Introduction
Alzheimer's disease (AD) has become the fourth
leading lethal disease among the elderly following cancer, heart
disease and stroke. AD is an age-related neurodegenerative
disorder, which is typically characterized by the deposition of
β-amyloid plaques, neurofibrillary tangles (NFTs) and neuronal loss
(1). These pathological
characteristics of the disease lead to the progressive loss of
memory, which causes cognitive dysfunction. The neurotoxicity of
amyloid-β (Aβ) peptides has been widely accepted to be responsible
for the pathogenesis of AD (2).
In fact, both in vitro and in vivo findings have
demonstrated that Aβ fragments promote a marked neuro-inflammatory
response, accounting for the synthesis of various cytokines and
pro-inflammatory mediators (3,4).
It is believed that the inflammatory process, once initiated, may
contribute independently to neuronal dysfunction and cell death
(5). The nuclear receptors known
as peroxisome proliferator-activated receptors (PPARs), which
antagonize the effects of the pro-inflammatory transcription
factor, nuclear factor-κB (NF-κB), regulate the expression of many
genes which encode proteins that play a decisive role in the
process of inflammation (6). The
three PPAR isotypes, PPAR-α, PPAR-β/δ and PPAR-γ, are expressed in
all cell types in the brain (7).
Numerous studies have described the neuroprotective properties of
PPAR-α and PPAR-γ agonists in different models of neurological
diseases, and propose PPAR-dependent mechanisms for their mode of
action. The efficiency of PPAR-β/δ agonists has previously been
reviewed in animal models of neurodegenerative diseases (8). However, the biology of PPAR-β/δ in
the brain is less understood compared to PPAR-α and PPAR-γ. Some
scholars have proposed that PPAR-γ is an opportunistic therapeutic
target in patients with mild cognitive impairment (MCI)/AD and
concomitant insulin dysregulation; the co-morbidity of insulin
resistance is shared by both AD and diabetes (9–11).
Indeed, PPAR-γ agonists, such as rosiglitazone (RSG) have been
shown to improve cognitive function in some patients with
early-stage AD, as well as in several animal models of AD (12–14).
A number of researchers have demonstrated the
involvement of the cyclic guanosine monophosphate (cGMP) pathway in
learning and memory (15–17). Of note, sildenafil (Viagra), a
specific phosphodiesterase 5 (PDE5) inhibitor, has been shown to
increase cGMP levels by inhibiting its degradation and is widely
used as the selective drug for the treatment of erectile
dysfunction and pulmonary hypertension. It has recently been
proposed as a molecule for use in the treatment a variety of
disorders, including AD and aging (18). In addition, the age-related
decline of cognitive functions is thought to be associated with an
increase in neuronal apoptosis (19), a process of programmed cell death
that may result in pathological processes, such as degeneration
(20,21). Many proteins are involved in the
process of apoptosis, such as Bcl-2 family members, caspases and
many more (22,23). Caspase-3 stimulates the formation
of Aβ by affecting amyloid precursor protein (APP), a single
transmembrane protein, via the cleavage of protease to generate Aβ
(24). To date, specific
treatment for AD is unavailable. Thus, it is urgent to further
explore novel treatment strategies for AD.
Hydrogen sulfide (H2S) is a well known
gasotransmitter along with nitric oxide (NO) and carbon monoxide
(CO) (25). H2S is
primarily produced in the brain from the cysteine precursor by the
cystathionine β-synthase (CBS) and cystathione γ-lyase (CGL)
enzymes (26). CBS, is highly
distributed in the hippocampus (27). 3-Mercaptopyruvate
sulfurtransferase (3MST) is a third enzyme also responsible for the
generation of endogenous H2S (28,29). H2S has been gradually
confirmed to be a new type of neuromodulator involved in multiple
physiological nerve functions. It has been previously demonstrated
that H2S exerts a variety of effects (including
antioxidant, anti-inflammatory and anti-apoptotic effects) in
animal models or neuronal and glial cells in AD, Parkinson's
disease and other diseases (30–33). The levels of H2S are
markedly decreased in patients with AD. Moreover, there is an
association between the levels of H2S and the severity
of AD (34). Recent data have
demonstrated that exogenous H2S significantly improves
spatial learning and memory impairment induced by
Aβ25–35, and exerts anti-inflammatory and anti-apoptotic
effects (35). These findings
suggest the possible involvement of H2S in attenuating
the pathogenesis of AD. However, the possible and corresponding
molecular mechanisms of action of H2S as an
anti-inflammatory and anti-apoptotic agent in a rat model of
Aβ25–35-induced neurotoxicity have not yet been fully
elucidated.
Therefore, the present study was designed to
investigate the effects of NaHS on Aβ25–35-induced
neurotoxicity and further explore its underlying mechanisms of
action.
Materials and methods
Animals
Healthy male SPF Sprague-Dawley (SD) rats (weighing
220 to 250 g) were obtained from the Animal Center of the Third
Military Medical University (Chongqing, China) (certificate no.
SCXK20020003). The animals were maintained under a 12 h light/dark
cycle in temperature (23 ±1°C) and humidity (relative,
60%)-controlled rooms and allowed free access to food and water.
All experiments were performed according to the National Institutes
of Health Guidelines for Humane Use and Care (Eighth Edition), and
the Current Guide for the Care and Use of Laboratory Animals under
a protocol approved by Zunyi Medical University Animal Studies
Committee.
Experimental design and treatment
Forty-two rats were randomly assigned to 3 groups as
follows: the sham-operated group, the Aβ25–35 group and
the Aβ25–35 + NaHS group (n=14 rats per group).
Aβ25–35 was purchased from Sigma-Aldrich (St. Louis, MO,
USA), dissolved in sterilized saline at a concentration of 2
µg/µl, and then incubated at 37°C for 7 days prior to
injection in order to allow aggregation. The animals were
intraperitoneal injected with chloral hydrate (40 mg/kg) anesthesia
and placed in a stereotaxic device (SR-6N; Narishige, Tokyo,
Japan). Aggregated Aβ25–35 was injected into the rats in
accordance with a previously pubished protocol (36). A midline incision was made on the
head skin of the rats following routine sterilization, exposing the
periosteum, and then, using a 5 µl microsyringe injector,
Aβ25–35 was injected into the bilateral CA1 subregion at
the following coordinates: 3.3 mm posterior to thye bregma, 2 mm
lateral to the sagittal suture, 3 mm beneath the surface of brain.
Rats in the sham-operated group were injected with normal
sterilized saline. The rats were injected with 5 µl
Aβ25–35 or 5 µl sterilized normal saline in each
bilateral CA1 subregion at a rate of 1 µl/min. The needle
was left for 5 min after injection. NaHS (Sigma-Aldrich) was
continuously intraperitoneally injected at a dose of 5 mg/kg for 15
days. Rats in the sham-operated and Aβ25–35 group were
administrated the same volume of normal saline.
Nissl staining
Four rats from each group were randomly selected and
were anesthetized and sacrificed by intracardiac perfusion with 0.1
M phosphate buffer containing 0.4% heparin. The brains were
carefully removed following decapitation and transferred into
ice-cold 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.38),
and fixed in 4% paraformaldehyde for 48 h, and then embedded in
paraffin. The conventional paraffin-embedded tissue sections were
stained with toluidine blue (Solarbio, Beijing, China). The Nissl
bodies were stained blue-purple under a light microscope (KS300;
Zeiss-Kontron, Göttingen, Germany). Neurons in the hippocampus from
each group were counted as previously described (37). Neurons in the area of the CA1
region of the hippocampus were counted using 5 equally spaced
coronal sections passing through the hippocampus for each
brain.
