1. Introduction
Since the late 1990s, the availability of public,
large databases containing growing information about genes, gene
products (RNAs and proteins), genomes and molecular functions has
radically changed the traditional approach to gene discovery and
characterization. Combining the deposited data about informational
molecules (1,2) obtained from multiple species is a
straightforward method to gain rapid knowledge about the structure
of an organism's genes and gene products, which in turn may be used
to obtain clues as to the function of each individual gene. While
this possibility has allowed the generation of an amount of data
incomparable to what was obtained by classic molecular biology
methods used in the pre-genomic era (3), the fact that the quality and degree
of the information available for an individual gene may tend to
decrease is less evident. For example, if we consider the
characterization of the messenger RNA (mRNA) expressed by a human
locus, through the 1980s and 1990s it was typical to obtain
accurate information about the total mRNA size and tissue
distribution by northern blot analysis and about the transcription
initiation sites by S1 nuclease mapping, primer extension and
run-off assays (4). In later
years, mRNA full-length sequences were obtained by tailored
experiments designed for polymerase chain reaction (PCR)
amplification of DNA complementary to RNA (cDNA) ends [rapid
amplification of cDNA ends (RACE)], alternative splicing
information by cDNA in vivo and in vitro cloning of
individual RNA isoforms, and protein sequences by in vitro
translation and polypeptide biochemical analysis. Indeed, genes
were usually studied on a one-by-one basis, and there was the
possibility to cross-check data made available through different
methods (5). An example would be
the comparison of the mRNA length deduced from northern blotting
(taking into account the polyadenylated tail) and the one of the
isolated cDNA (6), or the
comparison of the molecular weight of a known protein (7) and the one of the polypeptide
predicted to be encoded by the open reading frame (ORF)/coding
sequence (CDS) of its relative cDNA.
New large-scale methods cannot always reach the
resolution of previous ones; therefore, while they set a new
standard in the methods used in genetics, more detailed analysis
aimed at characterizing each individual gene remains necessary in
order to avoid incomplete or erroneous knowledge of the gene
structure and function. However, the genome-scale information has
been in turn invaluable in effectively directing further
investigations needed for each genomic locus using classical
methods. This has been shown in particular for the human genome, by
a large corpus of millions of short sequences (a few hundred base
pairs in length) which has been derived by partial, single-pass
sequencing of the cDNA clones from RNA of specific tissues
(8). These have accumulated in
the expressed sequence tag (EST) database since its creation >20
years ago (9). A variety of
EST-based methods (10,11) were then used for the rapid in
silico cloning of genes (12), determining differential gene
expression (13), characterizing
alternative forms of transcripts derived from alternative splicing
(14,15), and defining at least one complete
ORF (16) for each mRNA. This
last point is a well-known issue in molecular biology and genomics,
with relevant consequences for the prediction of the gene product
structure and function, and will be analyzed in detail in this
review.
2. The 5′ end mRNA artifact
According to the classic molecular biology central
dogma, the final effector of the genetic information is the protein
(a chain of amino acids) encoded from a given gene; thus it is
crucial to know the basic, primary structure of the protein (its
amino acid sequence). A landmark in this field was the sequencing
of the two amino acid chains composing human insulin by Sanger
(17). Polypeptide sequencing has
the advantage of determining the natural primary structure of the
polypeptide chain, and in particular the actual first amino acid of
the sequence, thanks to the ability of fluorodinitrobenzene to
react with the N-terminal amino group at one end of the chain. Key
subsequent advancements were the recognition that, due to the
colinearity of nucleic acids and proteins and to the mechanisms of
mRNA translation, amino terminal amino acids are encoded by the 5′
end of the mRNA (18). Therefore,
when Sanger et al proposed a new effective method to
sequence DNA (19), it became
evident that it was much more convenient to sequence the nucleic
acids rather than the proteins, and that the amino acid sequence of
gene products could be conveniently deduced from the nucleotide
sequence of the relative cloned cDNA. This change of experimental
paradigm led to 'reverse genetics' (20), the passage from nucleic acid
sequences to their functions rather than the contrary as in classic
genetics and has had the fundamental consequence that actually,
since the late 1970s, the vast majority of protein sequences were
no longer directly determined, but were predicted following
sequencing of the relative cDNAs according to rules for recognition
of the start codon (first-AUG rule, optimal sequence context) and
the genetic code (21).
While this advancement greatly sped up the pace of
the availability of protein sequences, it should be kept in mind
that all standard experimental methods for the cloning of cDNA are
affected by a potential inability to effectively clone the 5′
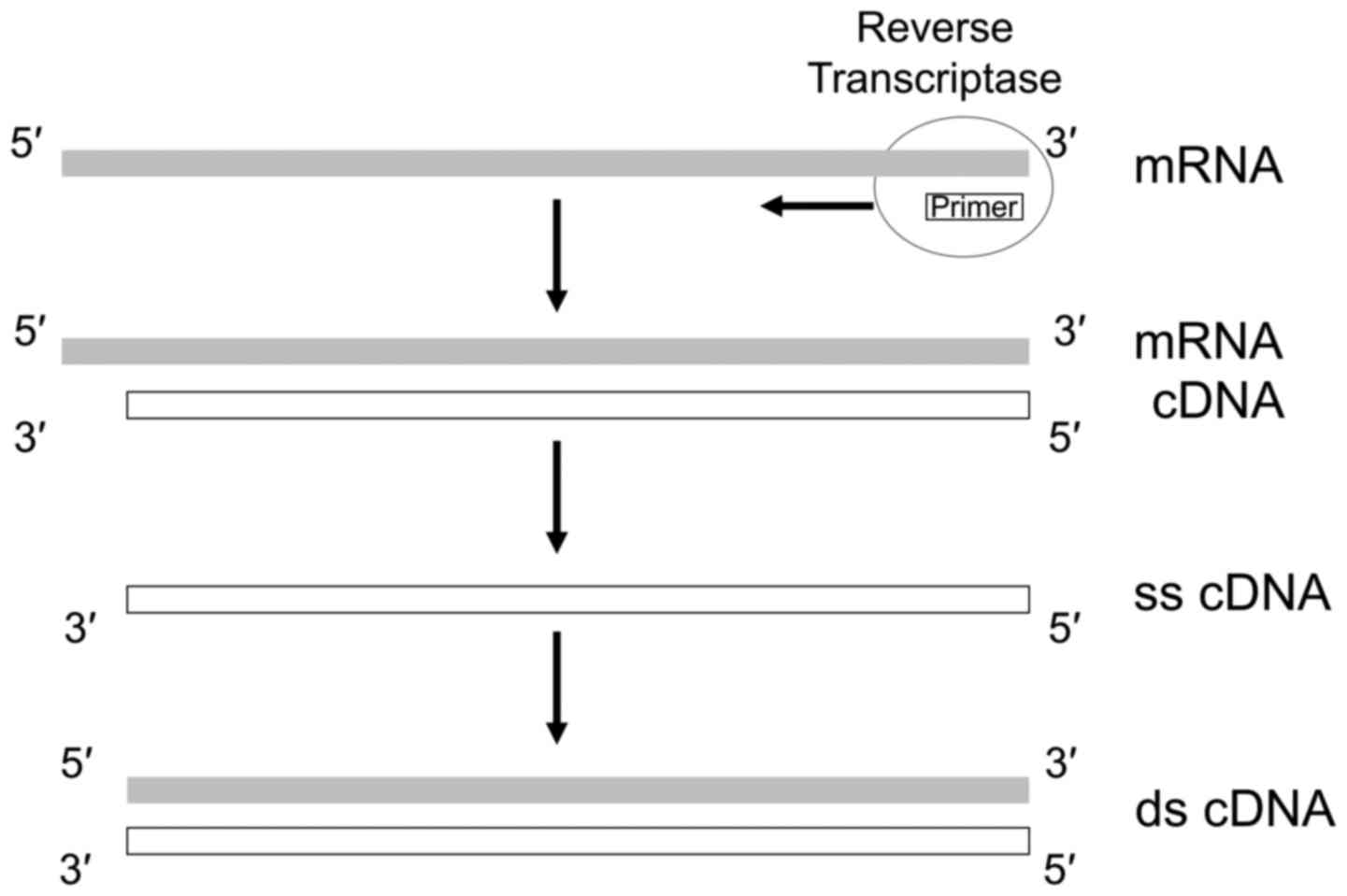
region of mRNA in its completeness (22). This is due to the reverse
transcriptase (RT) failure to extend first-strand cDNA along the
full length of the mRNA template toward its 5′ end (22) (Fig.
