Introduction
Successful pregnancy requires normal placental
function from adequate placentation and placental vascularization.
It may increase proportionally to the organ volume to maintain the
number of placental vessels throughout the gestation. Therefore,
the study of placental vascularization can enhance our
understanding of the physiopathological process of increased
resistance in umbilical arteries associated with placental
insufficiency. Abnormal placentation causes inadequate
utero-placental blood flow (1),
resulting in a number of pregnancy complications, including
preeclampsia, a leading cause of maternal and perinatal mortality
and morbidity (2), and
intrauterine growth restriction (IUGR) (3,4).
During normal pregnancy, women experience changes in
hormone levels. The levels of both estrogen and progesterone are
increased during pregnancy. The blocking of estrogen production
results in pregnancy loss in animal models, suggesting that
estrogen plays a role in the maintenance of healthy pregnancy. It
has been suggested that in addition to its anti-inflammatory effect
(5), estrogen also stimulates
endothelial cell function and angiogenesis, in particular in vessel
formation [reviewed in (6)].
There is growing evidence to suggest that lower levels of estrogen
and an increase in progesterone are associated with the
pathogenesis of preeclampsia (7–9).
However, whether estrogen plays an important role in the regulation
of vascular growth in the placenta during pregnancy has not yet
been fully investigated.
The biological effects of estrogen are usually
mediated by estrogen receptors (ERs)α and β (10,11). However, previous studies have
suggested that mediating the function of estrogen can be achieved
not only through ERα and ERβ. G-protein-coupled receptor 30
(GPR30), identified as a novel estrogen receptor in 2005 has been
suggested to mediate the action of estrogen (12,13). Recently, we also reported that
GPR30 is involved in the development of preeclampsia (14,15). GPR30 is a specific receptor for
17β-estradiol, a form of estrogens (16), and is expressed in endothelial
cells. It is a regulator of the inflammatory response in
endothelial cells (5).
Abnormal placentation causes placental ischemia and
hypoxia, and it is well known that hypoxia/reoxygenation (H/R) may
be a potential mechanism which contributes to the development of
preeclampsia (17). Therefore, we
undertook this in vitro study to investigate the effects of
estrogen on endothelial cell tube formation, as well as the
potential mechanisms responsible for these effects.
Materials and methods
Reagents
17-β-estradiol (E2; ab120657), a general ER agonist,
and monoclonal anti-GPR30 (ab154069) antibody were purchased from
Abcam (Cambridge, MA, USA). The selective GPR30 agonist, G1, the
selective GPR30 inhibitor, G15, and the specific PI3K inhibitor,
wortmannin, were purchased from Sigma-Aldrich, St. Louis, MO, USA
(G6798, G6748 and W1628, respectively). Anti-endothelial nitric
oxide synthase (eNOS; Cat. no. 5880), monoclonal
anti-phosphorylated (p-)PI3K (p85; Cat. no. 4228), monoclonal
anti-p-eNOS Ser1177 (Cat. no. 9570) and monoclonal
anti-p-Akt (p-Akt Ser473; Cat. no. 4060) antibodies were
purchased from Cell Signaling Technology Inc. (Danvers, MA,
USA).
Cell culture under H/R conditions
Human umbilical vein endothelial cells (HUVECs) were
purchased from Biomics Biotechnologies Co., Ltd. (Nantong, China)
and cultured in Medium-1640 (Gibco Life Technologies, Beijing,
China) with 10% (v/v) fetal bovine serum (FBS) (Gibco Life
Technologies) at 37°C in 5% CO2 in air. The HUVECs were
seeded and grown to 80% confluence under H/R conditions, as
previously described (18–21).
In brief, to decrease the influence of stimulation by serum
mitogens, the HUVECs were incubated for 12 h in low-serum medium
which was supplemented with 1% FBS prior to exposure to H/R. The
HUVECs were then incubated in a hypoxic environment (5%
CO2, 94% N2 and 1% O2) in a
tri-gas cell culture incubator (Thermo Fisher Scientific Oxoid,
Ltd., Basingstoke, UK) for 4 h, and subsequently moved to a
normoxic incubator with 5% CO2 in air with normal
culture medium (10% FBS) for a further 18 h. The oxygen
concentration inside the tri-gas incubator was monitored by an
oxygen analyser (Vascular Technology Inc., Nashua, NH, USA).
In some experiments, the HUVECs were individually
pre-treated with 17-β-estradiol (E2) (100 nM) or G1 (1 mM) or G15
(2 mM) for 1 h (5,22), and then exposed to 4 h of hypoxia
followed by 18 h of reoxygenation.
Immunofluorescence
The immunofluorescence staining of GPR30 and p-eNOS
in the HUVECs was performed as previously described (23). Following treatment, the HUVECs
were fixed in 4% formaldehyde (Aladdin, Shanghai, China) and
blocked with 10% normal goat serum (Sigma-Aldrich), then incubated
with anti-GPR30 antibody (1:80 dilution) or anti-p-eNOS
Ser1177 antibody (1:25 dilution). A fluorescein
isothiocyanate-conjugated goat anti-rabbit antibody (1:50 dilution;
sc-2012; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) was
then used. The nuclei were stained with propidium iodide (3 mg/ml;
Santa Cruz Biotechnology, Inc.). Images were acquired using a
confocal microscope (FV10i; Olympus Corp., Tokyo, Japan).
