Introduction
Obesity and its related metabolic disorder,
non-insulin-dependent type 2 diabetes mellitus, are prevalent and
affect more than 150 million people in Western countries (1). The incidence of type 2 diabetes
mellitus continues to increase and has reached a proportion at an
epidemic level which severely affects public healthcare and costs
both human lives and financial cost (2,3).
Generally, the high-fat content in typical Western diets has been
recognized as an important factor contributing to obesity and its
related insulin resistance in type 2 diabetes mellitus (4,5).
Previous studies indicate that defective insulin secretion in type
2 diabetes mellitus is caused by islet β cell dysfunction and
reduced islet β cell mass (6–8).
Long-term hyperglycemia or/and hyperlipidemia due to a high-fat
diet (HFD) cause glucotoxicity and lipotoxicity in β pancreatic
cells further leading to the development of metabolic dysfunctions
(6–8). Thus, development of therapies
targeting the improvement of islet β cell function has become a
central subject in the field related to the treatment of type 2
diabetes mellitus.
Owing to the rapid progress of genetics and omics
techniques, numerous discoveries of molecular compounds influencing
the function and development of islet β cells were elucidated
(9–12). Among all these regulators, PDX-1
is a pancreatic and duodenal homeobox-1 transcription factor that
modulates the development and differentiation of pancreatic islet β
cells (13). It has been
confirmed that both genetic and acquired reductions in PDX-1
expression in humans and in animal models have been involved in the
causes of the onset of type 2 diabetes mellitus, hepatic diabetes
mellitus, and dysfunction in β pancreatic cells (14–16). In adults, expression of PDX-1 is
maintained in the duodenal epithelium and in insulin-secreting
islet β cells, where it may activate transcription of insulin genes
(17,18). In mouse embryos, PDX-1 expression
precedes insulin and glucagon expression (12). As reported by Stoffers et
al focusing on intrauterine growth retardation due to diseases
in adulthood, mRNA levels of PDX-1 are inhibited in more than 50%
of IUGR fetuses (19). Therefore,
upregulation of PDX-1 levels in islet β cells may lead to the
improvement of cell function and facilitate the treatment of type 2
diabetes mellitus.
Glucagon-like peptide 1 (GLP-1) is a peptide of 30
amino acids and is produced by L cells of the intestinal mucosa
(20). The molecule responds to
food intake and plays a major role in the control of postprandial
metabolism by augmenting nutrient-induced insulin release,
inhibiting glucagon secretion, and reducing endogenous glucose
production (21,22). Studies in normal weight cases
showed that peripheral administration of GLP-1 decreased food
intake and suppressed appetite (23,24). A study based on mice fed with an
HFD also revealed that GPL-1 treatment reduced endogenous insulin
resistance via activation of central GLP-1 receptors (25). Although multiple studies
demonstrated the beneficial effects of GLP-1 on HFD-induced type 2
diabetes mellitus are validated by multiple studies, the underlying
mechanism involved in this treatment process is only partially
revealed. Considering the key role of PDX-1 in the onset of type 2
diabetes mellitus and dysfunction of islet β cells, we conducted a
comprehensive study to assess the interaction between GLP-1 and
PDX-1, which may provide novel insight into the treatment
strategies for type 2 diabetes mellitus.
In the present study, C57/BL6 mice were fed with a
HFD to mimic the type 2 diabetes mellitus condition in vivo.
Then the effect of GLP-1 administration on glucose tolerance,
insulin release, and glucose-dependent insulinotropic polypeptide
(GIP) level was detected for assessment of islet β cell function.
Moreover, the effect of GLP-1 administration on the activation of
PDX-1-related signaling transduction was also quantified to profile
the interaction between the two indicators.
Materials and methods
Chemicals and animals
Liraglutide (GLP-1 receptor agonist) (cat. no.
J20110020) and human insulin Novolin (cat. no. J20120026) were both
purchased from Novo Nordisk (Oslo, Norway). Eight-week-old C57/BL6
mice were provided by the Laboratory Animal Center, West China
Hospital, Sichuan West China School of Medicine (Chengdu, China)
and maintained in cages at room temperature (20–25°C) with a
constant humidity (55±5%) with free access to food and water in a
12:12-h light/dark cycle. All animal experiments were conducted in
accordance with the Institutional Animal Ethics Committee and
Animal Care Guidelines for the Care and Use of Laboratory Animals
of Sichuan West China School of Medicine.
HFD treatment and animal grouping
Thirty-two mice were randomly divided into four
groups (8 for each group) with different administrations: i)
control group, mice fed with standard diet (7.8% fat, 41.9% protein
and 50.3% carbohydrate, total energy of 2.18 kcal/g) for 12 weeks;
ii) HFD group, mice fed with HFD (61% fat, 15% protein and 25%
carbohydrate, 0.2% cholesterol, total energy of 5.28 kcal/g) + high
glucose water (2% fructopyranose plus 2.5% glucose) ad
libitum for 12 weeks; iii) liraglutide group, mice fed with HFD
(61% fat, 15% protein and 25% carbohydrate, 0.2% cholesterol, total
energy of 5.28 kcal/g) + high glucose water (2% fructopyranose plus
2.5% glucose) ad libitum for 12 weeks and GPL-1 (0.6 mg/kg
body weight/day) was subcutaneously injected into mice from the 8th
week of the experiment; iv) liraglutide paired group, mice in this
group were fed with an HFD but were calorically restricted to
follow the weight trajectory of mice in the liraglutide group. The
group was set up to eliminate the possibility that the effect of
liraglutide-induced GLP-1 activation was exerted through the
influence on the body weight of the treated mice. Body weight of
mice in each group was measured every two weeks until the
completion of the different administrations.
Oral glucose tolerance test (OGTT) and
insulin release test
Upon completion of the 12-week treatment, mice
ingested a solution containing 2 g/kg of dextrose after a 5-h
overnight fast. Thereafter, tail venous blood samples of mice were
obtained with a sterile syringe at 0, 30, 60, 90 and 120 min for
determination of plasma glucose using the Glucose Oxidase Activity
Assay kit (cat. no. MAK097; Sigma-Aldrich, St. Louis, MO, USA)
according to the manufacturer's instructions. The plasma insulin
levels at corresponding time-points were measured using Mouse
Insulin ELISA kit (cat. no. ab100578) according to the
manufacturer's instructions.
Hyperinsulinemic euglycemic clamp
assay
For the hyper-insulinemic euglycemic clamp
experiment, mice were previously fasted for 16 h and then sedated
with 1% pentobarbital (cat. no. P3761; Sigma-Aldrich). Basal blood
glucose (BBG) levels of mice in the different groups were measured
with blood sampled from the tail vein using the Glucose Oxidase
Activity Assay kit (cat. no. MAK097; Sigma-Aldrich) according to
the manufacturer's instructions. Then a T-branch pipe was placed at
the jugular vein of the mice. Insulin (25 mU/kg/min) and glucose
[basal glucose infusion rate (GIR), 360 mg/kg/h] were continuously
dripped into the mice through one branch of the T-branch pipe for
120 min. Blood glucose content was measured every 10 min until a
level of BBG ± 0.05 mmol/l (stable blood glucose). During the
experimental, GIR was adjusted according to the stable blood
glucose. When the blood glucose level was lower than 5 mmol/l,
infusion of insulin was stopped.