1), an operation whose success depends on the natural
processivity of the enzyme, as well as its quality, the integrity
of the RNA, the secondary structures assumed by the 5′ region of
the mRNA hampering the RT progression and the reaction conditions
(23).
It should be highlighted here that, due to the
intrinsic functional mechanisms of the polymerases able to generate
DNA copies of mRNAs, cDNA is typically obtained through a primer
starting polymerization from the 3′ region of the mRNA [e.g., a
poly(dT) oligonucleotide pairing with the poly(dA) tail present in
the vast majority of mRNAs]. This implies that a cDNA collection is
by definition enriched in the 3′ regions of the mRNAs, and
consequently it is expected that the prediction of the amino acid
sequence at the carboxy terminus of the gene product is more
accurate than the one at the amino terminus. This problem was
recognized early on, in the publication of the first sequenced
human cDNA, the one for the β chain of hemoglobin in 1977 when the
5′-untranslated region (UTR) was the last region to be reported in
December (24) following previous
descriptions of 3′-UTR in April (25) and CDS in July (26): 'cloning cDNA has proven to be a
most valuable technique for sequencing mRNA (27,28). During the construction of
double-stranded cDNA, however, a considerable number of
5′-non-coding region sequences are lost. The independent sequencing
of this region will therefore be a necessary step to complete our
knowledge of the primary structure of any mRNA' (24); Okayama and Berg clearly wrote in
1982: 'obtaining cloned cDNAs with complete 5′-UTR and
protein-coding sequences is rare, particularly if the mRNA codes
for a large protein. Although such truncated cDNAs are still useful
as hybridization probes, they cannot direct the synthesis of
complete proteins after their introduction into bacterial or
mammalian cells via appropriate expression vectors' (23).
A flourishing of reports in the 1980s presented the
determination of the often called 'cDNA full-length sequence' for
many human genes. For the reasons discussed, the concept of the
'full-length sequence' becomes de facto equivalent to the
one of 'completeness of mRNA sequence at its 5′ end' and remains an
open issue in molecular biology as cDNA incompletely representing
the 5′ end of the relative mRNA may lead to the incorrect
assignment of the first AUG codon. In these cases, should an
additional upstream AUG - in frame with the previously determined
one - have been identified in a more complete mRNA 5′ end, it would
have been considered the actual translation start codon, thus
extending the predicted amino terminus sequence of the product.
Assignment of the inexact start codon leads to a series of
subsequent relevant errors in the experimental study of the
relative cDNA. We therefore introduced the term '5′ end mRNA
artifact' to refer to the incorrect assignment of the first
translation codon (AUG sequence) in an mRNA, due to the incomplete
determination of its 5′ end sequence (29).
From the experimental point of view, the recognition
of this technical issue, although often without systematic
investigation of its possible consequences for genome annotation,
has led to the development of several methods to determine the full
length mRNA sequence on a large scale. Some were based on the
presence of the 'cap' at the true 5′ end of the mRNA [reviewed in
(30)], such as 5′ cap trapping
(31) and cap analysis of gene
expression (CAGE) (32).
Systematic empirical annotation of a set of transcript products by
5′ RACE (33) has also been
employed, as well as after the introduction of microarray-based
platforms, hybridization of RNA on high-density resolution tiling
arrays (34). However, these
techniques were found to be experimentally labor-intensive and they
have not been routinely applied.
Concurrently, the growing incorporation of
information derived from individual cDNA and large-scale sequencing
projects, including those specifically designed to characterize
mRNA 5′ end (31,35,36), led to a continuous refinement and
improvement of completeness at the 5′ region of deposited and
verified mRNA reference sequences (e.g., RefSeq, https://www.ncbi.nlm.nih.gov/refseq/),
as also regarding the corresponding protein-coding sequences.
Therefore, it became possible to exploit the data from EST or other
large-scale RNA sequencing projects to verify if sequence analysis
could be optimized to reveal the extension of the 5′ region of
known mRNAs and possibly the consequential redefinition of the
amino acid sequence of the encoded products.
The recent availability of massive RNA-sequencing
(RNA-Seq) methods for the generation of transcriptome sequence
databases (37) offers a new
potential tool to deal with the issue, although to date it appears
not to have been systematically used to this aim. Moreover,
information about sequences possibly extending the knowledge of the
5′ end of mRNA is not easily derivable from RNA-Seq data in
comparison with the EST-based approach, due to short sequence reads
typically obtained by this technique, as well as difficulty in
building full-length transcript structures.
Furthermore, a ribosome footprinting profiling
strategy based upon high-throughput sequencing of
ribosome-protected mRNA fragments has been developed, enabling the
genome-wide investigation of translation (38). This technique, used in combination
with initiation-specific translation inhibitors, allows the
identification of translation initiation with subcodon or even
single-nucleotide resolution and was successfully exploited in
order to predict also additional upstream AUG codons (39–41).
Finally, we should note the existence of ORFs and
out-of-frame AUGs located in the 5′-UTR, upstream of the main
coding region (42). These
situations are different from the artifact reported herein as they
do not extend the known coding region, but are implicated in the
regulation of gene expression by modulating mRNA stability and
translation (42,43).
