Introduction
Tumor metastasis is a major clinical challenge that
accounts for the vast majority of cancer-related deaths. Breast
cancer patients have high rates of development of metastatic
disease even following successful primary tumor resection and
adjuvant therapy. An estimated 30% of node-negative breast cancer
patients and a larger percentage of node-positive breast cancer
patients develop metastatic disease despite receiving standard
therapy (1). Approximately 90% of
breast cancer deaths are caused by the local invasion and distant
metastasis of tumor cells (2).
The path of metastatic colonization is a complex and multi-faceted
process. To develop metastasis, primary cancer cells must invade
and escape the physical barriers at the primary site.
Epithelial-mesenchymal transition (EMT) is a process
whereby cancer cells lose their epithelial properties to acquire a
mesenchymal phenotype and become motile and invasive, which is
linked to metastasis (3–5). The expression of the inter-cellular
epithelial adhesion molecule E-cadherin is decreased, and markers
of mesenchymal cells, such as α-smooth muscle actin (α-SMA),
N-cadherin and vimentin, are upregulated during EMT (3). EMT has also been connected to the
induction of cancer stem cells and drug resistance, suggesting that
EMT may underlie many biological processes of cancer development
(4). Transforming growth factor-β
(TGF-β) is secreted by many cell types and directly stimulates the
production of the extracellular matrix and microenvironment by both
normal and cancer cells (6). As
tumors progress, TGF-β induces neoplastic cell invasiveness and
metastasis by promoting EMT in many cancer cell types (7). Reactive oxygen species (ROS) are
radicals, ions or molecules that have a single unpaired electron in
their outermost shell and are constantly generated inside cells by
dedicated enzyme complexes or as by-products of redox reactions
(8). A recent study suggested
that ROS play an important role in TGF-β-induced EMT. A significant
increase was found to occur in intracellular ROS upon TGF-β
stimulation (9). The release of
TGF-β-dependent ROS is responsible for the phosphorylation of
Smad2, p38 MAPK and extracellular signal-regulated kinase 1/2
(ERK1/2) and accounts for the upregulation of α-SMA and fibronectin
and the repression of E-cadherin (9).
The cellular defense system against oxidative stress
is composed of a subset of antioxidant proteins. Heme oxygenase-1
(HMOX-1) is a microsomal enzyme that is induced in response to
cellular stress and diverse oxidative stimuli (10). The enzymatic activity of HMOX-1
produces CO, ferrous iron and biliverdin. Therefore, HMOX-1 can
reduce oxidative stress, attenuate inflammatory responses and lower
the rate of apoptosis (10).
HMOX-1 can also inhibit the migration and invasion of prostate
cancer cells and renal tubular epithelial cells (11). Additionally, the biological
activity of HMOX-1 reduces ROS generation (10). However, the effect of HMOX-1 on
EMT, which plays a critical role in the metastasis of breast
cancer, requires further research.
In this study, we found that the HMOX-1 inducer
hemin inhibited ROS production in the MCF-7 breast cancer cell
line. Furthermore, we observed that hemin inhibited the migration,
invasion and EMT of MCF-7 breast cancer cells. These results show
that HMOX-1 may function as an important player in breast cancer
metastasis.
Materials and methods
Cell culture
The MCF-7 human breast cancer cell line was
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1%
penicillin and 1% streptomycin (both from Gibco, Karlsruhe,
Germany) in a 5% CO2 atmosphere at 37°C.
Transfection of siRNAs
The HMOX-1 siRNA was synthesized by RiboBio
Biotechnology (Guangzhou, China). The HMOX-1 siRNA sequences were
sense, 5′-CCAGCAACAAAGUGCA AGAdTdT-3′ and antisense,
3′-dTdTGGUCGUUGUUUCACG UUCU-5′. Breast cancer cells were
transfected with 50 nM of siRNA for 8 h using the RNAiMAX
transfection agent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions.
Reverse transcription-quantitative
(real-time) polymerase chain reaction (RT-qPCR) for mRNA
quantification
Total RNA was extracted from the cells using TRIzol
(Invitrogen), and cDNA was synthesized from 1,000 ng of total RNA
using the PrimeScript RT reagent kit (Takara, Dalian, China)
following the manufacturer's instructions. Quantitative PCR was
performed on the Bio-Rad CFX 96 real-time PCR machine (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) using SYBR®
Premix Ex Taq™ II (Tli RNaseH Plus) (Takara). The primer sequences
were as follows: HMOX-1-F, 5′-TACCACATCTAT GTGGCCCTG-3′ and
HMOX-1-R, 5′-TGGCTGGTGTGTA GGG GAT-3′, and glyceraldehyde
3-phosphate dehydrogenase (GAPDH)-F,
5′-GCACCGTCAAGGCTGAGAAC-3′ and GAPDH-R,
5′-TGGTGAAGACGCCAGTGGA-3′. Data analysis was performed using the
comparative Ct method with the Bio-Rad Manager 2.1 software
(Bio-Rad Laboratories, Inc.).
Western blotting
The cells were lysed in RIPA lysis buffer (Cell
Signaling Technology, Boston, MA, USA) supplemented with protease
inhibitor (Roche, Basel, Switzerland). The total protein
concentration was determined using a BCA kit (Keygen, Nanjing,
China). Equal amounts of protein (35 μg) for each group were
separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel
electrophoresis, transferred to polyvinylidene difluoride (PVDF)
membranes (Millipore, Bedford, MA, USA), and blotted using
anti-HMOX-1 (ab52947; Abcam, Cambridge, UK), anti-E-cadherin
(#3195), anti-vimentin (#5741) or anti-β-tubulin (#2128) antibodies
(Cell Signaling Technology). The bands were visualized using the
Luminol reagent (Thermo Pierce, Waltham, MA, USA) and imaged using
the GE ImageQuant Las 4000 Mini (GE Healthcare, Fairfield, CT,
USA).