Terminal
deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL)
staining
Cells undergoing apoptosis induced by
Aβ25–35 were detected by TUNEL staining using an In
Situ Cell Death Detection kit, Fluorescein (Roche Applied
Science, Indianapolis, IN, USA), according to the manufacturer's
instructions. In order to block endogenous peroxidase activity, the
sections were immersed in 3% H2O2 for 15 min
in the dark. After being washed 3 times in phosphate-buffered
saline (PBS) for 5 min each, the sections were treated with
proteinase K solution (20 µg/ml in 10 mM Tris/HCl, pH 7.6)
at 37°C for 15 min. They were then incubated for 60 min at 37°C
with TUNEL reaction mixture. The sections were then washed again
and incubated for 30 min at 37°C with converter-POD. The sections
were rinsed in PBS, treated with DAB substrate solution and washed
again with PBS. The sections were viewed and counted under a light
microscope (BX43; Olympus Corporation, Tokyo, Japan).
Enzyme-linked immunosorbent assay (ELISA)
for the detection of PDE5
The content of PDE5 in the hippocampus was measured
by ELISA. Six rats from each group were randomly selected and
sacrificed, and the right hippocampus was collected for ELISA.
Hippocampal tissues were homogenized (1:5, w:v) in 0.01 M PBS (pH
7.4) and centrifuged (3,000 rpm at 4°C for 20 min), as previously
described (38). The supernatant
was stored at −80°C for subsequent determination. The protein
levels of homogenate samples were analyzed using the BCA protein
assay kit (Biocolor Biotechnology, Shanghai, China). PDE5 (Shanghai
Jiang Lai Biotechnology Co., Ltd., Shanghai, China) was quantified
in these samples using the PDE5 ELISA kit according to the
manufacturer's instructions.
Western blot analysis
The protein expression of PPAR-α (ab8934), PPAR-β
(ab137724), PPAR-γ (ab19481) and active + pro-caspase-3 (ab47131)
(all from Abcam, Cambridge, UK), p-NF-κB p65 (#3033), NF-κB p65
(#8242) and IκB-α (#9242) (all from Cell Signaling Technology,
Danvers, MA, USA) and β-actin (AF0003; Beyotime Biotechnology,
Nanjing, China) was analyzed by western blot analysis. Three rats
from each group were sacrificed and the right hippocampal tissues
were dissected and immediately frozen at −80°C. The frozen tissues
were sliced into small sections and homogenized on ice in cold
radioimmunoprecipitation assay (RIPA) lysis buffer (150 mM NaCl,
0.5% deoxycholate, 1% NP-40, 0.1% sodium dodecyl sulfate, 2 mM
phenylmethylsulfonyl fluoride and 50 mM Tris-hydrochloric acid, pH
7.4) containing protease and phosphatase inhibitor cocktail.
Following homogenization, the dissolved proteins were gathered by
centrifugation for 30 min at 10,000 × g. The supernatant was
collected and the protein concentration was then determined using
the BCA protein assay kit (Biocolor Biotechnology). The protein (30
µg) was then separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto PVDF membranes (Millipore Trading Co., Ltd.,
Bedford, MA, USA). Blotting membranes were incubated with 3% bovine
serum albumin (BSA) in Tris-buffered saline with Tween-20 (TBST)
(10 mM Tris, 150 mM NaCl, 0.05% Tween-20, pH 7.5) and then probed
with a primary antibody against PPAR-α (1:2,000), PPAR-β (1:3,000),
PPAR-γ (1:2,000), p-NF-κB p65 (1;1,000), NF-κB p65 (1:1,000), IκB-α
(1;1,000), active and pro-caspase-3 (1:2,000) and β-actin (1:5,000;
Beyotime Institute of Biotechnology) at 4°C overnight. After
washing, the membranes were incubated with appropriate horseradish
peroxidase-coupled secondary antibodies for 2 h at room
temperature. The blots were then revealed using the ECL select kit
(Beyotime Institute of Biotechnology) and exposed to Gel Imaging
(Bio-Rad, Hercules, CA, USA).
Statistical analysis
All data are presented as the means ± standard error
of the mean (SEM). One-way analysis of variance (ANOVA) was used to
examine statistical comparisons between groups. Post hoc
comparisons were performed by LSD with equal variances, and by
Dunnett's T3 with unequal variances. All analyses were performed
using SPSS 16.0 software. In all cases, a value of P<0.05 was
considered to indicate a statistically significant difference.
Results
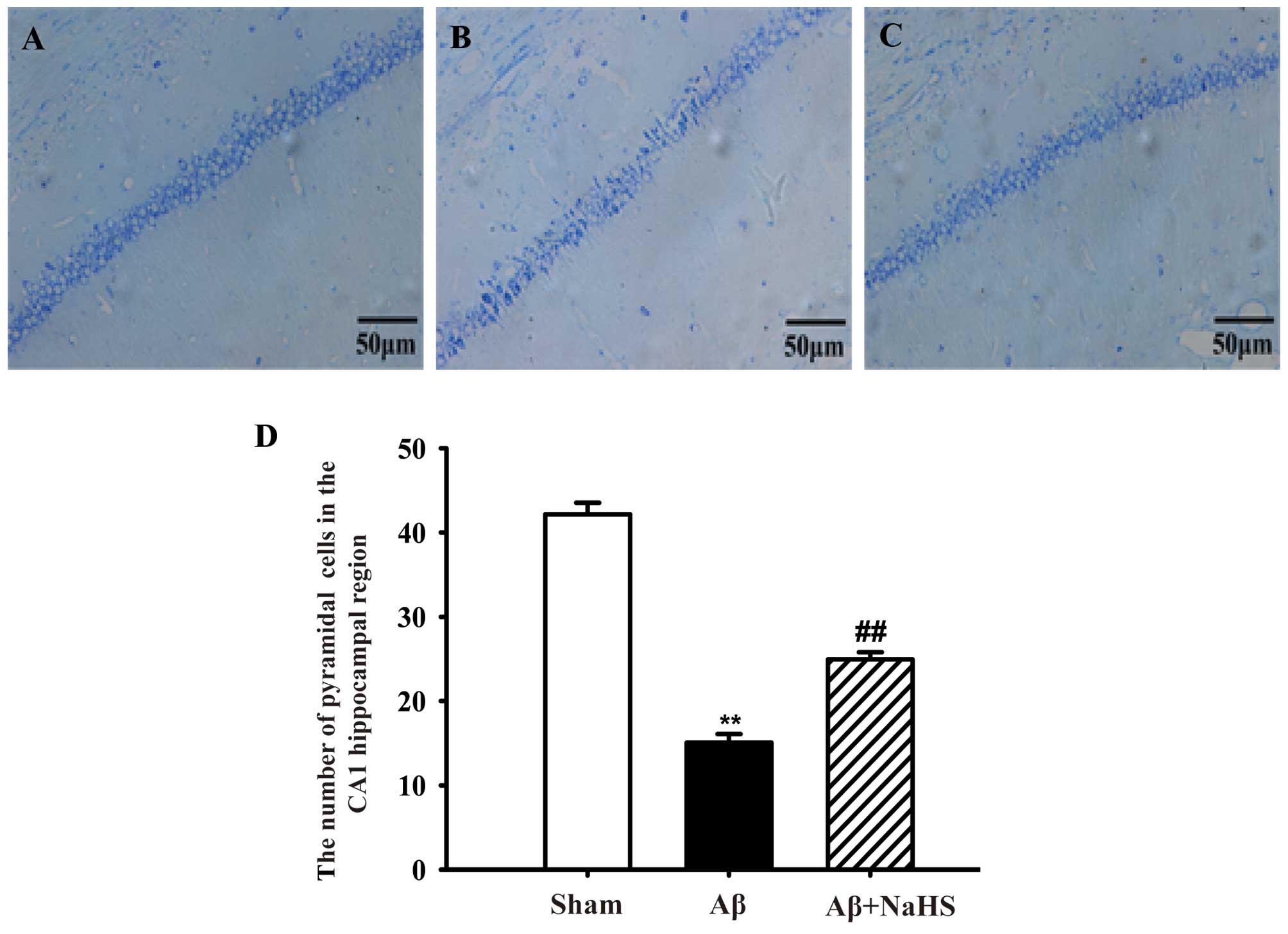
NaHS attenuates
Aβ25–35-induced neuronal cell death in the hippocampus
of rats
Nissl staining was utilized to evaluate the effects
of NaHS on Aβ25–35-induced neuronal cell death in the
hippocampus. Healthy neurons in the CA1 region in the hippocampus
were observed in the sham-operated group. The pyramidal layer of
cells was neatly and closely arranged and the structure was clear.