Fasting insulin and GIP level
Upon completion of hyperinsulinemic euglycemic clamp
assay, mice were fasted for 16 h to determine the fasting insulin
and GIP level. Insulin level was determined using the Mouse Insulin
ELISA kit according to the manufacturer's instructions. The GIP
level was also determined using the GIP ELISA kit (cat. no.
RAB0209; Sigma-Aldrich) according to the manufacturer's
instructions. Upon completion of measurement of fasting insulin and
GIP levels, the mice were sacrificed using air embolism methods and
their pancreatic tissues were harvested and preserved at −80°C for
further histological and molecular analyses.
Hematoxylin and eosin (H&E) staining
and TUNEL staining
The histological changes in sections of the pancreas
from the different groups were observed using routine H&E
staining and the results were detected under a microscope at ×20
magnification. Cell apoptotic rates in the pancreatic tissues were
determined with TUNEL staining using DeadEnd™ Fluorometric TUNEL
system (cat. no. G3250; Promega, Madison, WI, USA) and the results
were detected using fluorescent microscopy at ×200
magnification.
Immunofluorescent microscopy
Expression of the insulin antibody and glucagon
antibody in the pancreatic tissues was detected with
immunofluorescent microscopy. Briefly, the treated cells were
seeded into 24-well chambers, washed with phosphate-buffered saline
(PBS), and fixed with 4% parafor-maldehyde for 15 min. Then the
cells were permeabilized with 0.5% Triton X-100 for 30 min. After
being washed with PBS for three cycles, the cells were blocked in
10% goat serum for 15 min. Primary rabbit polyclonal antibodies to
insulin (1:200, cat. no. EPR17359) and glucagon (1:200, cat. no.
EP3070) (both from Abcam, Cambridge, MA, USA) were then added and
the cells were incubated overnight at 4°C in 1% goat serum.
Staining was performed by incubating the cells with fluorescein
isothiocyanate secondary antibody for 1 h. After incubation with
the secondary antibody (1:1,000), the cells were washed and then
stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min at room
temperature. After extensive washing with PBS, the slides were
fixed and imaged with fluorescent microscopy at ×40
magnification.
Reverse transcription-quantitative PCR
(RT-qPCR)
For RT-qPCR detection, whole RNA in pancreatic
tissues from the different groups was extracted using RNA Simple
Total RNA kit according to the manufacturer's instructions (cat.
no. DP419; Tiangen Biotech Co., Ltd., Beijing, China). β-actin was
selected as the internal reference gene. Then the whole RNA was
reversely transcribed to cDNA templates using Super M-MLV reverse
transcriptase (cat. no. RP6502; BioTeke Corporation, Beijing,
China). The final qPCR reaction mixture of volume 20 µl
consisted of 10 µl of SYBR-Green Master Mix, 0.5 µl
of each primer (PDX-1 forward, 5′-CCCCAGTTTACAAGCTCGCT-3′
and reverse, 5′-CTCGGTTCCATTCGGGAAAGG-3′; MafA forward,
5′-AGGAGGAGGTCATCCGACTG-3′ and reverse,
5′-CTTCTCGCTCTCCAGAATGTG-3′; β-actin forward,
5′-CATTGCTGACAGGATGCAGA-3′ and reverse,
5′-CTGCTGGAAGGTGGACAGTGA-3′), 1 µl of the cDNA template, and
8 µl of RNase-free H2O. Amplification parameters
were as follows: denaturation at 94°C for 2 min, followed by 40
cycles at 94°C for 20 sec, 58°C for 20 sec and 72°C for 20 sec.
Relative expression levels of the targeted molecules were
calculated with DataAssist software version 3.0 (Applied
Biosystems; Life Technologies, Carlsbad, CA, USA) according to the
2−ΔΔcq method.
Western blotting
The whole protein product in the different groups
was extracted using the Total Protein Extraction kit according to
the manufacturer's instructions (cat. no. WLA019; Wanleibio,
Shenyang, China). GAPDH was used as the internal reference.
Concentration of protein samples was determined using the BCA
method. Twenty microliters of protein (40 µg) was subject to
10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE). Targeted proteins were transferred onto polyvinylidene
difluoride (PVDF) sheets and the membranes were washed with TTBS
for 5 min before incubation in skim milk powder solution for 1 h.
Primary antibodies against PDX-1 (1:1,000, cat. no. 5679P; Cell
Signaling Technology, Inc., Danvers, MA, USA), MafA (1:1,000, cat.
no. ab26405; Abcam) (26), p-JAK2
(1:2,000, cat. no. 8082) (30),
p-STAT3 (1:5,000, cat. no. 9145) (27) and Bax (1:1,500, cat. no. BS2538)
(all from Cell Signaling Technology, Inc., Boston, MA, USA), Bcl-2
(1:1,000, cat. no. BS1511) and GAPDH (1:10,000, cat. no. ab9485)
(both from Abcam) were incubated with membranes at 4°C overnight.
After four washes using TTBS, secondary HRP goat anti-rabbit IgG
antibodies (1:20,000, cat. no. BA1054; Boster Biological
Technology, Ltd., Wuhan, China) were added into the mixture and
incubated with the membranes for 45 min at 37°C. After an
additional six washes with TTBS, the blots were developed using
Beyo ECL Plus reagent and the results were detected in the gel
imaging system. The relative expression levels of BDNF in the
different samples were calculated with Gel-Pro Analyzer (Media
Cybernetics Inc., Rockville, MD, USA).
Statistical analysis
All the data are expressed as the mean ± SD.
Two-group data were compared using the Student's t-test or paired
Student's t-test. Significance was accepted at P<0.05. All the
statistical analysis and graph manipulation were conducted using
GraphPad Prism 6 (GraphPad Software, Inc., San Diego, CA, USA).
