbraio MA, Reeder DW, Kemp BE, Yaspelkis BB III and Hawley JA:
Tissue-specific effects of rosiglitazone and exercise in the
treatment of lipid-induced insulin resistance. Diabetes.
56:1856–1864. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Djaouti L, Jourdan T, Demizieux L, Chevrot
M, Gresti J, Vergès B and Degrace P: Different effects of
pioglitazone and rosiglitazone on lipid metabolism in mouse
cultured liver explants. Diabetes Metab Res Rev. 26:297–305. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Morán-Salvador E, López-Parra M,
García-Alonso V, Titos E, Martínez-Clemente M, González-Périz A,
López-Vicario C, Barak Y, Arroyo V and Clària J: Role for PPARγ in
obesity-induced hepatic steatosis as determined by hepatocyte and
macrophage-specific conditional knockouts. FASEB J. 25:2538–2550.
2011. View Article : Google Scholar
|
|
30
|
Sugii S, Olson P, Sears DD, Saberi M,
Atkins AR, Barish GD, Hong SH, Castro GL, Yin YQ, Nelson MC, et al:
PPARgamma activation in adipocytes is sufficient for systemic
insulin sensitization. Proc Natl Acad Sci USA. 106:22504–22509.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hevener AL, He W, Barak Y, Le J,
Bandyopadhyay G, Olson P, Wilkes J, Evans RM and Olefsky J:
Muscle-specific Pparg deletion causes insulin resistance. Nat Med.
9:1491–1497. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Norris AW, Chen L, Fisher SJ, Szanto I,
Ristow M, Jozsi AC, Hirshman MF, Rosen ED, Goodyear LJ and Gonzalez
FJ: Muscle-specific PPARgamma-deficient mice develop increased
adiposity and insulin resistance but respond to thiazolidinediones.
J Clin Invest. 112:608–618. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Puigserver P, Rhee J, Donovan J, Walkey
CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D
and Spiegelman BM: Insulin-regulated hepatic gluconeogenesis
through FOXO1-PGC-1alpha interaction. Nature. 423:550–555. 2003.
View Article : Google Scholar
|
|
34
|
Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S,
Befroy D, Romanelli AJ and Shulman GI: Mechanism of hepatic insulin
resistance in non-alcoholic fatty liver disease. J Biol Chem.
279:32345–32353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Samuel VT, Liu ZX, Wang A, Beddow SA,
Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S and Shulman GI:
Inhibition of protein kinase Cepsilon prevents hepatic insulin
resistance in nonalcoholic fatty liver disease. J Clin Invest.
117:739–745. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Deushi M, Nomura M, Kawakami A, Haraguchi
M, Ito M, Okazaki M, Ishii H and Yoshida M: Ezetimibe improves
liver steatosis and insulin resistance in obese rat model of
metabolic syndrome. FEBS Lett. 581:5664–5670. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nomura M, Ishii H, Kawakami A and Yoshida
M: Inhibition of hepatic Niemann-Pick C1-like 1 improves hepatic
insulin resistance. Am J Physiol Endocrinol Metab. 297:E1030–E1038.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Henkel J, Neuschäfer-Rube F,
Pathe-Neuschäfer-Rube A and Püschel GP: Aggravation by
prostaglandin E2 of interleukin-6-dependent insulin resistance in
hepatocytes. Hepatology. 50:781–790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen J, Liu D, Bai Q, Song J, Guan J, Gao
J, Liu B, Ma X and Du Y: Celecoxib attenuates liver steatosis and
inflammation in non-alcoholic steatohepatitis induced by high-fat
diet in rats. Mol Med Rep. 4:811–816. 2011.PubMed/NCBI
|
|
40
|
Boden G, She P, Mozzoli M, Cheung P,
Gumireddy K, Reddy P, Xiang X, Luo Z and Ruderman N: Free fatty
acids produce insulin resistance and activate the proinflammatory
nuclear factor-kappaB pathway in rat liver. Diabetes. 54:3458–3465.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tosello-Trampont AC, Landes SG, Nguyen V,
Novobrantseva TI and Hahn YS: Kupffer cells trigger nonalcoholic
steatohepatitis development in diet-induced mouse model through
tumor necrosis factor-α production. J Biol Chem. 287:40161–40172.
2012. View Article : Google Scholar : PubMed/NCBI
|