3. Systematic identification of incomplete
5′ end region in human known mRNAs
The theoretical possibility that the presence of a
more precise knowledge of the mRNA 5′ end sequence may lead to
consequential correction of the previously accepted predicted
product appeared in several reports in the form of anecdotal
evidence randomly found for single genes that were under detailed
investigation. For example, mRNA CDS was extended in this way for
RANBP9/RanBPM gene (RAN binding protein 9, on 6p23),
where the study performed by Nishitani et al (44) allowed the addition of 230 new
amino acids. In the case of nuclear factor, erythroid 2-like 3
(NFE2L3) gene (on 7p15.2), the corresponding #AB010812.1
mRNA sequence of 2,174 bp in length derived from Kobayashi et
al (45) was replaced by the
sequence #AF134891.1 of 2,618 bp, leading to the addition of 294
new amino acids to the predicted protein. The study performed by
Nomura et al (46) for
SP2 gene (Sp2 transcription factor, on 17q21.32) allowed the
release of the #D28588.1 mRNA sequence entry recording a CDS of
3,288 bp leading to the addition of 111 new amino acids compared to
the previous #M97190 entry of 2,063 bp provided by Kingsley and
Winoto (47). The coding nature
of these extensions was also supported by very high similarity with
the respective murine hortologs (29). These and other similar reports
suggested that a high-throughput approach was desirable to discover
all the incompletenesses in the CDSs (Table I).
 | Table IMain published results of systematic
search for completeness of mRNA 5′ CDS region. |
Table I
Main published results of systematic
search for completeness of mRNA 5′ CDS region.
| Ref. | Year | Organism | Method | mRNAs | Extended 5′
CDSa |
|---|
| (35) | 2000 | H.
sapiens | Oligo-capping | 954 | 68 (7.1%) |
| (29) | 2003 | H.
sapiens | Manual and
automated sequence analysis | 13,124 | 556 (4.2%) |
| (53) | 2007 | D.
rerio | Automated sequence
analysis | 8,528 | 285 (3.3%) |
| (39) | 2011 | Mouse embrionic
stem cells | Ribosome
footprinting profiling and support vector machine (SVM)-based
machine learning strategy | 4,994 | 570 (11.4%) |
| (54) | 2012 | H.
sapiens | Fully automated
sequence analysis | 18,665 | 477 (2.6%) |
| (40) | 2012 | H.
sapiens | Ribosome
footprinting profiling and neural network prediction | 5,062 | 6 AUG (0.1%) and
540 non-AUG (10.7%) |
| (55) | 2014 | M.
musculus | Fully automated
sequence analysis | 20,221 | 351 (1.7%) |
| (41) | 2014 | H.
sapiens | Ribosome
footprinting profiling and manual analysis | 1,255 | 17 (1.4%) |
| | M.
musculus | Ribosome
footprinting profiling and manual analysis | 930 | 4 AUG (0.4%) and 13
non-AUG (1.4%) |
Regarding our group, as a first approach to the
issue, due to our interest in an integrated route to identifying
new pathogenesis-based therapeutic approaches for trisomy 21 (Down
syndrome) (48,49), we focused on the known,
well-characterized genes present in the original map of human
chromosome 21 (Hsa21), manually analyzing 109 RefSeq mRNA sequences
catalogued as 'category: known' by Hattori et al (50), and linked to at least one
published report, for the presence of an in-frame stop codon
upstream of the described ATG. In 49 cases, the finding of such a
stop codon allowed the exclusion of the possibility that the
recorded 5′-UTR sequence may actually be part of a longer CDS
(51). The sequence of the
remaining 60 mRNAs in which bases in the 5′-UTR could on the
contrary be consistent with the presence of translated codons was
systematically aligned with sequences available in databanks using
Basic Local Alignment Search Tool (BLAST software, http://www.ncbi.nih.gov/BLAST/), leading to the
discovery of a total of 20 genes for which EST (or also non-EST RNA
sequences) homology suggested the existence of mRNAs more complete
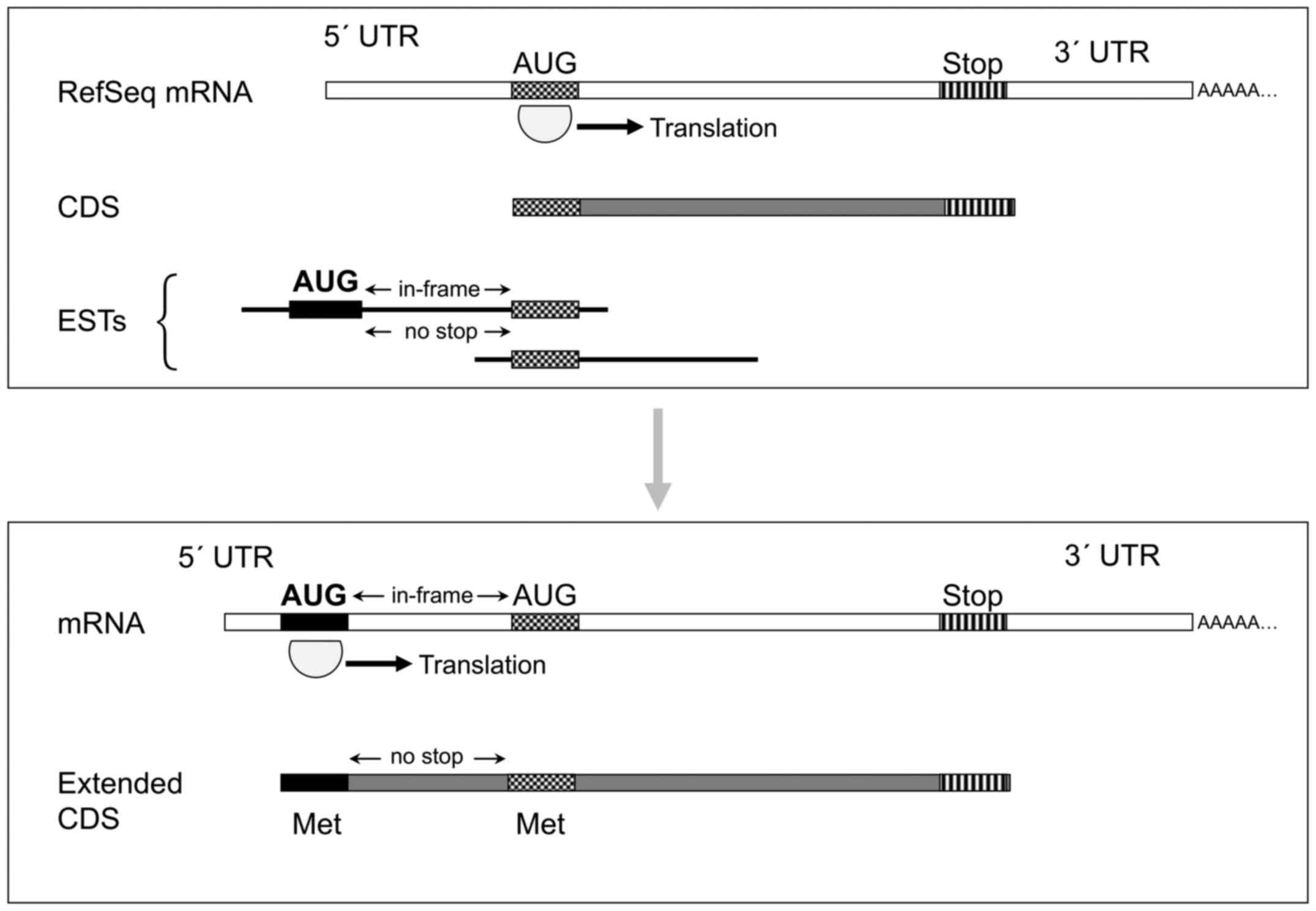
at 5′ terminus. They putatively encode for protein products longer
at their amino terminus, due to the presence of a previously
unknown start codon in frame with and upstream of the described one
(Fig. 2). Experimental evidence
for the existence of these transcripts was finally obtained,
following RT-PCR and sequencing, for five loci: down syndrome
critical region 1 (DSCR1) [now regulator of calcineurin 1
(RCAN1)], disco interacting protein 2 homolog A
(DIP2A; KIAA0184), URB1 ribosome biogen-esis 1
homolog (URB1; KIAA0539I), SON DNA binding protein
(SON) and trefoil factor 3 (TFF3) (29). In these cases, both of the
following conditions occurred: an extension of described exon 1
predicted new coding codons upstream of the known AUG; and a novel
AUG was present upstream of these codons, in frame with the
previously described AUG and without any intervening stop codon.