However, following the injection of Aβ25–35, typical
neuropathological changes were observed, including the pyknosis of
the pyramidal layer of cells and appreciable neuronal cell loss or
disappearance. However, treatment with NaHS reduced neuronal
morphological impairment compared to exposure to Aβ25–35
alone (Fig. 1). On the whole, our
results suggest that the administration of NaHS attenuates
Aβ25–35-induced neuronal loss in the hippocampus of
rats.
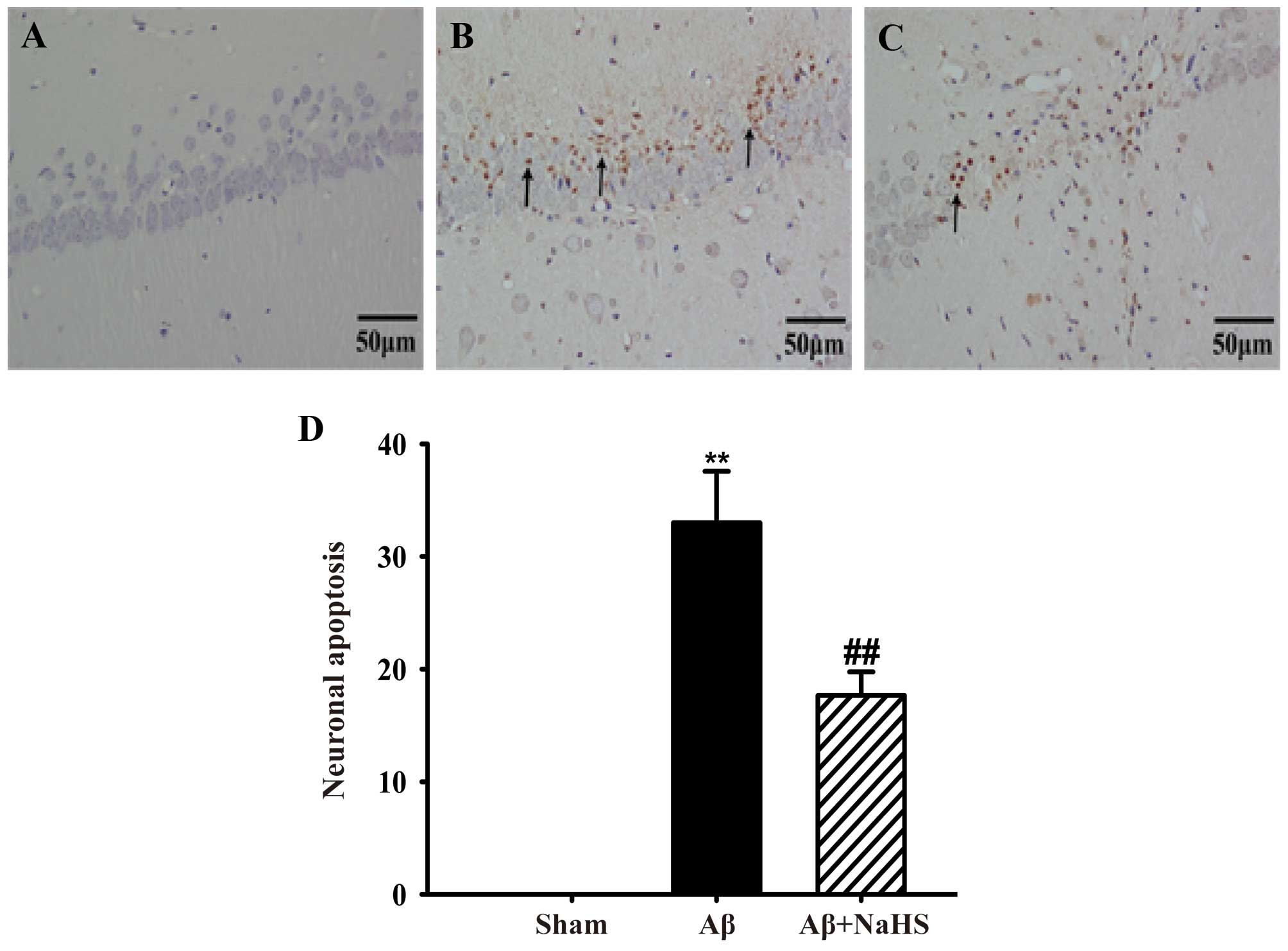
NaHS suppresses
Aβ25–35-induced cell apoptosis in the hippocampus of
rats
Aβ25–35-induced cell apoptosis in the
hippocampus of rats was detected by TUNEL staining. There was no
TUNEL reaction in the hippocampus of the rats from the
sham-operated group and examination revealed morphologically normal
neurons. There was an increase in the number of TUNEL-positive
pyramidal neurons after the Aβ25–35 injection. However,
treatment with NaHS markedly reduced the number of TUNEL-positive
neurons (Fig. 2). These results
indicate that NaHS suppresses Aβ25–35-induced cell
apoptosis in the hippocampus of rats.
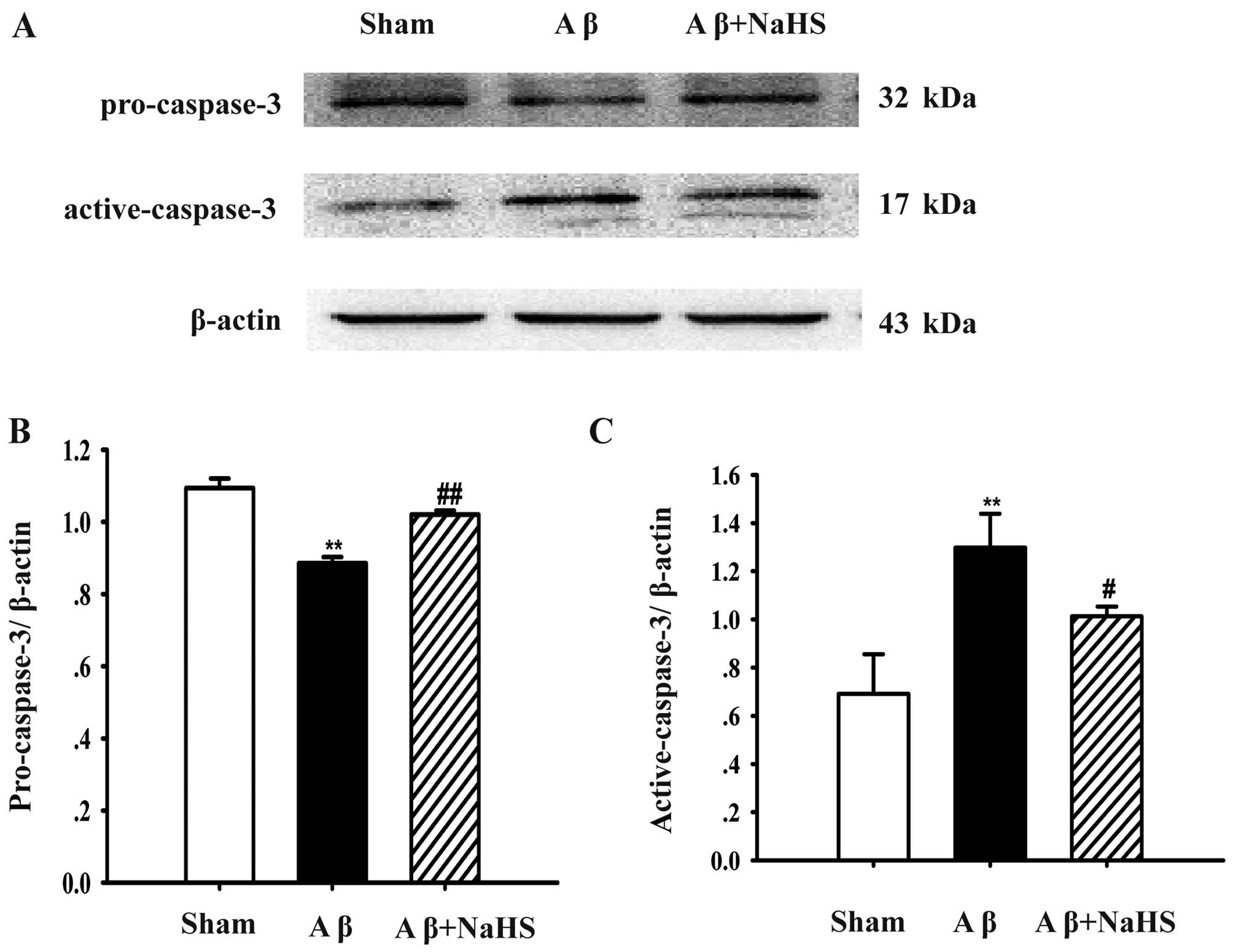
NaHS inhibits the activation of caspase-3
in the hippocampus of rats
To further examine the protective effects of NaHS
against Aβ25–35-induced apoptosis, the protein levels of
pro-caspase-3 and active-caspase-3 were examined by western blot
analysis. The protein level of pro-caspase-3 was decreased after
the Aβ25–35 injection compared with the sham-operated
group (P<0.01; Fig. 3).