Results
Administration of liraglutide improved
the HFD-induced insulin resistance in the C57/BL6 mice
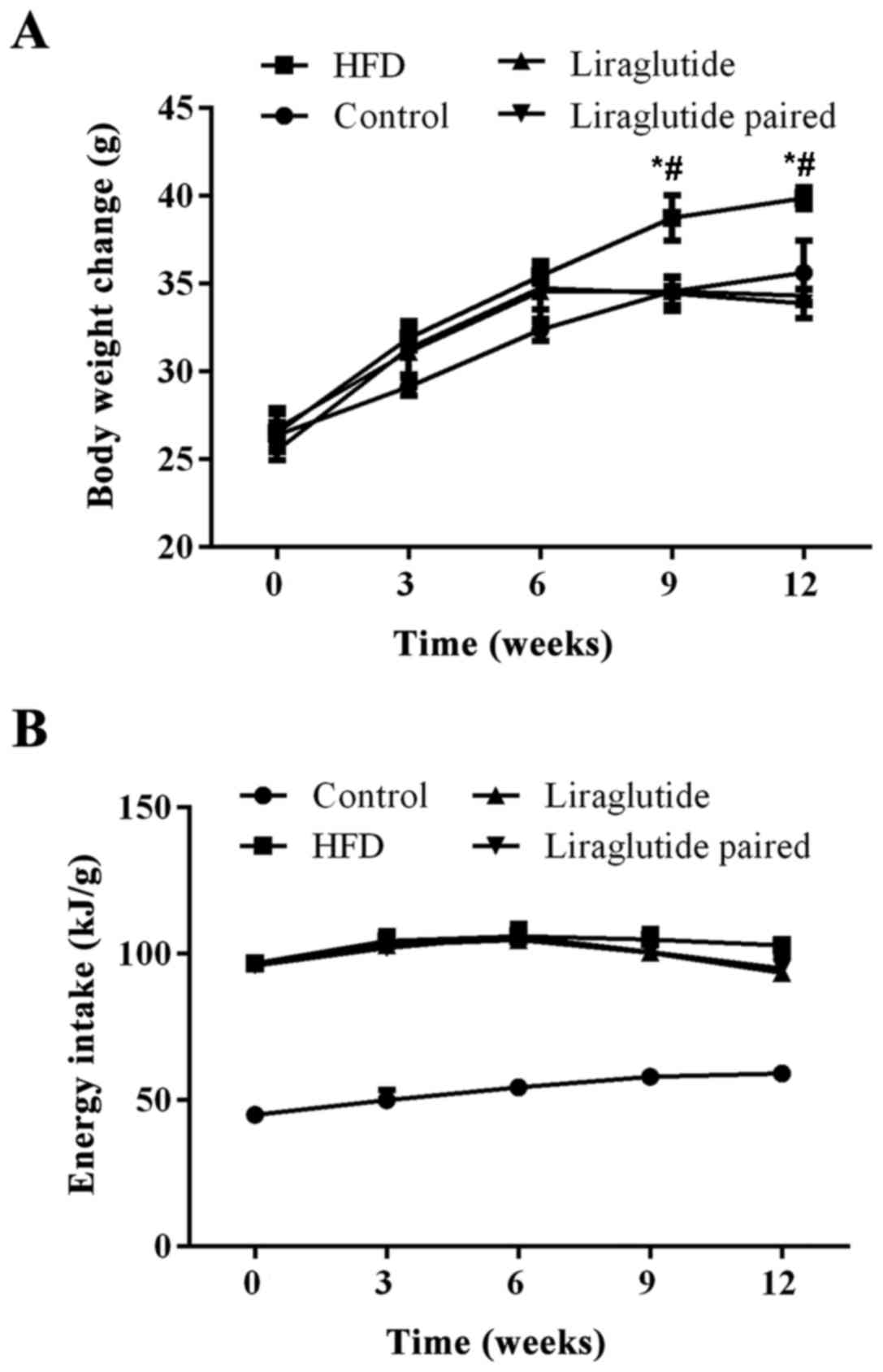
The body weight and energy intake changes in the
different groups during 12 weeks are shown in Fig. 1. HFD consumption increased the
body weight of the mice in the HFD group compared with control and
liraglutide groups (P<0.05), while administration of liraglutide
confounded the effect of the HFD on body weight changes in the mice
from the 9th week of the experiment (P<0.05) (Fig. 1).
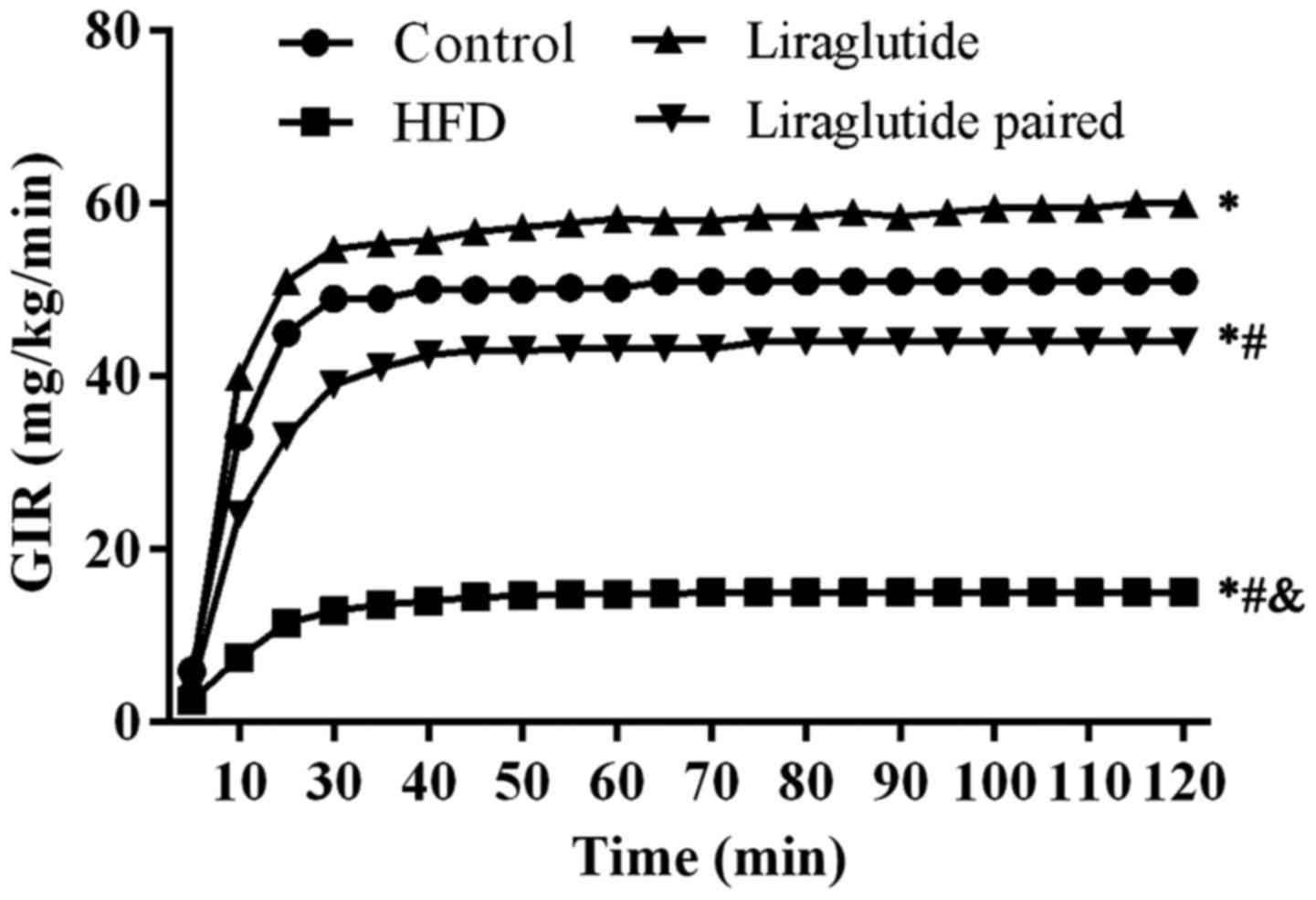
To assess the insulin resistance induced by the HFD,
a hyperinsulinemic euglycemic clamp assay was conducted. As shown
in Fig. 2, the GIR value in the
HFD group was lower than the other three groups (representing
stronger insulin resistance), and the difference was statistically
significant (P<0.05). Admi nistration of liraglutide
significantly increased the GIR value compared with GIR values in
the HFD and liraglu-tide paired groups (representing weaker insulin
resistance). However, GIR values in the liraglutide group were even
significantly higher than those in the control group (P<0.05)
(Fig. 2). Taken together, the
results of the hyperinsulinemic euglycemic clamp assay showed that
the insulin resistance induced by HFD could be ameliorated by
treatment of liraglutide, but the optimal amount for practical
application of liraglutide should be further assessed. Furthermore,
considering no significant difference could be detected between the
HFD and liraglutide paired groups, it was concluded that the
insulin resistance induced by HFD was attributed to the composition
of the diet instead of the amount of the diet.