This thus suggests that, following the rules of translation
initiation [reviewed by Kozak (21)], the actual CDS should be
considered as the one included between the novel 'first-AUG' and
the known stop (Fig. 2). It was
observed that no known mechanism hampers the possibility that the
newly identified start codon is not the point of actual translation
as the use of 'internal' AUGs, enabling additional initiation
events at downstream AUG codons in some mRNAs may occur only in
three well-defined circumstances (21): re-initiation, which does not apply
to the mRNAs investigated by this approach, as the newly determined
AUG is not part of a small upstream ORF separated from the main ORF
by a stop codon; context-dependent leaky scanning, which may be
excluded as we considered the concordance with the Kozak sequence
(21,52) for the novel AUGs, observing full
(sometimes better) compatibility with the use of the novel AUG
(29); a third mechanism, that is
the use of internal ribosome entry site (IRES) sequence modules,
adopted only by some known viral mRNAs.
These positive results suggested to extend the
approach to the whole set of human RefSeq mRNAs known at the time
(n=13,124), following automation by a simple program to detect the
presence or the absence of an in-frame stop in the described 5′-UTR
of an mRNA. The percentage of the latter type of mRNA in the set
(51%) was very similar to the one found for the Hsa21 gene set
(55%), thus estimating that, in proportion, the CDS of 556 known
human mRNAs might be incomplete at the 5′ end (29).
This approach required manual curation to analyze in
detail, by sequence comparison, any mRNA candidate to have an
incomplete CDS at 5′ region. An improvement of the algorithm was
then published and applied with success to zebrafish [see below
(53)], showing that the
automated detection of putative additional bases at the known 5′
end of a set of mRNAs following elaboration of multiple results of
sequence comparison analysis (by BLAST tool) was possible. Some
technical limitations of the used environment made the
implementation of this pipeline difficult for the much more
numerous human sequences which hampered progress in this direction
for a while. Further improvement of the automated EST-based
approach (5′_ORF_Extender 2.0, freely available at http://apollo11.isto.unibo.it/software/)
finally made the systematic identification (Fig. 2) of CDSs at the 5′ end of all
human known mRNAs possible, parsing >7 million BLAT alignments
and thus finding 477 human loci out of 18,665 analyzed (Table I), with an extension of their RNA
5′ CDS identified in detail (54). In addition, in this study, a
proof-of-concept confirmation was obtained by in vitro
cloning and sequencing for some example genes: GNB2L1 [now
receptor for activated C kinase 1 (RACK1)], glutaminyl-tRNA
synthetase (QARS) and tyrosyl-DNA phosphodiesterase 2
(TDP2) cDNAs. On the other hand, a list of 20,775 human
mRNAs where the presence of an in-frame stop codon upstream of the
known start codon indicates completeness of the CDS at 5′ end in
the current form was generated (54). This approach could also be aimed
at the different 5′-UTR sequence identification, but the length of
the bases aligned upstream of the novel AUG is usually too short to
allow this type of investigation. In addition, should the length be
long enough, the analysis would require an ad hoc algorithm able to
discriminate mRNA isoforms of this type, including mapping of the
newly determined 5′-UTR to the genome to derive the alternative
transcription/splicing events responsible for the different 5′-UTR
sequences.
While this review is more focused on human mRNAs for
the possible repercussion in medicine, it should be noted that
similar results are to be expected for the genomes of other
organisms due to the sharing of common molecular techniques, whose
limitations are at the basis of the artifact. Actually, studies on
two of the most commonly used model organisms for the investigation
of the human genome, Danio rerio (zebrafish) and Mus
musculus (domestic mouse) have confirmed this expectation. A
novel proposed automated approach (5′_ORF_Extender 1.0) was able to
systematically compare available ESTs with all the zebrafish
experimentally determined mRNA sequences, identify additional
sequence stretches at 5′ region and scan for the presence of all
conditions needed to define a new, extended putative ORF. The tool
identified 285 (3.3%) mRNAs with putatively incomplete ORFs at the
5′ region and, in three example selected cases (selt1a,
unc119.2 and nppa or selenoprotein T 1a, unc-119
lipid binding chaperone B homolog 2 and natriuretic peptide A,
respectively), the extended coding region at 5′ end was
experimentally demonstrated (53). As regards the mouse mRNAs, the
application of the improved method used for human transcripts
(54) showed that in 351 mouse
loci, out of 20,221 analyzed, an extension of the mRNA 5′-coding
region could be identified. Experimental confirmation was obtained
by in vitro cloning and sequencing for adenomatosis
polyposis coli 2 (Apc2) and MAP kinase-interacting
serine/threonine kinase 2 (Mknk2) cDNAs and a list of 16,330
mouse mRNAs with estimated complete CDS at 5′ end was provided
(55). Remarkably, 82% of the
results were original and have not been identified by the
annotation pipelines used in the main mouse genome databases and
genome browser (55). The
diffusion of the 5′ end mRNA artifact may thus be considered
approximately constant from lower vertebrates to humans because the
methods used to characterize the relative mRNAs are the same or
very similar (Table I).
The identification of the most upstream definable
start codon does not exclude that a downstream AUG codon may also
be used by the ribosome, a phenomenon known as alternative
translation (56). It has been
shown that alternative translation start sites tend to be conserved
in eukaryotic genomes, providing a functional mechanism under
selection for increased efficiency of translation and/or for
obtainment of different N-terminal protein variants (57). It has also already been noted that
this type of analysis cannot formally exclude that the extended ORF
may derive from alternative transcription starting site (due to
alternative promoter usage) and/or splicing of the investigated
locus (53). However, it reveals
in any case that additional coding sequences not previously
identified exist, as may be confirmed by phylogenetic comparison at
the amino acid level (53). As in
the case of any other computer prediction, further investigation is
required, in silico but especially in vitro, for a
fine characterization of the putative model.
While the published approaches have considered
algorithms assuming that the start codon has an AUG sequence, it
should be noted that in a minor percentage of mRNA CDSs the start
codon may have alternative sequences, particularly CUG, UUG, GUG,
ACG, AUA and AUU (58). Actually,
recent experiments have confirmed this phenomenon and suggested
that it may be more frequent than was previously assumed.
Therefore, when the use of a non-AUG codon is known or suspected,
further analysis not included in standard pipelines should be
performed in individual cases to identify in frame upstream non-AUG
start codons which may also be responsible of encoding proteins
longer that the ones previously described.