However, treatment with NaHS increased the expression of
pro-caspase-3 in contrast to the Aβ25–35 group
(P<0.05). On the contrary, the Aβ25–35 injection
markedly increased the level of active-caspase-3 compared with the
sham-operated group (P<0.01). The administration of NaHS
significantly inhibited the protein level of active-caspase-3
compared with the Aβ25–35 group (P<0.01). Taken
together, our results indicate that NaHS prevents the
Aβ25–35-induced the activation of pro-caspase-3, and
thereafter decreases the level of active-caspase-3 in the
hippocampus.
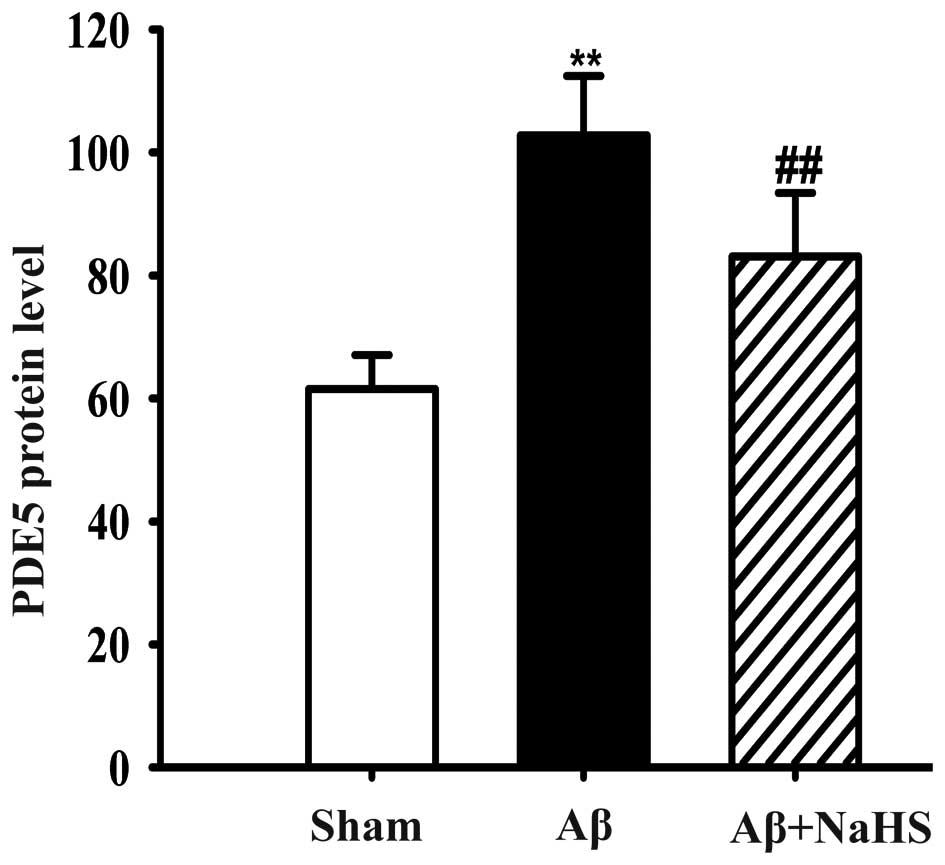
NaHS decreases the protein content of
PDE5 in the hippocampus of rats
The protein content of PDE5 in the hippocampus of
the rats was detected by ELISA. The rats in the Aβ25–35
group had a higher PDE5 protein level compared with the rats in the
sham-operated group (P<0.01; Fig.
4). By contrast, treatment with NaHS significantly reduced the
PDE5 protein level in the hippocampus of the rats compared to
exposure to Aβ25–35 alone (P<0.01; Fig. 4). Our data thus indicate that NaHS
inhibits PDE5 protein expression in the hippocampus induced by
Aβ25–35.
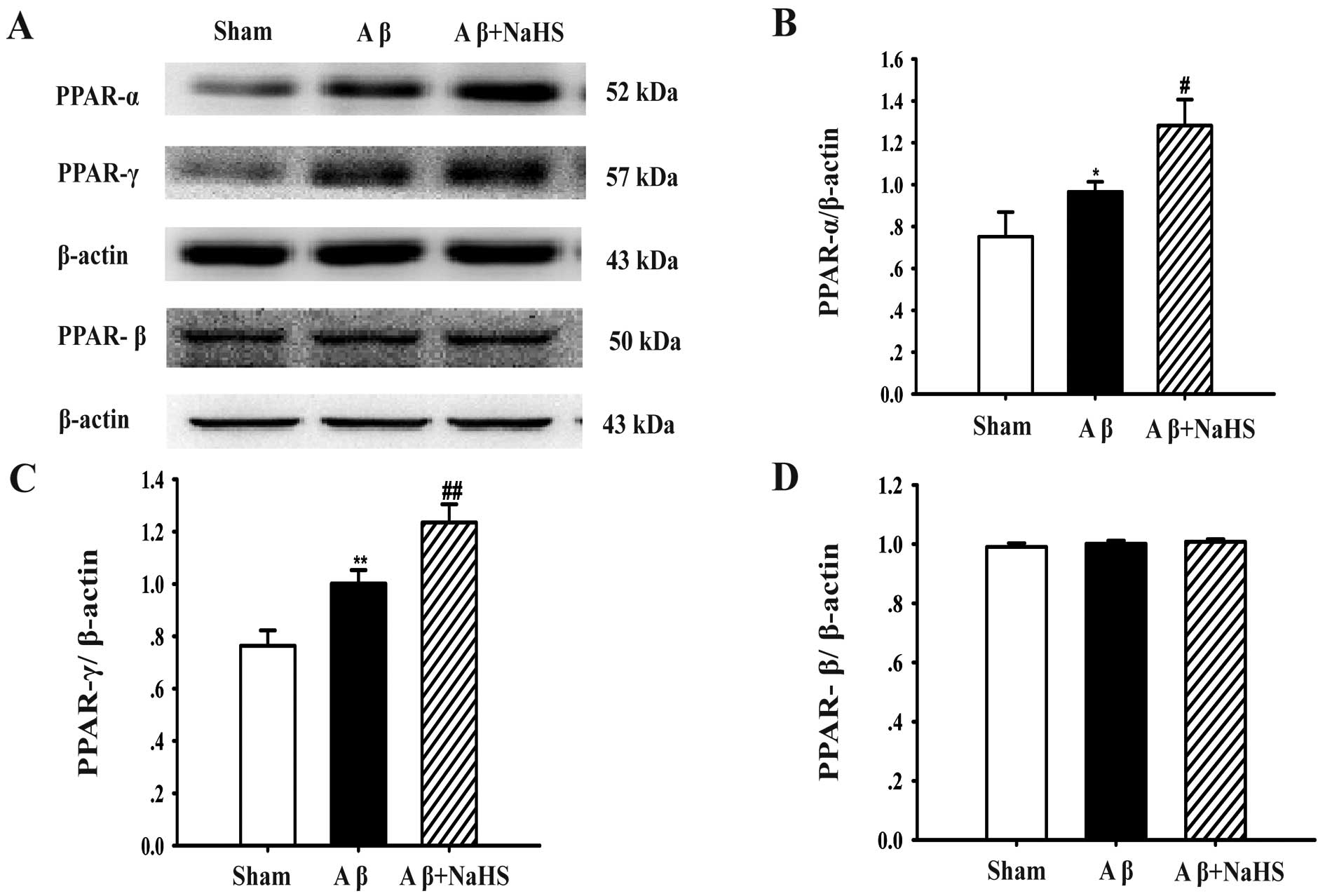
NaHS upregulates the expression of PPAR-α
and PPAR-γ, but not that of PPAR-β in the hippocampus
To determine whether the expression of PPARs is
associated with the protective effects of NaHS against
Aβ25–35-induced neurotoxicity, the protein levels of
PPAR-α, PPAR-β and PPAR-γ were determined by western blot analysis.