Administration of liraglutide ameliorates
the HFD-induced islet β cell dysfunction and delays insulin release
in the C57/BL6 mice
Based on the results of the OGTT assay, HFD
treatment decreased the glucose tolerance in the experimental mice
(Fig. 3A) with the highest plasma
insulin level in all four groups (Fig. 3B), which indicated impairments in
islet β cells and a delayed release of insulin. However, treatment
with liraglutide reversed the abnormal glucose tolerance induced by
the HFD, inferring a pancreas-protecting effect of liraglutide
(Fig. 3A).
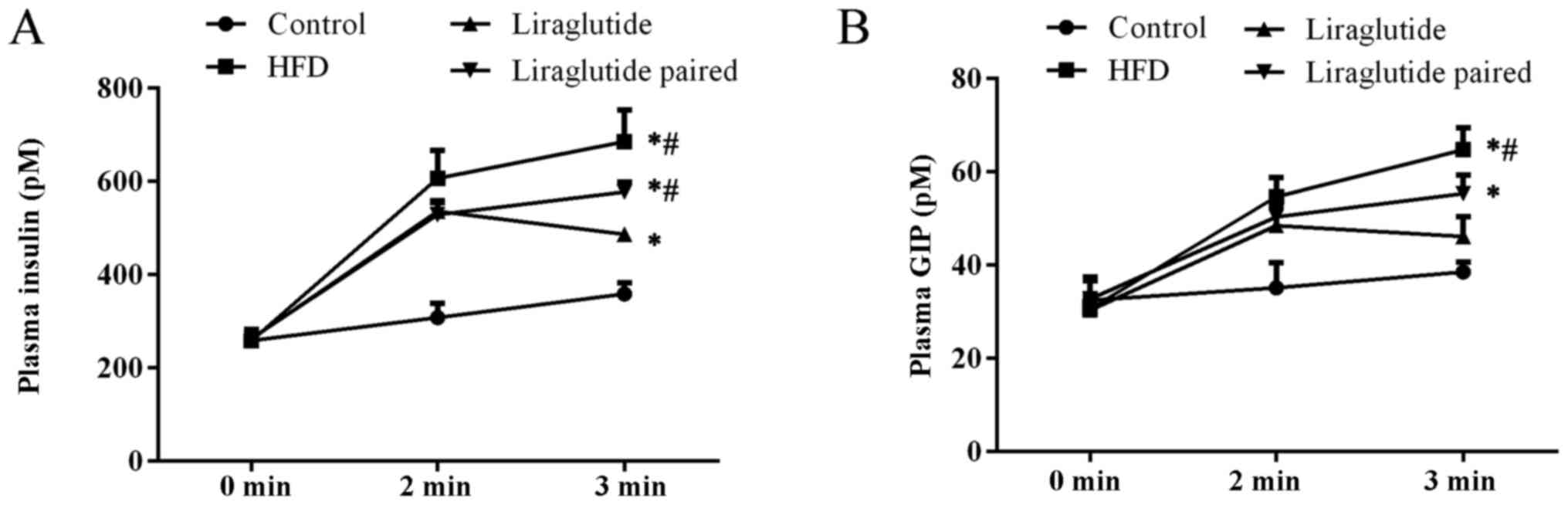
Measurement of fasting insulin also confirmed damage
to islet β cell function (Fig.
4A). HFD markedly increased the fasting plasma insulin level
when compared with control group at the last sampling point, and
the differences were both statistically significant (P<0.05).
The treatment of HFD also significantly increased the plasma GIP
level in the HFD and liraglutide paired groups when compared with
the other two groups (Fig. 4B)
(P<0.05), representing augmentation of islet α cell function in
the model animals. The increasing level of fasting insulin and GIP
level in the HFD and liraglutide paired groups may be the key
reason that HFD induced insulin resistance in mice. Similarly to
the results of the OGTT assay, liraglutide administration confound
the effect of HFD on the abnormal function of the pancreas
(Fig. 4).
Additionally, it was shown by the above data that
although mice in the liraglutide paired group exhibited a similar
changing pattern in body weight to the liraglutide group, the
induced insulin, fasting insulin, and fasting GIP levels were all
significantly different from those in the liraglutide group
(P<0.05), which evidently confirmed our hypothesis that the
effect of liraglutide on the pancreas was not attributed to its
effect on body weight.