Western blot analysis
The relative activation levels of eNOS and Akt in
the HUVECs following treatment were measured by western blot
analysis as described (18,24). Briefly, the HUVECs were
homogenised in RIPA buffer (50 mM Tris, 150 mM NaCl, 1% sodium
deoxycholate, 0.1% SDS, 1% Nonident P40 substitute, protease
inhibitor, 1 mM phenylmethanesulfonylfluoride). All the samples (10
µl) were loaded on 6–10% SDS-PAGE gels and electrophoresed
then transferred onto nitrocellulose membranes. Non-specific
binding was blocked by incubating membranes in 5% non-fat milk for
1 h and the membranes were then incubated with monoclonal
anti-GPR30 (1:1,000 dilution) or monoclonal anti-p-PI3K (p85)
(1:500 dilution) or polyclonal anti-PI3K (p85) (1:500 dilution;
sc-292114; Santa Cruz Biotechnology, Inc.) or monoclonal anti-p-Akt
(p-Akt Ser473) (1:1,000 dilution) or polyclonal anti-Akt
(1:500 dilution; sc-5298; Santa Cruz Biotechnology, Inc.) or
monoclonal anti-p-eNOS (Ser1177) (1:1,000 dilution) or
monoclonal anti-eNOS (1:1,000 dilution) antibodies. After washing
with PBS-T, the membranes were incubated with goat anti-mouse
(1:2,000 dilution; ZB-2305; ZSGB-BIO, Beijing, China) or goat
anti-rabbit (1:2,000 dilution; ZB-2301; ZSGB-BIO) secondary
antibodies for 1 h at room temperature. After washing with PBS-T,
the membranes were then incubated with streptavidin-conjugated
horseradish peroxidase (1:3,000) for 1 h at room temperature. After
washing with (PBS-T), the membranes were incubated with Amersham™
ECL™ Prime Western blotting detection reagent. β-actin (1:500
dilution; A1978; Sigma-Aldrich) was used as a loading control.
In vitro tube formation assay
The in vitro tube formation assay in the
HUVECs was performed as previously described (18,25). Matrigel (BD Biosciences, Franklin
Lakes, NJ, USA) was thawed at 4°C overnight and then diluted with
serum-free medium at a ratio of 1:2; the mixture was distributed
into a 96-well plate (65 µl/well) and incubated at 37°C for
2 h. The HUVECs (1.0×104), subjected to a variety of
pre-treatments, were added to wells in triplicate under H/R
conditions (hypoxia for 4 h and reoxygenation for 18 h) or culture
in normal oxygen for 22 h. Following treatment, the cultures were
captured from each well using a microscope (Olympus Corp.) (5 for
each, ×100 magnification). The total pipe length of the tube-like
structure was calculated by using image-Pro Plus software (version
6.0; NIH image). Tracks of HUVECs organised into networks of
cellular cords were counted and averaged in 5 randomly selected
view fields (×100 magnification). The tube formation indexes were
expressed as tube length (mm)/mm2 area.
Statistical analysis
All the experiments in this study were performed at
least 3 times and the data are expressed as the means ± standard
deviation (SD). Statistical analyses were performed using Graph Pad
Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA). Differences
between 2 groups were analysed by independent t-test assuming,
while differences between multi-groups were analysed by one-way
ANOVA. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
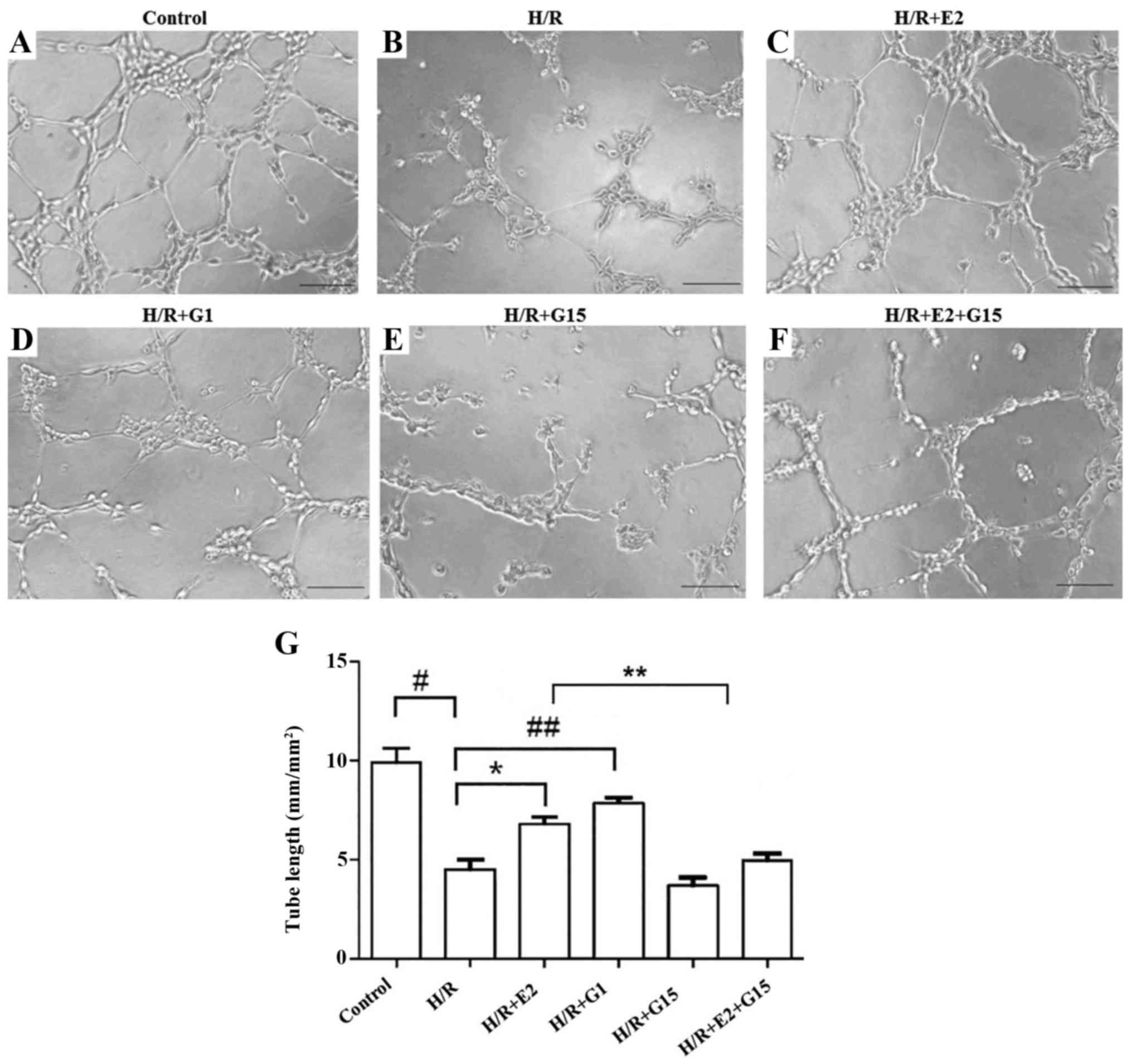
A previous study suggested that estrogen has an
effect on endothelial cell angiogenesis (6). In this study, to further investigate
the effects of estrogen on endothelial cell tube formation, the
total pipe length of the tube-like structure in HUVECs that had
been treated with 17-β-estradiol (E2) or the selective GPR30
agonist, G1, was measured after 22 h of culture under H/R
conditions (Fig. 1A–F). The total
pipe length of the tube-like structure in the HUVECs that had been
cultured under H/R conditions was significantly reduced compared to
the HUVECs that were cultured under normoxic control conditions
(Fig. 1G; P=0.003). This
reduction was reversed by pre-treatment with 17-β-estradiol (E2)
(Fig. 1G; P=0.017) or G1
(Fig. 1G; P=0.003). However, the
protective effects of 17-β-estradiol (E2) on tube formation were
inhibited by G15 (Fig. 1G;
P=0.021).