The PPAR-α level in the Aβ25–35 group was higher than
that in the sham-operated group (P<0.05), but PPAR-α expression
significantly increased further when the rats were treated with
NaHS (P<0.05; Fig. 5A and B).
As regards the PPAR-β protein level, there was no significant
difference between the Aβ25–35, sham and
Aβ25–35 + NaHS groups (Fig. 5A and D). As regards PPAR-γ protein
expression, Aβ25–35 injection significantly increased
the protein level compared with the sham-operated group
(P<0.01). Treatment with NaHS further enhanced the expression of
PPAR-γ compared to exposure to Aβ25–35 alone (P<0.01;
Fig. 5A and C). These results
suggest that NaHS attenuates Aβ25–35-induced
neurotoxicity by upregulating the expression of PPAR-α and PPAR-γ,
but it does not affect the protein level of PPAR-β.
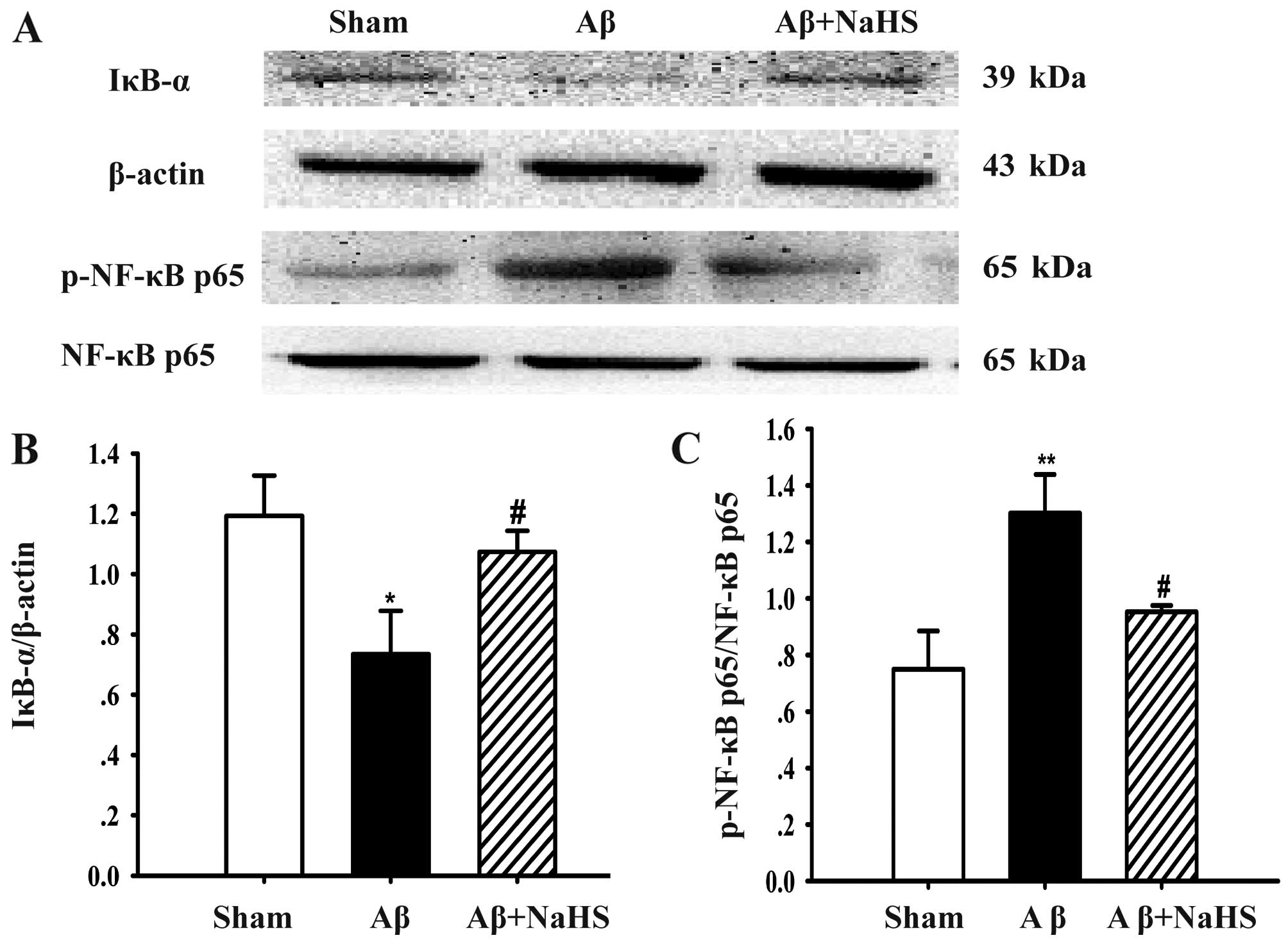
NaHS blocks the degradation of IκB-α and
suppresses NF-κB p65 phosphorylation
To further explore the molecular mechanisms
underlying the agonistic effects of NaHS, the protein levels of
IκB-α and NF-κB p65 phosphorylation were examined by western blot
analysis. There was a marked decrease in IκB-α protein expression
after the Aβ25–35 injection, whereas treatment with NaHS
induced a significant increase in the protein expression of IκB-α
(Fig. 6A and B), a primary member
of the IκB family. It was found that Aβ25–35 injection
into the hippocampus markedly enhanced the level of phosphorylated
NF-κB p65 (P<0.01). However, treatment with NaHS significantly
decreased the Aβ25–35-induced NF-κB p65 phosphorylation
(P<0.05; Fig. 6A and C). On
the whole, these findings demonstrate that NaHS blocks IκB-α
degradation and the activation of NF-κB p65 induced by
Aβ25–35.
Discussion
It is widely recognized that the formation and
deposition of Aβ is one of the main typical pathological
characteristics of AD the brain. Aβ is a 40–42 amino acid peptide
fragment derived by proteolysis from the integral membrane protein
known as Aβ precursor protein (39). The neurotoxicity of Aβ, including
different Aβ fragments, has been widely reported.
