Introduction
Given their unique locations, vascular endothelial
cells (ECs) appear to be one of the primary targets of
hypoxia/reoxygenation (H/R) injury (1,2).
The generation of and substantially increased levels of reactive
oxygen species (ROS) (3), have
been widely implicated in EC injury (4,5).
Xanthine oxidoreductase (XOR) exists as two distinct enzyme forms:
xanthine dehydrogenase (XDH) and xanthine oxidase (XO). XDH
requires NAD+ to reduce hypoxanthine to xanthine. XO
requires O2 for purine oxidation, thereby generating ROS
(6). XO has been identified as a
source of ROS in atherosclerosis (7), coronary artery disease (8) and heart failure (9). An in vivo study showed that
XDH expression is increased during H/R in rat kidneys (10). Another in vitro study
demonstrated that the XDH-to-XO conversion is stimulated by
hydrogen peroxide and calcium (Ca2+) in bovine aortic
endothelial cells (11).
Furthermore, the p38 mitogen-activated protein kinase (p38 MAPK),
janus kinase 2 (JAK2) and signal transducers and activators of
transcription (STAT) signaling pathways are reportedly involved in
the process of XO activation and XDH-to-XO conversion during
hypoxia in pulmonary microvascular endothelial cells (12,13).
Hepatocyte growth factor (HGF) has been found to
promote survival, proliferation and morphogenesis by activating its
receptor cMet. HGF enhances the migration of epithelial cells
following acute kidney injury (14). HGF has also been found to regulate
neovascularization in developing fat pads (15). In our previous study, we
demonstrated that HGF may inhibit XO production and activation by
reducing the cytosolic Ca2+ concentration increased in
response to H/R; thus, HGF protects cardiac microvascular
endothelial cells (CMECs) from H/R-induced ROS production and
H/R-induced cell apoptosis (16).
However, the signaling mechanisms through which HGF regulates
cytosolic Ca2+ concentrations and XO activation in CMECs
under conditions of H/R remain to be elucidated. In the present
study, we examined the signaling pathway through which HGF
regulates Ca2+ concentrations and the activation of XO
during H/R in primary cultured rat CMECs.
Materials and methods
Isolation and culture of CMECs
A total of 80 five-to-seven day old Sprague-Dawley
(SD) rats weighing 12–16 g, were purchased from the Experimental
Animal Center of the Chinese PLA General Hospital (Beijing, China).
Ethics approval was obtained from the Ethics Committee of the
Experimental Animal Center of the Chinese PLA General Hospital
(approval no. SCXK20120001). All procedures were performed
according to the National Institutes of Health Guideline for the
Care and Use of Laboratory Animals. The rats were sacrificed with
an overdose of isoflurane and the hearts were removed. The enzyme
dissociation method based on the one described by Nishida et
al was used (17). The cells
were collected and resuspended in Dulbecco's modified Eagle's
medium (DMEM) (#12100046; Gibco, Grand Island, NY, USA)
supplemented with 20% fetal bovine serum (FBS) (#YS-OS-001091;
HyClone, Logan, UT, USA) and then seeded in 25 cm2
polystyrene flasks. Cell purity was identified by morphological
(18) and immunohistochemical
characteristics (4): the CMEC
monolayer displayed a uniform 'cobblestone' morphology and positive
immunohistochemical assays of factor VIII (#ab61910; Abcam,
Cambridge, MA, USA) and CD31 (#sc71873; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) (>95%).
H/R procedure and drug treatment
The H/R procedure was achieved by subjecting the
cells to 4 h of hypoxia and 1 h of reoxygenation. For hypoxic
exposure, the cells were incubated in D-Hank's solution (in mM:
136.89 NaCl, 5.37 KCl, 4.166 NaHCO3, 0.44
KH2PO4, 0.338 Na2HPO4,
pH 7.3–7.4 at 37°C) saturated with 95% N2 and 5%
CO2. The pH was adjusted to 6.8 to mimic ischemic
conditions. The cells were placed in a hypoxic incubator (Invivo2;
Ruskinn Technology Ltd., Pencoed, UK) that was equilibrated with
95% N2 and 5% CO2. Ambient O2
levels in the incubator were monitored by an O2 analyzer
(series-2000; Alpha Omega Instruments, Cumberland, RI, USA).
Following hypoxic exposure, the culture medium was rapidly replaced
with DMEM together with 1% FBS to initiate the reoxygenation
procedure (19–21).
For drug treatment, the CMECs were pre-incubated
with 30 µM AG490 (#S1509), 30 µM SB203580 (#S1863), 1
µM LY294002 (#S1737) (all from Beyotime Biotech, Jiangsu,
China) or 10 or 20 ng/ml HGF (#294-HG-005; R&D Systems, Inc.,
Minneapolis, MN, USA) for 30 min prior to exposure to hypoxic
conditions.
Measurement of ROS generation
Following the different treatments, the CMECs were
incubated with 2 mM DCFH-DA (#287810; Sigma-Aldrich, St. Louis, MO,
USA) for 20 min at 37°C in a 5% CO2 incubator. The cells
were washed and resuspended in phosphate-buffered saline (PBS) at a
concentration of 1×106 cells/ml. DCF fluorescence was
analyzed using a flow cytometer (Becton-Dickinson, Mountainview,
CA, USA) at the excitation and emission wavelengths of 514 and 525
nm, respectively. Untreated cells served as controls. The amount of
ROS was calculated as the fold-increase in DCF fluorescence
compared with the controls (22).
