Introduction
Visfatin, also known as pre-B-cell colony-enhancing
factor and nicotinamide phosphoribosyltransferase (Nampt), was
originally cloned in 1994 and is a multifaceted molecule (1). As a nicotinamide adenine
dinucleotide (NAD) biosynthetic enzyme, visfatin determines the
activity of NAD-consuming enzymes, such as sirtuins and influences
a variety of metabolic responses, prompting increasing attention in
the literature.
Visfatin plays an important role in regulating
insulin secretion, as it functions as an immunomodulatory cytokine
and is involved in the inflammatory responses (2). Visfatin has also been implicated in
the pathogenesis of various metabolic disorders, with increased
plasma concentrations of visfatin reported in overweight/obese
subjects, as well as in pateints with type 2 diabetes mellitus,
metabolic syndrome, atherosclerosis and cardiovascular diseases
(3–7). In atherosclerosis, visfatin has been
implicated causatively by inducing oxidative stress and
inflammation in endothelial cells, which subsequently damages
vascular endothelial function (8). Visfatin also participates in the
inflammatory process in rat spleen by modulating macrophages and
inflammatory cytokines (9). In
addition, treatment with visfatin in vitro has been shown to
promote the proliferation of vascular smooth muscle cells (10) and to increase the endothelial
expression of vascular cell adhesion molecule-1 (VCAM-1),
intercellular adhesion molecule-1 (ICAM-1) and E-selectin, all of
which are considered as biomarkers of endothelial dysfunction
(11). Thus, these cell types are
implicated as both the targets and cellular sources of visfatin.
Finally, a pathological role for visfatin was also reported by a
study showing that the administration of visfatin impairs
microvascular endothelium-dependent relaxation through a mechanism
involving NADPH oxidase stimulation (12). Despite these in vitro and
in vivo data, the mechanisms underlying the function of
visfatin remain controversial.
Endothelial progenitor cells (EPCs) are progenitor
cells that can migrate to the peripheral circulation to
differentiate into mature endothelial cells. In the case of vessel
impairment or tissue ischemia, EPCs mobilize to peripheral blood
circulation, specifically home in to the damaged or ischemic
tissue, and then differentiate further into mature endothelial
cells (13). Thus, the
restoration and regeneration of endothelial cells depends not only
on the proliferation and migration of mature endothelial cells, but
also on the circulating EPC levels. Indeed, the decrease in the
number of circulating EPCs has been used to independently predict
atherosclerotic progression and poor prognosis in patients with
coronary heart disease (14,15). It is also known that apoptosis
plays a crucial role in the loss of EPCs, with data suggesting
several factors that may be associated with traditional and the
disease-related risk of eliciting apoptosis in EPCs, including
C-reactive protein, oxidized low-density lipoprotein (LDL) and
interferon-α (16–18).
Emerging evidence has established a potential link
between adipokines and vascular dysfunction, while our previous
studies have correlated EPC dysfunction in obese rats with serum
visfatin levels (19), with
higher levels of serum visfatin and a lower level of circulating
EPCs in the obese population (7).
However, the specific mechanisms remain unclear, and few studies
have specifically investigated visfatin and EPCs, and the related
mechanisms of action. In the present study, we investigated whether
visfatin plays a role in promoting EPCs apoptosis and aimed to
elucidate the mechanisms underlying such effects.
Materials and methods
Isolation and culture of EPCs
This study was approved by the Ethics Committee of
Hebei General Hospital. Informed consent was obtained from all
subjects. Umbilical vein blood mononuclear cells (MNCs) of healthy
pregnant woman donors were isolated by Ficoll density gradient
centrifugation (Dakewe, Shenzhen, China). After washing with
phosphate-buffered saline (PBS), the MNCs were resuspended in
EGM-2MV medium (Lonza, Walkersville, MD, USA), supplemented with
20% fetal bovine serum, 1% Pen/Strep, 0.04% hydrocortisone, 0.1%
heparin and 0.1% ascorbic acid in the presence of insulin-like
growth factor-1 (IGF-1; 50 ng/ml), epidermal growth factor (EGF; 10
ng/ml), fibroblast growth factor (FGF; 50 ng/ml), vascular
endothelial growth factor (VEGF; 50 ng/ml) and fibronectin (10
µg/ml). Six-well tissue culture plates pre-coated with
fibronectin (Solarbio, Beijing, China) and the cells were seeded at
a density of 2×106 cells/ml and cultured in a 5%
CO2 incubator at 37°C. After 48 h of culture,
non-adherent cells were removed by washing with PBS, and EGM-2MV
medium was added to each well. The medium was changed every 2 days
until treatment. The cells were observed daily under an inverted
microscope (TS100F; Nikon, Tokyo, Japan).
Immunophenotyping of EPCs
Immunophenotyping of cultured cells was detected by
flow cytometry. Briefly, the EPCs were washed twice with PBS and
incubated with EPC-specific markers for 30 min at 4°C in the dark.
The following FITC- or PE-conjugated monoclonal antibodies (mAbs)
were used as EPCs markers: anti-human CD34 (555748) and anti-human
CD309 (554680), also termed kinase insert domain receptor (KDR)
(both from Becton-Dickinson, Franklin Lakes, NJ, USA).
Isotype-matched irrelevant mAbs were used as negative controls. The
cells were electronically gated according to their light scattering
properties to exclude cell debris.
CD309+CD34+ double-positive cells were
considered as EPCs. Data were detected on a FACSCanto flow
cytometer (Becton-Dickinson) and analysed using SPSS 13.0 software
(IBM, Armonk, NY, USA).
Uptake of Dil-acetylated low-density
lipoprotein and staining for Ulex europaeus lectin
The EPCs were marked by cellular uptake of
DiI-labeled acetylated LDL (DiI-ac-LDL; Molecular Probes, Eugene,
OR, USA) and binding of FITC-conjugated lectin from Ulex
europaeus agglutinin (FITC-Lectin-UEA-1; Sigma-Aldrich, St.
Louis, MO, USA). After 10 days in culture, the attached cells were
incubated with Dil-ac-LDL (2.5 µg/ml) in EGM-2MV medium for
3 h at 37°C. The cells were then fixed with 2% paraformaldehyde for
10 min and incubated for 1 h with FITC-Lectin-UEA-1 (10
µg/ml). The cells were then observed under an Olympus
Fluoview FV1000 laser scanning confocal microscope (OLFV-34CMXE;
Olympus, Tokyo, Japan). Orange cells positive for both DiI-ac-LDL
and FITC-Lectin-UEA-1 were identified as EPCs.
Treatment of EPCs with visfatin
After being cultured for 7 days, the adherent EPCs
were serum-starved for 24 h, and then incubated with human
recombinant visfatin (Peprotech, Rocky Hill, NJ, USA) at various
concentrations (0, 50, 100 and 150 ng/ml) for 48 h. In some
experiments, the EPCs were pretreated with visfatin inhibitor
(FK866, 10 µM; Sigma-Aldrich) or nuclear factor-κB (NF-κB)
inhibitor [BAY11-7085 (referred to as BAY11), 5 µM; Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA] 1 h prior to
incubation with visfatin at various concentrations for 48 h.