Migration and invasion assays of MCF-7
breast cancer cells
The migratory ability of the MCF-7 cells was
determined using a wound-healing assay. Briefly, MCF-7 cells were
treated with hemin (20 μM; Sigma-Aldrich, St. Louis, MO,
USA) for 72 h and then seeded into 6-well plates (60,000
cells/well). When the cells were almost 100% confluent, the
monolayer was wounded using a sterilized 200-μl disposable
pipette tip to scratch a wound in each well. Then, the cells were
treated with TGF-β1 (10 ng/ml; Peprotech, Rocky Hill, NJ, USA). The
scratch wounds were visualized under a microscope (Zeiss Axio
Observer Z1; Zeiss, Jena, Germany).
The cell invasion assay was performed in the BD
BioCoat™ Matrigel™ Invasion Chamber (8-μm pore size) (BD
Bioscience, Franklin Lakes, NJ, USA). MCF-7 cells were treated with
hemin (20 μM) for 72 h. Then, the cells were added to the
upper chambers with 200 μl of serum-free DMEM medium, and
the lower chambers were filled with 500 μl of DMEM medium
supplemented with TGF-β1 (10 ng/ml). After 12 h, non-migrating
cells were removed from the upper chamber, and the migrating cells
adhering to the lower surface of the membrane were fixed with 4%
formaldehyde and quantified by 0.1% crystal violet staining.
Immunofluorescence assay
The cells were seeded into 24-well plates and
treated with TGF-β1 (10 ng/ml) for 5 days. The cells were washed in
phosphate-buffered saline (PBS), fixed in 4% formaldehyde for 15
min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked
with 0.1% BSA for 1 h. Then, the cells were incubated with an
anti-E-cadherin (#3195) or anti-vimentin (#5741) antibody (Cell
Signaling Technology) overnight at 4°C. After washing with PBS 3
times, the cells were incubated with a fluorescent-conjugated
secondary antibody (red, A11008; green, A11010; Life Technologies,
Grand Island, NY, USA) for 1 h at room temperature and stained with
4′,6-diamidino-2-phenylindole (DAPI; Roche) for 10 min. Images were
acquired using a fluorescence microscope (Carl Zeiss Axio Observer
Z1, Jena, Germany).
Fluorescent ROS assay
ROS generation was analyzed after staining the cells
with the fluorescent probe dichlorofluorescein-DA (DCFDA)