Small interfering RNA (siRNA)
transfection experiments
The transient transfection of CMECs with 50 nM siRNA
oligonucleotide was performed using Lipofectamine RNAiMAX reagent
(#13778030; Invitrogen, Dublin, Ireland). The cells were seeded in
6-well plates (2×105 cells/well) for these studies. The
following siRNA sequences were used: rat XDH siRNA,
5′-CCACCUCCAAGAUUCAUAUTT-3′; rat phosphoinositide 3-kinase (PI3K)
siRNA, 5′-GCAGCCAGCU CUGAUAAUATT-3′; rat JAK2 siRNA,
5′-GCCCUAAGGACUUCAACAATT-3′ and rat p38 MAPK siRNA,
5′-GGACCUCCUUAUAGACGAATT-3′. These siRNAs and their non-targeting
sequences (negative controls) were synthesized by GenePharma Co.,
Ltd. (Shanghai, China). After 48 h of transfection, the CMECs were
subjected to the H/R procedure. Finally, the cells were harvested
for other experiments.
Measurement of cytosolic
Ca2+
The CMECs were loaded with fluo-3 (#F23915;
Invitrogen, Carlsbad, CA, USA) in 1% working solution at 37°C for
30 min. The cells were washed three times with Ca2+-free
PBS to remove extracellular fluo-3 AM, and then resuspended in PBS
at a concentration of 1×106 cells/ml. The cells were
analyzed by flow cytometry (Becton-Dickinson) at an excitation
wavelength of 488 nm, and an emission wavelength of 530 nm
(23). The untreated cells served
as controls.
Western blot analysis
The CMECs were homogenized in RIPA lysis buffer
(#P0013C; Beyotime) containing 1X Phosphatase Inhibitor Cocktail
(#5870S; Cell Signaling Technology, Beverly, MA, USA) and 1
µg/ml each of aprotinin (A1153; Sigma-Aldrich) and leupeptin
(#L2884; Sigma-Aldrich). Forty micrograms of protein was separated
by sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE), transferred to polyvinylidene difluoride (PVDF)
membranes, and then probed with antibodies for XO (#ab109235;
Abcam), phospho-PI3 kinase p85 (Tyr458)/p55 (Tyr199) (#4228), PI3
kinase p85 (19H8) (#4257), phospho-p38 MAP kinase (Thr180/Thr182)
(#4631), p38 MAPK (D13E1) (#8690), JAK2 (D2E12) (#3230) and
phospho-JAK2 (Tyr1007/1008) (C80C3) (#3776) (all from Cell
Signaling Technology, Beverly, MA, USA). The same membranes were
reprobed with an antibody for tubulin (#AT819; Beyotime). The
blotting film was quantified using a scanner and a densitometry