Administration of liraglutide inhibits
HFD-induced islet β cell apoptosis and islet α cell hyperplasia in
C57/BL6 mice
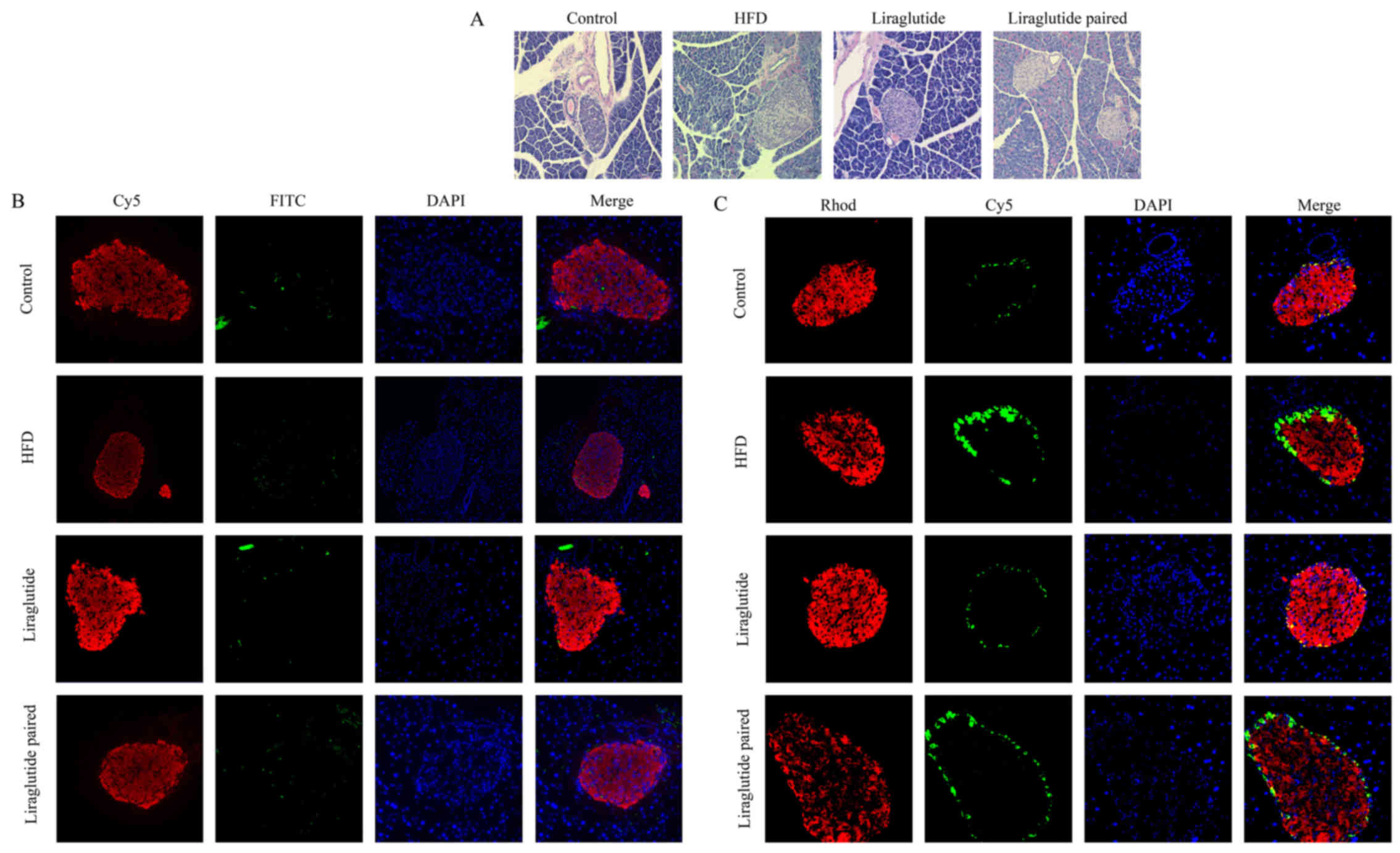
Following H&E staining, nuclei in pancreatic
tissues were stained blue and the cytoplasm was stained red. The
degree of staining varied with ages of the cells. Based on the
above criteria, a comparison between control and HFD groups
revealed severe injury due to HFD. In the HFD and liraglutide
paired groups, the volumes of pancreas islet obviously increased
and the infiltration of inflammatory cells was clearly observed
(Fig. 5A). Administration with
liraglutide alleviated the damage in which numbers of aged cells
decreased in the liraglutide compared with that in HFD and
liraglutide paired groups (Fig.
5A) and no obvious proliferation of pancreas islet could be
detected in the liraglutide group (Fig. 5A). Concomitant with the results of
the H&E staining, the apoptotic cell numbers in the HFD and
liraglutide paired groups were also increased by HFD, and the
apoptosis process was also inhibited by liraglutide treatment
(Fig. 5B). FITC (TUNEL-positive)
stains green, Cy5 (insulin-positive) stains red, and DAPI stains
blue. As illustrated by the immunofluorescent detection, islet β
cells (insulin-positive) were stained red and islet α cells
(glucagan-positive) were stained green. In Fig. 5C, the number of islet β cells
(stained red) was reduced in the HFD and liraglutide paired groups
compared with the control group, and admi nistration of liraglutide
increased the number of islet β cells to a relatively normal level
(Fig. 5C). In contrast, the
number of islet α cells (stained green) was clearly increased by
HFD and decreased by liraglutide administration, which was
consistent with the augmentation of islet α cell function as shown
above (Fig. 5C).
The protective effect of
liraglutide-induced activation of GLP-1 is exerted through the
activation of PDX-1
To further explore the mechanism through which GLP-1
exerts its function on the pancreas, the expression of PDX-1 was
detected at both the mRNA and protein levels. As illustrated in
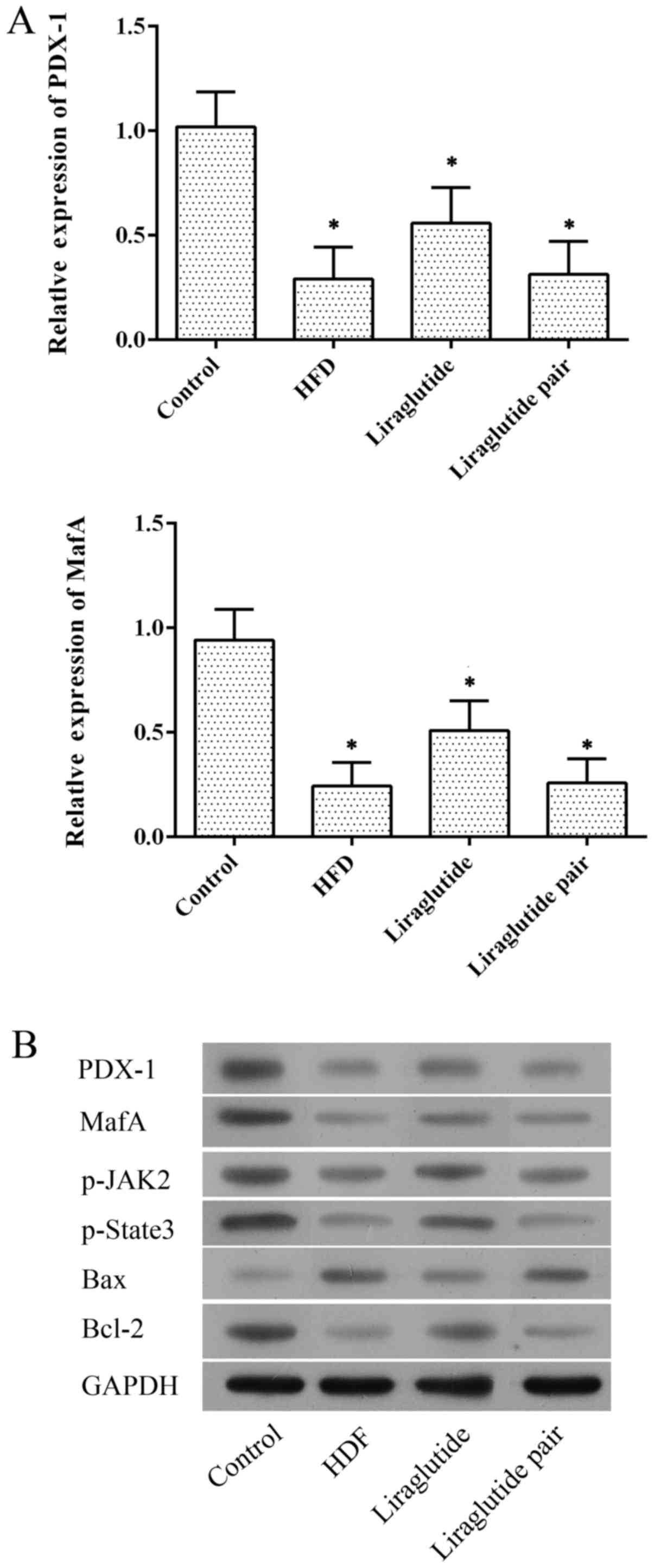
Fig. 6A, at the mRNA level,
expression of PDX-1 and MafA was inhibited in the HFD
