Introduction
Cardiovascular disease is the leading cause of
morbidity and mortality in the modern world. It is now evident that
cardiomyocyte apoptosis plays a key role in the pathogenesis of
various cardiovascular diseases, including myocardial infarction
(1), ischemia/reperfusion injury
(2), dilated cardiomyopathy
(3) and end-stage heart failure
(4). Apoptosis is a highly
regulated program of cell death and can be activated in
cardiomyocytes by multiple stressors, including cytokines (5), oxidative stress (6) and DNA damage (7). Previous studies have demonstrated
that the inhibition of apoptosis exerts cardioprotective effects
and can prevent the development of heart failure (8,9).
Lactate is produced as the result of glycolysis in
the cytosol and is balanced by oxidation in the mitochondria. This
process of the intracellular lactate shuttle is mediated by
monocarboxylate transporter 1 (MCT1) on the mitochondrial membrane
and plays an important role in the energy metabolism of
cardiomyocytes (10,11). However, it is now known that
lactate is produced continuously under fully aerobic conditions.
Lactate functions as an important signaling molecule to regulate
intracellular reactive oxygen species (ROS) the generation and the
lactate signaling cascade (12).
Under normal physiological conditions, low levels of ROS upregulate
the expression of MCT1 by activating several transcription factors,
which, in turn, facilitate the transport of lactate into the
mitochondria for oxidative metabolism (13). However, under pathological
conditions, the intracellular lactate concentration increases
several fold and promotes the excessive generation of ROS. High
levels of ROS can cause oxidative stress and mitochondrial damage,
which lead to the activation of mitochondrial-dependent apoptosis
(14). A significant association
has been identified between the lactate signaling cascade and
cardiovascular diseases, such as myocardial infarction (15), ischemic/reperfusion injury
(16), atrial fibrillation
(17) and heart failure (18). Therefore, targeting the regulation
of the lactate signaling cascade may be a promising therapeutic
approach for the treatment of multiple cardiovascular diseases.
Asiatic acid (AA) is a triterpenoid derived from
Centella asiatica. Pharmacologically, it is used as an
antioxidant (19) and
anti-inflammatory (20) agent,
and has been shown to exert neuroprotective (21) and hepatoprotective (22) effects. Recently, Si et al
(23) reported that AA attenuated
cardiac hypertrophy by blocking transforming growth factor-β1
(TGF-β1) mediated hypertrophic signaling. Zhang et al
(24) reported that AA prevented
C2-ceramide-induced neuronal injury by inhibiting
mitochondrial-dependent apoptosis. Therefore, in this study, we
hypothesized that AA may inhibit cardiomyocyte apoptosis. To
examine this hypothesis, we investigated the anti-apoptotic effects
and mechanisms of action of AA using an in vitro model of
lactate-induced cardiomyocyte apoptosis.
Materials and methods
Reagents
The following reagents were obtained from the
indicated commercial sources. Purified natural product of AA (97%),
dimethyl sulfoxide (DMSO), sodium lactate and
2′,7′-dichlorofluorescein-diacetate (DCFH-DA) were obtained from
Sigma-Aldrich (St. Louis, MO, USA).
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide
(JC-1) was obtained from BD Biosciences (San Jose, CA, USA). The
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI)
staining kit and the in situ cell death detection kit were
obtained from Roche Applied Science (Mannheim, Germany). Dulbecco's
modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were
obtained from Gibco-BRL Life Technologies, Inc. (Carlsbad, CA,
USA). The cell counting kit-8 (CCK-8) assay kit was obtained from
Dojindo Laboratories (Kumamoto, Japan). The anti-cleaved caspase-9
(9509S), anti-cleaved caspase-3 (9661S), anti-cytochrome c
oxidase polypeptide IV (COX4; 4844S) and anti-β-actin (4967S)
primary antibodies, and HRP-conjugated secondary antibody (7074S)
were obtained from Cell Signaling Technology (Danvers, MA, USA).
The anti-cytochrome c primary antibody (sc-13560) was
obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
The anti-MCT1 primary antibody (AB3540P) was obtained from Chemicon
International (Temecula, CA, USA). The cytoplasmic and
mitochondrial protein extraction kits were obtained from Keygen
Biotechnology (Nanjing, China). Unless otherwise indicated, all
chemicals and materials were purchased from Sigma-Aldrich.
Cell culture and treatment
Primary cultures of ventricular myocytes were
prepared from neonatal Sprague-Dawley rats. All animal care and
experimental protocols complied with the Guide for the Care and Use
of Laboratory Animals, published by the US National Institutes of
Health (NIH Publication no. 85–23, revised 1996) and were approved
by the Animal Ethics Committee of First Hospital Affiliated to
Soochow University. All efforts were made to minimize animal
suffering and the number of animals used. As previously described
(23), 20 neonatal Sprague-Dawley
rats (1–2 days of age, weighing, 5–8 g; from the Experimental
Animal Center of Soochow University, Suzhou, China) were
anesthetized by isoflurane. The hearts were then immediately
removed under aseptic conditions and washed in Ca2+- and
Mg2+-free phosphate-buffered saline (PBS). After the
atria and aorta were excised, the ventricles were minced and
enzymatically digested with 0.125% trypsin (Gibco BRL Life
Technologies, Inc.) and 0.1% collagenase I (Sigma-Aldrich). The
liberated cells were collected by centrifugation at 200 × g for 5
min at room temperature and incubated in a 100-mm culture dish for
90 min at 37°C in an incubator with 5% CO2 air.
Non-adherent cells were harvested as cardiomyocytes. They were
seeded at a density of 1×106 cells/ml and cultured in
DMEM supplemented with 10% FBS and 0.1% penicillin/streptomycin at
37°C in an incubator with 5% CO2. The medium was changed
twice daily to maintain lactate at low concentrations in all groups
for 2 days prior to experimentation. There were 4 experimental
groups: i) the control group, ii) the AA group, iii) the lactate
group and iv) the lactate + AA group. The cells were incubated with
AA (20 µM) for 24 h prior to stimulation with lactate (20
mM) for 24 h. Untreated cells served as the controls. AA was
freshly prepared as a stock solution in DMSO and diluted with
sterile double-distilled water [0.1% (v/v) DMSO]. DMSO was present
at equal concentrations (0.03%) in all groups. Sodium lactate was
dissolved in sterile double-distilled water.