program (ImageJ; https://imagej.nih.gov/ij/index.html) (24). To quantify the phosphor-specific
signal in the activated samples, the background was subtracted and
the band was normalized to the amount of tubulin or total target
protein in the lysate.
Statistical analysis
Statistical comparisons were performed using the
paired, two-tailed Student's t-test for experiments consisting of
two groups only, with one-way ANOVA and a multiple comparison
method for experiments consisting of more than two groups. A
p<0.05 was considered to indicate a statistically significant
difference. Data are presented as the means ± SE.
Results
XO plays a key role in the H/R-induced
production of ROS
In our previous study, the production of ROS
following H/R was significantly attenuated by allopurinol (20 and
40 µmol/l), an inhibitor of XO (16). In the present study, the
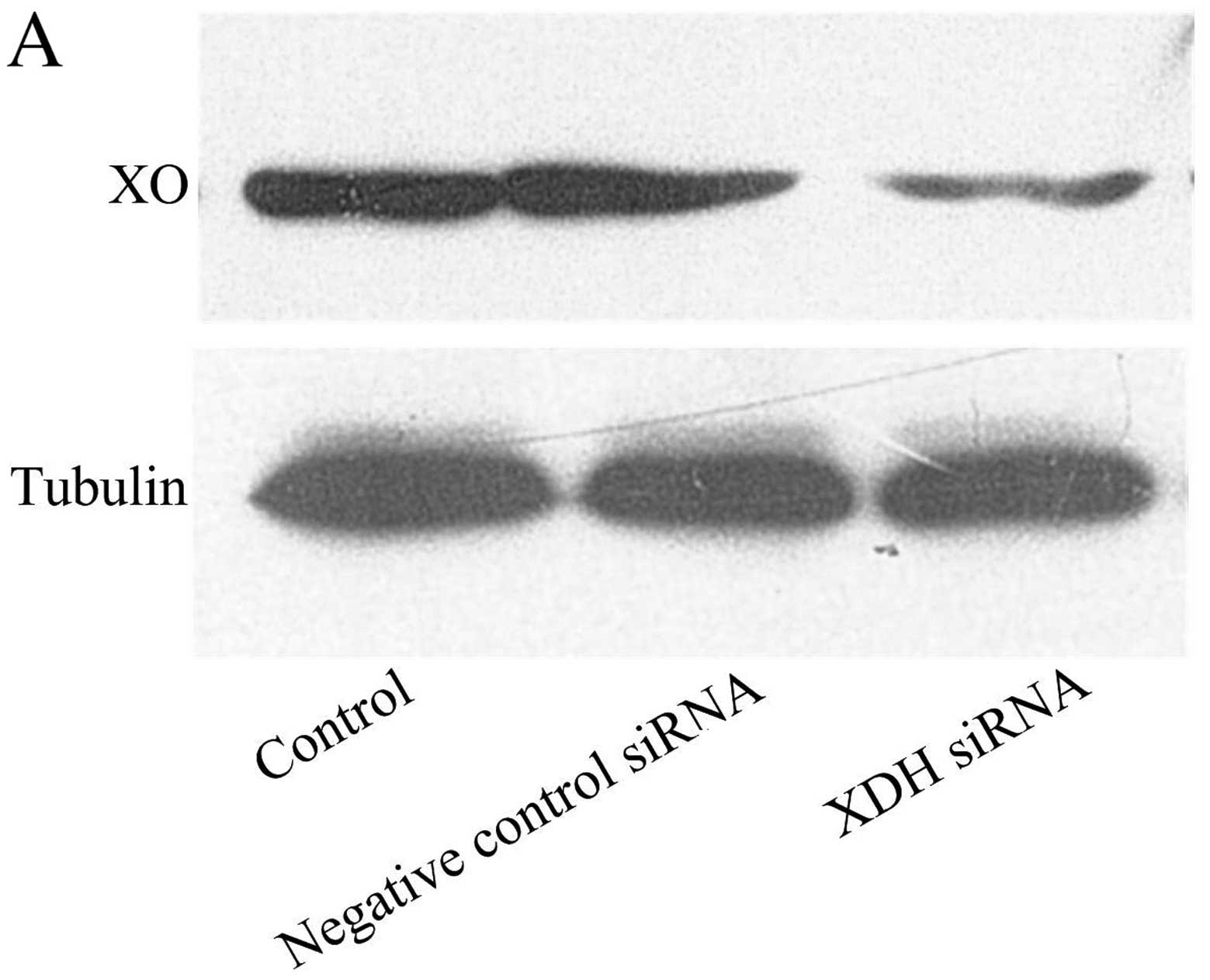
expression of XO was knocked down by XDH siRNA (Fig. 1A). Four hours of hypoxia increased
intracellular DCF fluorescence compared with normoxia (control
group). The transfection of XDH siRNA attenuated the increased
production of ROS following H/R (Fig.
1B and C).
PI3K and JAK2 pathways are involved in
the production and activation of XO
To determine whether PI3K, p38 MAPK or JAK2
signaling pathways are involved in the production and activation of
XO, we examined the effect of PI3K inhibitor LY294002, p38 MAPK
inhibitor SB203580 and JAK2 inhibitor AG490 on the production and
activation of XO. Pre-treatment with LY294002 and AG490 inhibited
H/R-mediated XO production (Fig. 2A
and B). The phosphorylation of PI3K and JAK2 was significantly
increased following H/R as compared with the normoxia controls
(Fig. 2C and D). However, the
phosphorylation of p38 MAPK was not found to be increased after H/R
(Fig. 2C and D). ROS production
following H/R was also partly blocked by LY294002 and AG490,
respectively (Fig. 2E and F).
These data indicated that XO activation induced by H/R in CMECs is
mediated through the PI3K and JAK2 signaling pathways rather than
the p38 MAPK signaling pathway.
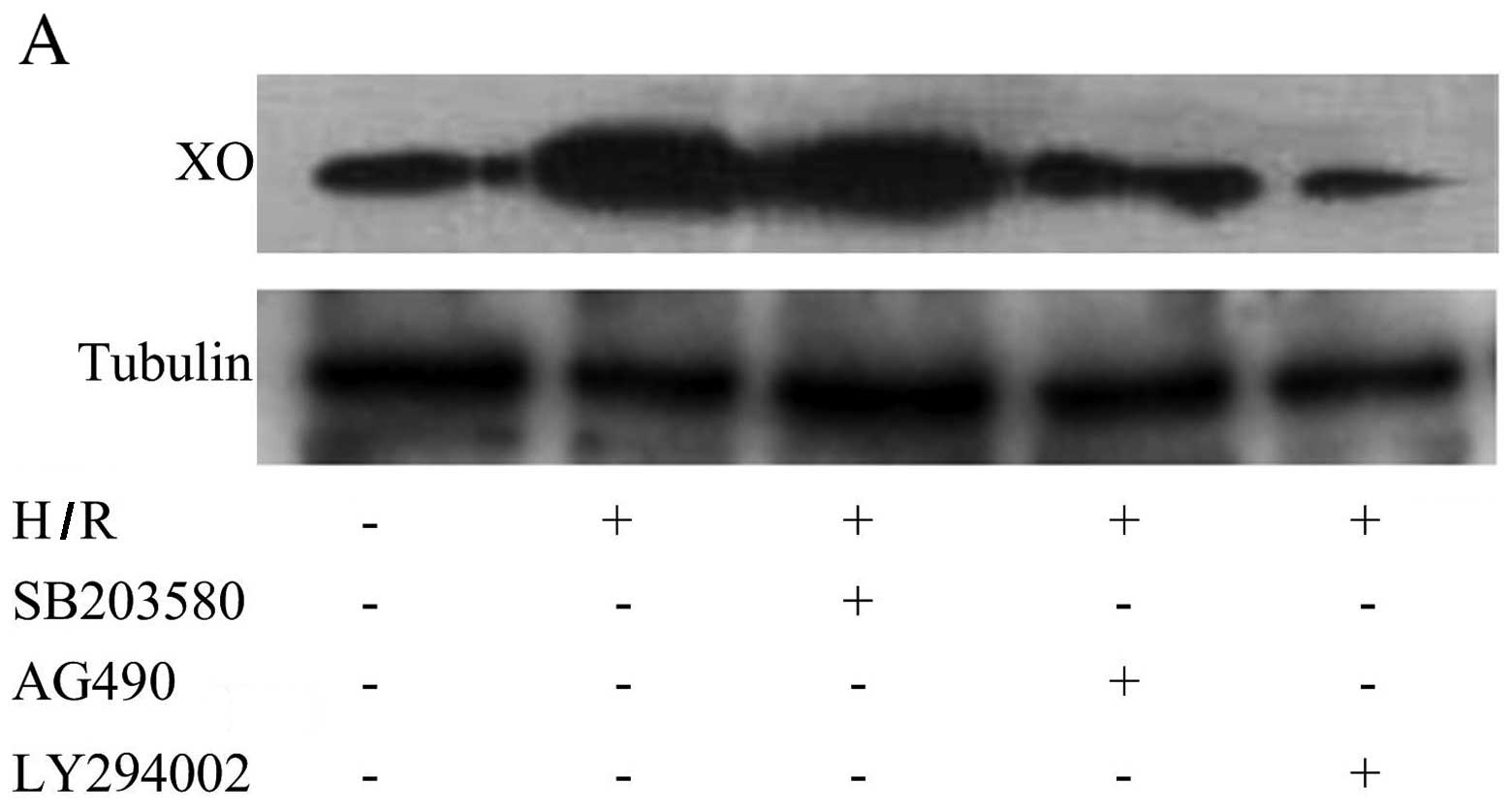 | Figure 2PI3K and JAK2 signaling pathways are
involved in the production and activation of xanthine oxidase (XO).