and liraglutide paired groups while they were significantly
upregulated by liraglutide administration (P<0.05). Similar
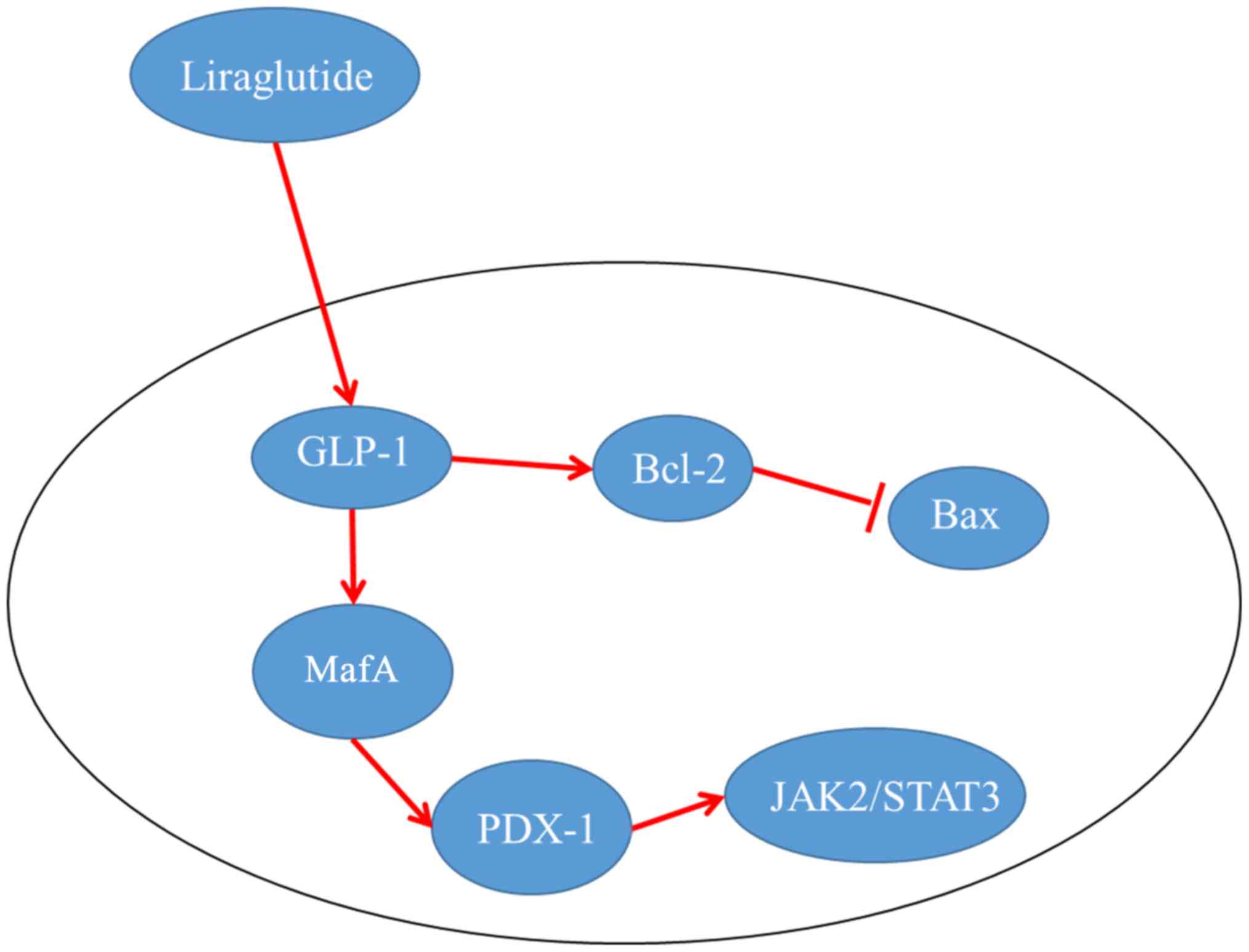
results were also detected at the protein level (Figs. 6B and 7). Such results offer a possible
explanation that the pancreas protective effect of
liraglutide-induced activation of GLP-1 may be exerted through the
activation of PDX-1, which contributed to islet β cell repair and
insulin sensitivity. The protective effect of liraglutide was
further confirmed by enhanced expression of MafA, p-JAK2 and
p-Stat3, which indicated restoration of pancreatic function.
Moreover, administration of liraglutide also clearly suppressed the
expression of pro-apoptotic factor, Bax, compared with that noted
in the HFD and liraglutide paired groups (Figs. 6B and 7) and increased the expression of
anti-apoptotic factor Bcl-2, which corresponded to the
anti-apoptotic effect of liraglutide as illustrated by TUNEL
staining.
Discussion
Although a high-fat diet (HFD) is not common in all
human populations, it is evidently recognized as a contributor to
elevated blood triglyceride levels, which is validated as a risk
factor for clinical acute pancreatitis, as are hyperlipidemia and
obesity (28). In addition, HFD
induced hyperinsulinemia and insulin resistance are two major
characteristics of type 2 diabetes (1). An HFD has been proven to generate
defective insulin secretion in animal models and can lead to fatty
infiltration in the pancreas (29). Previous studies have demonstrated
that HFD-induced insulin resistance in type 2 diabetes is
attributed to the dysfunction and mass loss of islet β cells
(6–8). In the present study, HFD was
employed to induce pancreatic injury and insulin resistance in
C57/BL6 mice. Thereafter, the HFD-fed rats were administered
liraglutide to ameliorate the impairments due to HFD. It was found
that administration of liraglutide to HFD-fed mice restored the
loss of islet β cells and inhibited the proliferation of islet α
cells, which contributed to the repair of pancreatic function.
Moreover, detection at the molecular level indicated that the
effect of liraglutide-induced activation of GLP-1 on pancreatic
function may be exerted through the activation of PDX-1.