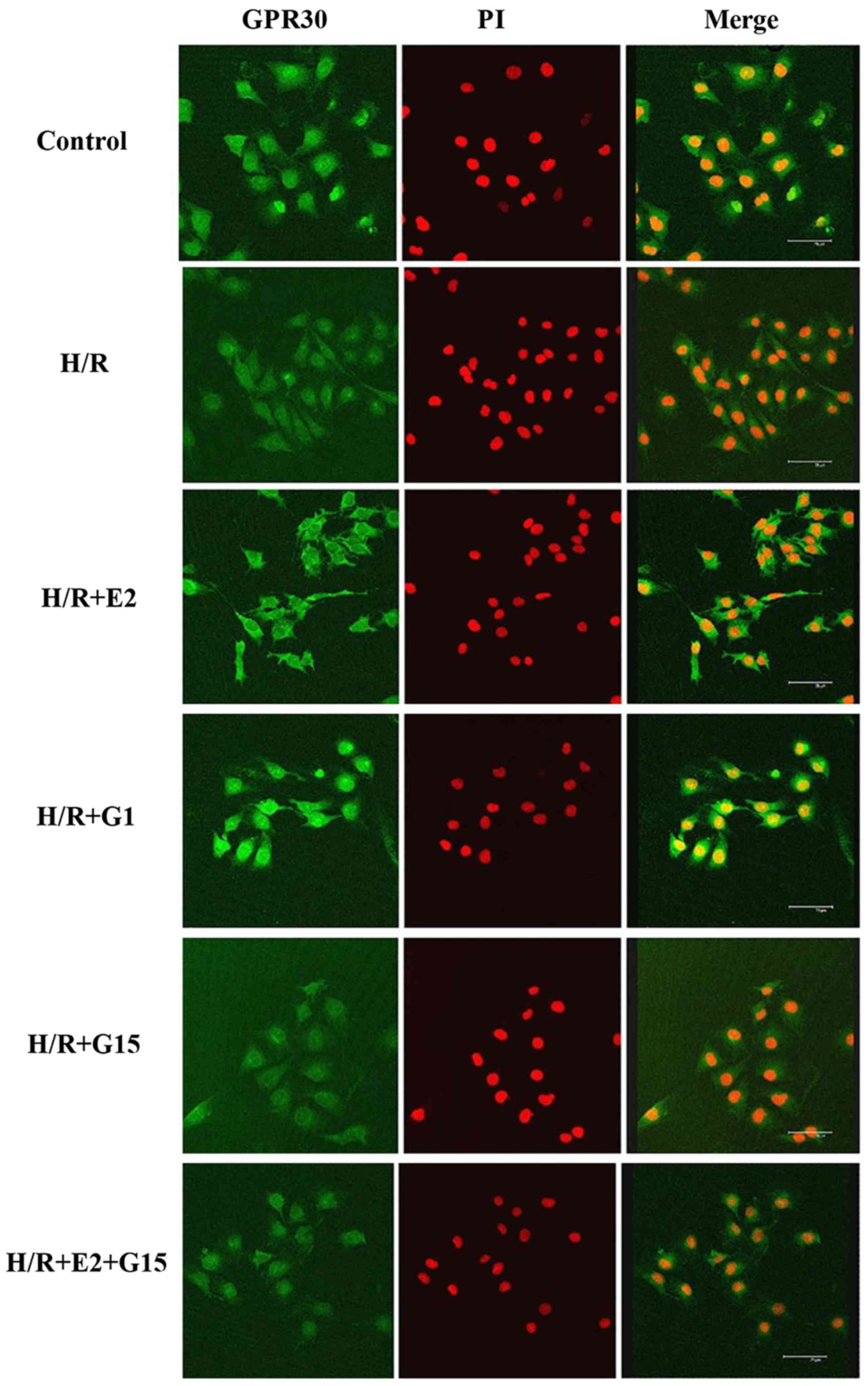
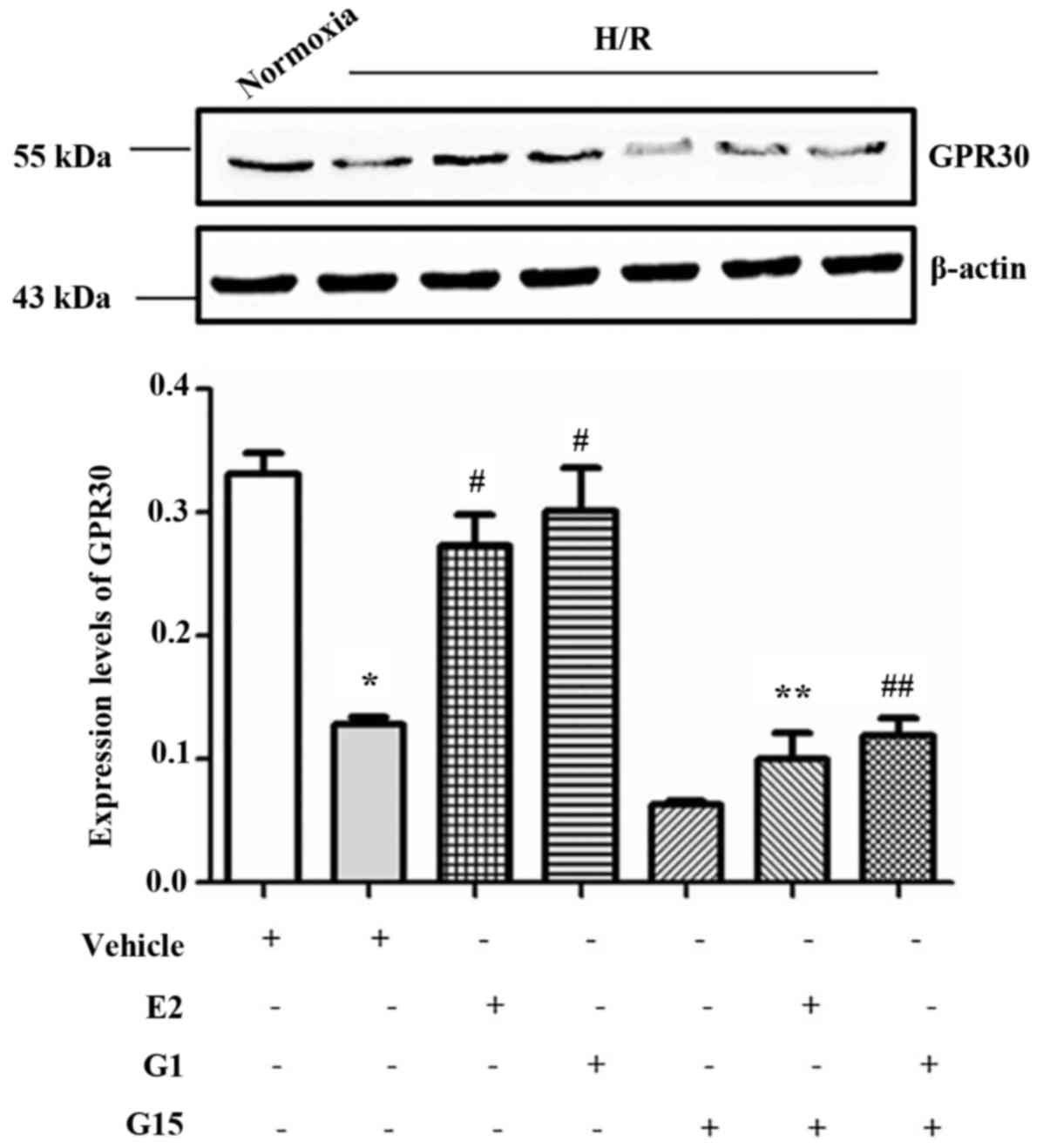
In order to investigate whether GPR30, one of the
estrogen receptors, is involved in the protective effects of
17-β-estradiol (E2) on endothelial cell tube formation, GPR30
expression in HUVECs was measured in the presence or absence of
17-β-estradiol (E2) or G1 in culture under H/R conditions. The
expression of GPR30 was significantly decreased under H/R
conditions compared with that under normoxic conditions (Fig. 2). However, this reduction of GPR30
expression in the HUVECs induced by H/R conditions was
significantly reversed when the cells were pre-treated with
17-β-estradiol (E2) or G1 (Fig.
2). Howeveer, the effects of 17-β-estradiol (E2) and G1 were
inhibited by treatment with the specific GPR30 inhibitor, G15
(Fig. 2). The decrease in GPR30
expression in the HUVECs was also confirmed by western blot
analysis (Fig. 3;
P<0.001).