(A) Representative western blot of XO and tubulin expression in
cardiac microvascular endothelial cells (CMECs) following
hypoxia/reoxygenation (H/R). LY294002 and AG490 significantly
decreased the increase in the XO protein level induced by H/R. (B)
Summary data (n=3 biological replicates) of western blot analysis
of XO and tubulin expression in CMECs following H/R. LY294002,
SB203580 and AG490 treatment group compared with H/R group.
*p<0.05 vs. H/R group. PI3K and JAK2 signaling
pathways are involved in the production and activation of xanthine
oxidase (XO). (C) Representative western blots of phosphorylated
(p-)PI3K, total (t-)PI3K, p-JAK2, t-JAK2, p-p38 MAPK and t-p38 MAPK
expression in cardiac microvascular endothelial cells (CMECs)
following hypoxia/reoxygenation (H/R). H/R activates the PI3K and
JAK2 signaling pathways but not the p38 MAPK signaling pathway. (D)
Summary data (n=3 biological replicates) of western blot analysis
of p-PI3K, t-PI3K, p-JAK2, t-JAK2, p-p38 MAPK and t-p38 MAPK
expression in CMECs. **p<0.01 vs. control group. (E)
Representative data for flow cytometric analysis of DCFH-DA-stained
CMECs following H/R. LY294002, SB203580 and AG490 treatment groups
compared with H/R group. (F) Summary data (n=3 biological
replicates) of flow cytometric analysis of DCFH-DA-stained CMECs
following H/R. LY294002 (1 µM), SB203580 (30 µM) and
AG490 (30 µM) treatment groups compared with H/R group.
*p<0.05 vs. H/R group; **p<0.01 vs. H/R
group. |
PI3K siRNA and JAK2 siRNA downregulate
the production and activation of XO
To further confirm the involvement of PI3K and JAK2
signaling pathways in the production and activation of XO following
H/R, CMECs were transfected with either PI3K siRNA, JAK2 siRNA or
p38 MAPK siRNA to introduce knockdown (Fig. 3A). When the expression of PI3K and
JAK2 was inhibited by their respective siRNAs, the H/R-induced
increase in XO production was downregulated (Fig. 3B and C). ROS production following
H/R was also partly blocked by PI3K siRNA and JAK2 siRNA,