Flow cytometry
The apoptosis of the EPCs was evaluated by flow
cytometry. The EPCs were fluorescently labeled by the addition of
500 µl of binding buffer, 5 µl of Annexin V-FITC and
5 µl of propidium iodide (Miltenyi Biotec, Bergisch
Gladbach, Germany). Following incubation in the dark at room
temperature for 15 min, the cells were subjected to flow cytometric
analysis. A minimum of 10,000 cells in the gated region was
analyzed using a BD FACSCalibur Flow Cytometer (Becton-Dickinson).
The esults were presented by the percentage of total cells.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the EPCs using TRIzol
reagent (Invitrogen, Carlsbad, CA, USA). RNA was
reverse-transcribed into cDNA according to the instructions of the
Easy Script First Strand cDNA Synthesis Super Mix kit (TransGen
Biotech, Beijing, China). Specific primers designed for the
amplification of caspase-3, Bax, Bcl-2, NF-κB and glyceraldehyde
3-phosphate dehydrogenase (GAPDH) were verified by NCBI Blast;
primer sequences are listed in Table
I. Reactions were carried out on an ABI PRISM 7300 PCR system
(Applied Biosystems, Foster City, CA, USA) using SYBR-Green I
GoTaq® qPCR Master Mix (Promega, Madison, WI, USA). PCR
reactions were performed in a total of 25 µl as follows: an
initial cycle at 95°C for 5 min, followed by 40 cycles of 95°C for
15 sec, 58°C for 20 sec and 72°C for 30 sec. The gene expression
was analyzed in duplicate and normalized against GAPDH. The results
of each gene are expressed as relative expression using the ΔCt
method.
 | Table ISequences of primers used for
RT-qPCR. |
Table I
Sequences of primers used for
RT-qPCR.
| Genes | Forward
(5′→3′) | Reverse
(5′→3′) |
|---|
| GADPH |
TGAACGGGAAGCTCACTGG |
GCTTCACCACCTTCTTGATGTC |
| Caspase-3 |
TGGACTGTGGCATTGAGAC |
AATAACCAGGTGCTGTGGAGT |
| Bax |
GGCTGGACATTGGACTTCCT |
CCACAAAGATGGTCACGGT |
| Bcl-2 |
GTGGATGACTGAGTACCTGAAC |
CGCATCTCGGACCTGTG |
| NF-κB |
CAAGGCAGCAAATAGACGAG |
TGTTGAGAGTTAGCAGTGAGGC |
Western blot analysis
The cells were washed twice with ice-cold PBS and
cellular proteins from EPCs under various treatments were prepared
with lysis buffer [1% NP-40, 150 mM NaCl, 50 mM Tris (pH 8.0), 0.1%
aprotinin, 0.1% leupeptin, 0.035% pepstain A and 100 µg/ml
PMSF] supplemented with protease inhibitor cocktail tablets (Roche
Diagnostics, Mannheim, Germany) and solubilized for 30 min at 4°C.
The samples were centrifuged at 11,600 × g for 30 min at 4°C.
Protein concentrations were determined via Nanojob (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Protein samples were then
denatured in glyceraldehyde 3-phosphate dehydrogenase (SDS) sample
buffer (125 mM Tris-HCl, pH 6.8, 50% glycerol, 2% SDS, 5%
β-mercaptoethanol and 0.01% bromophenol blue) for 10 min at 100°C.
Equal amounts of proteins were separated by 10% SDS-PAGE and
transferred onto polyvinylidene fluoride (PVDF) membranes
(Millipore, Billerica, MA, USA). Subsequently, 5% non-fat milk in
Tris-buffered saline was used for blocking for 4 h at room
temperature. After blocking, the membranes were incubated overnight
at 4°C with appropriately diluted rabbit anti-human primary
antibodies to caspase-3 (sc-2048; Santa Cruz Biotechnology, Inc.),
Bax (50599-2-Ig; Proteintech, Chicago, IL, USA), Bcl-2 (2870; Cell
Signaling Technology, Danvers, MA, USA), NF-κB (#21013; Signalway
Antibody, Baltimore, MD, USA), ICAM-1 (ab53013) and interleukin
(IL)-6 (ab32530) (both from Abcam, Cambridge, MA, USA), or mouse
anti-human β-actin (66009-1-Ig; Proteintech), followed by
incubation with the relevant secondary antibodies [caspase-3, Bax,
Bcl-2, NF-κB, ICAM, IL-6: anti-rabbit IgG (L3012-1); β-actin:
anti-mouse IgG (L3032-2); all from Signalway Antibody] for 2 h at
room temperature. The immunoreactive proteins were visualized by
the enhanced chemiluminescence (ECL) detection system. β-actin
served as an internal control protein.
Statistical analysis
All experiments were repeated at least 3 times. Data
are presented as the means ± SD and were analyzed using the SPSS
statistical package (SPSS 13.0; IBM). Data were analyzed by
homogeneity testing for variance. Statistical significance was
determined by ANOVA (post-hoc used Student-Newman-Keuls test) for
comparison of normally distributed data with homogeneous variance
relevant groups. P-values <0.05 were considered to indicate
statistically significant differences.