Aβ25–35 is the shorter toxic fragment corresponding to
amino acids 25–35, which encompasses the β sheet of the full
protein (40). In the present
study, we injected Aβ25–35 into the hippocampus of the
rats to induce neurodegenerative changes and neurotoxicity. The
rats in the Aβ25–35 + NaHS group were treated with NaHS
at the dose of 5 mg/kg once daily intraperitoneally as previous
reported (35,41). Nissl staining was applied to
observe the neurons in the hippocampus and Nissl bodies are one of
the characteristic structures of neurons. Our results revealed that
the Aβ25–35 injection induced neuronal cell death,
whereas treatment with NaHS significantly diminished neuronal cell
death. Apoptosis is a means of neuronal death; thus, in present
study, TUNEL staining was adapted to confirm the neuronal cell
death induced by Aβ25–35. There were many apoptotic
cells which were stained dark brown in the Aβ25–35
group, whereas in the NaHS group, the number of apoptotic cells was
decreased. These results indicate that NaHS suppresses
Aβ25–35-induced apoptosis. Apoptosis is a basic
physiological process in different biological systems. Previous
studies have shown that neuronal apoptosis is a critical factor
leading to neuronal loss, and neuronal loss in AD is intimately
linked with apoptosis (42). Aβ
is the core component of senile plaques (SP) in the AD-affected
brain. The abnormal deposition of Aβ is an important cause of AD.
As an initiation factor of apoptosis, Aβ can induce mitochondrial
dysfunction caused by the extrinsic pathway of apoptosis. It has
been shown in in vitro experiments that Aβ activates
caspases and then induces apoptosis only in the presence of the
functional electron transport chain of the mitochondria (43).
The caspase family plays a very important role in
mediating the process of apoptosis, where caspase-3 is responsible
for the proteolytic cleavage of many major proteins in a number of
apoptotic signaling pathways. Normally, caspase-3 acts as a zymogen
(pro-caspase-3, 32 kDa) that is active and presents in the
cytoplasm. Pro-caspase-3 can be activated by the Fas/FasL pathway
(44) and also through the
activation of the granzyme B pathway in the cytotoxic effects of
CTL cells (45). The active
enzyme is shown to consist of two subunits of 17 and 12 kDa,
originated from the precursor protein by cleavage at Asp-28-Ser-29
and Asp-175-Ser-176 by using electrospray MS and N-terminal
sequence analysis (46). The two
subunits comprise the active-caspase-3. Thus, in this study, the
protein levels of pro-caspase-3 and active-caspase-3 were examined
to further investigate the protective effects of NaHS against
Aβ25–35-induced apoptosis. Indeed, in the present study,
Aβ25–35 increased the levels of active-caspase-3,
whereas treatment with NaHS decreased the protein expression of
active-caspase-3. However, the protein expression of pro-caspase-3
is contrary to active-caspase-3. It is confirmed that
Aβ25–35 induces the apoptosis of hippocampal neuronal
cells by an enhanced caspase signaling pathway. On the other hand,
treatment with NaHS reversese these apoptotic changes.
ELISA was applied to determine whether
H2S attenuates memory impairment by inhibiting PDE5 in
the central nervous system (CNS). In our study, the expression of
PDE5 was significantly increased after the Aβ25–35
injection, whereas treatment with NaHS decreased the level of PDE5
in the hippocampus. Evidence suggests that endogenous
H2S can act both as a vasodilator and a vasoconstrictor
according to its concentration, and it also has a promoting effect
on erectile function (47,48).
The occurrence of erectile dysfunction in aged rats is related to
the disruption of the H2S pathway and the deficiency of
androgen in vivo (49). It
is widely accepted that PDE5 inhibitors, such as vardenafil,
sildenafil and tadalafil, are appropriate for the treatment of
erectile dysfunction. A number of studies have demonstrated that
PDE5 inhibitors can restore memory impairment in different models
of AD. For example, sildenafil has been shown to decrease
beta-secretase 1 (BACE1) and cathepsin B levels and reduce APP
amyloidogenic processing in SAMP8 mice (50) and to attenuate the age-related
impairment of synaptic plasticity and memory by restoring CREB
phosphorylation (51). Overall,
PDE5 inhibitors can attenuate age-related memory impairment and
cognitive dysfunction in physiological animal models of AD through
a variety of central and peripheral mechanisms. Of note,
H2S has also been identified as an endogenous inhibitor
of PDE5, able to enhance cGMP and cAMP levels in vessels (52). This suggests that NaHS may be a
PDE5 inhibitor although its underlying mechanisms of action remain
to be elucidated.
There is evidence to indicate that type 2 diabetes
mellitus (T2DM) enhances the risk of developing AD (53–55). PPARs belong to the family of
ligand-dependent nuclear hormone receptor transcription factors.
Three isotypes have been identified, including PPAR-α, PPAR-β/δ and
PPAR-γ in various species. The present study also demonstrated the
effect of NaHS on the expression of PPAR-α, PPAR-β and PPAR-γ in
the hippocampus of rats with neurotoxicity induced by
Aβ25–35. Our results demonstrated that NaHS enhanced the
expression of PPAR-α and PPAR-γ in the hippocampus of both the
sham-operated and Aβ-treated animals. Consistent with the PPAR-γ
elevation in the AD-affected brain, our data revealed an increase
in both the expression and transcriptional activity of PPAR-α and
PPAR-γ in the hippocampus of Aβ-inoculated rats compared with the
sham-operated group. Since PPAR-γ is a transcription factor with
well-established neuroprotective features (56), its activation may serve as an
adaptive response to protect neurons against the deleterious
effects of Aβ. Our results were consistent with those of another
study which demonstrated that WIN55212-2 exerts neuroprotective and
anti-inflammatory effects against Aβ-induced damage by increasing
the PPAR-γ level (57). It has
been shown that both a PPAR-γ agonist (ciglitazone) and a PPAR-α
agonist (WY 14.643) are able to protect neurons by modulating
mitochondrial fusion and fission, leading to a better response of
neurons to oxidative stress in neurodegenerative disorders, such as
AD (58). However, it is not
possible to determine whether NaHS acts as a PPAR agonist by
observing the upregulation of PPAR-α and PPAR-γ.
The inflammatory reaction induced by Aβ deposition,
leading to the activation of microglia and astroglia, and the
subsequent release of inflammatory cytokines (IL-β, TNF-α and COX-2
and so on), plays a significant role in the pathological processing
of AD. PPAR-γ has been shown to inhibit the expression of IL-1β,
TNF-α and other inflammation-related mediators (59), although the potential mechanisms
responsible for these effects are not yet fully understood. These
factors may be situated downstream of the NF-κB signaling pathway;
as a result, the suppressive effect on the pro-inflammatory genes
of PPAR-γ is through the antagonism of the actions of NF-κB
(60). NF-κB is well known as a
key regulator that upregulates the expression of many
pro-inflammatory cytokines and inducible effector enzymes linked to
the inflammatory process. NF-κB remains inactivated by being
coupled with an inhibitory protein, IκB. NF-κB p65 is widely
studied among its several protein subtypes. The degradation of IκB
is followed by the translocation of NF-κB p65 and subsequent
liberation (61). In this study,
it was found that the degradation of IκB-α and NF-κB p65
phosphorylation were enhanced after the Aβ25–35
injection. However, treatment with NaHS decreased the degradation
of IκB-α and restrained NF-κB p65 phosphorylation in rats with
Aβ25–35-induced neurotoxicity. Therefore, this study
suggests that NaHS may act as an anti-inflammatory mediator. These
findings are consistent with those of a previous study showing that
NF-κB and its nuclear translocation can prevent Aβ-induced toxicity
and apoptosis (62).
In conclusion, the present study demonstrated that
NaHS attenuated Aβ25–35-induced neuronal death and
suppressed apoptosis in the rat hippocampus. The underlying
mechanisms are, at least partly due to the inhibition of the
protein content of PDE5 and the upregulation of PPAR-α and PPAR-γ
expression. Hence, NaHS may prove to be beneficial in the treatment
of AD.