respectively (Fig. 3D and E)
However, p38 MAPK siRNA did not exert similar effects. These data
further confirmed that the production and activation of XO induced
by H/R in CMECs is mediated through the PI3K and JAK2 signaling
pathways rather than the p38 MAPK signaling pathway.
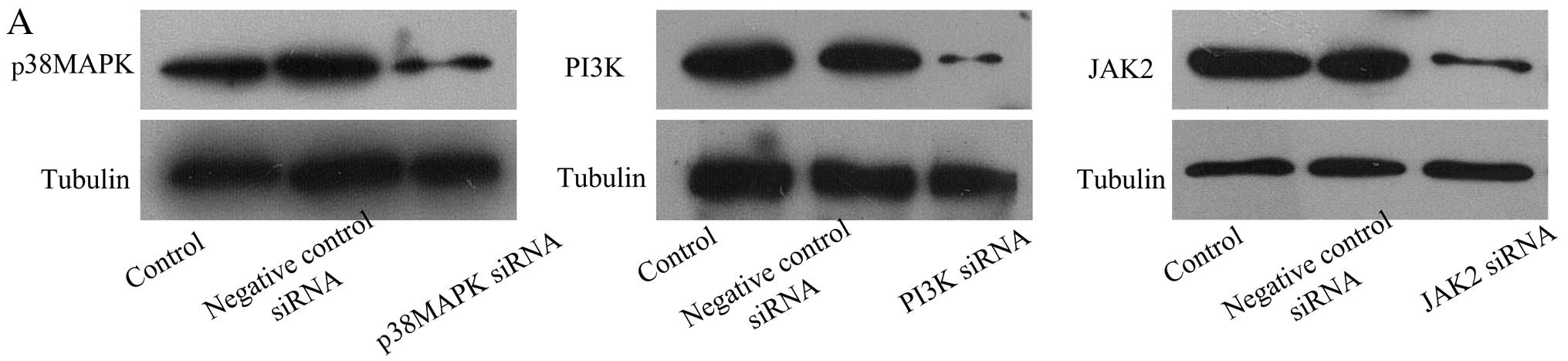 | Figure 3PI3K siRNA and JAK2 siRNA
downregulate the production and activation of xanthine oxidase
(XO). (A) Representative western blots of p38 MAPK, PI3K, JAK2 and
tubulin expression in cardiac microvascular endothelial cells
(CMECs). (B) Representative western blot of XO and tubulin
expression in CMECs following hypoxia/reoxygenation (H/R). PI3K
siRNA-, p38 MAPK siRNA-, JAK2 siRNA- and negative control
siRNA-transfected groups compared with H/R group. (C) Summary data
(n=3 biological replicates) for western blot analysis of XO and
tubulin expression in CMECs following H/R. PI3K siRNA-, p38 MAPK
siRNA-, JAK2 siRNA- or negative control siRNA-transfected groups
compared with H/R group. **p<0.01 vs. H/R group. (D)
Representative flow cytometric analysis of DCFH-DA-stained CMECs
after H/R. PI3K siRNA-, p38 MAPK siRNA-, JAK2 siRNA- or negative
control siRNA-transfected groups compared with H/R group. (E)
Summary data (n=3 biological replicates) for flow cytometric
analysis of DCFH-DA-stained CMECs after H/R. PI3K siRNA-, p38 MAPK
siRNA-, JAK2 siRNA- or negative control siRNA-transfected groups
compared with H/R group. **p<0.01 vs. H/R group. |
HGF inhibits JAK2 activation but not PI3K
activation
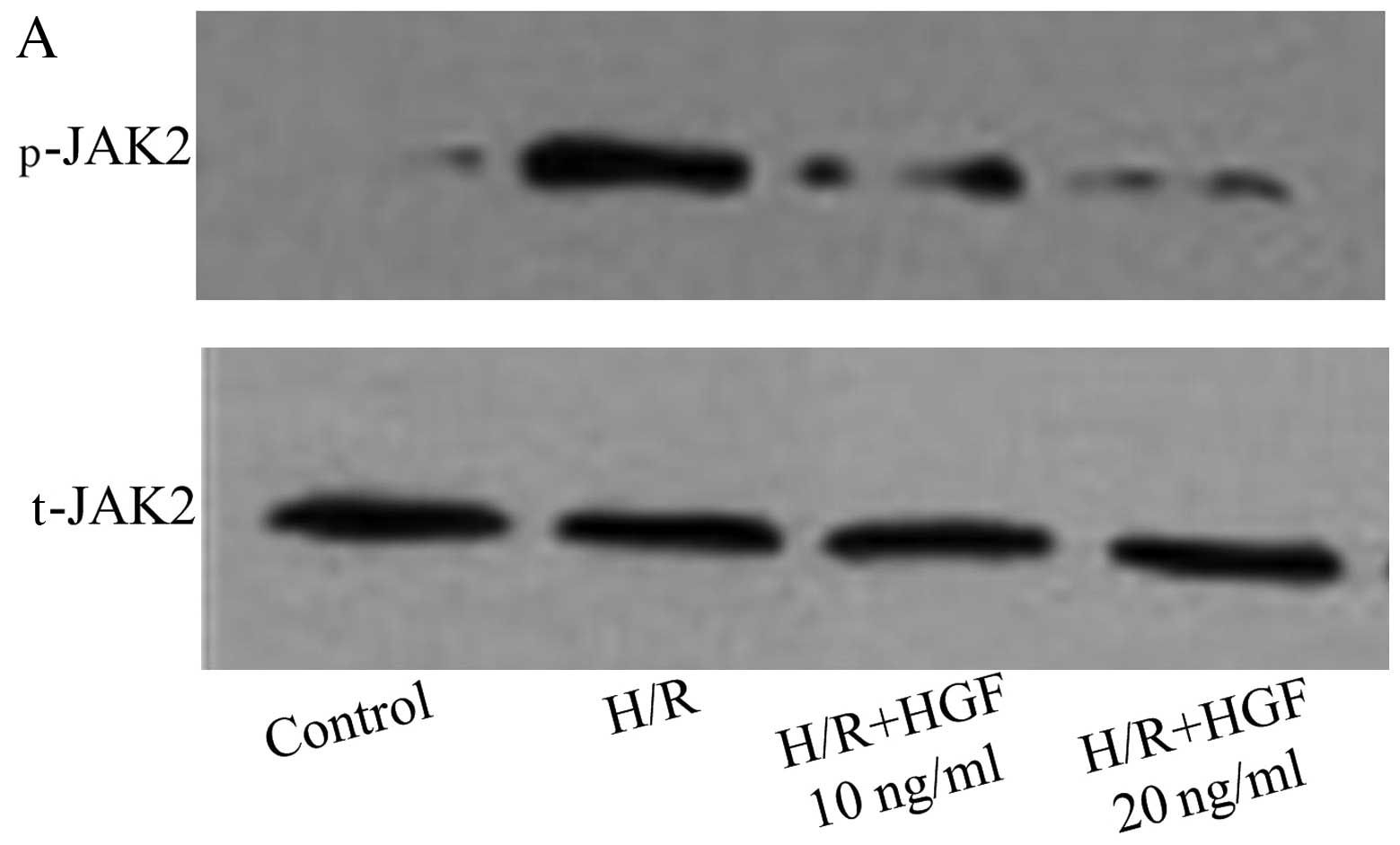
The phosphorylation of JAK2 and PI3K was evaluated
by western blot analysis in CMECs pre-treated with HGF (10 and 20
ng/ml). The phosphorylation of JAK2 induced by H/R was inhibited by
HGF (Fig. 4A and B). However, the
phosphorylation of PI3K induced by H/R was unaffected by HGF
(Fig. 4C and D). These findings
suggest that HGF inhibited the activation and production of XO
through the JAK2 signaling pathway.