Results
Culture of EPCs
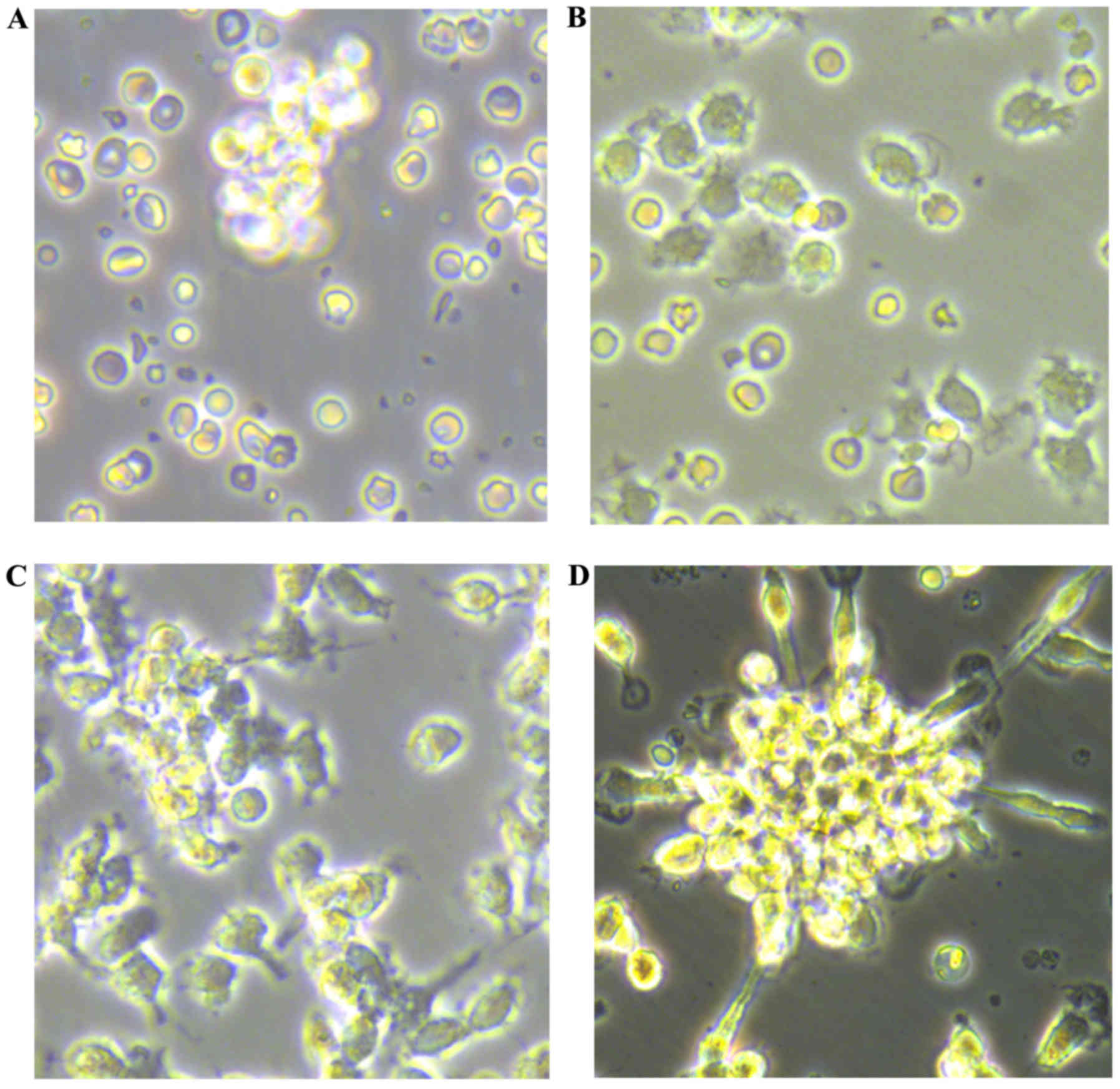
Newly isolated umbilical vein blood MNCs were round
in shape when suspended in medium (Fig. 1A). After 48 h of plating, the
cells were partly attached (Fig.
1B), with the adherent cells gradually elongating into a
spindle shape (Fig. 1C). At 7–10
days after plating, the adherent cells grew as colonies. A colony
of EPCs was defined as a central core of round cells with elongated
sprouting cells at the periphery (Fig. 1D). The cells were round, triangle,
oval or irregular. After 10 days, the cultured EPCs were linearly
grown.
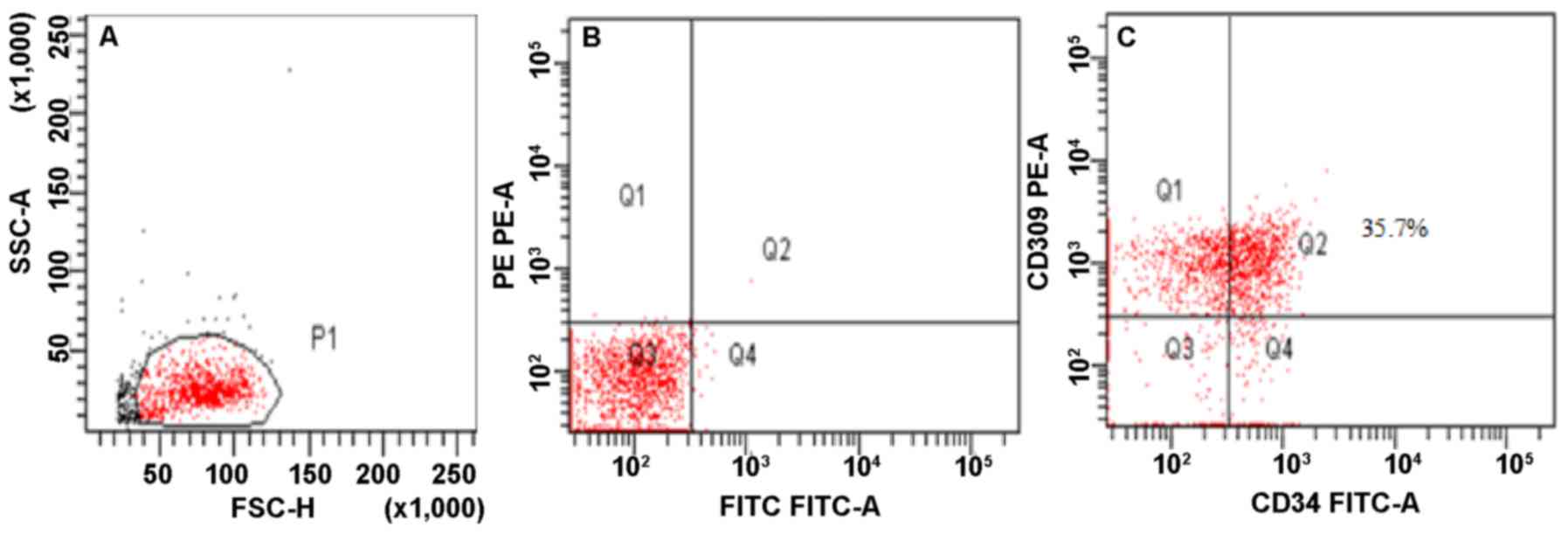
Immunophenotyping of EPCs
By flow cytometry, CD309+CD34+
double-positive cells were determined as EPCs (Fig. 2).
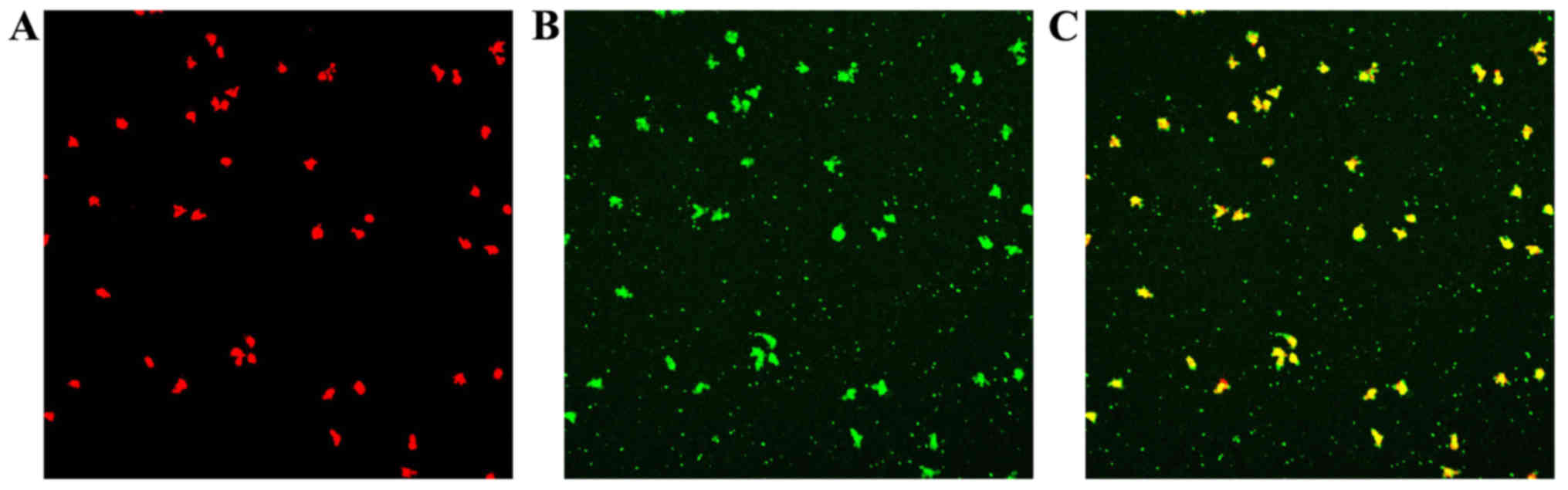
Uptake of Dil-ac-LDL and staining for
FITC-Lectin-UEA-1
Following the uptake of Dil-ac-LDL and the binding
of FITC-Lectin-UEA-1, double-stained EPCs were observed under a
laser scanning confocal microscope (Fig. 3).