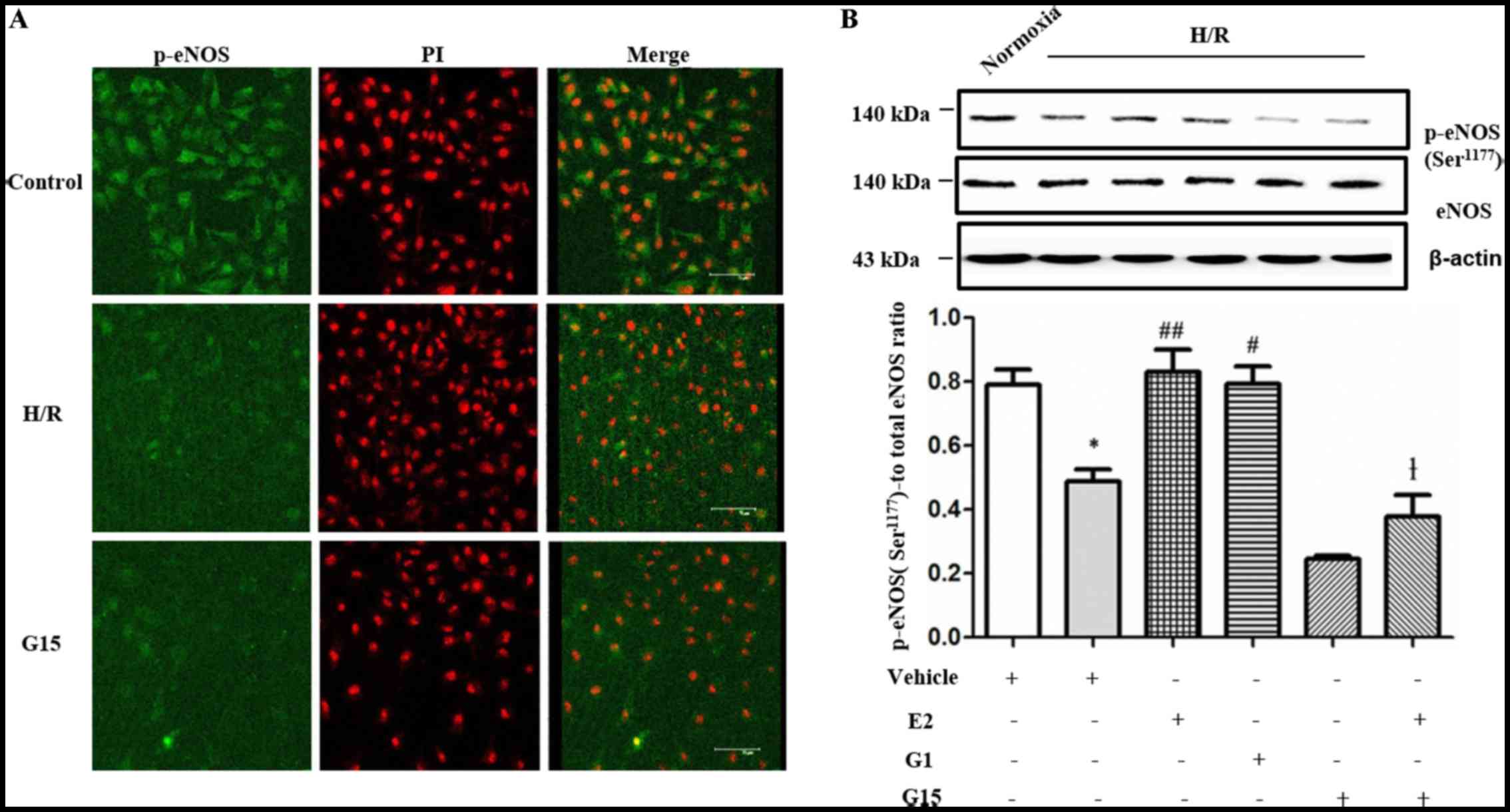
It has previously been suggested that GPR30
activates eNOS. The activation of eNOS is involved in angiogenesis
(26–28). In this study, we further
investigated whether eNOS is involved in tube formation of
endothelial cells in vitro under H/R conditions.
Immunofluorescence revealed that the levels of p-eNOS
(Ser1177) in the HUVECs were decreased under H/R
conditions and following treatment with G15 compared with the
control group (Fig. 4A). The
decrease in the expression of p-eNOS at Ser1177 in the
HUVECs under H/R conditions was also confirmed by western blot
analysis (Fig. 4B; P=0.0132).
However, the decrease in p-eNOS expression was reversed by
pre-treatment with 17-β-estradiol (E2) (Fig. 4B; P=0.0052) or the selective GPR30
agonist, G1 (Fig. 4B; P=0.0123).
However, the effects of 17-β-estradiol (E2) were inhibited by
treatment with G15 (Fig. 4B;
P=0.0005).
We further investigated the estrogen-mediated
phosphorylation of PI3K (p85) and Akt (Ser473) in the
HUVECs under H/R conditions. The expression of p-PI3K (p85) and
p-Akt (Ser473) in the HUVECs was significantly increased
by pre-treatment with either G1 (Fig.
5A and B; P=0.0001 and P=0.0027, respectively) or
17-β-estradiol (E2) (Fig. 5A and
B; P=0.0001). This effect of 17-β-estradiol (E2) was inhibited
by treatment with G15 (Fig. 5A and
B; P<0.0001 and P=0.0098).