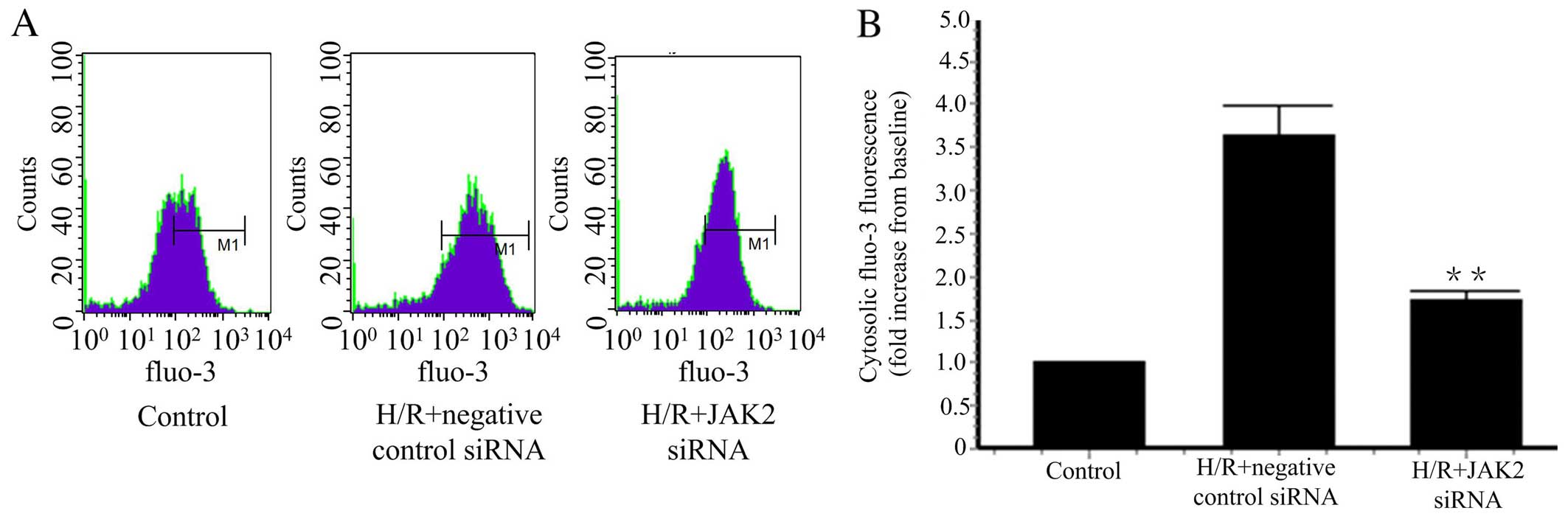
JAK2 siRNA downregulates the
concentration of cytosolic calcium
In our previous study, we found that HGF inhibits XO
activation by reducing cytosolic Ca2+ concentrations
induced by H/R (16). We further
studied whether the JAK2 signaling pathway regulates cytosolic
Ca2+ concentrations. JAK2 knockdown was achieved by JAK2
siRNA, and resulted in a reduction in the cytosolic Ca2+
concentration induced by H/R (Fig.
5). Taken together, these findings suggest that HGF reduced
cytosolic Ca2+ concentrations by inhibiting JAK2
phosphorylation.
Discussion
Excessive oxidative stress is believed to be an
important contributor to H/R injury. In our previous study, we
revealed that HGF protects CMECs from H/R-induced apoptosis by
reducing ROS production (16).
The findings of the present study indicate that H/R increased DCF
oxidation. DCF detects H2O2 but does not
detect superoxide (25). However,
H2O2 accounts for 90% of ROS production under
hypoxic conditions and superoxide accounts for 10% (26). DCF oxidation appears to be a
reliable method for the detection of cellular ROS production
(25,27). Potential sources of ROS include XO
(6), NADPH oxidase (1), the mitochondrial respiratory chain
(21), and the metabolic cascade
of arachidonic acid (28). XO is
the major source of ROS in the rat jugular venous (29) and rat pulmonary circulation
(27) following H/R. The
mitochondrial respiratory chain is the major source of ROS in
embryonic chick cardiomyocytes (21) and human umbilical vein ECs
(HUVECs) (30). Thus, we
hypothesized that the major source of ROS following H/R is both
species-specific and organ-specific. When XOR expression was
knocked down by XDH siRNA, H/R-induced ROS production in CMECs was
also attenuated. XO accounts for, at least part of, the ROS
production induced by H/R in CMECs.
The signaling pathways involved in the production
and activation of XO are controversial under different
circumstances in different cell types. p38 MAPK and CK2 have been
found to be involved in the activation of XO following hypoxia in
rat pulmonary microvascular ECs (RPMECs) (12). JAKs and STATs are involved in the
hypoxia-mediated activation of XO in lung microvascular ECs
(LMVECs) (13). The
phosphorylation of PI3K increases ROS production during hypoxia in
endothelial progenitor cells (31) and in mouse pulmonary microvascular
ECs (PMVECs) (25). In the
present study, AG490 and LY294002 partially blocked the increase in
ROS production following H/R. The PI3K and JAK2 signaling pathway
is significantly activated after H/R whereas the p38 MAPK signaling
pathway is unaffected. The pre-treatment of CMECs with AG490 and
LY294002 markedly attenuated XO protein levels. The pre-treatment
of CMECs with SB203580 did not have the above-mentioned effect.