(Sigma-Aldrich). The cells were incubated with or without hemin
following TGF-β1 treatment. Then, the cells were loaded with DCFDA
(20 mM) in Hank's Balanced Salt Solution (HBSS; Gibco, Karlsruhe,
Germany) at 37°C for 30 min in the dark. After washing with HBSS,
the DCFDA fluorescence of each well was measured at 485 nm
(excitation) and 528 nm (emission) with a Multi-Mode Microplate
Reader (BioTek, Winooski, VT, USA).
Statistical analysis
The data are expressed as the mean ± SD. Differences
between the treatment groups and the control group were assessed
with Student's t-test using GraphPad Prism version 5.0 (GraphPad,
San Diego, CA, USA). Statistically significant differences are
indicated as P<0.05, P<0.01 and P<0.001.
Results
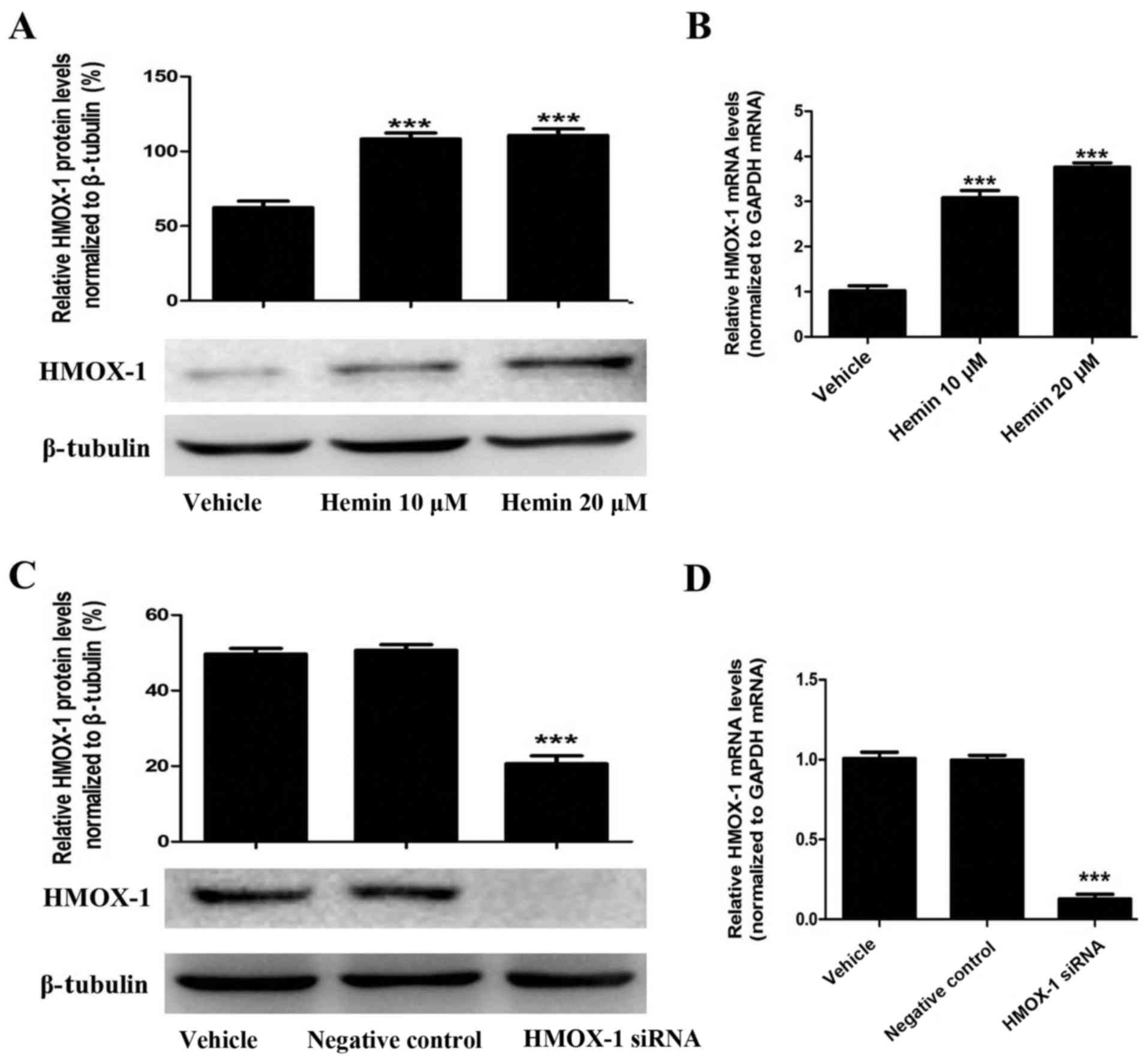
Hemin induces HMOX-1 expression in the
MCF-7 breast cancer cell line
To evaluate the effect of HMOX-1 on TGF-β-induced
EMT, we treated MCF-7 cells with an HMOX-1 inducer (hemin, 20
μM) and then examined whether hemin induced HMOX-1
expression using RT-qPCR and western blotting. Hemin treatment
significantly upregulated HMOX-1 mRNA and protein expression in the
MCF-7 cells (Fig. 1A and B).
Then, we confirmed that the HMOX-1 siRNA knocked down hemin-induced
HMOX-1 expression by RT-qPCR and western blotting (Fig. 1C and D).
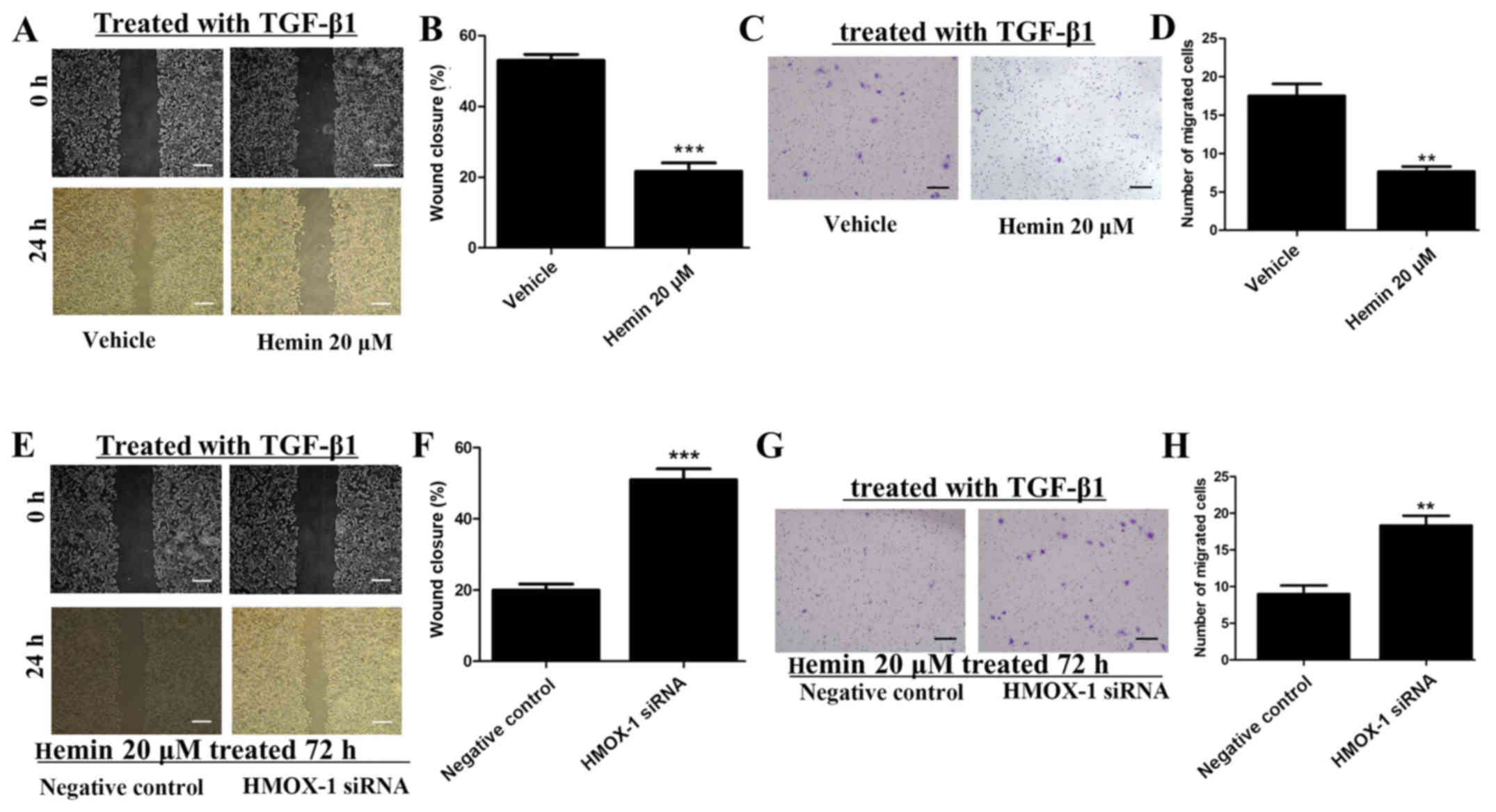
Hemin inhibits the migration and invasion
of TGF-β-treated MCF-7 cells
Wound-healing and cell invasion assays were
performed to investigate the effect of HMOX-1 on the migration and
invasion of TGF-β1-treated MCF-7 cells after incubating the cells
with or without 20 μM of hemin. Hemin significantly
inhibited the migratory ability of the MCF-7 cells in the
wound-healing assay (Fig. 2A and
B). Additionally, upregulation of HMOX-1 by hemin significantly
inhibited the invasiveness of the MCF-7 cells (Fig. 2C and D). MCF-7 cells were treated
with hemin (20 μM) for 72 h and then transfected with the
HMOX-1 siRNA or a negative control. Then, we induced the migration
and invasion of the MCF-7 cells with TGF-β1 (10 ng/ml). The HMOX-1
siRNA attenuated the inhibitory effect of hemin on the MCF-7 cells
and promoted the migration (Fig. 2E
and F) and invasion (Fig. 2G and