Cell viability assay
Cardiomyocyte viability was measured by CCK-8 assay
according to the manufacturer's instructions. In brief,
cardiomyocytes were initially cultured at a density of
1×104 cells/well in 96-well plates. The cells were then
incubated with various concentrations of AA (5–30 µM) for 24
h or exposed to various concentrations of lactate (10–40 mM) for 24
h at 37°C. CCK-8 solution (10 µl) was subsequently added to
each well and the culture was incubated for 4 h at 37°C. The
absorbance at 450 nm was measured using a micro-plate reader
(Bio-Rad Laboratories, Hercules, CA, USA). All measurements were
performed in triplicate, and cell viability was calculated as a
percentage.
Determination of intracellular ROS
levels
The levels of intracellular ROS were measured by
flow cytometry using DCFH-DA as the probe. Cardiomyocytes were
incubated with lactate for 24 h after being pre-treated with or
without AA for 24 h. The cells were then harvested and incubated
with DCFH-DA (10 µmol/l) diluted in serum-free DMEM at 37°C
for 30 min. Subsequently, the cells were washed with PBS 3 times
and analyzed by flow cytometry at an excitation wavelength of 488
nm and an emission wavelength of 530 nm. Data analysis was
performed using a Kaluza flow cytometer (Beckman Coulter, Brea, CA,
USA).
Mitochondrial membrane potential (Δψm)
assay
Δψm was determined by flow cytometry and visualized
by fluorescence microscopy using the fluorescent probe, JC-1. JC-1
accumulates in the mitochondria with a normal Δψm, leading to the
formation of JC-1 aggregates and the emission of red fluorescence.
However, JC-1 leaks from the mitochondria into the cytoplasm as a
monomer when the Δψm is depolarized, resulting in a decrease in red
fluorescence and an increase in green fluorescence. Following
treatment, the cardiomyocytes were washed with PBS and incubated in
fresh medium containing JC-1 (10 µg/ml) for 20 min at 37°C.
The stained cells were visualized under a fluorescence microscope
(Nikon, Tokyo, Japan). In addition, the cells in each group were
also collected and resuspended in fresh medium containing JC-1 (10
µg/ml) and then incubated for 20 min at 37°C. The ratio of
red and green fluorescence was analyzed using a Kaluza flow
cytometer.
Annexin V-FITC/PI apoptosis assay
Cell apoptosis was evaluated using an Annexin
V-FITC/PI staining kit. In brief, the cardiomyocytes in each group
were harvested and washed 3 times with PBS, and then resuspended in
binding buffer. A total of 1×106 cells/ml was
transferred to a flow tube and then stained with 2 µl of
Annexin V-FITC and 2 µl of PI for 15 min at room temperature
in the dark. The samples were analyzed using a Kaluza flow
cytometer. Annexin V+/PI− and Annexin
V+/PI+ cells were considered as apoptotic
cells in the early and late phase, respectively.
Terminal
deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL)
staining
For the detection of apoptosis, the cardiomyocytes
were stained using the TUNEL technique with an in situ cell
death detection kit according to the manufacturer's instructions.
Briefly, the cultured cell smears were fixed with 4%
paraformaldehyde in PBS for 1 h at room temperature and then
permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate for 2
min on ice. The cells were then incubated with the TUNEL reaction
mixture for 1 h at 37°C. After rinsing with PBS, cells were stained
with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min and visualized
under a fluorescence microscope. The number of TUNEL-positive
nuclei (green fluorescence) was expressed as a percentage of total
nuclei (blue fluorescence).
Western blot analysis
Cytoplasmic and mitochondrial proteins were isolated
from the cardiomyocytes by differential centrifugation using the
mitochondrial fractionation kit according to the manufacturer's
instructions. Briefly, the harvested cardiomyocytes were
resuspended in ice-cold cytoplasmic lysis buffer for 10 min and
homogenized on ice by an ultrasonic homogenizer. The homogenate was
centrifuged at 800 × g for 10 min at 4°C. The collected supernatant
was centrifuged at 10,000 × g for 20 min at 4°C to obtain the
cytoplasmic protein (supernatant) and the mitochondrial fraction
(pellet). The mitochondrial fraction was then resuspended in the
mitochondria lysis buffer on ice for 20 min. The supernatant
(mitochondrial protein) was collected by centrifugation at 10,000 ×
g for 10 min at 4°C. The protein concentrations were determined by
the bicinchoninic acid (BCA) method. The proteins (20–30 µg)
were separated by 10–15% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto polyvinylidine
difluoride membranes. After blocking in 5% (v/v) non-fat dry milk
in Tris-buffered saline (TBS) with 0.1% Tween-20 (TBST) at room
temperature for 2 h, the membranes were then incubated with primary
antibodies overnight at 4°C. After washing with TBST, the membranes
were incubated with HRP-conjugated secondary antibodies at room
temperature for 1 h. Blots were developed using ECL reagents
(Thermo Scientific, Rockford, IL, USA) and were then exposed using
the digital imaging system (Bio-Rad Laboratories). The intensity of
each band was analyzed using Image Lab 2.0 software (Bio-Rad
Laboratories). The intensity of mitochondrial MCT1 was normalized
to that of COX4. The intensities of cytoplasmic cleaved caspase-9,
cleaved caspase-3 and cytochrome c were normalized to those
of β-actin.