Visfatin induces the apoptosis of
EPCs
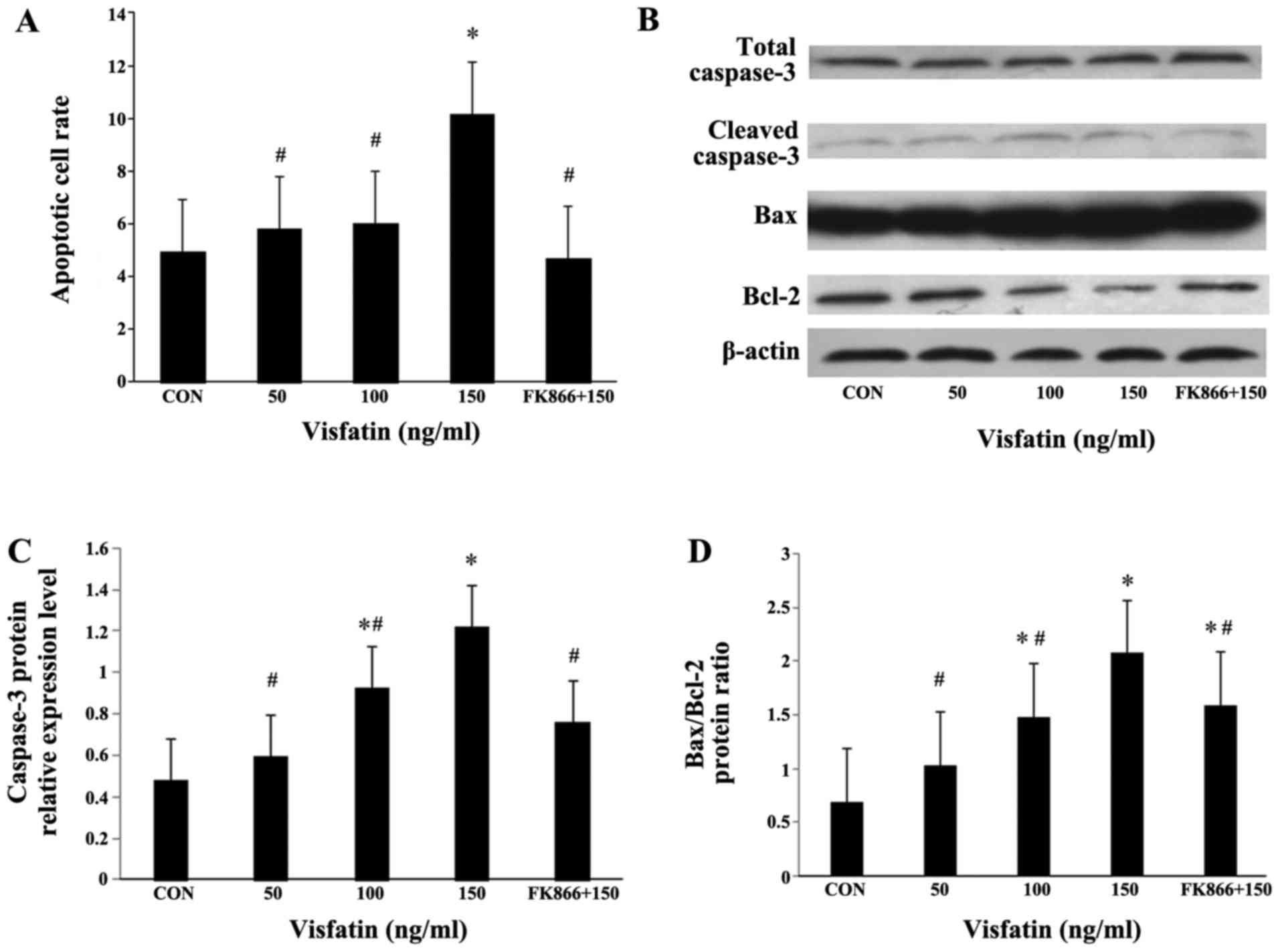
Recombinant human visfatin induced a dose-dependent
and significant increase in EPC apoptosis, and this effect was
completely abrogated by pre-treatment with FK866 (a visfatin
inhibitor) (Fig. 4A). As
expected, visfatin significantly increased the expression levels of
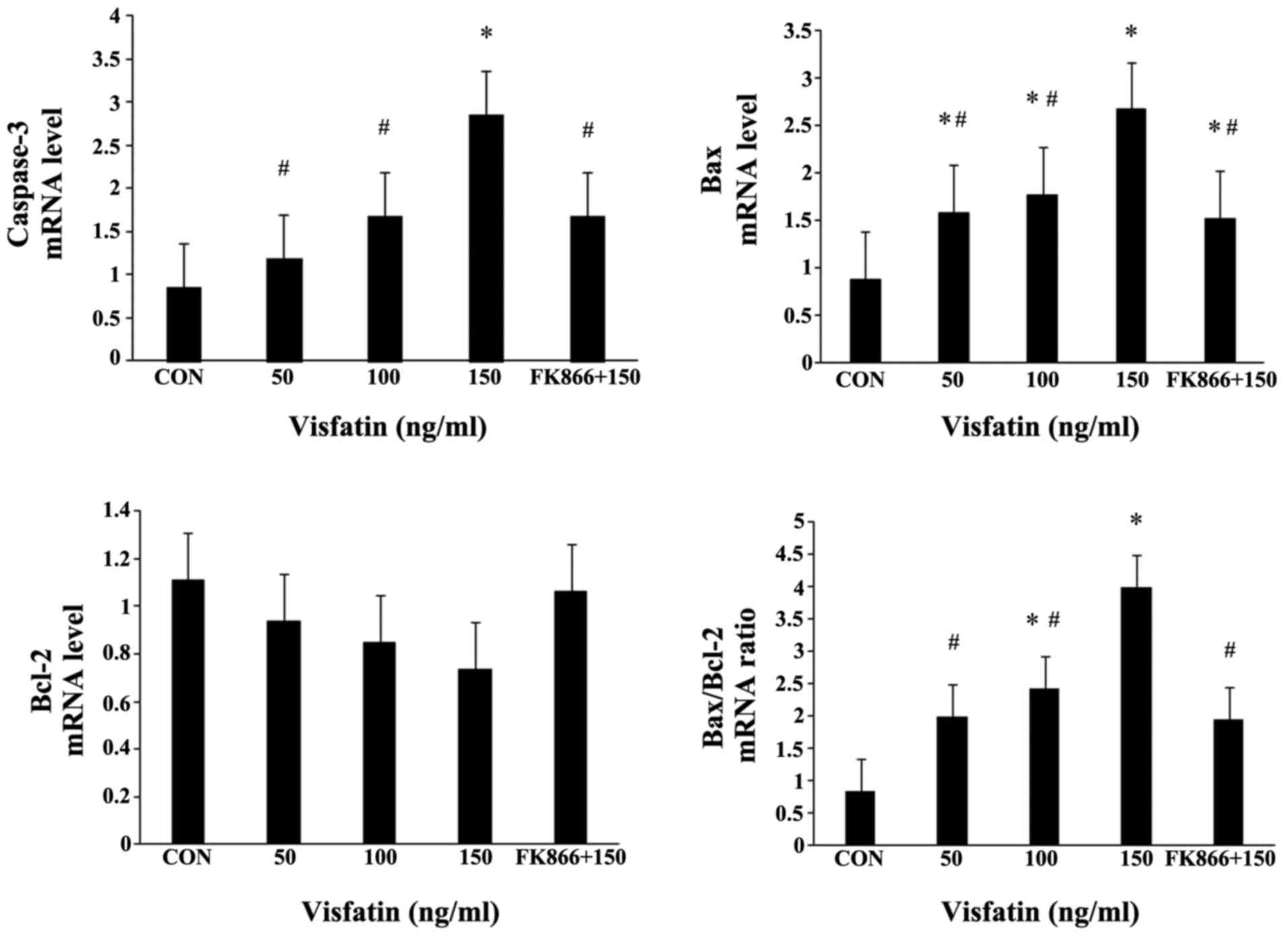
cleaved caspase-3 in a dose-dependent manner at both the mRNA and
protein level, and pre-treatment with FK866 markedly suppressed the
increased expression of cleaved caspase-3 (Fig. 4B and C, and Fig. 5). The Bax/Bcl-2 expression ratio
is critical for the induction of apoptosis; thus, we examined the
expression of Bax and Bcl-2 in EPCs by both RT-qPCR and western
blot analysis. Following treatment with visfatin, Bax expression
was significantly upregulated at both the mRNA and protein level;