Furthermore, when the knockdown of JAK2 or PI3K was achieved by
siRNA, increases in the XO protein levels and ROS production were
greatly attenuated. These data show that the PI3K and JAK2
signaling pathways are involved in the upregulation and activation
of XO following H/R. However, HGF inhibits the activation of the
JAK2 signaling pathway but not the PI3K signaling pathway. In our
previous study, we reported that HGF inhibits the activation and
production of XO by reducing cytosolic Ca2+
concentrations in CMECs after H/R (16). Thus, HGF inhibits XO activation by
inhibiting JAK2 signal pathway. In the present study, JAK2
knockdown by JAK2 siRNA significantly reduced cytosolic
Ca2+ concentrations. This finding is in agreement with
the results of a previous study which demonstrated that AG490, the
JAK2 signal inhibitor, blocked an
H2O2-induced increase in intracellular
Ca2+ in U937 cells (32). It has been recognised that XO
activation is regulated by cytosolic Ca2+, and an
unidentified Ca2+-dependent protease is involved in the
cleavage of XDH to XO (11). A
heat-liable protease that cleaves XDH to XO has also been found in
the mitochondrial intermembrane space (33). In our previous study, when CMECs
were pre-treated with BAPTA-AM, a cell permeable calcium chelator,
the H/R-induced activation of XO was blocked. HGF prevents JAK2
activation, reduces cytosolic Ca2+ concentrations and in
turn, inhibits XO activation in CMECs following H/R.
In conclusion, these findings suggest a novel
mechanism whereby HGF regulates H/R-induced XO activation in CMECs.
The upregulation and activation of XO as well as increased ROS
production following H/R primarily involve the PI3K and JAK2
signaling pathways but not the p38 MAPK signaling pathway. HGF
inhibits the activation of JAK2. The knockdown of JAK2 attenuated
cytosolic Ca2+ concentrations in CMECs following H/R.
Thus, HGF inhibits XO activation by inhibiting JAK2 activation and
reducing cytosolic Ca2+ concentrations in CMECs
following H/R. HGF may exhibit protective and therapeutic effects
against H/R injury in H/R-related diseases.
Abbreviations:
|
ECs
|
endothelial cells
|
|
H/R
|
hypoxia/reoxygenation
|
|
HGF
|
hepatocyte growth factor
|
|
XO
|
xanthine oxidase
|
|
CMECs
|
cardiac microvascular endothelial
cells
|
|
XDH
|
xanthine dehydrogenase
|
|
ROS
|
reactive oxygen species
|
|
siRNA
|
small interfering RNA
|
Acknowledgments
The present study was supported by grants (no.
81070185 and 81102079) from the National Natural Science Foundation
of China.
References
|
1
|
Yu G, Peng T, Feng Q and Tyml K: Abrupt
reoxygenation of microvascular endothelial cells after hypoxia
activates ERK1/2 and JNK1, leading to NADPH oxidase-dependent
oxidant production. Microcirculation. 14:125–136. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang T, Yang D, Fan Y, Xie P and Li H:
Epigallocatechin-3-gallate enhances ischemia/reperfusion-induced
apoptosis in human umbilical vein endothelial cells via AKT and
MAPK pathways. Apoptosis. 14:1245–1254. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dhar-Mascareño M, Cárcamo JM and Golde DW:
Hypoxia-reoxygenation-induced mitochondrial damage and apoptosis in
human endothelial cells are inhibited by vitamin C. Free Radic Biol
Med. 38:1311–1322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pearlstein DP, Ali MH, Mungai PT, Hynes
KL, Gewertz BL and Schumacker PT: Role of mitochondrial oxidant
generation in endothelial cell responses to hypoxia. Arterioscler
Thromb Vasc Biol. 22:566–573. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jang H-J, Koo BK, Lee HS, Park JB, Kim JH,
Seo MK, Yang HM, Park KW, Nam CW, Doh JH and Kim HS: Safety and
efficacy of a novel hyperaemic agent, intracoronary nicorandil, for
invasive physiological assessments in the cardiac catheterization
laboratory. Eur Heart J. 34:2055–2062. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meneshian A and Bulkley GB: The physiology
of endothelial xanthine oxidase: from urate catabolism to
reperfusion injury to inflammatory signal transduction.
Microcirculation. 9:161–175. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Landmesser U, Spiekermann S, Preuss C,
Sorrentino S, Fischer D, Manes C, Mueller M and Drexler H:
Angiotensin II induces endothelial xanthine oxidase activation:
role for endothelial dysfunction in patients with coronary disease.
Arterioscler Thromb Vasc Biol. 27:943–948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Spiekermann S, Landmesser U, Dikalov S,
Bredt M, Gamez G, Tatge H, Reepschläger N, Hornig B, Drexler H and
Harrison DG: Electron spin resonance characterization of vascular
xanthine and NAD(P)H oxidase activity in patients with coronary
artery disease: Relation to endothelium-dependent vasodilation.
Circulation. 107:1383–1389. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Berry CE and Hare JM: Xanthine
oxidoreductase and cardiovascular disease: molecular mechanisms and
pathophysiological implications. J Physiol. 555:589–606. 2004.
View Article : Google Scholar
|
|
10
|
Sulikowski T, Domanski L, Ciechanowski K,
Adler G, Pawlik A, Safranow K, Dziedziejko V, Chlubek D and
Ciechanowicz A: Effect of trimetazidine on xanthine oxidoreductase
expression in rat kidney with ischemia-reperfusion injury. Arch Med
Res. 39:459–462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McNally JS, Saxena A, Cai H, Dikalov S and
Harrison DG: Regulation of xanthine oxidoreductase protein
expression by hydrogen peroxide and calcium. Arterioscler Thromb
Vasc Biol. 25:1623–1628. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kayyali US, Donaldson C, Huang H,
Abdelnour R and Hassoun PM: Phosphorylation of xanthine
dehydrogenase/oxidase in hypoxia. J Biol Chem. 276:14359–14365.