H) of the hemin-treated MCF-7 cells.
Hemin inhibits the TGF-β-induced EMT in
MCF-7 cells
The morphological changes characteristic of cells
undergoing EMT are accompanied by a shift in gene expression from
an epithelial to a mesenchymal repertoire. To determine whether
HMOX-1 inhibits this shift, both the expression and cellular
distribution of selected EMT markers were investigated by
immunofluorescence staining and western blotting in the
TGF-β1-treated MCF-7 cells. The MCF-7 cells exhibited decreased
E-cadherin expression and higher expression of the mesenchymal
marker vimentin after TGF-β1 treatment (10 ng/ml) (Fig. 3B–D). These changes were reversed
by sequentially treating the cells with 20 μM of hemin.
These results implied that HMOX-1 inhibited the EMT progression in
MCF-7 cells. Our data showed that HMOX-1 prevented EMT changes in
the MCF-7 cells. To confirm this finding, we performed a
loss-of-function study in which the HMOX-1 siRNA was used to verify
the effect of HMOX-1 on EMT. After exposure to hemin (20 μM)
for 72 h, the cells were transfected with the HMOX-1 siRNA or a
negative control RNA and then treated with TGF-β1 (10 ng/ml) for 5
days and subjected to western blotting and immunofluorescence
assays. The results of both the western blotting and
immunofluorescence assays showed that hemin inhibited the
TGF-β-induced EMT in MCF-7 cells through the upregulation of
E-cadherin and downregulation of vimentin. In contrast, diminished
E-cadherin expression and increased vimentin expression were
observed in the HMOX-1 siRNA-treated cells compared to the vehicle
group (Fig. 3F–H). These results
indicated that the HMOX-1 siRNA attenuated the inhibitory effect of
hemin on the TGF-β-induced EMT in the MCF-7 cells.
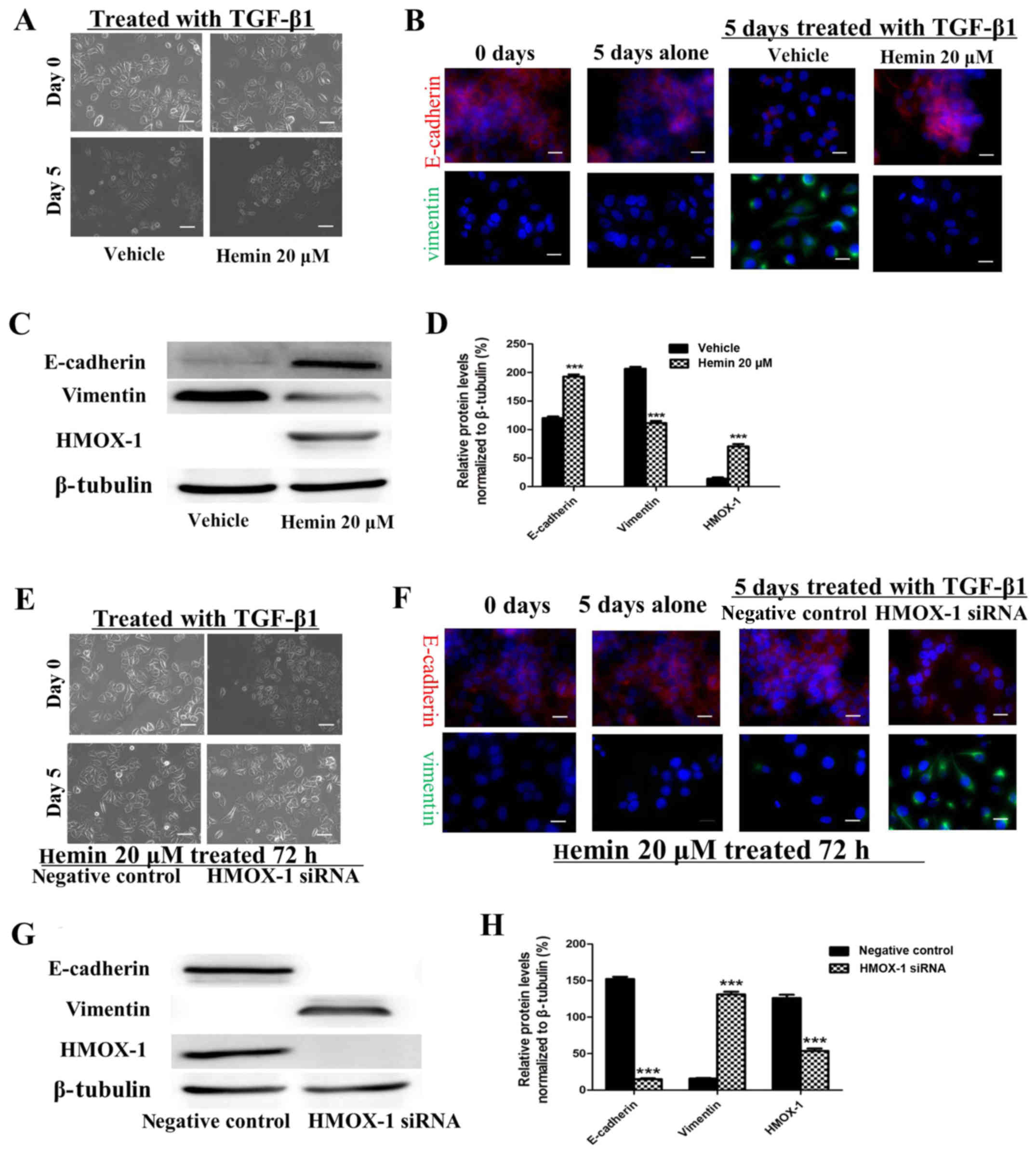 | Figure 3Hemin inhibits the transforming growth
factor-β1 (TGF-β1)-induced epithelial-mesenchymal transition (EMT)
of the MCF-7 breast cancer cell line. MCF-7 cells were exposed to
hemin (20 μM, 72 h) or vehicle and then treated with TGF-β
(10 ng/ml) and subjected to the respective assays. (A)
Representative photomicrograph showing that hemin inhibited the
change in morphology from epithelial to mesenchymal. Scale bar, 100