however, the protein expression of Bcl-2 was decreased in a
dose-dependent manner (P>0.05); these effects were all abolished
by pre-treatment with FK866 (Figs.
4 and 5). The mRNA expression
of Bcl-2 in the EPCs exhibited no statistically significant change,
although there was a decreased trend following treatment with
visfatin. As a result, the ratio of Bax/Bcl-2 expression was
markedly augmented in the EPCs (Figs.
4 and 5).
Pro-inflammatory mediators are involved
in the apoptosis of EPCs induced by visfatin
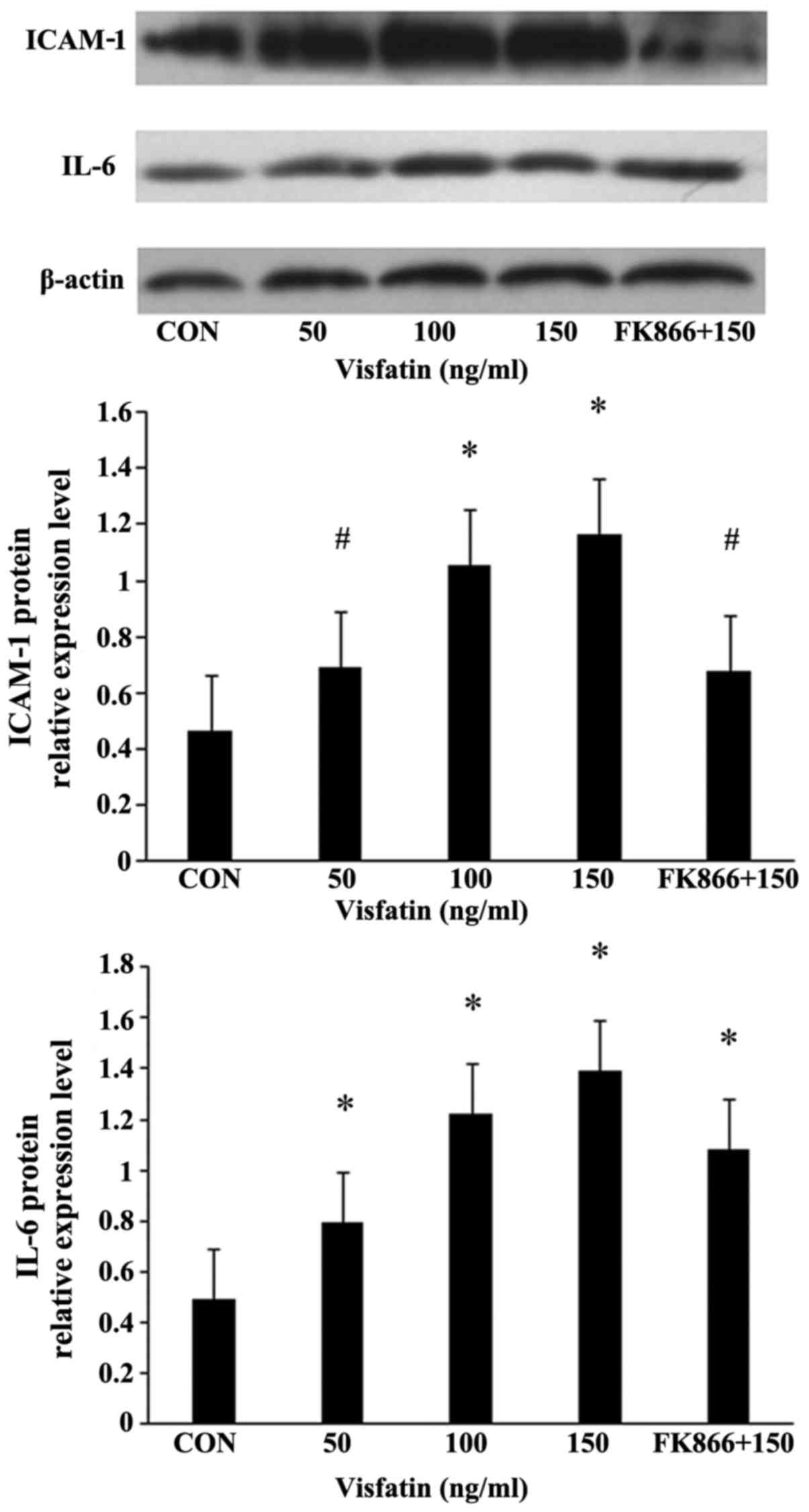
Treatment of the EPCs with various concentrations of
visfatin resulted in a dose-dependent and significant increase in
the protein expression of ICAM-1 and IL-6, as determined by western
blot analysis (Fig. 6). FK866
suppressed these upregulatory effects, indicating the involvement
of pro-inflammatory mediators in the apoptosis of EPCs induced by
visfatin.