Statistical analysis
All results are presented as the mean ± standard
deviation (SD). The GraphPad Prism 5.01 software (GraphPad
Software, Inc., CA, USA) and the PASW Statistics 18.0 (SPSS Inc.,
Fayetteville, NC, USA) packages were used. Differences among groups
were tested by one-way ANOVA. Comparisons between 2 groups were
performed by an unpaired two-tailed Student's t-test. A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
AA protects cardiomyocytes against
lactate-induced damage
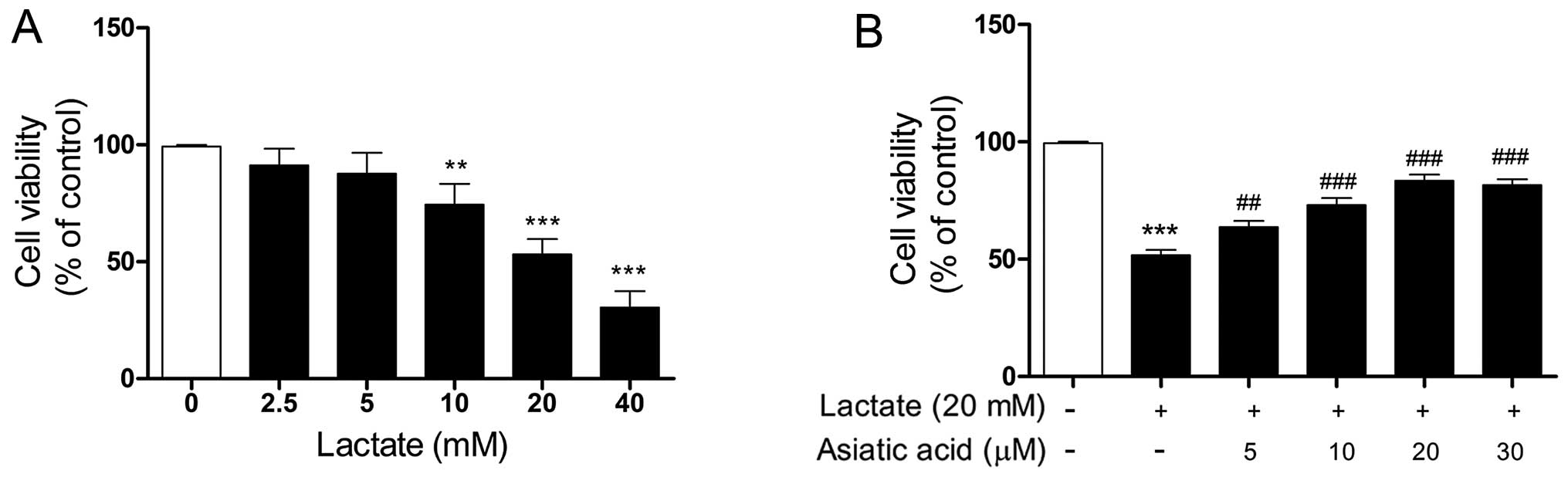
The effect of lactate on cell viability was first
evaluated. As shown in Fig. 1A,
cells were treated with increasing concentrations of lactate for 24
h. Lactate impaired cell viability in a concentration-dependent
manner over the tested concentration range (10–40 mM). We used 20
mM lactate for 24 h in our subsequent experiments, as this
concentration of lactate caused significant cell damage and reduced
cell viability by approximately 50% relative to the control cells.
We then evaluated the effects of AA on lactate-induced cell damage.
The cardiomyocytes were treated with lactate (20 mM) for 24 h in
the presence or absence of AA (5–30 µM). No noticeable
changes were observed in the viability of the cardiomyocytes
treated with AA (5–30 µM) for 24 h, as previously described
by Si et al (23). The
cytotoxic effects of lactate on cardio-myocytes were significantly
attenuated by pre-treatment with AA (5–30 µM) for 24 h, and
AA at 20 µM provided maximal protection against
lactate-induced damage (Fig.
1B).
AA inhibits the lactate-induced increase
in intracellular ROS levels
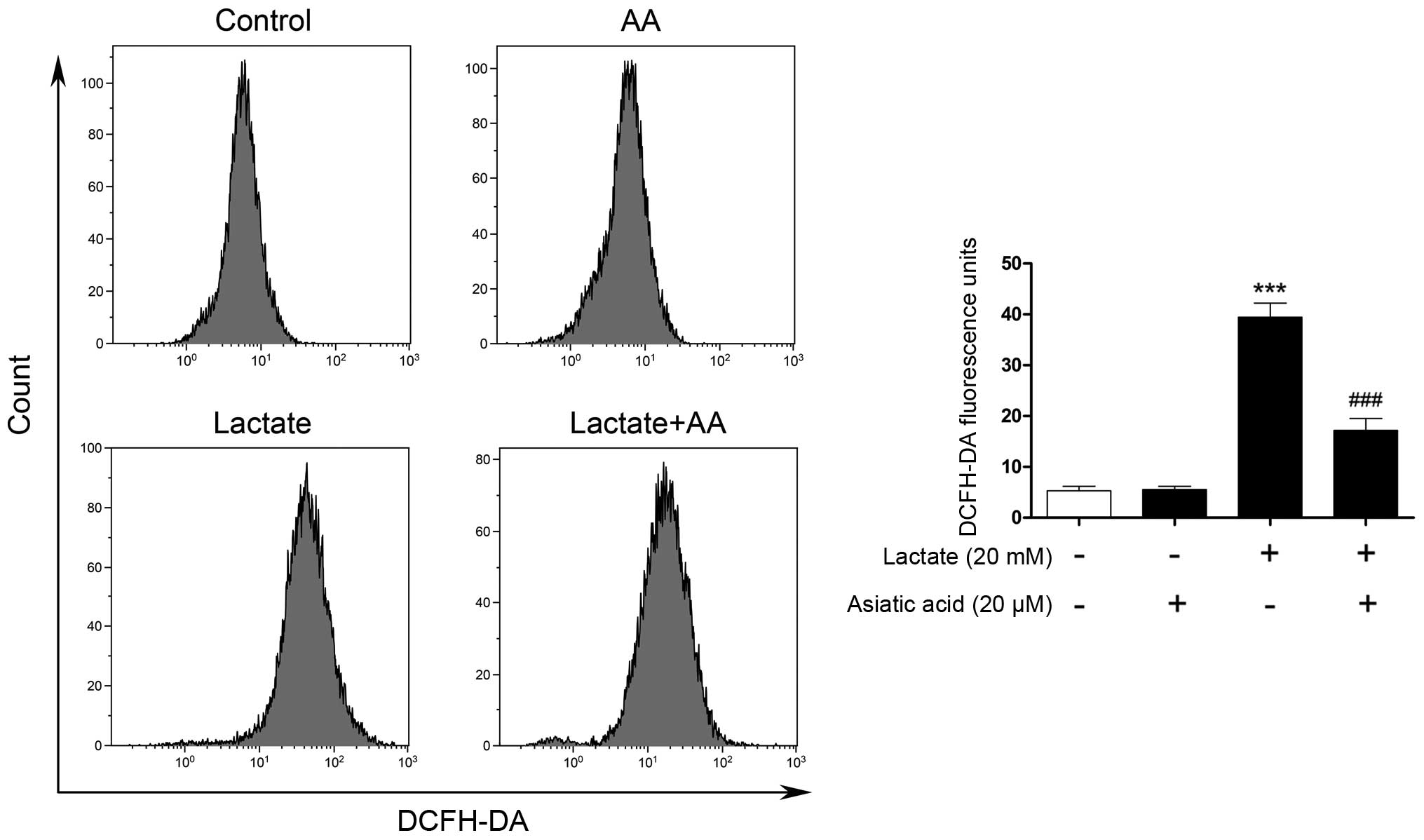
The intracellular ROS levels were measured by flow
cytometry using DCFH-DA as the probe. As shown in Fig. 2, when the cardiomyocytes were
exposed to 20 mM lactate for 24 h, the intracellular ROS levels
were substantially increased compared with those of the untreated
cells, revealing that lactate enhanced intracellular ROS
generation. Pre-treatment with AA attenuated the excess generation
of ROS caused by lactate. However, AA (20 µM) alone did not
affect the intracellular ROS levels.