4. Consequences of 5′ end mRNA artifact in
biology and medicine
The 5′ end mRNA artifact is expected, and
demonstrated, to cause a chain of consequences in biomedical
research, that will be now listed and discussed (Table II). The first obvious issue
associated with the artifact is the negative consequence on the
study of product structure and function (59). The possibility that vast amounts
of studies are based on incorrect starting data is real. For
instance, it occurred in the functional characterization of a
polypeptide expressed from its predicted incomplete DNA (60) and in a functional study of the
cytokine interleukin 16 (IL16) (61), where the product appears to be
expressed from an incomplete cDNA (Table II).
| Table IIPossible consequences of incomplete
determination of mRNA 5′ CDS region for example human genes. |
Table II
Possible consequences of incomplete
determination of mRNA 5′ CDS region for example human genes.
| Symbol | Ref. | AAsa | Ref. 2 |
|---|
| At protein
level | | | | |
| Errors in
determining the 3D protein structure | ALDOC | (59) | 87 | (54) |
| Prediction of an
incomplete polypeptide | QARS | (60) | 18 | (54) |
| Production of an
incomplete polypeptide | IL16 | (61) | 47 | (54) |
| Lack of
description of functional protein domains | SON | http://www.ncbi.nlm.nih.gov/gene/6651 | 968 | (29) |
| Errors in
identifying protein localization | RANBP9/RanBPM | (63) | 230 | (44) |
| Failure to predict
alternative polypeptides | UMOD | http://www.ncbi.nlm.nih.gov/gene/7369 | 49 or 28 | (54) |
| Errors in
identifying ortholog products | DSCR1.1 | (66) | 55 | (29) |
|
| Symbol | Ref. | ntsa | Ref. 2 |
|
| At cDNA level | | | | |
| Failure to screen
the complete CDS for mutations | ADAR | http://omim.org/entry/146920 | 48 | (54) |
| Incomplete cDNA in
two-hybrid test for function | DSCR1 | (65) | 55 | (29) |
| Potential errors
in designing morpholino oligos | unc-119.2
(Danio rerio) | (77) | 58 | (53) |
| At gene structure
level | | | | |
| Failure to
identify the full extension of the gene/labeling of genic regions
as intergenic space | DIP2A | (71) | 82,895 | (29) |
| Failure to
identify actual promoter regions | TFF3 | (72) | 170 | (29) |
The recording of protein sequences incomplete at
their amino terminus in the genomic databases may also cause the
failure to identify functionally remarkable protein domain
sequences (Table II); in
particular, sequences located at the amino terminus of proteins may
be represented by signal peptide sequences directing delivery of
the protein to its final destination (62,63) and may also affect its half-life
(64).
In addition, there is the possibility to
underestimate alternative splicing at the 5′ terminus of genes and
to not predict the corresponding alternative protein gene products
(Table II). The statement in the
classic article by Okayama and Berg still holds true: 'indeed, it
was comparison between cloned cDNAs and their genomic counterparts
that uncovered the existence of intervening sequences and splicing'
(23). Moreover, the design of a
mutation screening aimed at identifying pathological variations in
the coding sequences could be affected by the incomplete knowledge
of the CDS, a circumstance that could occasionally explain the
failure to find expected mutations in candidate or established
disease genes, and could possibly lead to inaccurate
genotype/phenotype correlations (Table II). From a functional point of
view, the new amino acid sequence could be responsible for new
interactions. The possibility of designing molecules with
pharmacological activity based on binding to proteins expressed as
bait in a two-hybrid test from incomplete cDNAs (65) emphasizes the importance of knowing
the actual primary structure of the protein. Finally, the presence
of a truncated protein sequence in the genomic databases may also
be at the origin of a chain of errors in the prediction of
orthologs in other species. In particular, the genome annotation
pipelines will tend to propagate the truncated sequence in the
predicted model proteins. For instance, the error in determination
of the highly similar corresponding murine DSCR1 ortholog (66) underlines that a bias deriving from
the original human incomplete data negatively affected the modeling
of the murine DSCR1 product sequence.
Due to the complex structure of the human loci
(67–70), errors in establishing an accurate
cDNA sequence may also finally cause drawbacks in the study of
genomic organization of a gene due to the tight connections between
DNA and RNA (Table II). If a
cDNA incomplete at its 5′ terminus is used to establish the genomic
structure of a locus, this could cause failure to recognize genomic
sequences as part of the locus (71). As a secondary consequence,
classification of a genic region as intergenic may keep the 'search
space' for novel genes artificially expanded (71). Due to the physical proximity of
the gene promoter region and the corresponding mRNA 5′ region, a
sequence supposed to be proximal to the transcription start site
and annotated as promoter could be actually part of a longer mRNA,
as was shown for TFF3 (72,29). This issue may further increase the
difficulty in identifying promoter sequences that do not have
regular start and stop signals or characteristic cross-species
conservations as the CDSs, and can even present with diverged
sequences among distant species, while being functionally conserved
(73). On the other hand, a
non-exact delimitation between 5′-UTR and CDS could lead to errors
in the knowledge of the 5′-UTR sequence itself and in the
interpretation of its role in the control of translation (74). Although this last class of
consequences does not directly affect the prediction of the CDS,
they should be considered as a further incentive to not
underestimate the relevance of this artifact due to the central
role of the 5′ terminus in gene expression regulation pathways. The
knowledge of the true mRNA end is also useful in designing probes
specific for this region that may be more variable between similar
loci or isoforms from the same locus rather than the central,
coding region. This is relevant regarding the possibility to
extract from publicly available microarray datasets quantitative
reference measures for the expression values of the whole
complement of the genes of both normal (75) or pathologic (76) transcriptomes. Exact knowledge of
mRNA 5′ region also affects the choice of morpholino
oligonucleotides, in particular in zebrafish (77), used in knockdown experiments
(Table II).
The artifact may also be a source for errors in
other types of genomic analysis, although in these cases the
consequences are expected not to be relevant, as the alteration of
calculations is likely to represent a small deviation, and not for
immediate medical application of these analyses [e.g., estimation
of codon usage at a genomic scale (78), although the knowledge of the whole
set of codons in a cDNA could affect the technology of the
production of the translated product in a host (79)].
5. Possible solutions for improving the
knowledge of the 5′-coding regions in mRNAs
Several methods have been described with the aim of
knowing with more precision the 5′ mRNA end, thus excluding that
its CDS may be incompletely predicted. The first were devised in
the 1990s and were based on experimental protocols exploiting the
capability of dedicated techniques to identify the first bases
transcribed from DNA or the first bases following the cap in mature
mRNAs. These methods have been cited in the Introduction section
and remain valid, although they were often labor-intensive and not
routinely used.
A second group of methods is based on computational
biology approaches, with the advantage of providing a first
systematic screening leading to exclusion of a relevant number of
genes as candidates for the 5′ end mRNA artifact. Due to the
availability of throughput results of an EST-based approach of this
type (54), it is advisable to
perform a simple first check against these results for a gene of
interest before assuming that the predicted product is the one
recorded in the current version of databases. Continuous refinement
over time of the human mRNA sequences has led to the current
estimation of 259 nucleotides as the mean 5′-UTR size (80), so there is the concrete
possibility that extended protein-coding sequences could actually
be hidden in longer 5′-UTRs. Further developments of the
computational analysis of high-throughput cDNA sequencing methods
(RNA-Seq) should also provide a means to increase the
characterization of whole sequences of human transcripts. Several
studies have been performed to implement RNA-Seq methods of
profiling mRNA 5′ ends in Drosophila melanogaster (81,82).