μm. (B) E-cadherin (red) and vimentin (green) expression was
visualized by immunofluorescence staining.
4′,6-Diamidino-2-phenylindole (DAPI) (blue) was used for nuclear
staining. Scale bar, 20 μm. (C and D) E-cadherin and
vimentin protein expression was analyzed by western blotting. MCF-7
cells were exposed to hemin (20 μM, 72 h), transfected with
the heme oxygenase-1 (HMOX-1) siRNA or negative control, and then
treated with TGF-β (10 ng/ml) and subjected to the respective
assays. (E) Representative photomicrograph showing the cell
morphology. Scale bar, 100 μm. (F) E-cadherin (red) and
vimentin (green) expression was visualized by immunofluorescence
staining. DAPI (blue) was used for nuclear staining. Scale bar, 20
μm. (G and H) E-cadherin and vimentin protein expression was
analyzed by western blotting. All the experiments were repeated at
least three times with similar results and one representative from
at least three independent experiments is shown. The results are
shown as the mean ± SD, ***P<0.001. |
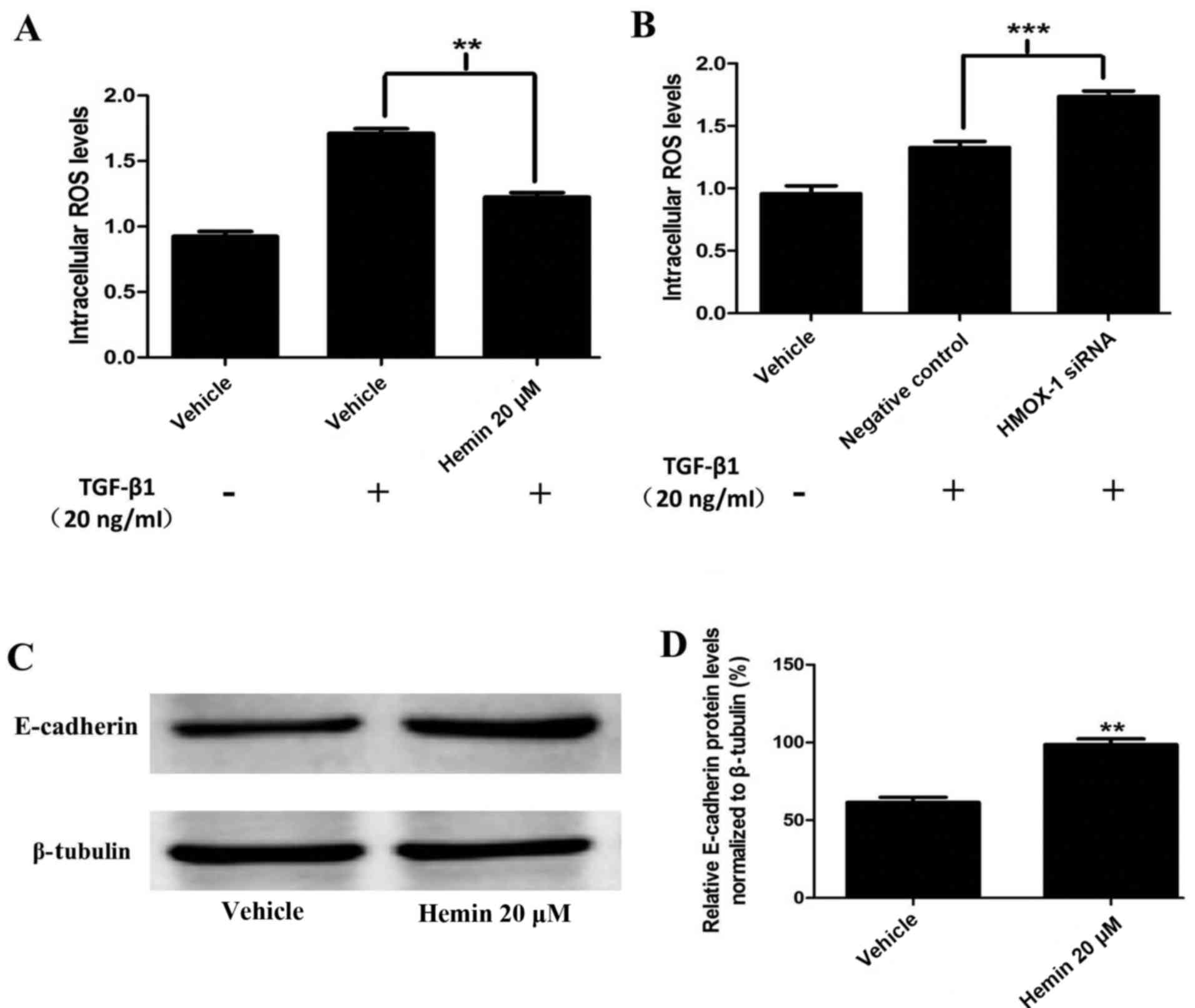
Hemin reduces ROS generation and induces
E-cadherin expression in MCF-7 cells
To determine the role of HMOX-1 as a
TGF-β-responsive ROS inhibitor, we treated the cells with TGF-β1
(20 ng/ml). ROS generation was measured using DCFDA and a
fluorescence spectrometer. The hemin-treated MCF-7 cells had
diminished ROS production compared to the control cells (Fig. 4A). These results were confirmed by
silencing HMOX-1 with the HMOX-1 siRNA. The HMOX-1
siRNA-transfected group showed increased TGF-β1-mediated ROS
generation compared to the negative control group (Fig. 4B). Our findings suggested that
HMOX-1 regulated the ROS levels in MCF-7 cells. Interestingly, we
observed that hemin induced E-cadherin protein expression in the
normal MCF-7 cells (Fig. 4C and
D).
Discussion
Metastasis is responsible for ~90% of
cancer-associated patient mortality (2). Although a great deal of effort has
been expended, progress in the research on metastasis is stagnating
due to the lack of effective tools to elucidate the complex network
of signaling pathways that drive this multistep process. EMT is a
phenotypic conversion linked to metastasis that was originally