AA prevents the lactate-induced reduction
in Δψm
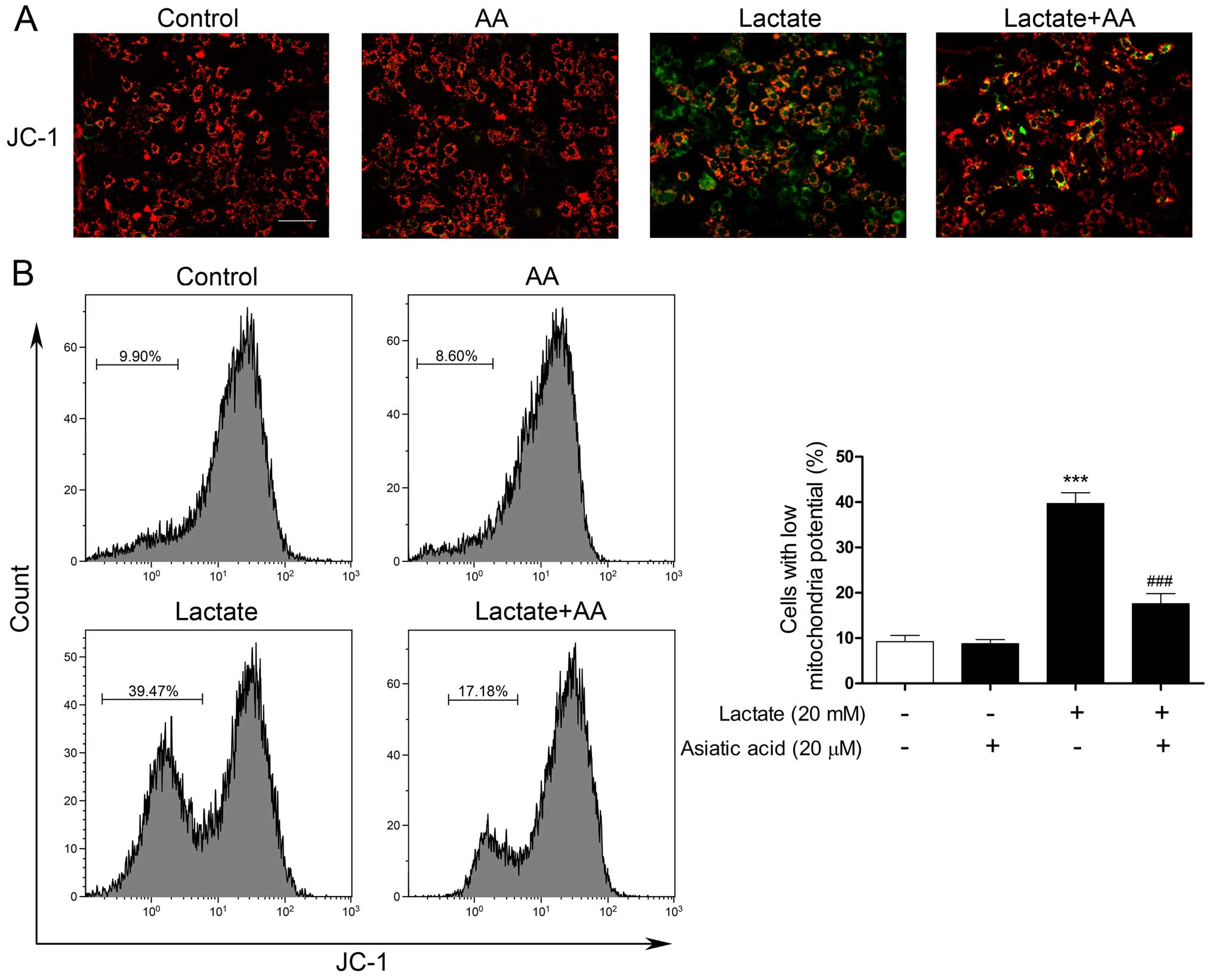
Δψm was examined by JC-1 staining and the loss of
Δψm was analyzed by both fluorescence microscopy and flow
cytometry. Stimulation with lactate resulted in a decrease in red
fluorescence and an increase in green fluorescence in the
cardiomyocytes, compared with the control cells (Fig. 3A). Pre-treatment with AA reversed
the dissipation of Δψm induced by lactate. Similarly, as shown by
flow cotymetry, the loss of Δψm was substantially increased when
the cardiomyocytes were exposed to 20 mM lactate for 24 h and the
lactate-induced loss of Δψm was counteracted by pre-treatment with
AA (Fig. 3B).