Finally, recent developments of proteomics research
open the way for a different, specular approach to the problem.
Knowledge of protein sequences obtained by massive analysis of
polypeptide nuclear magnetic resonance (NMR) or mass spectrometry
(MS) spectra, in particular oriented to N-terminal sequencing
(83,84), might be used for a reverse search
for genomic sequences predicted to be translated in the
corresponding identified protein sequences. This thus resembles the
first protein-toward-DNA experimental flow but at on a genomic
scale and largely based on computational methods.
In conclusion, we have presented evidence that
current methods of genomics research are subject to a possible
artifact regarding the exact determination of the mRNA 5′ region
sequence and the consequences that this may have on the annotation,
as well as on the experimental study of both genes and gene
products. While there are several strategies to deal with this
issue, the most important issue appears to bring this possibility
to the attention of the scientific community so that it is taken
into account when planning experiments in molecular biology and
genetics.
Acknowledgments
M.C.'s fellowship has been co-funded by a donation
from Fondazione Umano Progresso (Milano, Italy) and by a grant from
Fondazione del Monte di Bologna e Ravenna (Bologna, Italy).
M.C.P.'s fellowship has been co-funded by a donation from
Fondazione Umano Progresso and by donations following the
international fundraising initiative by Vittoria Aiello and
Massimiliano Albanese (Washington, DC, USA) - donors contributing
to this initiative are listed on the site: http://www.massimilianoalbanese.net/ds-research/?lang=en.
The fellowship for A.P. has been mainly funded by the Department of
Experimental, Diagnostic and Specialty Medicine (DIMES), University
of Bologna (Bologna, Italy) and co-funded by the Fondazione Umano
Progresso. We are grateful to Kirsten Welter for her expert
revision of the manuscript.
References
|
1
|
Borsani G, Ballabio A and Banfi S: A
practical guide to orient yourself in the labyrinth of genome
databases. Hum Mol Genet. 7:1641–1648. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pandey A and Lewitter F: Nucleotide
sequence databases: A gold mine for biologists. Trends Biochem Sci.
24:276–280. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baxevanis AD and Bateman A: The importance
of biological databases in biological discovery. Curr Protoc
Bioinformatics. 50:1.1.1–1.1.8. 2015. View Article : Google Scholar
|
|
4
|
Tropp BE: Molecular Biology: Genes to
Proteins. 3rd edition. Jones & Bartlett; Publishers, Sudbury,
MA: 2008
|
|
5
|
Sambrook J and Russel DW: Molecular
Cloning: A Laboratory Manual. 2. 3rd edition. Cold Spring Harbor
Laboratory Press, Cold Spring Harbor; NY: 2001
|
|
6
|
Vitale L, Casadei R, Canaider S, Lenzi L,
Strippoli P, D'Addabbo P, Giannone S, Carinci P and Zannotti M:
Cysteine and tyrosine-rich 1 (CYYR1), a novel unpredicted gene on
human chromosome 21 (21q21.2), encodes a cysteine and tyrosine-rich
protein and defines a new family of highly conserved
vertebrate-specific genes. Gene. 290:141–151. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang J, Lou X, Shen H, Zellmer L, Sun Y,
Liu S, Xu N and Liao DJ: Isoforms of wild type proteins often
appear as low molecular weight bands on SDS-PAGE. Biotechnol J.
9:1044–1054. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Adams MD, Kelley JM, Gocayne JD, Dubnick
M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF,
et al: Complementary DNA sequencing: Expressed sequence tags and
human genome project. Science. 252:1651–1656. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boguski MS, Lowe TM and Tolstoshev CM:
dbEST - database for 'expressed sequence tags'. Nat Genet.
4:332–333. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nagaraj SH, Gasser RB and Ranganathan S: A
hitchhiker's guide to expressed sequence tag (EST) analysis. Brief
Bioinform. 8:6–21. 2007. View Article : Google Scholar
|
|
11
|
Parkinson J and Blaxter M: Expressed
sequence tags: An overview. Methods Mol Biol. 533:1–12. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gill RW and Sanseau P: Rapid in silico
cloning of genes using expressed sequence tags (ESTs). Biotechnol
Annu Rev. 5:25–44. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Carulli JP, Artinger M, Swain PM, Root CD,
Chee L, Tulig C, Guerin J, Osborne M, Stein G, Lian J, et al: High
throughput analysis of differential gene expression. J Cell Biochem
Suppl. 30–31:286–296. 1998. View Article : Google Scholar
|
|
14
|
Sorek R, Shamir R and Ast G: How prevalent
is functional alternative splicing in the human genome? Trends
Genet. 20:68–71. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bonizzoni P, Rizzi R and Pesole G:
Computational methods for alternative splicing prediction. Brief
Funct Genomics Proteomics. 5:46–51. 2006. View Article : Google Scholar
|
|
16
|
Brent MR: Genome annotation past, present,
and future: How to define an ORF at each locus. Genome Res.
15:1777–1786. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sanger F: La structure de l'insuline. Bull
Soc Chim Biol (Paris). 37:23–35. 1955.In French.
|
|
18
|
Yanofsky C, Carlton BC, Guest JR, Helinski
DR and Henning U: On the colinearity of gene structure and protein
structure. Proc Natl Acad Sci USA. 51:266–272. 1964. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sanger F, Nicklen S and Coulson AR: DNA
sequencing with chain-terminating inhibitors. Proc Natl Acad Sci
USA. 74:5463–5467. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ruddle FH: The William Allan Memorial
Award address: Reverse genetics and beyond. Am J Hum Genet.
36:944–953. 1984.PubMed/NCBI
|
|
21
|
Kozak M: Pushing the limits of the
scanning mechanism for initiation of translation. Gene. 299:1–34.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sambrook J and Russel DW: Rapid
amplification of 5′ cDNA ends. Molecular Cloning: A Laboratory
Manual. 3. 3rd edition. Cold Spring Harbor Laboratory Press, Cold
Spring Harbor; NY: pp. 8.54–8.60. 2001
|
|
23
|
Okayama H and Berg P: High-efficiency
cloning of full-length cDNA. Mol Cell Biol. 2:161–170. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baralle F: Complete nucleotide sequence of
the 5′ noncoding region of human alpha-and beta-globin mRNA. Cell.
12:1085–1095. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Proudfoot NJ: Complete 3′ noncoding region
sequences of rabbit and human beta-globin messenger RNAs. Cell.
10:559–570. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Marotta CA, Wilson JT, Forget BG and
Weissman SM: Human beta-globin messenger RNA. III Nucleotide
sequences derived from complementary DNA. J Biol Chem.
252:5040–5053. 1977.PubMed/NCBI
|
|
27
|
Efstratiadis A, Kafatos FC and Maniatis T:
The primary structure of rabbit beta-globin mRNA as determined from
cloned DNA. Cell. 10:571–585. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ullrich A, Shine J, Chirgwin J, Pictet R,
Tischer E, Rutter WJ and Goodman HM: Rat insulin genes:
Construction of plasmids containing the coding sequences. Science.