defined as a morphological conversion occurring at specific sites
in embryonic epithelia to give rise to individual migratory cells
(3). Recent studies have found
that EMT plays a role in enhancing the invasive and metastatic
behaviors of tumor cells. During the EMT process, epithelial cells
gain mesenchymal properties and exhibit reduced intercellular
adhesion and increased motility. Sequentially, these cells break
through the basal membrane and migrate long distances (4). In this study, we showed that HMOX-1
inhibited TGF-β-induced EMT in the MCF-7 breast cancer cell
line.
HMOX-1 is an anti-inflammatory and antioxidant
protein. The role of HMOX-1 in cancer is controversial. Because
this protein is a potent inducer of vascular endothelial growth
factor (VEGF), which is a crucial factor that governs tumor
angiogenesis, HMOX-1 has also been recognized as an angiogenesis
exciter and a tumor-metastasis supporter (12). Alternatively, HMOX-1 has been
reported to suppress the invasive capacity of breast cancer cells
via downregulating matrix metallopeptidase 9 (MMP9) (13). In this study, we found that HMOX-1
was induced by hemin in the breast cancer MCF-7 cell line and
inhibited the invasion and migration of TGF-β-treated MCF-7 breast
cancer cells.
TGF-β is a primary inducer of EMT and plays a double
role in cancer. TGF-β inhibits the proliferation and induces the
apoptosis of cancer cells in the early stages of tumorigenesis.
However, this protein can also promote invasion and metastasis
during later tumor development (7). TGF-β induces EMT by activating
E-cadherin repressors and Smad2/3 (14). Additionally, TGF-β can induce EMT
via Smad-independent pathways, such as the phosphatidylinositol
3-kinase (PI3K), Akt, mitogen-activated protein kinase (MAPK) and
Rho family GTPase pathways (15).
Our data suggest a tumor-suppressor role for HMOX-1 in breast
cancer. HMOX-1 expression was increased via hemin stimulation, and
in turn the EMT induced by TGF-β was inhibited in the hemin-treated
breast cancer cells. The inhibitory effect of hemin on the
TGF-β-induced EMT was significantly attenuated by trans-fection of
the HMOX-1 siRNA into hemin-treated MCF-7 breast cancer cells.