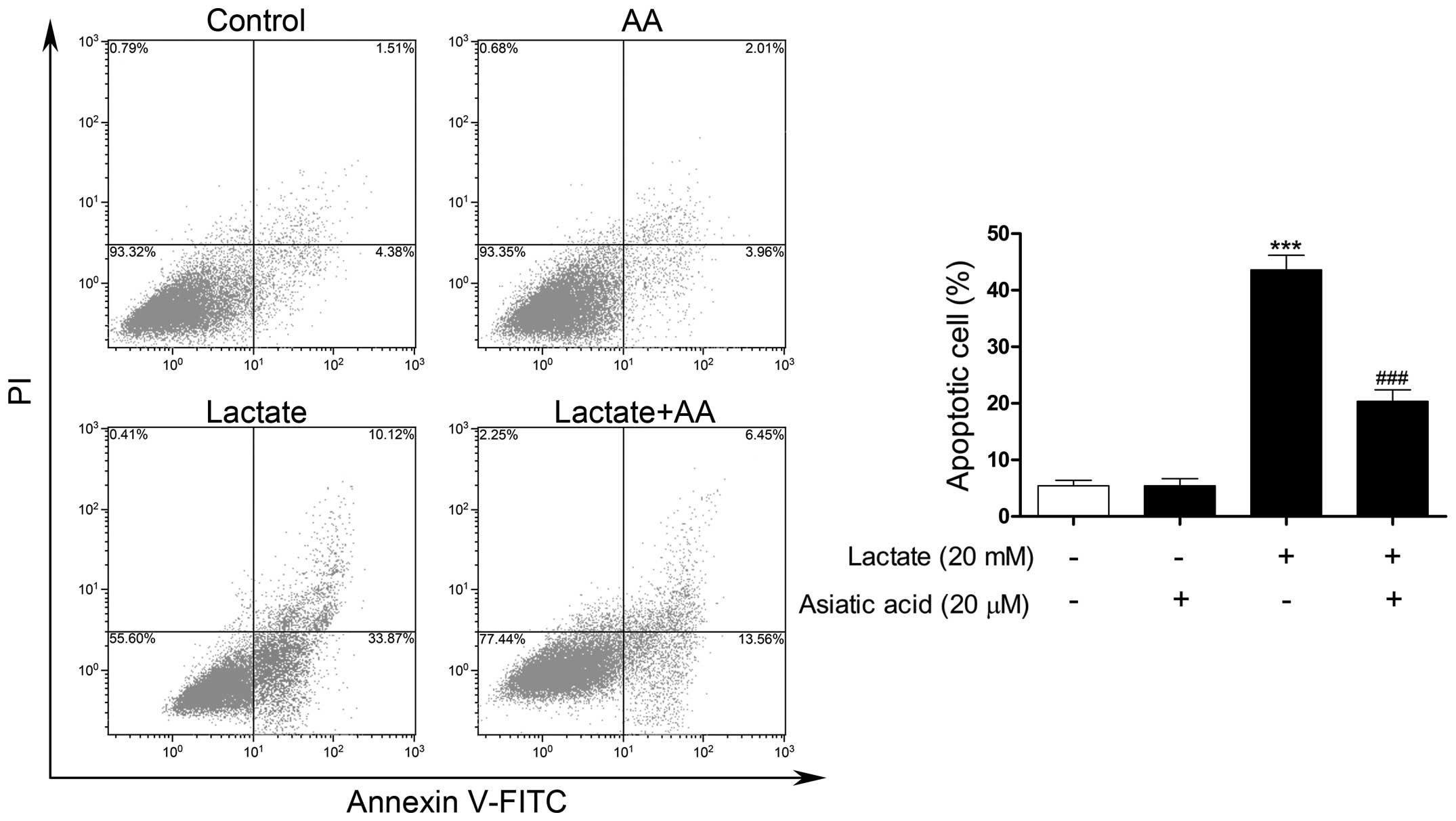
AA protects cardiomyocytes against
lactate-induced apoptosis
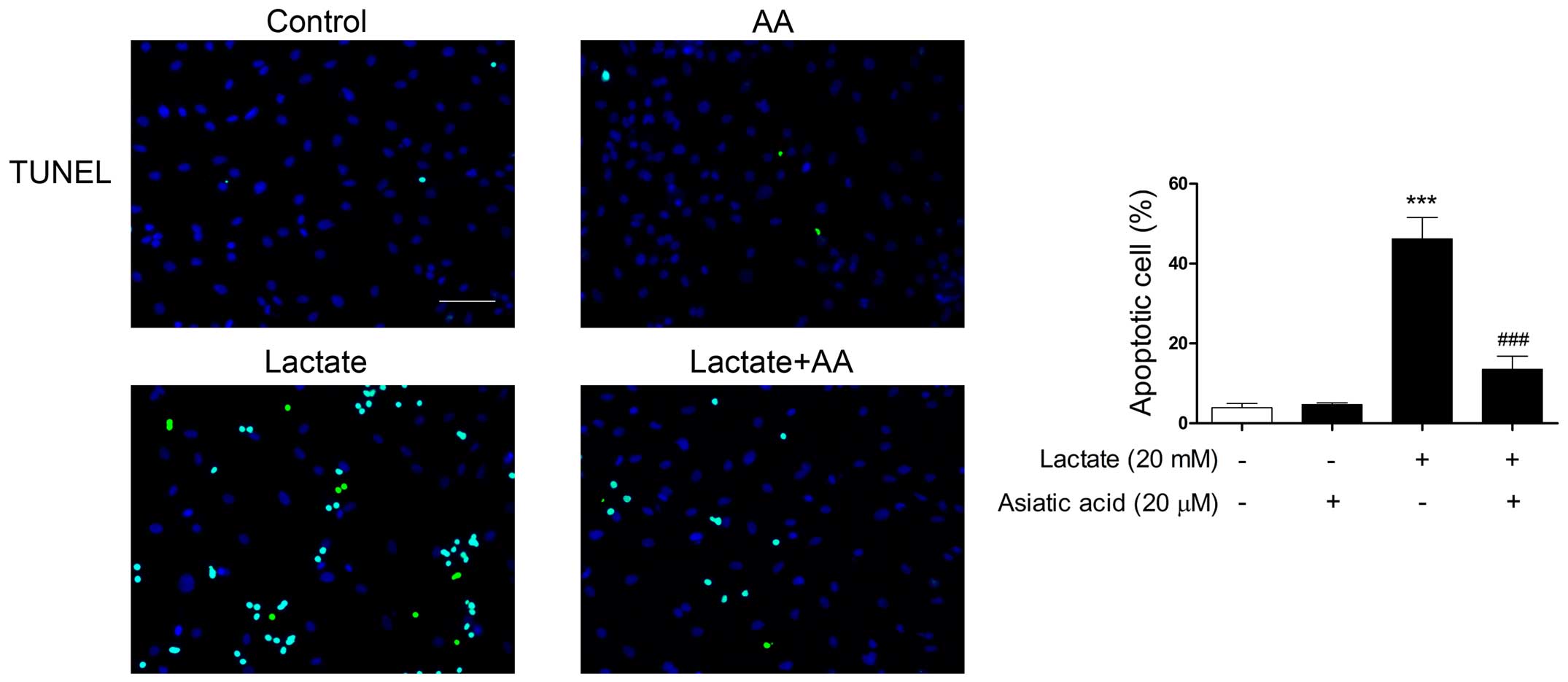
TUNEL-FITC staining was used to evaluate
morphological changes due to the apoptosis of cardiomyocytes.