It has been acknowledged that GLP-1 suppresses
glucagon release (30). Such
suppression is independent of the increased insulin secretion in
that it occurs even in cultured islet α cell lines (31). Furthermore, it has been reported
that GLP-1 treatment stimulates islet β cells without obvious
changes in other cell types (32). In the present study, the treatment
of HFD impaired islet β cell function and induced delayed insulin
release, islet β cell apoptosis, and islet α cell hyperplasia in
C57/BL6 mice, representing the increased compensatory secretion in
the pancreas. Such results were consistent with a previous study
(33), confirming the successful
induction of type 2 diabetes features in the present study. To
reverse damage to the pancreas and islet β cells, HFD-fed mice were
subcutaneously injected with liraglutide, a type of GLP-1 receptor
agonist. The administration of liraglutide significantly confounded
the effect of HFD on the experimental animals. Firstly, the glucose
tolerance of the mice in the liraglutide group was improved
compared with that in the HFD group. Moreover, the damage to the
structure and function of islet β cells was clearly ameliorated,
which could be further supported by the decreased insulin
resistance in the hyperinsulinemic euglycemic clamp assay.
Liraglutide treatment not only facilitated the function of recovery
of islet β cells but also inhibited the excess action of islet α
cells. As shown in the liraglutide group, the number of islet α
cells and secretion of GIP were both reduced by administration of
liraglutide. Although the above results could provide compelling
evidence of the effect of GLP-1 on HFD-induced pancreatic injury,
the mechanism through which GLP-1 takes action was partially
revealed.
Previous research (34) indicated that liraglutide
administration markedly influences the body weight of the treated
subjects. Whether the effect of liraglutide was exerted through
this pattern should be comprehensively explored. To fulfill such
purpose, a liraglutide paired group was established in the present
study. Mice in this group were fed with the HFD as in the HFD
group, but the calorie level was restricted to follow the weight
trajectory of mice in the liraglutide group. By establishing the
liraglutide paired group, it was found that although the decreased
level of body weight could influence the results of some assays
(difference between HFD and liraglutide paired groups was
statistically insignificant), the overall effect of decreased body
weight of the experimental mice had no impact on the outcomes of
most experiments. Therefore, the effect of liraglutide-induced
activation of GLP-1 was not attributed to its effect on the body
weight of the experimental animals.
To further study the mechanism of GLP-1 in
alleviating impairments due to HFD, the expression of PDX-1 was
detected. PDX-1 is a critical regulator of islet β cell growth and
function in both fetal and developmental stages (13). Even a modest decrease in the
release of the molecule can impair the compensatory response to
insulin resistance (16,35). In insulinoma cells and in islets
from adult rodents, it stimulates insulin and somatostatin
transcription (36,37). It has been verified that
development of type 2 diabetes is associated with progressive
epigenetic silencing of PDX-1 (13). Thus, activation of PDX-1 may be a
potential treatment strategy for prevention of type 2 diabetes. In
the present study, administration of liraglutide contributed to the
upregulation of PDX-1 both at the mRNA and protein level. The
restoration of PDX-1 level was concomitant with the amelioration of
the structure and function of the pancreas. The effect of GLP-1 on
PDX-1 was previously revealed by Wang et al (38). The authors found that GLP-1
regulated the amount of PDX-1 in whole cell extracts and in nuclear
extracts. Moreover, the effect of GLP-1 on nuclear PDX-1 was
additive with glucose. Except for expression of PDX-1, levels of
the other three markers of pancreatic function, including MafA,
STAT3 and JAK2 were all enhanced by liraglutide (Fig. 7), which confirmed the protective
effect of liraglutide-induced activation of GLP-1 on the pancreas
(26,27).
In conclusion, the findings outlined in the present
study demonstrated that GLP-1 could ameliorate impairments on the
pancreas due to HFD. Administration of GLP-1 receptor agonist
reinforced the capacity of insulin to stimulate glucose disposal by
restoring the function and number of islet β cells, and inhibited
the hyperplasia and redundant function of islet α cells. Moreover,
the effect of GLP-1 was exerted through its activating effect on
PDX-1 rather than its effect on a decrease in body weight, which
supplemented the mechanism by which GLP-1 functions.
References
|
1
|
Knauf C, Cani PD, Ait-Belgnaoui A, Benani
A, Dray C, Cabou C, Colom A, Uldry M, Rastrelli S, Sabatier E, et
al: Brain glucagon-like peptide 1 signaling controls the onset of
high-fat diet-induced insulin resistance and reduces energy
expenditure. Endocrinology. 149:4768–4777. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cummings DE and Schwartz MW: Genetics and
pathophysiology of human obesity. Annu Rev Med. 54:453–471. 2003.
View Article : Google Scholar
|
|
3
|
Alberti KG, Zimmet P and Shaw J; IDF
Epidemiology Task Force Consensus Group: The metabolic syndrome - a
new worldwide definition. Lancet. 366:1059–1062. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
French SA, Story M and Jeffery RW:
Environmental influences on eating and physical activity. Annu Rev
Public Health. 22:309–335. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wyatt SB, Winters KP and Dubbert PM:
Overweight and obesity: prevalence, consequences, and causes of a
growing public health problem. Am J Med Sci. 331:166–174. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Poitout V and Robertson RP: Minireview:
secondary beta-cell failure in type 2 diabetes - a convergence of
glucotoxicity and lipotoxicity. Endocrinology. 143:339–342.
2002.PubMed/NCBI
|
|
7
|
Prentki M, Joly E, El-Assaad W and Roduit
R: Malonyl-CoA signaling, lipid partitioning, and
glucolipotoxicity: role in β-cell adaptation and failure in the
etiology of diabetes. Diabetes. 51(Suppl 3): S405–S413. 2002.
View Article : Google Scholar
|
|
8
|
Kaiser N, Leibowitz G and Nesher R:
Glucotoxicity and beta-cell failure in type 2 diabetes mellitus. J
Pediatr Endocrinol Metab. 16:5–22. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakae J, Biggs WH III, Kitamura T, Cavenee
WK, Wright CV, Arden KC and Accili D: Regulation of insulin action
and pancreatic beta-cell function by mutated alleles of the gene
encoding forkhead transcription factor Foxo1. Nat Genet.
32:245–253. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sussel L, Kalamaras J, Hartigan-O'Connor
DJ, Meneses JJ, Pedersen RA, Rubenstein JL and German MS: Mice
lacking the homeodomain transcription factor Nkx2.2 have diabetes
due to arrested differentiation of pancreatic beta cells.
Development. 125:2213–2221. 1998.PubMed/NCBI
|
|
11
|
Withers DJ, Burks DJ, Towery HH, Altamuro
SL, Flint CL and White MF: Irs-2 coordinates Igf-1
receptor-mediated beta-cell development and peripheral insulin
signalling. Nat Genet. 23:32–40. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Offield MF, Jetton TL, Labosky PA, Ray M,
Stein RW, Magnuson MA, Hogan BL and Wright CV: PDX-1 is required
for pancreatic outgrowth and differentiation of the rostral
duodenum. Development. 122:983–995. 1996.PubMed/NCBI
|
|
13
|
Park JH, Stoffers DA, Nicholls RD and
Simmons RA: Development of type 2 diabetes following intrauterine
growth retardation in rats is associated with progressive
epigenetic silencing of Pdx1. J Clin Invest. 118:2316–2324.