Acknowledgments
This study was supported by the following grants:
the Program for Changjiang Scholars and Innovative Research Team in
University, China (grant no. IRT1197); the Science and Technology
Department of Guizhou Province of outstanding youth science and
technology talent capital (grant no. 201326); the National Natural
Science Foundation of China (grant no. 81360489); and the
Outstanding Youth Science and Technology Talent Capital of Guizhou
Province (grant no. 201326).
References
|
1
|
Selkoe DJ: Alzheimer's disease is a
synaptic failure. Science. 298:789–791. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Deshpande A, Mina E, Glabe C and Busciglio
J: Different conformations of amyloid β induce neurotoxicity by
distinct mechanisms in human cortical neurons. J Neurosci.
26:6011–6018. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mrak RE and Griffin WST: Interleukin-1,
neuroinflammation, and Alzheimer's disease. Neurobiol Aging.
22:903–908. 2001. View Article : Google Scholar
|
|
4
|
Kim H, Youn K, Ahn MR, Kim OY, Jeong WS,
Ho CT and Jun M: Neuroprotective effect of loganin against
Aβ25–35-induced injury via the NF-κB-dependent signaling pathway in
PC12 cells. Food Funct. 6:1108–1116. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Block ML and Hong JS: Microglia and
inflammation-mediated neurodegeneration: Multiple triggers with a
common mechanism. Prog Neurobiol. 76:77–98. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ricote M and Glass CK: PPARs and molecular
mechanisms of transrepression. Biochim Biophys Acta. 1771:926–935.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Heneka MT and Landreth GE: PPARs in the
brain. Biochim Biophys Acta. 1771:1031–1045. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schnegg CI and Robbins ME: Neuroprotective
mechanisms of PPARδ: Modulation of oxidative stress and
inflammatory processes. PPAR Res. 2011:3735602011. View Article : Google Scholar
|
|
9
|
Biessels GJ, Staekenborg S, Brunner E,
Brayne C and Scheltens P: Risk of dementia in diabetes mellitus: A
systematic review. Lancet Neurol. 5:64–74. 2006. View Article : Google Scholar
|
|
10
|
Geldmacher DS, Fritsch T, McClendon MJ and
Landreth G: A randomized pilot clinical trial of the safety of
pioglitazone in treatment of patients with Alzheimer disease. Arch
Neurol. 68:45–50. 2011. View Article : Google Scholar
|
|
11
|
Sato T, Hanyu H, Hirao K, Kanetaka H,
Sakurai H and Iwamoto T: Efficacy of PPAR-γ agonist pioglitazone in
mild Alzheimer disease. Neurobiol Aging. 32:1626–1633. 2011.
View Article : Google Scholar
|
|
12
|
Pedersen WA, McMillan PJ, Kulstad JJ,
Leverenz JB, Craft S and Haynatzki GR: Rosiglitazone attenuates
learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol.
199:265–273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Escribano L, Simón AM, Gimeno E,
Cuadrado-Tejedor M, López de Maturana R, García-Osta A, Ricobaraza
A, Pérez-Mediavilla A, Del Río J and Frechilla D: Rosiglitazone
rescues memory impairment in Alzheimer's transgenic mice:
Mechanisms involving a reduced amyloid and tau pathology.
Neuropsychopharmacology. 35:1593–1604. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rodriguez-Rivera J, Denner L and Dineley
KT: Rosiglitazone reversal of Tg2576 cognitive deficits is
independent of peripheral gluco-regulatory status. Behav Brain Res.
216:255–261. 2011. View Article : Google Scholar
|
|
15
|
Prickaerts J, Steinbusch HW, Smits JF and
de Vente J: Possible role of nitric oxide-cyclic GMP pathway in
object recognition memory: Effects of 7-nitroindazole and
zaprinast. Eur J Pharmacol. 337:125–136. 1997. View Article : Google Scholar
|
|
16
|
Ota KT, Pierre VJ, Ploski JE, Queen K and
Schafe GE: The NO-cGMP-PKG signaling pathway regulates synaptic
plasticity and fear memory consolidation in the lateral amygdala
via activation of ERK/MAP kinase. Learn Mem. 15:792–805. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wincott CM, Abera S, Vunck SA, Tirko N,
Choi Y, Titcombe RF, Antoine SO, Tukey DS, DeVito LM, Hofmann F, et
al: cGMP-dependent protein kinase type II knockout mice exhibit
working memory impairments, decreased repetitive behavior, and
increased anxiety-like traits. Neurobiol Learn Mem. 114:32–39.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Puzzo D, Loreto C, Giunta S, Musumeci G,
Frasca G, Podda MV, Arancio O and Palmeri A: Effect of
phosphodiesterase-5 inhibition on apoptosis and beta amyloid load
in aged mice. Neurobiol Aging. 35:520–531. 2014. View Article : Google Scholar
|
|
19
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pollack M, Phaneuf S, Dirks A and
Leeuwenburgh C: The role of apoptosis in the normal aging brain,
skeletal muscle, and heart. Ann NY Acad Sci. 959:93–107. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reix S, Mechawar N, Susin SA, Quirion R
and Krantic S: Expression of cortical and hippocampal
apoptosis-inducing factor (AIF) in aging and Alzheimer's disease.
Neurobiol Aging. 28:351–356. 2007. View Article : Google Scholar
|
|
22
|
Galbán S and Duckett CS: XIAP as a
ubiquitin ligase in cellular signaling. Cell Death Differ.
17:54–60. 2010. View Article : Google Scholar :
|
|
23
|
Eckelman BP, Salvesen GS and Scott FL:
Human inhibitor of apoptosis proteins: Why XIAP is the black sheep
of the family. EMBO Rep. 7:988–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zheng H and Koo EH: Biology and
pathophysiology of the amyloid precursor protein. Mol Neurodegener.
6:272011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gadalla MM and Snyder SH: Hydrogen sulfide
as a gasotransmitter. J Neurochem. 113:14–26. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kamoun P: Endogenous production of
hydrogen sulfide in mammals. Amino Acids. 26:243–254. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abe K and Kimura H: The possible role of
hydrogen sulfide as an endogenous neuromodulator. J Neurosci.
16:1066–1071. 1996.PubMed/NCBI
|
|
28
|
Enokido Y, Suzuki E, Iwasawa K, Namekata
K, Okazawa H and Kimura H: Cystathionine β-synthase, a key enzyme
for homocysteine metabolism, is preferentially expressed in the
radial glia/astrocyte lineage of developing mouse CNS. FASEB J.
19:1854–1856. 2005.PubMed/NCBI
|
|
29
|
Shibuya N, Mikami Y, Kimura Y, Nagahara N
and Kimura H: Vascular endothelium expresses 3-mercaptopyruvate
sulfurtransferase and produces hydrogen sulfide. J Biochem.
146:623–626. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kimura Y and Kimura H: Hydrogen sulfide
protects neurons from oxidative stress. FASEB J. 18:1165–1167.
2004.PubMed/NCBI
|
|
31
|
Yin WL, He JQ, Hu B, Jiang ZS and Tang XQ:
Hydrogen sulfide inhibits MPP(+)-induced apoptosis in PC12 cells.
Life Sci. 85:269–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee SW, Hu YS, Hu LF, Lu Q, Dawe GS, Moore
PK, Wong PT and Bian JS: Hydrogen sulphide regulates calcium
homeostasis in microglial cells. Glia. 54:116–124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kida K, Yamada M, Tokuda K, Marutani E,
Kakinohana M, Kaneki M and Ichinose F: Inhaled hydrogen sulfide
prevents neurodegeneration and movement disorder in a mouse model
of Parkinson's disease. Antioxid Redox Signal. 15:343–352. 2011.