NF-κB mediates visfatin-induced EPC
apoptosis
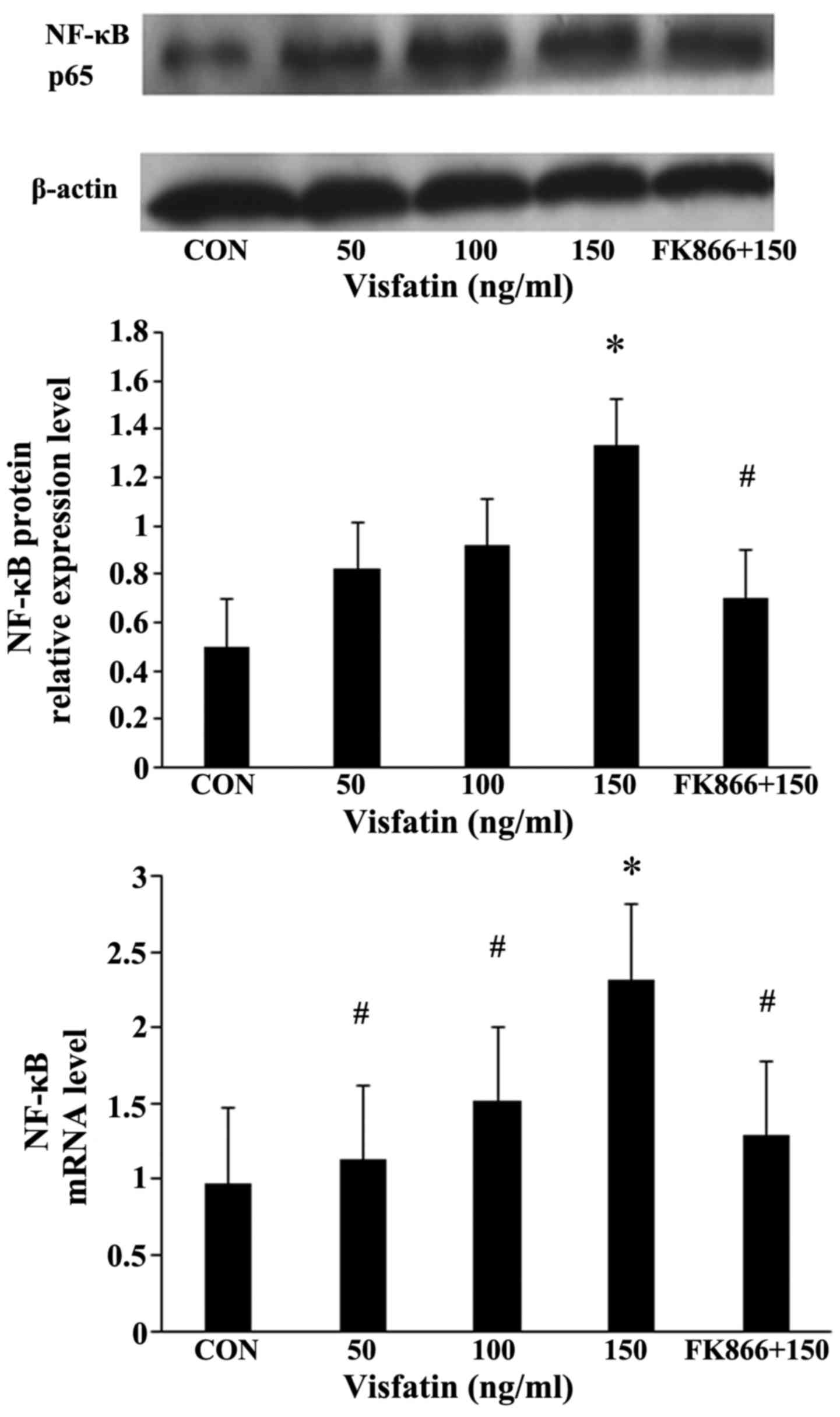
Compared with the EPCs not incubated with visfatin,
the expression of NF-κB in the EPCs following treatment with
various concentrations of visfatin increased in a significant and
dose-dependent manner at both the mRNA and protein level, an effect
that was inhibited by pre-treatment with FK866 (Fig. 7).
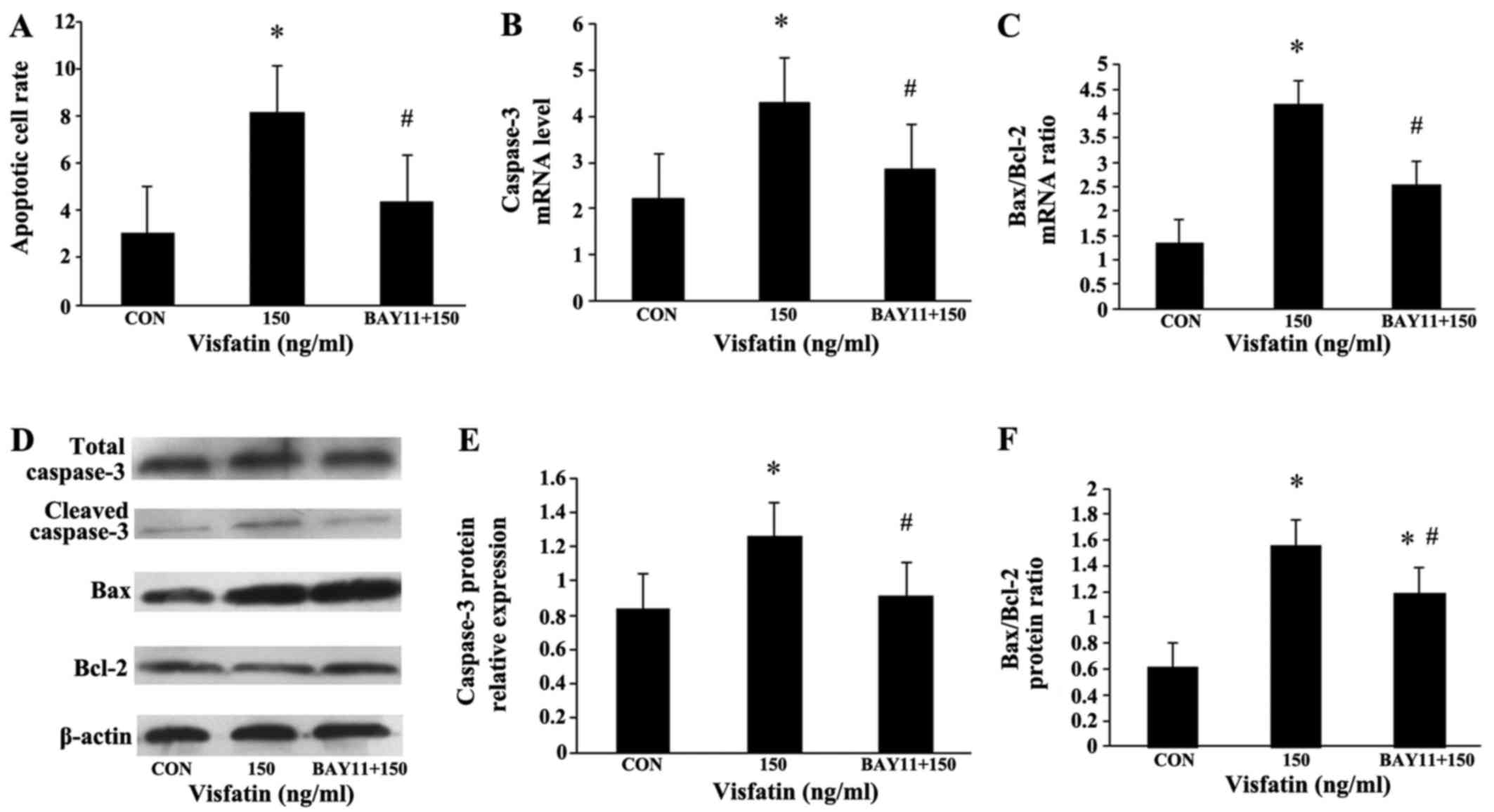
The EPCs were additionally pre-incubated with NF-κB
inhibitor (BAY11-7085, 5 µM) for 1 h, and then treated with
visfatin (150 ng/ml) for 48 h. Compared with the
visfatin-alone-treated group, BAY11 significantly diminished the
increase in EPC apoptosis induced by visfatin. In addition, the
increased expression of caspase-3 and Bax, and the decreased
expression of Bcl-2 were all abolished in the BAY11-treated cells
(Fig. 8), suggesting that NF-κB
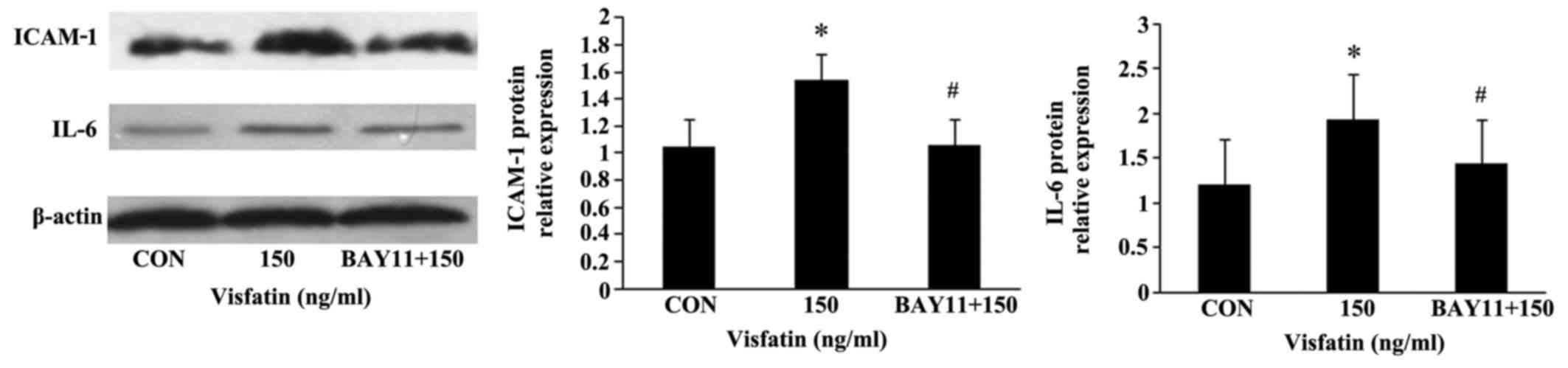
may mediate visfatin-induced EPC apoptosis. Moreover, BAY11 also
abolished the promoting effects of visfatin on the protein
expression of ICAM-1 and IL-6 in the EPCs (Fig. 9), indicating that visfatin
promotes EPC apoptosis by regulating ICAM-1 and IL-6 expression
through NF-κB.
Discussion
In the present study, we provide evidence that
visfatin promotes the apoptosis of EPCs, as determined by flow
cytometry, and that the pro-apototic effects were inhibited by
pre-treatment with FK866, a visfatin inhibitor.
The number of EPCs in the blood circulation are
regarded as an indicator of endothelial function and cardiovascular
disease prognosis (20).
Following vessel impairment and tissue ischemia, EPCs can originate
from the bone marrow and home into ischemic locations where they
participate in the re-endothelialization of impaired blood vessels
(13). Therefore, EPCs play an
important role in maintaining the completeness of endothelial
structure and the normal function of the vascular endothelium.
Several chronic factors have been implicated in mediating the
apoptosis and dysfunction of EPCs, such as gluco-lipotoxicity,
hyperglycemia, hyperlipidemia and oxidative stress (21). Apoptosis is a multi-step process