2008.PubMed/NCBI
|
|
14
|
Brissova M, Blaha M, Spear C, Nicholson W,
Radhika A, Shiota M, Charron MJ, Wright CV and Powers AC: Reduced
PDX-1 expression impairs islet response to insulin resistance and
worsens glucose homeostasis. Am J Physiol Endocrinol Metab.
288:E707–E714. 2005. View Article : Google Scholar
|
|
15
|
Kulkarni RN, Jhala US, Winnay JN,
Krajewski S, Montminy M and Kahn CR: PDX-1 haploinsufficiency
limits the compensatory islet hyperplasia that occurs in response
to insulin resistance. J Clin Invest. 114:828–836. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Holland AM, Góñez LJ, Naselli G, Macdonald
RJ and Harrison LC: Conditional expression demonstrates the role of
the homeodomain transcription factor Pdx1 in maintenance and
regeneration of beta-cells in the adult pancreas. Diabetes.
54:2586–2595. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guz Y, Montminy MR, Stein R, Leonard J,
Gamer LW, Wright CV and Teitelman G: Expression of murine STF-1, a
putative insulin gene transcription factor, in beta cells of
pancreas, duodenal epithelium and pancreatic exocrine and endocrine
progenitors during ontogeny. Development. 121:11–18.
1995.PubMed/NCBI
|
|
18
|
Peshavaria M, Gamer L, Henderson E,
Teitelman G, Wright CV and Stein R: XIHbox 8, an endoderm-specific
Xenopus homeodomain protein, is closely related to a mammalian
insulin gene transcription factor. Mol Endocrinol. 8:806–816.
1994.PubMed/NCBI
|
|
19
|
Stoffers DA, Desai BM, DeLeon DD and
Simmons RA: Neonatal exendin-4 prevents the development of diabetes
in the intrauterine growth retarded rat. Diabetes. 52:734–740.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Holst JJ: Glucagonlike peptide 1: a newly
discovered gastrointestinal hormone. Gastroenterology.
107:1848–1855. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Holst JJ, Ørskov C, Nielsen OV and
Schwartz TW: Truncated glucagon-like peptide I, an
insulin-releasing hormone from the distal gut. FEBS Lett.
211:169–174. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kreymann B, Williams G, Ghatei MA and
Bloom SR: Glucagon-like peptide-1 7–36: a physiological incretin in
man. Lancet. 2:1300–1304. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Flint A, Raben A, Astrup A and Holst JJ:
Glucagon-like peptide 1 promotes satiety and suppresses energy
intake in humans. J Clin Invest. 101:515–520. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gutzwiller JP, Göke B, Drewe J, Hildebrand
P, Ketterer S, Handschin D, Winterhalder R, Conen D and Beglinger
C: Glucagon-like peptide-1 is a physiologic regulator of food
intake in humans. Gut. 44:81–86. 1999. View Article : Google Scholar
|
|
25
|
Parlevliet ET, de Leeuw van Weenen JE,
Romijn JA and Pijl H: GLP-1 treatment reduces endogenous insulin
resistance via activation of central GLP-1 receptors in mice fed a
high-fat diet. Am J Physiol Endocrinol Metab. 299:E318–E324.
2010.PubMed/NCBI
|
|
26
|
Zhang C, Moriguchi T, Kajihara M, Esaki R,
Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, et
al: MafA is a key regulator of glucose-stimulated insulin
secretion. Mol Cell Biol. 25:4969–4976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Corcoran RB, Contino G, Deshpande V,
Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA
and Bardeesy N: STAT3 plays a critical role in KRAS-induced
pancreatic tumorigenesis. Cancer Res. 71:5020–5029. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tsuang W, Navaneethan U, Ruiz L, Palascak
JB and Gelrud A: Hypertriglyceridemic pancreatitis: presentation
and management. Am J Gastroenterol. 104:984–991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cedernaes J, Alsiö J, Västermark A,
Risérus U and Schiöth HB: Adipose tissue stearoyl-CoA desaturase 1
index is increased and linoleic acid is decreased in obesity-prone
rats fed a high-fat diet. Lipids Health Dis. 12:22013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ritzel R, Orskov C, Holst JJ and Nauck MA:
Pharmacokinetic, insulinotropic, and glucagonostatic properties of
GLP-1 [7–36 amide] after subcutaneous injection in healthy
volunteers. Dose-response-relationships. Diabetologia. 38:720–725.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matsumura T, Itoh H, Watanabe N, Oda Y,
Tanaka M, Namba M, Kono N, Matsuyama T, Komatsu R and Matsuzawa Y:
Glucagonlike peptide-1(7–36)amide suppresses glucagon secretion and
decreases cyclic AMP concentration in cultured In-R1-G9 cells.
Biochem Biophys Res Commun. 186:503–508. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Friedrichsen BN, Neubauer N, Lee YC, Gram
VK, Blume N, Petersen JS, Nielsen JH and Møldrup A: Stimulation of
pancreatic beta-cell replication by incretins involves
transcriptional induction of cyclin D1 via multiple signalling
pathways. J Endocrinol. 188:481–492. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schwasinger-Schmidt T, Robbins DC,
Williams SJ, Novikova L and Stehno-Bittel L: Long-term liraglutide
treatment is associated with increased insulin content and
secretion in β-cells, and a loss of α-cells in ZDF rats. Pharmacol
Res. 76:58–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nauck MA, Hompesch M, Filipczak R, Le TD,
Zdravkovic M and Gumprecht J; NN2211-1499 Study Group: Five weeks
of treatment with the GLP-1 analogue liraglutide improves glycaemic
control and lowers body weight in subjects with type 2 diabetes.
Exp Clin Endocrinol Diabetes. 114:417–423. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Del Guerra S, Lupi R, Marselli L, Masini
M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, et
al: Functional and molecular defects of pancreatic islets in human
type 2 diabetes. Diabetes. 54:727–735. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Petersen HV, Serup P, Leonard J, Michelsen
BK and Madsen OD: Transcriptional regulation of the human insulin
gene is dependent on the homeodomain protein STF1/IPF1 acting
through the CT boxes. Proc Natl Acad Sci USA. 91:10465–10469. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Miller CP, McGehee RE Jr and Habener JF:
IDX-1: a new homeodomain transcription factor expressed in rat
pancreatic islets and duodenum that transactivates the somatostatin
gene. EMBO J. 13:1145–1156. 1994.PubMed/NCBI
|
|
38
|
Wang X, Cahill CM, Piñeyro MA, Zhou J,
Doyle ME and Egan JM: Glucagon-like peptide-1 regulates the beta
cell transcription factor, PDX-1, in insulinoma cells.
Endocrinology. 140:4904–4907. 1999. View Article : Google Scholar : PubMed/NCBI
|