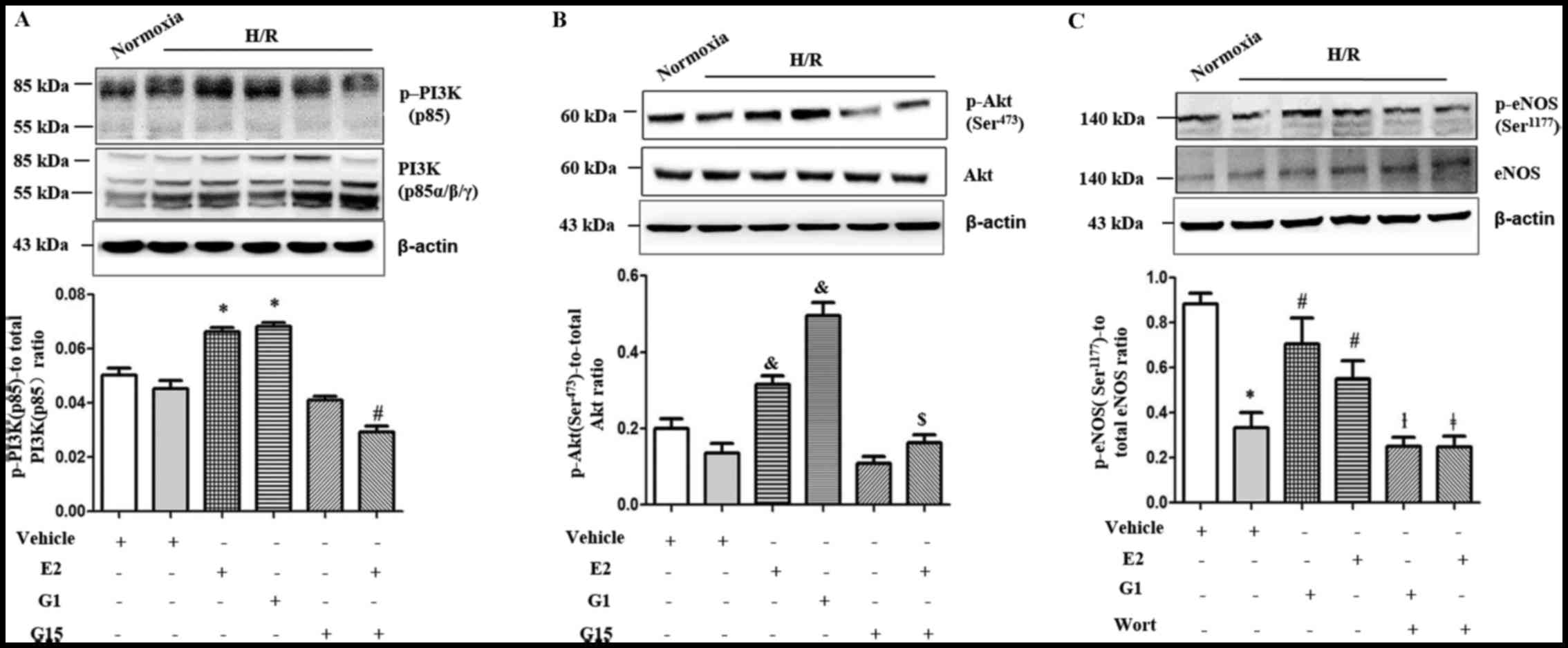 | Figure 5Western blot analysis demonstraing
that the levels of (A) p-PI3K (p85) and (B) p-Akt
(Ser473) were significantly increased in the presence of
E2 (A and B, *P=0.0001 and &P=0.0001,
respectively) or G1 (A and B, *P=0.0001 and
&P= 0.0027, respectively) in human umbilical vein
endothelial cells (HUVECs) under hypoxia-reoxygenation (H/R)
conditions. The effect was blocked by treatment with G15 (A and B,
#P<0.0001 and $P=0.0098, respectively).
Western blot analysis also demonstrated that the levels of (C)
p-eNOS (Ser1177) were significantly decreased under H/R
conditions in HUVECs compared to normoxic conditions
(*P= 0.0111). This decrease was significantly increased
in the presence of E2 or G1 in HUVECs under H/R conditions
(#P<0.0043 and P= 0.0104). This effect was
significantly blocked by treatment with wortmannin (Wort, 100 nM)
(**P<0.0001, ##P<0.0001).
eNOS, endothelial nitric oxide synthase; E2, 17-β-estradiol. |
We then investigated whether the PI3K/Akt signalling
pathway mediates GPR30-dependent eNOS activation in HUVECs under
H/R conditions. The effects of 17-β-estradiol (E2) and G1 on eNOS
phosphorylation in the HUVECs were blocked by the PI3K inhibitor,
wortmannin (Wort, 100 nM) (Fig.
5C; P<0.0001).
Discussion
During human pregnancy, the placenta is supplied
with maternal blood via the uterine spiral arteries. The fetus
requires an increasing supply of oxygen and nutrients, suggesting
that uterine spiral artery remodelling is necessary for a
successful pregnancy. However, the failure of spiral artery
remodelling impacts the oxygen concentration analogous to
hypoxia-reperfusion within the placental environment. A decreased
feto-placental perfusion and restricted oxygen delivery causes
placental insufficiency, resulting in a number of complications of
pregnancy, such as preeclampsia and IUGR. H/R is a secondary to
intermittent perfusion of the intervillous space and plays an
important role in placental development (17). Studies have suggested that H/R
causes apoptotic changes in syncytiotrophoblasts, which is another
characteristic feature of the preeclamptic placenta (29,30). During normal pregnancy, women
experience changes in hormone levels and the levels of both
estrogen and progesterone are increased. Lower levels of estrogen
have been reported in women with preeclampsia (7–9).
However, whether estrogen has an effect on placental vasculature is
unknown.
Placental vasculature expands in both maternal and
fetal placental tissue during pregnancy (31). Endothelial cell tube formation
assay is a common tool to study angiogenesis. In this in
vitro study, we found that the supplementation of
17-β-estradiol (E2), a form of estrogen reversed the failure of
endothelial cell tube formation induced by H/R. Another study
demonstrated that estrogen, such as 17-β-estradiol (E2) plays a
role in the modulation of endothelial cell function and in the
promotion of angiogenesis (6).
That study also suggested that the effect of 17-β-estradiol (E2) on
angiogenesis may also apply to in vivo (6). A recent study further suggested that
17-β-estradiol (E2) was able to protect cardiomyocytes against H/R
injury (32). To understand the
specific mechanisms of estrogen by which receptors improve
endothelial cell tube formation, in this in vitro study, we
found that the expression of GPR30, a novel estrogen receptor was
significantly reduced in endothelial cells under H/R conditions.
However, this decreased expression of GPR30 was reversed by the
supplementation of 17-β-estradiol (E2) or a selective GPR30 agonist
(G1) in endothelial cells under H/R conditions. GPR30 is associated
with the protective effects of estrogen in breast cancer (12,13). A recent study also suggested that
the GPR30 agonist, G1, improves cerebral microvascular function
following H/R injury in animal model (33). Taken together, our data suggest
that 17-β-estradiol (E2) may have a similar function as the GPR30
agonist, G1. 17-β-estradiol (E2) is involved in the regulation of
endothelial cell tube formation in H/R injury and its protective
effects are associated with estrogen receptor, GPR30, in
endothelial cells.