Recent observations suggest that ROS play an
important role in TGF-β-induced EMT (16–18). Upon TGF-β stimulation, a
significant increase was found to occur in intracellular ROS.
TGF-β-dependent ROS release was demonstrated to be responsible for
the phosphorylation of Smad2 and p38 MAPK, the upregulation of
α-SMA and fibronectin and the downregulation of E-cadherin
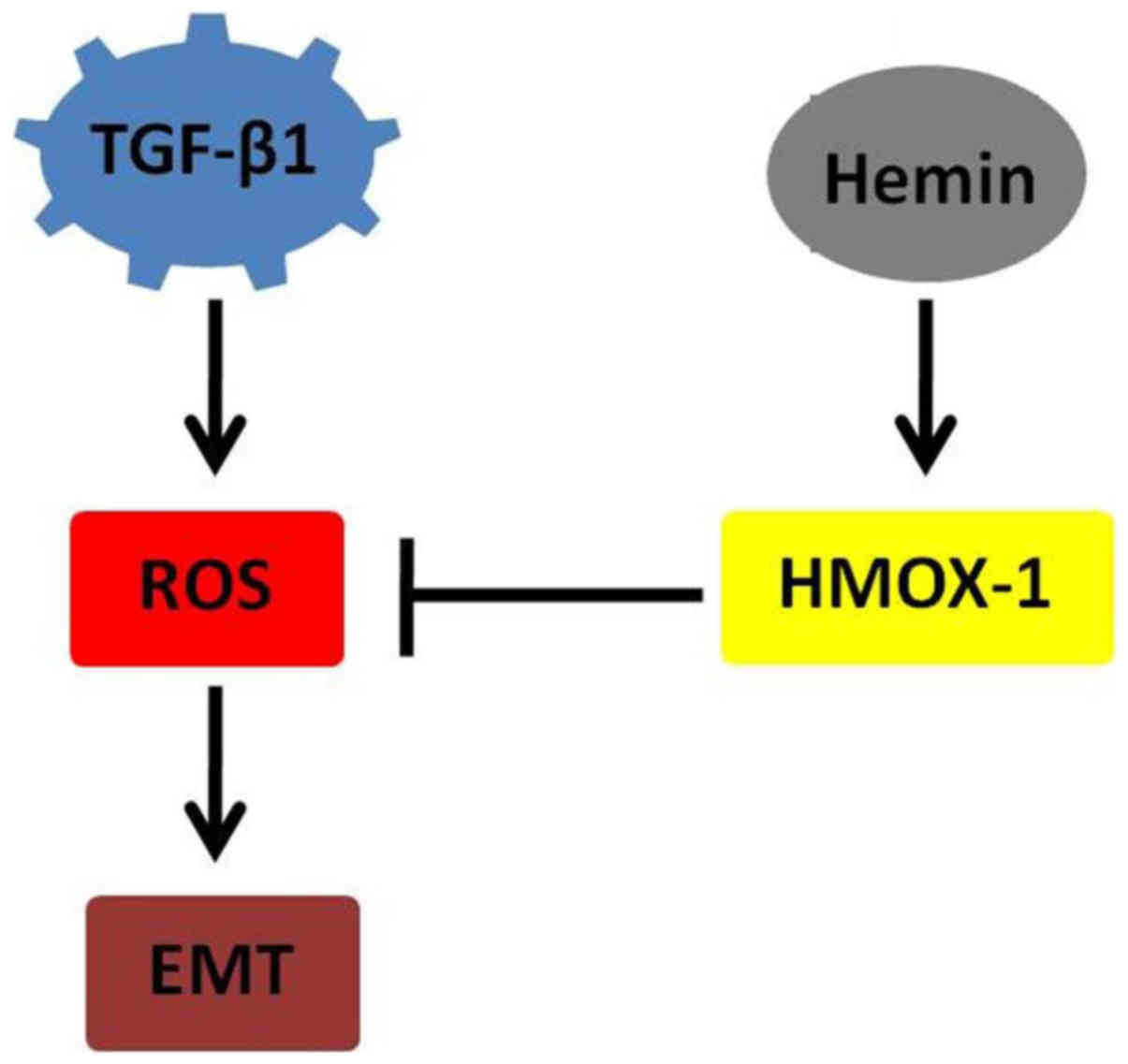
(13). Therefore, HMOX-1 may
impact the EMT process by regulating the release of ROS and their
dependent pathways (Fig. 5).
Thus, modulating HMOX-1 levels may be a strategy through which to
regulate TGF-β-induced EMT.
Interestingly, we found that HMOX-1 enhanced
E-cadherin expression in normal MCF-7 cells. Epithelial cells
communicate with their neighboring cells through the extension of
either lamellipodia or filopodia, which couple to the intracellular
cytoskeleton, trigger specific signaling pathways, and contribute
to cell adhesion (19,20). E-cadherin plays a major role in
intercellular adhesion. Additionally, the role of E-cadherin as a
tumor suppressor has been observed in a variety of cancers,
including prostate, ovarian, gastric and breast cancers (21–24). Cancer cells with low E-cadherin
expression levels have been suggested to be more invasive, although
the signaling pathways involved in this modulation are unknown.
Future study should focus on the potential molecular partners with
which HMOX-1 interacts to induce the cellular morphological
changes.
In conclusion, we found that HMOX-1 inhibited
TGF-β-induced EMT by regulating ROS release in MCF-7 cells. Our
results imply that HMOX-1 plays an antitumor role in the breast and
acts as a target for cancer therapy.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 81172337).