196:1313–1319. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Casadei R, Strippoli P, D'Addabbo P,
Canaider S, Lenzi L, Vitale L, Giannone S, Frabetti F, Facchin F,
Carinci P, et al: mRNA 5′ region sequence incompleteness: A
potential source of systematic errors in translation initiation
codon assignment in human mRNAs. Gene. 321:185–193. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Harbers M: The current status of cDNA
cloning. Genomics. 91:232–242. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carninci P, Kvam C, Kitamura A, Ohsumi T,
Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, et al:
High-efficiency full-length cDNA cloning by biotinylated CAP
trapper. Genomics. 37:327–336. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kodzius R, Kojima M, Nishiyori H, Nakamura
M, Fukuda S, Tagami M, Sasaki D, Imamura K, Kai C, Harbers M, et
al: CAGE: Cap analysis of gene expression. Nat Methods. 3:211–222.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Frohman MA, Dush MK and Martin GR: Rapid
production of full-length cDNAs from rare transcripts:
Amplification using a single gene-specific oligonucleotide primer.
Proc Natl Acad Sci USA. 85:8998–9002. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Denoeud F, Kapranov P, Ucla C, Frankish A,
Castelo R, Drenkow J, Lagarde J, Alioto T, Manzano C, Chrast J, et
al: Prominent use of distal 5′ transcription start sites and
discovery of a large number of additional exons in ENCODE regions.
Genome Res. 17:746–759. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Suzuki Y, Ishihara D, Sasaki M, Nakagawa
H, Hata H, Tsunoda T, Watanabe M, Komatsu T, Ota T, Isogai T, et
al: Statistical analysis of the 5′ untranslated region of human
mRNA using 'Oligo-Capped' cDNA libraries. Genomics. 64:286–297.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Porcel BM, Delfour O, Castelli V, De
Berardinis V, Friedlander L, Cruaud C, Ureta-Vidal A, Scarpelli C,
Wincker P, Schächter V, et al: Numerous novel annotations of the
human genome sequence supported by a 5′-end-enriched cDNA
collection. Genome Res. 14:463–471. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Metzker ML: Sequencing technologies - the
next generation. Nat Rev Genet. 11:31–46. 2010. View Article : Google Scholar
|
|
38
|
Ingolia NT, Ghaemmaghami S, Newman JR and
Weissman JS: Genome-wide analysis in vivo of translation with
nucleotide resolution using ribosome profiling. Science.
324:218–223. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ingolia NT, Lareau LF and Weissman JS:
Ribosome profiling of mouse embryonic stem cells reveals the
complexity and dynamics of mammalian proteomes. Cell. 147:789–802.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fritsch C, Herrmann A, Nothnagel M,
Szafranski K, Huse K, Schumann F, Schreiber S, Platzer M, Krawczak
M, Hampe J, et al: Genome-wide search for novel human uORFs and
N-terminal protein extensions using ribosomal footprinting. Genome
Res. 22:2208–2218. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Van Damme P, Gawron D, Van Criekinge W and
Menschaert G: N-terminal proteomics and ribosome profiling provide
a comprehensive view of the alternative translation initiation
landscape in mice and men. Mol Cell Proteomics. 13:1245–1261. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Iacono M, Mignone F and Pesole G: uAUG and
uORFs in human and rodent 5′ untranslated mRNAs. Gene. 349:97–105.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Barbosa C, Peixeiro I and Romão L: Gene
expression regulation by upstream open reading frames and human
disease. PLoS Genet. 9:e10035292013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nishitani H, Hirose E, Uchimura Y,
Nakamura M, Umeda M, Nishii K, Mori N and Nishimoto T: Full-sized
RanBPM cDNA encodes a protein possessing a long stretch of proline
and glutamine within the N-terminal region, comprising a large
protein complex. Gene. 272:25–33. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kobayashi A, Ito E, Toki T, Kogame K,
Takahashi S, Igarashi K, Hayashi N and Yamamoto M: Molecular
cloning and functional characterization of a new Cap'n' collar
family transcription factor Nrf3. J Biol Chem. 274:6443–6452. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nomura N, Nagase T, Miyajima N, Sazuka T,
Tanaka A, Sato S, Seki N, Kawarabayasi Y, Ishikawa K and Tabata S:
Prediction of the coding sequences of unidentified human genes. II
The coding sequences of 40 new genes (KIAA0041-KIAA0080) deduced by
analysis of cDNA clones from human cell line KG-1. DNA Res.
1:223–229. 1994. View Article : Google Scholar
|
|
47
|
Kingsley C and Winoto A: Cloning of GT
box-binding proteins: A novel Sp1 multigene family regulating
T-cell receptor gene expression. Mol Cell Biol. 12:4251–4261. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Strippoli P, Pelleri MC, Caracausi M,
Vitale L, Piovesan A, Locatelli C, Mimmi MC, Berardi AC, Ricotta D,
Radeghieri A, et al: An integrated route to identifying new
pathogenesis-based therapeutic approaches for trisomy 21 (Down
Syndrome) following the thought of Jérôme Lejeune. Sci Postprint.
1:e000102013. View Article : Google Scholar
|
|
49
|
Pelleri MC, Cicchini E, Locatelli C,
Vitale L, Caracausi M, Piovesan A, Rocca A, Poletti G, Seri M,
Strippoli P, et al: Systematic reanalysis of partial trisomy 21
cases with or without Down syndrome suggests a small region on
21q22.13 as critical to the phenotype. Hum Mol Genet. 25:2525–2538.
2016.PubMed/NCBI
|
|
50
|
Hattori M, Fujiyama A, Taylor TD, Watanabe
H, Yada T, Park HS, Toyoda A, Ishii K, Totoki Y, Choi DK, et al
Chromosome 21 mapping and sequencing consortium: The DNA sequence
of human chromosome 21. Nature. 405:311–319. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Reymond A, Camargo AA, Deutsch S,
Stevenson BJ, Parmigiani RB, Ucla C, Bettoni F, Rossier C, Lyle R,
Guipponi M, et al: Nineteen additional unpredicted transcripts from
human chromosome 21. Genomics. 79:824–832. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pesole G, Gissi C, Grillo G, Licciulli F,
Liuni S and Saccone C: Analysis of oligonucleotide AUG start codon
context in eukariotic mRNAs. Gene. 261:85–91. 2000. View Article : Google Scholar
|
|
53
|
Frabetti F, Casadei R, Lenzi L, Canaider
S, Vitale L, Facchin F, Carinci P, Zannotti M and Strippoli P:
Systematic analysis of mRNA 5′ coding sequence incompleteness in
Danio rerio: An automated EST-based approach. Biol Direct.
2:342007. View Article : Google Scholar
|
|
54
|
Casadei R, Piovesan A, Vitale L, Facchin
F, Pelleri MC, Canaider S, Bianconi E, Frabetti F and Strippoli P:
Genome-scale analysis of human mRNA 5′ coding sequences based on
expressed sequence tag (EST) database. Genomics. 100:125–130. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Piovesan A, Caracausi M, Pelleri MC,
Vitale L, Martini S, Bassani C, Gurioli A, Casadei R, Soldà G and
Strippoli P: Improving mRNA 5′ coding sequence determination in the
mouse genome. Mamm Genome. 25:149–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kochetov AV, Sarai A, Rogozin IB, Shumny
VK and Kolchanov NA: The role of alternative translation start
sites in the generation of human protein diversity. Mol Genet
Genomics. 273:491–496. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bazykin GA and Kochetov AV: Alternative
translation start sites are conserved in eukaryotic genomes.