Following exposure to 20 mM lactate for 24 h, the percentage of
apoptotic nuclei was markedly increased compared with the untreated
nuclei. By contrast, pre-treatment with AA significantly reduced
the number of TUNEL-positive nuclei induced by lactate (Fig. 4). Apoptosis was also confirmed by
flow cytometry. Following exposure to 20 mM lactate for 24 h, the
percentage of apoptotic cells was significantly increased compared
with the untreated cells. By contrast, pre-treatment with AA
substantially reduced the rate of apoptosis by 54.51% compared with
lactate stimulation alone (Fig.
5).
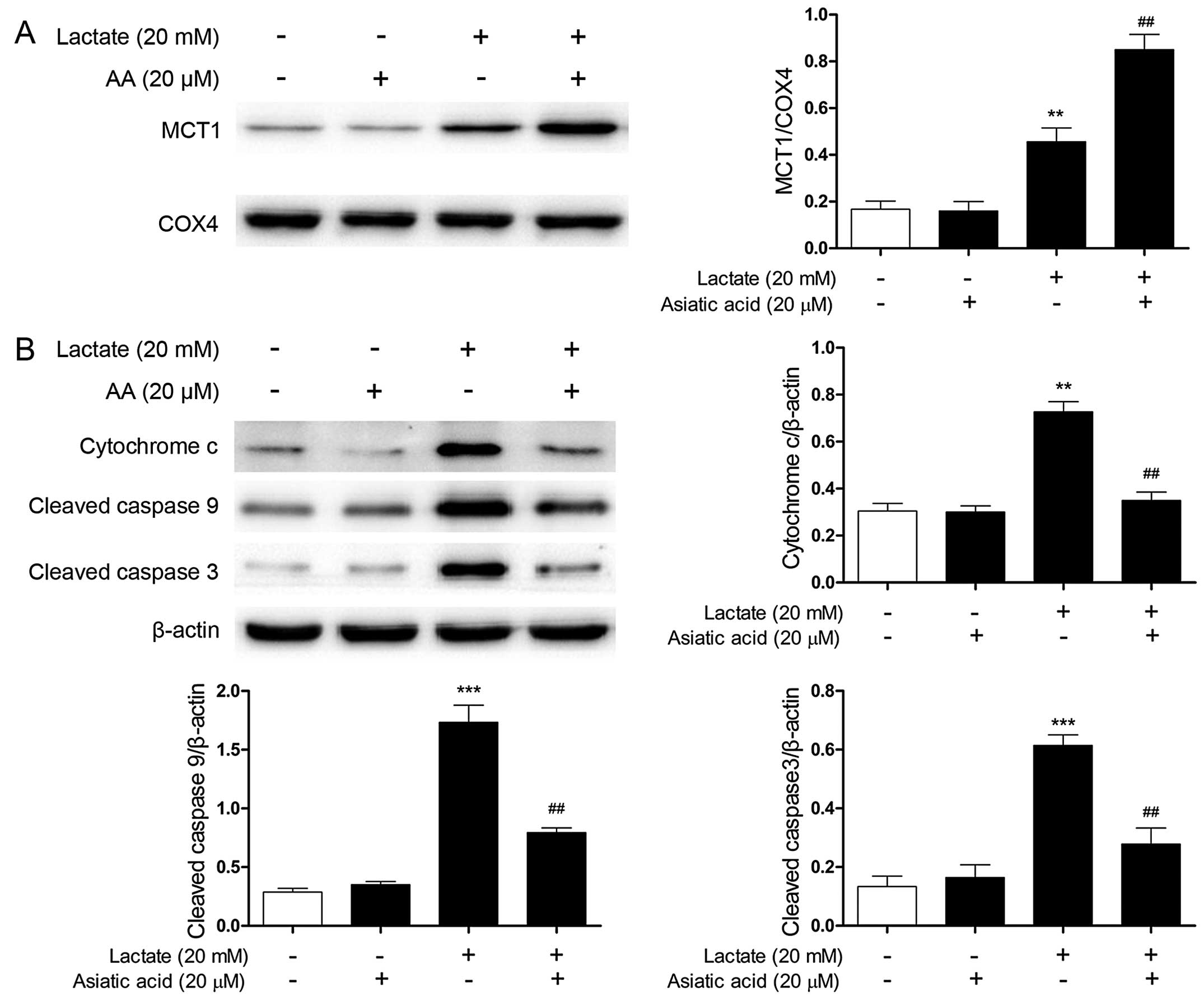
AA regulates the lactate-induced
signaling cascade
Following exposure to 20 mM lactate for 24 h, the
expression of mitochondrial MCT1 was significantly increased
compared with the untreated cells. Moreover, pre-treatment with AA
further increased the expression of mitochondrial MCT1 induced by
lactate (Fig. 6A). However,
treatment with AA (20 µM) alone had no effect on the
expression of mitochondrial MCT1.
With the collapse of Δψm, cytochrome c can
activate the caspase responsible for apoptosis when it is released
into the cytosol from the mitochondria. The expression levels of
cytoplasmic cytochrome c, cleaved caspase-9 and caspase-3
were markedly increased following stimulation with lactate
(Fig. 6B). By contrast,
pre-treatment with AA substantially reduced the lactate-induced
increase in the expression of cytoplasmic cytochrome c,
cleaved caspase-9 and caspase-3.
Discussion
In the present study, we examined the effect of AA
on lactate-induced cardiomyocyte apoptosis in vitro. We
found that AA protected the cardiomyocytes against lactate-induced
apoptosis by regulating the lactate signaling cascade, involving
the inhibition of oxidative stress and mitochondria-dependent
caspase activation, as well as the upregulation of mitochondrial
MCT1 expression. Our results indicated that AA is a suitable
candidate for the prevention and treatment of cardiomyocyte
apoptosis.