Reactive oxygen species (ROS) play an important role
in endothelial cell dysfunction (34) and multiple ROS systems are
activated during H/R, including NOS (35). A number of studies have suggested
that the activation of eNOS is involved in angiogenesis (26,36). Endogenous estrogens mediate
protective effects at least partially due to the activation of eNOS
and the bioactivity of NO is regulated by GPR30 in the
cardiovascular system. In addition, the protective effects of the
GPR30 agonist, G1, in endothelial cells following H/R injury, at
least partially also depends on eNOS (33). In this in vitro study,
consistent with other studies, we found that the decreased
expression of eNOS induced by H/R was reversed by the addition of
17-β-estradiol (E2) or the GPR30 agonist, G1, in endothelial cell
culture.
A previous study suggested that in addition to the
association with the expression of eNOS, GPR30 also mediated the
activation of Akt in endothelial cells (37) and is involved in cell
proliferation (38). In this
in vitro study, we also found that the expression of Akt was
decreased by H/R in endothelial cells; however, this decrease in
Akt expression was reversed by supplementation with either
17-β-estradiol (E2) or the GPR30 agonist, G1. We further found that
the activation of eNOS induced by 17-β-estradiol (E2) or the GPR30
agonist, G1, was inhibited by an inhibitor of PI3K/Akt
(wortmannin), suggesting the activation of eNOS induced by
17-β-estradiol (E2) is Akt-dependent in endothelial cells under H/R
conditions. Taken together our data suggest that both eNOS and
PI3K/Akt activation are involved in the prevention of endothelial
cell tube formation by 17-β-estradiol (E2), and the PI3K/Akt
signalling pathway regulates the activation of eNOS in the
prevention of endothelial cell tube formation by 17-β-estradiol
(E2).
In conclusion, to the best of our knowledge, in the
present in vitro study, we report for the first time that
the supplementation of estrogen 17-β-estradiol (E2) may prevent the
failure of endothelial cell tube formation induced by H/R. The
estrogen receptor, GPR30, is at least partially involved in this
protective effect through the activation of eNOS and the Akt
signalling pathway in endothelial cells. Lower levels of estrogen
are reported in complications of pregnancy, such as preeclampsia.
Therefore, our data suggest that increased levels of estrogen
during normal pregnancy promote placental vasculature development,
thus exerting positive effects.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (nos. 81370732 and 81571453).
References
|
1
|
Khong Y and Brosens I: Defective deep
placentation. Best Pract Res Clin Obstet Gynaecol. 25:301–311.
2011. View Article : Google Scholar
|
|
2
|
Sibai B, Dekker G and Kupferminc M:
Preeclampsia. Lancet. 365:785–799. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Roberts JM and Hubel CA: Is oxidative
stress the link in the two-stage model of preeclampsia? Lancet.
354:788–789. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hubel CA: Oxidative stress in the
pathogenesis of preeclampsia. Proc Soc Exp Biol Med. 222:222–235.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chakrabarti S and Davidge ST: G-protein
coupled receptor 30 (GPR30): A novel regulator of endothelial
inflammation. PLoS One. 7:e523572012. View Article : Google Scholar
|
|
6
|
Rubanyi GM, Johns A and Kauser K: Effect
of estrogen on endothelial function and angiogenesis. Vascul
Pharmacol. 38:89–98. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zeisler H, Jirecek S, Hohlagschwandtner M,
Knöfler M, Tempfer C and Livingston JC: Concentrations of estrogens
in patients with preeclampsia. Wien Klin Wochenschr. 114:458–461.
2002.PubMed/NCBI
|
|
8
|
Tamimi R, Lagiou P, Vatten LJ, Mucci L,
Trichopoulos D, Hellerstein S, Ekbom A, Adami HO and Hsieh CC:
Pregnancy hormones, preeclampsia, and implications for breast
cancer risk in the offspring. Cancer Epidemiol Biomarkers Prev.
12:647–650. 2003.PubMed/NCBI
|
|
9
|
Jobe SO, Tyler CT and Magness RR: Aberrant
synthesis, metabolism, and plasma accumulation of circulating
estrogens and estrogen metabolites in preeclampsia implications for
vascular dysfunction. Hypertension. 61:480–487. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Molvarec A, Vér A, Fekete A, Rosta K,
Derzbach L, Derzsy Z, Karádi I and Rigó J Jr: Association between
estrogen receptor alpha (ESR1) gene polymorphisms and severe
preeclampsia. Hypertens Res. 30:205–211. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maruyama A, Nakayama T, Sato N, Mizutani
Y, Furuya K and Yamamoto T: Association study using single
nucleotide polymorphisms in the estrogen receptor beta (ESR2) gene
for preeclampsia. Hypertens Res. 27:903–909. 2004. View Article : Google Scholar
|
|
12
|
Thomas P, Pang Y, Filardo EJ and Dong J:
Identity of an estrogen membrane receptor coupled to a G protein in
human breast cancer cells. Endocrinology. 146:624–632. 2005.
View Article : Google Scholar
|
|
13
|
Revankar CM, Cimino DF, Sklar LA,
Arterburn JB and Prossnitz ER: A transmembrane intracellular
estrogen receptor mediates rapid cell signaling. Science.