Nucleic Acids Res. 39:567–577. 2011. View Article : Google Scholar :
|
|
58
|
Ivanov IP, Firth AE, Michel AM, Atkins JF
and Baranov PV: Identification of evolutionarily conserved
non-AUG-initiated N-terminal extensions in human coding sequences.
Nucleic Acids Res. 39:4220–4234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Arakaki TL, Pezza JA, Cronin MA, Hopkins
CE, Zimmer DB, Tolan DR and Allen KN: Structure of human brain
fructose 1,6-(bis)phosphate aldolase: Linking isozyme structure
with function. Protein Sci. 13:3077–3084. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lamour V, Quevillon S, Diriong S, N'Guyen
VC, Lipinski M and Mirande M: Evolution of the Glx-tRNA synthetase
family: The glutaminyl enzyme as a case of horizontal gene
transfer. Proc Natl Acad Sci USA. 91:8670–8674. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Hermann E, Darcissac E, Idziorek T, Capron
A and Bahr GM: Recombinant interleukin-16 selectively modulates
surface receptor expression and cytokine release in macrophages and
dendritic cells. Immunology. 97:241–248. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Schatz G and Dobberstein B: Common
principles of protein translocation across membranes. Science.
271:1519–1526. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nakamura M, Masuda H, Horii J, Kuma K,
Yokoyama N, Ohba T, Nishitani H, Miyata T, Tanaka M and Nishimoto
T: When overexpressed, a novel centrosomal protein, RanBPM, causes
ectopic microtubule nucleation similar to gamma-tubulin. J Cell
Biol. 143:1041–1052. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Varshavsky A: The N-end rule: Functions,
mysteries, uses. Proc Natl Acad Sci USA. 93:12142–12149. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Rothermel B, Vega RB, Yang J, Wu H,
Bassel-Duby R and Williams RS: A protein encoded within the Down
syndrome critical region is enriched in striated muscles and
inhibits calcineurin signaling. J Biol Chem. 275:8719–8725. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Strippoli P, Petrini M, Lenzi L, Carinci P
and Zannotti M: The murine DSCR1-like (Down syndrome candidate
region 1) gene family: Conserved synteny with the human orthologous
genes. Gene. 257:223–232. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Vitale L, Frabetti F, Huntsman SA,
Canaider S, Casadei R, Lenzi L, Facchin F, Carinci P, Zannotti M,
Coppola D, et al: Sequence, 'subtle' alternative splicing and
expression of the CYYR1 (cysteine/tyrosine-rich 1) mRNA in human
neuroendocrine tumors. BMC Cancer. 7:662007. View Article : Google Scholar
|
|
68
|
Facchin F, Canaider S, Vitale L, Frabetti
F, Griffoni C, Lenzi L, Casadei R and Strippoli P: Identification
and analysis of human RCAN3 (DSCR1L2) mRNA and protein isoforms.
Gene. 407:159–168. 2008. View Article : Google Scholar
|
|
69
|
Facchin F, Vitale L, Bianconi E, Piva F,
Frabetti F, Strippoli P, Casadei R, Pelleri MC, Piovesan A and
Canaider S: Complexity of bidirectional transcription and
alternative splicing at human RCAN3 locus. PLoS One. 6:e245082011.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Casadei R, Pelleri MC, Vitale L, Facchin
F, Canaider S, Strippoli P, Vian M, Piovesan A, Bianconi E, Mariani
E, et al: Characterization of human gene locus CYYR1: A complex
multi-transcript system. Mol Biol Rep. 41:6025–6038. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Nagase T, Seki N, Ishikawa K, Tanaka A and
Nomura N: Prediction of the coding sequences of unidentified human
genes. V The coding sequences of 40 new genes (KIAA0161-KIAA0200)
deduced by analysis of cDNA clones from human cell line KG-1. DNA
Res. 3:17–24. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ribieras S, Lefèbvre O, Tomasetto C and
Rio MC: Mouse Trefoil factor genes: Genomic organization, sequences
and methylation analyses. Gene. 266:67–75. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Doglio L, Goode DK, Pelleri MC, Pauls S,
Frabetti F, Shimeld SM, Vavouri T and Elgar G: Parallel evolution
of chordate cis-regulatory code for development. PLoS Genet.
9:e10039042013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hinnebusch AG, Ivanov IP and Sonenberg N:
Translational control by 5′-untranslated regions of eukaryotic
mRNAs. Science. 352:1413–1416. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Caracausi M, Vitale L, Pelleri MC,
Piovesan A, Bruno S and Strippoli P: A quantitative transcriptome
reference map of the normal human brain. Neurogenetics. 15:267–287.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Pelleri MC, Piovesan A, Caracausi M,
Berardi AC, Vitale L and Strippoli P: Integrated differential
transcriptome maps of Acute Megakaryoblastic Leukemia (AMKL) in
children with or without Down Syndrome (DS). BMC Med Genomics.
7:632014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Manning AG, Crawford BD, Waskiewicz AJ and
Pilgrim DB: unc-119 homolog required for normal development of the
zebrafish nervous system. Genesis. 40:223–230. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Piovesan A, Vitale L, Pelleri MC and
Strippoli P: Universal tight correlation of codon bias and pool of
RNA codons (codonome): The genome is optimized to allow any
distribution of gene expression values in the transcriptome from
bacteria to humans. Genomics. 101:282–289. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Komar AA: The Yin and Yang of codon usage.
Hum Mol Genet. 25(R2): R77–R85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Piovesan A, Caracausi M, Antonaros F,
Pelleri MC and Vitale L: GeneBase 11: A tool to summarise data from
NCBI gene datasets and its application to an update of human gene
statistics. Database (Oxford). 2016. pii: baw153. 2016, View Article : Google Scholar
|
|
81
|
Ahsan B, Saito TL, Hashimoto S, Muramatsu
K, Tsuda M, Sasaki A, Matsushima K, Aigaki T and Morishita S:
MachiBase: A Drosophila melanogaster 5′-end mRNA transcription
database. Nucleic Acids Res. 37(Database): D49–D53. 2009.
View Article : Google Scholar
|
|
82
|
Machida RJ and Lin YY: Four methods of
preparing mRNA 5′ end libraries using the Illumina sequencing
platform. PLoS One. 9:e1018122014. View Article : Google Scholar
|
|
83
|
Helbig AO, Gauci S, Raijmakers R, van
Breukelen B, Slijper M, Mohammed S and Heck AJ: Profiling of
N-acetylated protein termini provides in-depth insights into the
N-terminal nature of the proteome. Mol Cell Proteomics. 9:928–939.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Doucet A and Overall CM: Amino-Terminal
Oriented Mass Spectrometry of Substrates (ATOMS) N-terminal
sequencing of proteins and proteolytic cleavage sites by
quantitative mass spectrometry. Methods Enzymol. 501:275–293. 2011.
View Article : Google Scholar : PubMed/NCBI
|