and an increasing number of genes have been identified to be
involved in the control or execution of apoptosis (22). There are two major pathways
through which apoptosis occurs; the extrinsic pathway and the
intrinsic pathway (23). Although
these two pathways may act independently of each other, they
converge at the level of caspase-3. As the most important member of
the caspase-family, caspase-3 is responsible for many biochemical
mechanisms driving apoptosis that lead to the cleavage of nuclear
and cytosolic material, chromatin condensation, fragmentation of
DNA, and disassembly into membrane-enclosed vesicles (24). Apoptosis is also regulated by
members of the Bcl-2 family of genes (25), with the effect of the
anti-apoptotic gene, Bcl-2, counteracted by the pro-apoptotic gene,
Bax. Indeed, the balance between these opposing genes may be the
key determinant of cell survival by regulating the apoptotic
process, whereby an increase in the Bax/Bcl-2 ratio leads to the
activation of caspase-3, and thus determines a cell's
susceptibility to undergo apoptosis (25,26). Therefore, we detected the
expression of apoptosis-related proteins in this study.
In this study, following treatment with visfatin, we
detected decreased levels of the anti-apoptotic gene, Bcl-2, and
increased expression levels of the pro-apoptotic gene, caspase-3,
as well as an increased Bax/Bcl-2 ratio; all these effects were
inhibited by FK866. This finding suggests that visfatin regulates
the expression of Bax/Bcl-2/caspase-3 in the process of EPC death,
which may explain, at least in part, the decreased number of
EPCs.
Due to its role in inflammation, visfatin has been
implicated in the pathogenesis of various metabolic disorders, such
as metabolic syndrome, type 2 diabetes mellitus and obesity
(6,7,27).
Our previous studies also revealed that visfatin treatment impaired
the migration and adhesion capacity of EPCs (19). A possible mechanism through which
visfatin exerts these effects lies in the inflammation induced by
visfatin, which may upregulate a series of inflammatory factors,
leading to the apoptosis of EPCs and resulted in the reduction of
their number and function (19).