References
|
1
|
Libson S and Lippman M: A review of
clinical aspects of breast cancer. Int Rev Psychiatry. 26:4–15.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Criscitiello C, André F, Thompson AM, De
Laurentiis M, Esposito A, Gelao L, Fumagalli L, Locatelli M,
Minchella I, Orsi F, et al: Biopsy confirmation of metastatic sites
in breast cancer patients: Clinical impact and future perspectives.
Breast Cancer Res. 16:2052014. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sharma RR, Pollock K, Hubel A and McKenna
D: Mesenchymal stem or stromal cells: A review of clinical
applications and manufacturing practices. Transfusion.
54:1418–1437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kupcova Skalnikova H: Proteomic techniques
for characterisation of mesenchymal stem cell secretome. Biochimie.
95:2196–2211. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akhurst RJ and Hata A: Targeting the TGFβ
signalling pathway in disease. Nat Rev Drug Discov. 11:790–811.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li B, Wen G, Zhao Y, Tong J and Hei TK:
The role of TGFBI in mesothelioma and breast cancer: Association
with tumor suppression. BMC Cancer. 12:2392012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brown DI and Griendling KK: Regulation of
signal transduction by reactive oxygen species in the
cardiovascular system. Circ Res. 116:531–549. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Krstić J, Trivanović D, Mojsilović S and
Santibanez JF: Transforming growth factor-beta and oxidative stress
interplay: Implications in tumorigenesis and cancer progression.
Oxid Med Cell Longev. 2015:6545942015. View Article : Google Scholar
|
|
10
|
Jozkowicz A, Was H and Dulak J: Heme
oxygenase-1 in tumors: Is it a false friend? Antioxid Redox Signal.
9:2099–2117. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gueron G, Giudice J, Valacco P, Paez A,
Elguero B, Toscani M, Jaworski F, Leskow FC, Cotignola J, Marti M,
et al: Heme-oxygenase-1 implications in cell morphology and the
adhesive behavior of prostate cancer cells. Oncotarget.
5:4087–4102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cherrington JM, Strawn LM and Shawver LK:
New paradigms for the treatment of cancer: The role of
anti-angiogenesis agents. Adv Cancer Res. 79:1–38. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen HW, Chao CY, Lin LL, Lu CY, Liu KL,
Lii CK and Li CC: Inhibition of matrix metalloproteinase-9
expression by docosahexaenoic acid mediated by heme oxygenase 1 in
12-O-tetradecanoylphorbol-13-acetate-induced MCF-7 human breast
cancer cells. Arch Toxicol. 87:857–869. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zavadil J, Cermak L, Soto-Nieves N and
Böttinger EP: Integration of TGF-beta/Smad and Jagged1/Notch
signalling in epithelial-to-mesenchymal transition. EMBO J.
23:1155–1165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vincent T, Neve EP, Johnson JR, Kukalev A,
Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL,
et al: A SNAIL1-SMAD3/4 transcriptional repressor complex promotes
TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol.
11:943–950. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang KH, Tian HY, Gao X, Lei WW, Hu Y,
Wang DM, Pan XC, Yu ML, Xu GJ, Zhao FK, et al: Ferritin heavy
chain-mediated iron homeostasis and subsequent increased reactive
oxygen species production are essential for epithelial-mesenchymal
transition. Cancer Res. 69:5340–5348. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lim SO, Gu JM, Kim MS, Kim HS, Park YN,
Park CK, Cho JW, Park YM and Jung G: Epigenetic changes induced by
reactive oxygen species in hepatocellular carcinoma: Methylation of
the E-cadherin promoter. Gastroenterology. 135:2128–2140.
2140.e1–2140.e8. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lochter A, Galosy S, Muschler J, Freedman
N, Werb Z and Bissell MJ: Matrix metalloproteinase stromelysin-1
triggers a cascade of molecular alterations that leads to stable
epithelial-to-mesenchymal conversion and a premalignant phenotype
in mammary epithelial cells. J Cell Biol. 139:1861–1872. 1997.
View Article : Google Scholar
|
|
19
|
Baum B and Georgiou M: Dynamics of
adherens junctions in epithelial establishment, maintenance, and
remodeling. J Cell Biol. 192:907–917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Herszterg S, Leibfried A, Bosveld F,
Martin C and Bellaiche Y: Interplay between the dividing cell and
its neighbors regulates adherens junction formation during
cytokinesis in epithelial tissue. Dev Cell. 24:256–270. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tomita K, van Bokhoven A, van Leenders GJ,
Ruijter ET, Jansen CF, Bussemakers MJ and Schalken JA: Cadherin
switching in human prostate cancer progression. Cancer Res.
60:3650–3654. 2000.PubMed/NCBI
|
|
22
|
Sawada K, Mitra AK, Radjabi AR, Bhaskar V,
Kistner EO, Tretiakova M, Jagadeeswaran S, Montag A, Becker A,
Kenny HA, et al: Loss of E-cadherin promotes ovarian cancer
metastasis via alpha 5-integrin, which is a therapeutic target.
Cancer Res. 68:2329–2339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang B, Peng ZH, Yu PW, Yu G and Qian F:
Expression and significance of Cx43 and E-cadherin in gastric
cancer and metastatic lymph nodes. Med Oncol. 28:502–508. 2011.
View Article : Google Scholar
|
|
24
|
Chao YL, Shepard CR and Wells A: Breast
carcinoma cells re-express E-cadherin during mesenchymal to
epithelial reverting transition. Mol Cancer. 9:1792010. View Article : Google Scholar : PubMed/NCBI
|