2001.PubMed/NCBI
|
|
13
|
Wang G, Qian P, Jackson FR, Qian G and Wu
G: Sequential activation of JAKs, STATs and xanthine
dehydrogenase/oxidase by hypoxia in lung microvascular endothelial
cells. Int J Biochem. Cell Biol. 40:461–470. 2008.
|
|
14
|
Reviriego-Mendoza MM and Santy LC: The
cytohesin guanosine exchange factors (GEFs) are required to promote
HGF-mediated renal recovery after acute kidney injury (AKI) in
mice. Physiol Rep. 3:e124422015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
White HM, Acton AJ, Kamocka MM and
Considine RV: Hepatocyte growth factor regulates neovascularization
in developing fat pads. Am J Physiol Endocrinol Metab.
306:E189–E196. 2014. View Article : Google Scholar :
|
|
16
|
Zhang Y, Hu S and Chen Y: Hepatocyte
growth factor suppresses hypoxia/reoxygenationinduced XO activation
in cardiac microvascular endothelial cells. Heart Vessels.
30:534–544. 2015. View Article : Google Scholar
|
|
17
|
Nishida M, Carley WW, Gerritsen ME,
Ellingsen O, Kelly RA and Smith TW: Isolation and characterization
of human and rat cardiac microvascular endothelial cells. Am J
Physiol. 264:H639–H652. 1993.PubMed/NCBI
|
|
18
|
Zhang Z, Li W, Sun D, Zhao L, Zhang R,
Wang Y, Zhou X, Wang H and Cao F: Toll-like receptor 4 signaling in
dysfunction of cardiac microvascular endothelial cells under
hypoxia/reoxygenation. Inflamm Res. 60:37–45. 2011. View Article : Google Scholar
|
|
19
|
Ladilov Y, Schäfer C, Held A, Schäfer M,
Noll T and Piper HM: Mechanism of Ca(2+) overload in endothelial
cells exposed to simulated ischemia. Cardiovasc Res. 47:394–403.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu G, Bolon M, Laird DW and Tyml K:
Hypoxia and reoxygenation-induced oxidant production increase in
microvascular endothelial cells depends on connexin40. Free Radic
Biol Med. 49:1008–1013. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Loor G, Kondapalli J, Iwase H, Chandel NS,
Waypa GB, Guzy RD, Vanden Hoek TL and Schumacker PT: Mitochondrial
oxidant stress triggers cell death in simulated
ischemia-reperfusion. Biochim Biophys Acta. 1813:1382–1394. 2011.
View Article : Google Scholar :
|
|
22
|
Kong R, Jia G, Cheng ZX, Wang YW, Mu M,
Wang SJ, Pan SH, Gao Y, Jiang HC, Dong DL and Sun B:
Dihydroartemisinin enhances Apo2L/TRAIL-mediated apoptosis in
pancreatic cancer cells via ROS-mediated up-regulation of death
receptor 5. PLoS One. 7:e372222012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Przygodzki T, Sokal A and Bryszewska M:
Calcium ionophore A23187 action on cardiac myocytes is accompanied
by enhanced production of reactive oxygen species. Biochim Biophys
Acta. 1740:481–488. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rasband WS: ImageJ. U.S National
Institutes of Health; Bethesda, MD: 1997–2012
|
|
25
|
Chatterjee S, Browning EA, Hong N, DeBolt
K, Sorokina EM, Liu W, Birnbaum MJ and Fisher AB: Membrane
depolarization is the trigger for PI3K/Akt activation and leads to
the generation of ROS. Am J Physiol Heart Circ Physiol.
302:H105–H114. 2012. View Article : Google Scholar :
|
|
26
|
Kelley EE, Khoo NK, Hundley NJ, Malik UZ,
Freeman BA and Tarpey MM: Hydrogen peroxide is the major oxidant
product of xanthine oxidase. Free Radic Biol Med. 48:493–498. 2010.
View Article : Google Scholar :
|
|
27
|
Saito S, Ogawa J and Minamiya Y: Pulmonary
reexpansion causes xanthine oxidase-induced apoptosis in rat lung.
Am J Physiol Lung Cell Mol Physiol. 289:L400–L406. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Krause GS, White BC, Aust SD, Nayini NR
and Kumar K: Brain cell death following ischemia and reperfusion: A
proposed biochemical sequence. Crit Care Med. 16:714–726. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ono T, Tsuruta R, Fujita M, Aki HS,
Kutsuna S, Kawamura Y, Wakatsuki J, Aoki T, Kobayashi C, Kasaoka S,
et al: Xanthine oxidase is one of the major sources of superoxide
anion radicals in blood after reperfusion in rats with forebrain
ischemia/reperfusion. Brain Res. 1305:158–167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Therade-Matharan S, Laemmel E, Duranteau J
and Vicaut E: Reoxygenation after hypoxia and glucose depletion
causes reactive oxygen species production by mitochondria in HUVEC.
Am J Physiol Regul Integr Comp Physiol. 287:R1037–R1043. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dai T, Zheng H and Fu GS: Hypoxia confers
protection against apoptosis via the PI3K/Akt pathway in
endothelial progenitor cells. Acta Pharmacol Sin. 29:1425–1431.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shimizu S, Yonezawa R, Hagiwara T, Yoshida
T, Takahashi N, Hamano S, Negoro T, Toda T, Wakamori M, Mori Y and
Ishii M: Inhibitory effects of AG490 on
H2O2-induced TRPM2-mediated Ca(2+) entry. Eur
J Pharmacol. 742:22–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saksela M, Lapatto R and Raivio KO:
Irreversible conversion of xanthine dehydrogenase into xanthine
oxidase by a mitochondrial protease. FEBS Lett. 443:117–120. 1999.
View Article : Google Scholar : PubMed/NCBI
|