In conventional wisdom, lactate is formed as the
result of oxygen insufficiency and utilized under fully aerobic
conditions in myocardial mitochondria. Lactate can be transported
across lipid bilayer membranes by monocarboxylate transport (MCT)
proteins (10). MCT1 is expressed
abundantly in rodent and human hearts, and is localized in
sarcolemmal and mitochondrial membranes (10,25,26). Moreover, the co-localization of
MCT1, lactate dehydrogenase (LDH), CD147 and COX in the
mitochondrial membrane has been demonstrated by Hashimoto et
al (27) and is described as
the mitochondrial lactate oxidation complex. As part of the
intracellular lactate shuttle mechanism, the presence of MCT1
allows mitochondrial lactate influx and oxidation. However, during
exercise or pathological conditions, such as hypoxia and ischemia,
heightened glycolysis causes a surge in lactate production. The
corresponding increased expression of MCT1 helps to attenuate
intracellular acidification. Previous studies have identified a
convincingly positive correlation between the dysfunction of
lactate metabolism and cardiovascular disorders. A maximal lactate
influx into cardiomyocytes and increased MCT1 expression has been
observed both in rat models of myocardial infarction (15) and volume overload (18). In patients with atrial
fibrillation, the upregulation of cytoplasmic lactate concentration
and mitochondrial MCT1 expression has also been confirmed in right
atrial appendage tissues (RAAs) (17). These findings reveal that the
excessive accumulation of lactate and the increased expression of
MCT1 concurrs in cardiovascular diseases. Conversely, the blockade
of MCT1 function by its competitive inhibitor in a mouse model of
ischemia/reperfusion injury has been shown to decrease cardiac
performance and increase the infarct size. In vitro, the
MCT1 inhibitor also increased the death of HL-1 cells stimulated
with hydrogen peroxide (28). In
the present study, we found that the expression of mitochondrial
MCT1 was significantly increased in lactate-stimulated
cardiomyocytes in vitro. Pre-treatment with AA further
increased the expression of mitochondrial MCT1 induced by lactate
and inhibited cardiomyocyte apoptosis. Taken together, these
findings indicate that lactate may have an independent effect on
the development of cardiovascular diseases.
Rather than an end-product of glycolysis during
oxygen insufficiency, we now know that lactate is produced
continuously under fully aerobic conditions (12). Previous studies have confirmed
that lactate is an important cell-signaling molecule involved in
the regulation of the cell redox state and related signaling
cascade. For instance, a low level of exogenous lactate can rapidly
activate ROS production, upregulate mitochondrial MCT1 expression
and increase mitochondrial biogenesis in L6 cells by activating
several transcription factors (29). However, in CHO cells,
lactate-induced cytosolic acidification has been shown to lead to
mitochondrial-dependent apoptosis, including the reduction of Δψm,
the release of cytochrome c, and an increase in caspase-3
enzymatic activity (30).
Moreover, the ROS levels and mitochondrial-dependent apoptotic
proteins were also increased consistent with the elevated lactate
concentration in RAAs in atrial fibrillation (17). Conversely, Kubasiak et al
(31) reported that hypoxia did
not induce cardiomyocyte apoptosis in the absence of lactate
acidosis. In the present study, we observed that stimulation with
lactate significantly increased intracellular ROS levels and
activated mitochondrial-dependent apoptosis, including the
reduction of Δψm, the release of cytochrome c, and the
upregulation of cleaved caspase-9 and caspase-3 expression. Our
data suggest that lactate signaling plays a key role in
mitochondrial energy metabolism and the apoptosis of
cardiomyocytes. Although the increased expression of mitochondrial
MCT1 facilitates lactate oxidation from the cytosol to the
mitochondria, lactic acidosis still occurs inevitably, thereby
resulting in oxidative stress and mitochondrial-dependent
apoptosis. However, pre-treatment with AA substantially attenuated
oxidative stress, preserved the Δψm, and inhibited cardiomyocyte
apoptosis induced by lactate.
In conclusion, to the very best of our knowledge,
the present study is the first to demonstrate that AA inhibits the
lactate-induced cardiomyocyte apoptosis. The mechanisms through
which AA inhibits this apoptotic process involve the regulation of
the lactate signaling cascade, including the inhibition of
oxidative stress and mitochondrial-dependent caspase activation, as
well as the upregulation of mitochondrial MCT1 expression, thereby
relieving lactate acidosis and inhibiting cardiomyocyte apoptosis.
Our results suggest that AA may be used as a therapeutic agent for
the prevention and treatment of cardiomyocyte apoptosis. Further
studies are warranted to determine its biological efficacy and
precise mechanisms of action in vitro and in
vivo.
Acknowledgments
This study was funded by the Jiangsu Province's
Outstanding Medical Academic Leader Program (grant no.
LJ201140).
References
|
1
|
Saraste A, Pulkki K, Kallajoki M,
Henriksen K, Parvinen M and Voipio-Pulkki LM: Apoptosis in human
acute myocardial infarction. Circulation. 95:320–323. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gao HK, Yin Z, Zhou N, Feng XY, Gao F and
Wang HC: Glycogen synthase kinase 3 inhibition protects the heart
from acute ischemia-reperfusion injury via inhibition of
inflammation and apoptosis. J Cardiovasc Pharmacol. 52:286–292.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aharinejad S, Andrukhova O, Lucas T,
Zuckermann A, Wieselthaler G, Wolner E and Grimm M: Programmed cell
death in idiopathic dilated cardiomyopathy is mediated by
suppression of the apoptosis inhibitor Apollon. Ann Thorac Surg.
86:109–114; discussion 114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Narula J, Haider N, Virmani R, DiSalvo TG,
Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW and Khaw BA:
Apoptosis in myocytes in end-stage heart failure. N Engl J Med.