View Article : Google Scholar :
|
|
34
|
Liu XQ, Liu XQ, Jiang P, Huang H and Yan
Y: Plasma levels of endogenous hydrogen sulfide and homocysteine in
patients with Alzheimer's disease and vascular dementia and the
significance thereof. Zhonghua Yi Xue Za Zhi. 88:2246–2249. 2008.In
Chinese. PubMed/NCBI
|
|
35
|
Xuan A, Long D, Li J, Ji W, Zhang M, Hong
L and Liu J: Hydrogen sulfide attenuates spatial memory impairment
and hippocampal neuroinflammation in β-amyloid rat model of
Alzheimer's disease. J Neuroinflammation. 9:2022012. View Article : Google Scholar
|
|
36
|
Laursen SE and Belknap JK:
Intracerebroventricular injections in mice. Some methodological
refinements. J Pharmacol Methods. 16:355–357. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Biagini G, D'Arcangelo G, Baldelli E,
D'Antuono M, Tancredi V and Avoli M: Impaired activation of CA3
pyramidal neurons in the epileptic hippocampus. Neuromolecular Med.
7:325–342. 2005. View Article : Google Scholar
|
|
38
|
Jin F, Gong Q-H, Xu Y-S, Wang LN, Jin H,
Li F, Li LS, Ma YM and Shi JS: Icariin, a phosphodiesterase-5
inhibitor, improves learning and memory in APP/PS1 transgenic mice
by stimulation of NO/cGMP signalling. Int J Neuropsychopharmacol.
17:871–881. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Amtul Z, Uhrig M and Beyreuther K:
Additive effects of fatty acid mixtures on the levels and ratio of
amyloid β40/42 peptides differ from the effects of individual fatty
acids. J Neurosci Res. 89:1795–1801. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaminsky YG, Marlatt MW, Smith MA and
Kosenko EA: Subcellular and metabolic examination of amyloid-β
peptides in Alzheimer disease pathogenesis: Evidence for
Abeta(25-35). Exp Neurol. 221:26–37. 2010. View Article : Google Scholar
|
|
41
|
Gong QH, Wang Q, Pan LL, Liu XH, Huang H
and Zhu YZ: Hydrogen sulfide attenuates lipopolysaccharide-induced
cognitive impairment: A pro-inflammatory pathway in rats. Pharmacol
Biochem Behav. 96:52–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yuan J and Yankner BA: Apoptosis in the
nervous system. Nature. 407:802–809. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Morais Cardoso S, Swerdlow RH and Oliveira
CR: Induction of cytochrome c-mediated apoptosis by amyloid β 25–35
requires functional mitochondria. Brain Res. 931:117–125. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhong B, Hu Z, Tan J, Lu T, Lei Q, Chen C
and Zeng L: Hsp20 protects against oxygen-glucose
deprivation/reperfusion-induced Golgi fragmentation and apoptosis
through Fas/FasL pathway. Oxid Med Cell Longev. 2015:6069342015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ben Safta T, Ziani L, Favre L, Lamendour
L, Gros G, Mami-Chouaib F, Martinvalet D, Chouaib S and Thiery J:
Granzyme B-activated p53 interacts with Bcl-2 to promote cytotoxic
lymphocyte-mediated apoptosis. J Immunol. 194:418–428. 2015.
View Article : Google Scholar
|
|
46
|
Nicholson DW, Ali A, Thornberry NA,
Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle
M, Lazebnik YA, et al: Identification and inhibition of the
ICE/CED-3 protease necessary for mammalian apoptosis. Nature.
376:37–43. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zuo C, Huang YM, Jiang R, Yang HF, Cheng B
and Chen F: Endogenous hydrogen sulfide and androgen
deficiency-induced erectile dysfunction in rats. Zhonghua Nan Ke
Xue. 20:605–612. 2014.In Chinese. PubMed/NCBI
|
|
48
|
Leonardi R and Alemanni M: The management
of erectile dysfunction: Innovations and future perspectives. Arch
Ital Urol Androl. 83:60–62. 2011.PubMed/NCBI
|
|
49
|
Srilatha B, Muthulakshmi P, Adaikan PG and
Moore PK: Endogenous hydrogen sulfide insufficiency as a predictor
of sexual dysfunction in aging rats. Aging Male. 15:153–158. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Orejana L, Barros-Miñones L, Jordan J,
Cedazo-Minguez A, Tordera RM, Aguirre N and Puerta E: Sildenafil
decreases BACE1 and cathepsin B levels and reduces APP
amyloidogenic processing in the SAMP8 mouse. J Gerontol A Biol Sci
Med Sci. 70:675–685. 2015. View Article : Google Scholar
|
|
51
|
Palmeri A, Privitera L, Giunta S, Loreto C
and Puzzo D: Inhibition of phosphodiesterase-5 rescues age-related
impairment of synaptic plasticity and memory. Behav Brain Res.
240:11–20. 2013. View Article : Google Scholar
|
|
52
|
Bucci M, Papapetropoulos A, Vellecco V,
Zhou Z, Pyriochou A, Roussos C, Roviezzo F, Brancaleone V and
Cirino G: Hydrogen sulfide is an endogenous inhibitor of
phosphodiesterase activity. Arterioscler Thromb Vasc Biol.
30:1998–2004. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ott A, Stolk RP, van Harskamp F, Pols HA,
Hofman A and Breteler MM: Diabetes mellitus and the risk of
dementia: The Rotterdam Study. Neurology. 53:1937–1942. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Leibson CL, Rocca WA, Hanson VA, Cha R,
Kokmen E, O'Brien PC and Palumbo PJ: Risk of dementia among persons
with diabetes mellitus: A population-based cohort study. Am J
Epidemiol. 145:301–308. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kivipelto M, Ngandu T, Fratiglioni L,
Viitanen M, Kåreholt I, Winblad B, Helkala EL, Tuomilehto J,
Soininen H and Nissinen A: Obesity and vascular risk factors at
midlife and the risk of dementia and Alzheimer disease. Arch
Neurol. 62:1556–1560. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Abdelrahman M, Sivarajah A and Thiemermann
C: Beneficial effects of PPAR-γ ligands in ischemia-reperfusion
injury, inflammation and shock. Cardiovasc Res. 65:772–781. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fakhfouri G, Ahmadiani A, Rahimian R,
Grolla AA, Moradi F and Haeri A: WIN55212-2 attenuates
amyloid-beta-induced neuroinflammation in rats through activation
of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology.
63:653–666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zolezzi JM, Silva-Alvarez C, Ordenes D,
Godoy JA, Carvajal FJ, Santos MJ and Inestrosa NC: Peroxisome
proliferator-activated receptor (PPAR) γ and PPARα agonists
modulate mitochondrial fusion-fission dynamics: Relevance to
reactive oxygen species (ROS)-related neurodegenerative disorders?
PLoS One. 8:e640192013. View Article : Google Scholar
|
|
59
|
Li AC, Binder CJ, Gutierrez A, Brown KK,
Plotkin CR, Pattison JW, Valledor AF, Davis RA, Willson TM, Witztum
JL, et al: Differential inhibition of macrophage foam-cell
formation and atherosclerosis in mice by PPARalpha, β/δ, and γ. J
Clin Invest. 114:1564–1576. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Berghe WV, Vermeulen L, Delerive P, De
Bosscher K, Staels B and Haegeman G: A paradigm for gene
regulation: Inflammation, NF-κB and PPAR. Adv Exp Med Biol.
544:181–196. 2003. View Article : Google Scholar
|
|
61
|
Bannon A, Zhang SD, Schock BC and Ennis M:
Cystic fibrosis from laboratory to bedside: The role of A20 in
NF-κB-mediated inflammation. Med Princ Pract. 24:301–310. 2015.
View Article : Google Scholar
|
|
62
|
Chong ZZ, Li F and Maiese K:
Erythropoietin requires NF-kappaB and its nuclear translocation to
prevent early and late apoptotic neuronal injury during β-amyloid
toxicity. Curr Neurovasc Res. 2:387–399. 2005. View Article : Google Scholar : PubMed/NCBI
|