307:1625–1630. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tong C, Feng X, Chen J, Qi X, Zhou L, Shi
S, Kc K, Stanley JL, Baker PN and Zhang H: G protein-coupled
receptor 30 regulates trophoblast invasion and its deficiency is
associated with preeclampsia. J Hypertens. 34:710–718. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Chen Z, Zhou X, Shi S, Qi H, Baker
PN and Zhang H: Imbalance between proliferation and
apoptosis-related impaired GPR30 expression is involved in
preeclampsia. Cell Tissue Res. 366:499–508. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Prossnitz ER, Arterburn JB and Sklar LA:
GPR30: A G protein-coupled receptor for estrogen. Mol Cell
Endocrinol. 265–266:138–142. 2007. View Article : Google Scholar
|
|
17
|
Hung TH, Skepper JN, Charnock-Jones DS and
Burton GJ: Hypoxia-reoxygenation: A potent inducer of apoptotic
changes in the human placenta and possible etiological factor in
preeclampsia. Circ Res. 90:1274–1281. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo X, Yao ZW, Qi HB, Liu DD, Chen GQ,
Huang S and Li QS: Gadd45α as an upstream signaling molecule of p38
MAPK triggers oxidative stress-induced sFlt-1 and sEng upregulation
in preeclampsia. Cell Tissue Res. 344:551–565. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dhar-Mascareño M, Cárcamo JM and Golde DW:
Hypoxia-reoxygenation-induced mitochondrial damage and apoptosis in
human endothelial cells are inhibited by vitamin C. Free Radic Biol
Med. 38:1311–1322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee SR and Lo EH: Interactions between p38
mitogen-activated protein kinase and caspase-3 in cerebral
endothelial cell death after hypoxia-reoxygenation. Stroke.
34:2704–2709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee SR and Lo EH: Induction of
caspase-mediated cell death by matrix metalloproteinases in
cerebral endothelial cells after hypoxia-reoxygenation. J Cereb
Blood Flow Metab. 24:720–727. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tian R, Wang Z, Shi Z, Li D, Wang Y, Zhu
Y, Lin W, Gui Y and Zheng XL: Differential expression of
G-protein-coupled estrogen receptor-30 in human myometrial and
uterine leiomyoma smooth muscle. Fertil Steril. 99:256–263. 2013.
View Article : Google Scholar
|
|
23
|
Otto C, Rohde-Schulz B, Schwarz G, Fuchs
I, Klewer M, Brittain D, Langer G, Bader B, Prelle K, Nubbemeyer R,
et al: G protein-coupled receptor 30 localizes to the endoplasmic
reticulum and is not activated by estradiol. Endocrinology.
149:4846–4856. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang Z, Bai B, Luo X, Xiao X, Liu X, Ding
Y, Zhang H, Gao L, Li J and Qi H: Downregulated Krüppel-like factor
8 is involved in decreased trophoblast invasion under
hypoxia-reoxygenation conditions. Reprod Sci. 21:72–81. 2014.
View Article : Google Scholar :
|
|
25
|
Marconcini L, Marchiò S, Morbidelli L,
Cartocci E, Albini A, Ziche M, Bussolino F and Oliviero S:
c-fos-induced growth factor/vascular endothelial growth factor D
induces angiogenesis in vivo and in vitro. Proc Natl Acad Sci USA.
96:9671–9676. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim KM, Kim NS, Kim J, Park JS, Yi JM, Lee
J and Bang OS: Magnolol suppresses vascular endothelial growth
factor-induced angiogenesis by inhibiting Ras-dependent
mitogen-activated protein kinase and phosphatidylinositol
3-kinase/Akt signaling pathways. Nutr Cancer. 65:1245–1253. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Batenburg WW, Jansen PM, van den Bogaerdt
AJ and J Danser AH: Angiotensin II-aldosterone interaction in human
coronary microarteries involves GPR30, EGFR, and endothelial NO
synthase. Cardiovasc Res. 94:136–143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li ZL, Liu JC, Liu SB, Li XQ, Yi DH and
Zhao MG: Improvement of vascular function by acute and chronic
treatment with the GPR30 agonist G1 in experimental diabetes
mellitus. PLoS One. 7:e387872012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Redman CW and Sargent IL: Placental
debris, oxidative stress and preeclampsia. Placenta. 21:597–602.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Allaire AD, Ballenger KA, Wells SR,
McMahon MJ and Lessey BA: Placental apoptosis in preeclampsia.
Obstet Gynecol. 96:271–276. 2000.PubMed/NCBI
|
|
31
|
Burton GJ, Charnock-Jones DS and Jauniaux
E: Regulation of vascular growth and function in the human
placenta. Reproduction. 138:895–902. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cong B, Xu Y, Sheng H, Zhu X, Wang L, Zhao
W, Tang Z, Lu J and Ni X: Cardioprotection of 17β-estradiol against
hypoxia/reoxygenation in cardiomyocytes is partly through
up-regulation of CRH receptor type 2. Mol Cell Endocrinol.
382:17–25. 2014. View Article : Google Scholar
|
|
33
|
Murata T, Dietrich HH, Xiang C and Dacey
RG Jr: G protein-coupled estrogen receptor agonist improves
cerebral microvascular function after hypoxia/reoxygenation injury
in male and female rats. Stroke. 44:779–785. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Granger DN, Kvietys PR and Perry MA:
Leukocyte - endothelial cell adhesion induced by ischemia and
reperfusion. Can J Physiol Pharmacol. 71:67–75. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zulueta JJ, Sawhney R, Yu FS, Cote CC and
Hassoun PM: Intracellular generation of reactive oxygen species in
endothelial cells exposed to anoxia-reoxygenation. Am J Physiol.
272:L897–L902. 1997.PubMed/NCBI
|
|
36
|
Song Y, Zhao XP, Song K and Shang ZJ:
Ephrin-A1 is up-regulated by hypoxia in cancer cells and promotes
angiogenesis of HUVECs through a coordinated cross-talk with eNOS.
PLoS One. 8:e744642013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rowlands DJ, Chapple S, Siow RC and Mann
GE: Equol-stimulated mitochondrial reactive oxygen species activate
endothelial nitric oxide synthase and redox signaling in
endothelial cells: Roles for F-actin and GPR30. Hypertension.
57:833–840. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kolkova Z, Casslén V, Henic E, Ahmadi S,
Ehinger A, Jirström K and Casslén B: The G protein-coupled estrogen
receptor 1 (GPER/GPR30) does not predict survival in patients with
ovarian cancer. J Ovarian Res. 5:92012. View Article : Google Scholar : PubMed/NCBI
|