335:1182–1189. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bryant D, Becker L, Richardson J, Shelton
J, Franco F, Peshock R, Thompson M and Giroir B: Cardiac failure in
transgenic mice with myocardial expression of tumor necrosis
factor-alpha. Circulation. 97:1375–1381. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sayen MR, Gustafsson AB, Sussman MA,
Molkentin JD and Gottlieb RA: Calcineurin transgenic mice have
mitochondrial dysfunction and elevated superoxide production. Am J
Physiol Cell Physiol. 284:C562–C570. 2003. View Article : Google Scholar
|
|
7
|
Wang J, Silva JP, Gustafsson CM, Rustin P
and Larsson NG: Increased in vivo apoptosis in cells lacking
mitochondrial DNA gene expression. Proc Natl Acad Sci USA.
98:4038–4043. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Crow MT, Mani K, Nam YJ and Kitsis RN: The
mitochondrial death pathway and cardiac myocyte apoptosis. Circ
Res. 95:957–970. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee Y and Gustafsson AB: Role of apoptosis
in cardiovascular disease. Apoptosis. 14:536–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Halestrap AP and Wilson MC: The
monocarboxylate transporter family - role and regulation. IUBMB
Life. 64:109–119. 2012. View
Article : Google Scholar
|
|
11
|
Brooks GA, Dubouchaud H, Brown M,
Sicurello JP and Butz CE: Role of mitochondrial lactate
dehydrogenase and lactate oxidation in the intracellular lactate
shuttle. Proc Natl Acad Sci USA. 96:1129–1134. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gladden LB: Lactate metabolism: A new
paradigm for the third millennium. J Physiol. 558:5–30. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bonen A: The expression of lactate
transporters (MCT1 and MCT4) in heart and muscle. Eur J Appl
Physiol. 86:6–11. 2001. View Article : Google Scholar
|
|
14
|
Hashimoto T and Brooks GA: Mitochondrial
lactate oxidation complex and an adaptive role for lactate
production. Med Sci Sports Exerc. 40:486–494. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jóhannsson E, Lunde PK, Heddle C, Sjaastad
I, Thomas MJ, Bergersen L, Halestrap AP, Blackstad TW, Ottersen OP
and Sejersted OM: Upregulation of the cardiac monocarboxylate
transporter MCT1 in a rat model of congestive heart failure.
Circulation. 104:729–734. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Halestrap AP, Wang X, Poole RC, Jackson VN
and Price NT: Lactate transport in heart in relation to myocardial
ischemia. Am J Cardiol. 80:17A–25A. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu J, Xu X, Si L, Xue L, Zhang S, Qin J,
Wu Y, Shao Y, Chen Y and Wang X: Intracellular lactate signaling
cascade in atrial remodeling of mitral valvular patients with
atrial fibrillation. J Cardiothorac Surg. 8:342013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Evans RK, Schwartz DD and Gladden LB:
Effect of myocardial volume overload and heart failure on lactate
transport into isolated cardiac myocytes. J Appl Physiol (1985).
94:1169–1176. 2003. View Article : Google Scholar
|
|
19
|
Pittella F, Dutra RC, Junior DD, Lopes MT
and Barbosa NR: Antioxidant and cytotoxic activities of Centella
asiatica (L) Urb. Int J Mol Sci. 10:3713–3721. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yun KJ, Kim JY, Kim JB, Lee KW, Jeong SY,
Park HJ, Jung HJ, Cho YW, Yun K and Lee KT: Inhibition of
LPS-induced NO and PGE2 production by asiatic acid via NF-kappa B
inactivation in RAW 264.7 macrophages: Possible involvement of the
IKK and MAPK pathways. Int Immunopharmacol. 8:431–441. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krishnamurthy RG, Senut MC, Zemke D, Min
J, Frenkel MB, Greenberg EJ, Yu SW, Ahn N, Goudreau J, Kassab M, et
al: Asiatic acid, a pentacyclic triterpene from Centella asiatica,
is neuroprotective in a mouse model of focal cerebral ischemia. J
Neurosci Res. 87:2541–2550. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang LX, He RH, Yang G, Tan JJ, Zhou L,
Meng XM, Huang XR and Lan HY: Asiatic acid inhibits liver fibrosis
by blocking TGF-beta/Smad signaling in vivo and in vitro. PLoS One.
7:e313502012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Si L, Xu J, Yi C, Xu X, Wang F, Gu W,
Zhang Y and Wang X: Asiatic acid attenuates cardiac hypertrophy by
blocking transforming growth factor-β1-mediated hypertrophic
signaling in vitro and in vivo. Int J Mol Med. 34:499–506.
2014.PubMed/NCBI
|
|
24
|
Zhang X, Wu J, Dou Y, Xia B, Rong W,
Rimbach G and Lou Y: Asiatic acid protects primary neurons against
C2-ceramide-induced apoptosis. Eur J Pharmacol. 679:51–59. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brooks GA, Brown MA, Butz CE, Sicurello JP
and Dubouchaud H: Cardiac and skeletal muscle mitochondria have a
monocarboxylate transporter MCT1. J Appl Physiol (1985).
87:1713–1718. 1999.
|
|
26
|
Butz CE, McClelland GB and Brooks GA: MCT1
confirmed in rat striated muscle mitochondria. J Appl Physiol
(1985). 97:1059–1066. 2004. View Article : Google Scholar
|
|
27
|
Hashimoto T, Hussien R and Brooks GA:
Colocalization of MCT1, CD147, and LDH in mitochondrial inner
membrane of L6 muscle cells: Evidence of a mitochondrial lactate
oxidation complex. Am J Physiol Endocrinol Metab. 290:E1237–E1244.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Martinov V, Rizvi SM, Weiseth SA, Sagave
J, Bergersen LH and Valen G: Increased expression of
monocarboxylate transporter 1 after acute ischemia of isolated,
perfused mouse hearts. Life Sci. 85:379–385. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hashimoto T, Hussien R, Oommen S, Gohil K
and Brooks GA: Lactate sensitive transcription factor network in L6
cells: Activation of MCT1 and mitochondrial biogenesis. FASEB J.
21:2602–2612. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jeong D, Kim TS, Lee JW, Kim KT, Kim HJ,
Kim IH and Kim IY: Blocking of acidosis-mediated apoptosis by a
reduction of lactate dehydrogenase activity through antisense mRNA
expression. Biochem Biophys Res Commun. 289:1141–1149. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kubasiak LA, Hernandez OM, Bishopric NH
and Webster KA: Hypoxia and acidosis activate cardiac myocyte death
through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci USA.
99:12825–12830. 2002. View Article : Google Scholar : PubMed/NCBI
|