Inflammation in EPCs has also been linked to pro-inflammatory
adhesion molecules, which can be inducers of leukocyte recruitment
(28). Vascular inflammation is a
common feature of vascular damage in a number of diseases and is a
complex process that is initiated by activation of the immune
system, leading to the increased expression of pro-inflammatory
cytokines (29,30). Previous studies have shown that
IL-6 is a potent pro-inflammatory cytokine involved in a number of
pathological processes of vascular inflammation and injury, such as
proliferative diabetic retinopathy and atherosclerosis, while IL-6
blockade can result in the prevention or therapeutic suppression of
disease development (31–34). ICAM-1 is linked to the development
of atherosclerosis and is well characterized for its roles in cell
adhesion and actin polymerization processes (35,36). In addition, both IL-6 and ICAM-1
can be stimulated by the activation of the NF-κB signaling pathway
(37,38).
In this study, we found that visfatin enhanced the
protein expression of ICAM-1 and IL-6 in the EPCs, and that this
effect was inhibited by pre-treatment with FK866 or BAY11-7085.
These results suggest that in cultured EPCs, the synthesis of IL-6
and ICAM-1 is modulated by visfatin, and our data in agreement with
those of a previous study, showing that visfatin enhances the
expression of adhesion molecules (ICAM-1 and VCAM-1), as well as
that of other inflammatory mediators through NF-κB activation in
human endothelial cells (38). We
also found that treatment with visfatin increased the expression of
NF-κB in the EPCs in a dose-dependent manner at both the mRNA and
protein level. Given the regulatory effects of visfatin on
Bcl-2/Bax/caspase-3, the present data showing that pre-treatment
with the inhibitors, FK866 or BAY11-7085, effectively suppressed
visfatin-induced apoptosis, as well as ICAM-1 and IL-6 protein
expression, support the notion that NF-κB in part mediates
visfatin-induced apoptosis. Taken together, the present data
suggest that visfatin is an influential upstream factor inducing
inflammation and the apoptosis of EPCs, and that its activity
results in decreased quantities of EPCs via the upregulation of
NF-κB. These findings are in line with those of previous studies
demonstrating the involvement of NF-κB in endothelial cell
dysfunction and impaired angiogenesis (38,39).
It has previously been reported that FK866
attenuates inflammatory responses and cellular apoptosis following
organ injury, ultimately leading to improved cell survival by
inhibiting NF-κB activation (40). As FK866 is a specific inhibitor of
visfatin, it is reasonable to speculate that visfatin induces
apoptosis, at least in part, through NF-κB activation.
However, different cells show different responses
following visfatin stimulation. Patel et al (41) demonstrated that the exogenous
expression of visfatin in PC3 prostate cancer cells increased tumor
cell proliferation and the expression of matrix metalloproteinase
(MMP)-2/9 at the molecular level. Yang et al (42) generated stable cell lines from
fibrosarcoma HT1080 and 293 cells which overexpress human visfatin
protein and stable cell lines with visfatin knockdown using siRNA,
and found that the stable visfatin-overexpressing cells were
substantially more resistant to apoptosis induced by methylmethane
sulfonate (MMS) than the control. FK866 also activates the
apoptotic cascade in cancer cells with the release of cytochrome
c and the activation of caspase-3 (43,44). In addition, differences in
metabolism have been noted between normal cells and cancer cells
(45). The different metabolic
pathways that cancer cells utilize to survive and proliferate under
stress environments (46) may be
explained by the discrepancy between normal cells and cancer cells
in response to visfatin and FK866.
Furthermore, the effect of visfatin seems also to
rely on dosage and the state of cellular activation. On the one
hand, circulating visfatin induces the cellular expression of
inflammatory cytokines, such as tumor necrosis factor-α (TNF-α),
IL-8, IL-6 and MMP-9 by activating the p38-MAPK signaling pathway
(47,48), in accordance with our data on
EPCs, whereby IL-6 was prominently upregulated by visfatin. On the
other hand, other studies have suggested that higher concentrations
of visfatin result in the upregulation of anti-inflammatory
mediators, such as IL-10 and IL-1 receptor antagonists (9,11).
It is known that visfatin has dual functions in that it acts
extracellularly as a pro-inflammatory cytokine and intracellularly
as an enzyme catalyzing the rate-limiting step of the NAD salvage
pathway from nicotinamide. Specifically, Rongvaux et al
(49) demonstrated that visfatin,
as an enzyme catalyzing the condensation of nicotinamide with
phosphoribosyl pyro-phosphate, participated in cellular resistance
to genotoxic or oxidative stress, and it conferred to cells of the
immune system the ability to survive during stressful situations.
In addition, visfatin enables proliferating human endothelial cells
to resist the oxidative stress indcued by high glucose, and to
productively utilize excess glucose to support replicative
longevity and angiogenic activity via SIRT1 (50), while it also induces angiogenesis
via the MAPK and PI3K/Akt signaling pathways (51). The reason for such discrepancies
in the published findings surrounding visfatin is unclear. The
doses and species used or exposure times are different among the
studies and may have an effect that generates the different
results.
To date, the mechanisms of action of visfatin as a
cytokine remain unclear and numerous questions remain to be
answered. However, our data indicate that visfatin induces EPC
apoptosis by increasing the expression of pro-inflammatory
mediators partly through the regulation of NF-κB. To fully
understand the physiological machanism of visfatin and given the
clear importance of this molecule to several physiological and
pathological processes, further research is required.
Abbreviations:
|
EPCs
|
endothelial progenitor cells
|
|
NF-κB
|
nuclear factor-κB
|
|
ICAM-1
|
intercellular adhesion molecule-1
|
|
IL-6
|
interleukin-6
|
|
Nampt
|
nicotinamide
phosphoribosyltransferase
|
|
NAD
|
nicotinamide adenine dinucleotide
|
|
VCAM-1
|
vascular cell adhesion molecule-1
|
|
MNCs
|
mononuclear cells
|
|
KDR
|
kinase insert domain receptor
|
|
DiI-acLDL
|
DiI-labeled acetylated LDL
|
|
FITC-Lectin-UEA-1
|
fluorescein isothiocyanate-conjugated
lectin from Ulex europaeus agglutinin
|
|
PVDF
|
polyvinylidene fluoride
|
|
ECL
|
enhanced chemiluminescence
|
Acknowledgments
This study was supported by grants from the Hebei
Natural Science Foundation of China (no. H2014307035). We are
indebted to Chao Wang, Hanying Xing, Zhihua Wang and Jingci Yang
for assistance and technical advice.
References
|
1
|
Samal B, Sun Y, Stearns G, Xie C, Suggs S
and McNiece I: Cloning and characterization of the cDNA encoding a
novel human pre-B-cell colony-enhancing factor. Mol Cell Biol.
14:1431–1437. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garten A, Petzold S, Schuster S, Körner A,
Kratzsch J and Kiess W: Nampt and its potential role in
inflammation and type 2 diabetes. Handb Exp Pharmacol. 203:147–164.
2011. View Article : Google Scholar
|
|
3
|
Kadoglou NP, Sailer N, Moumtzouoglou A,
Kapelouzou A, Tsanikidis H, Vitta I, Karkos C, Karayannacos PE,
Gerasimidis T and Liapis CD: Visfatin (nampt) and ghrelin as novel
markers of carotid atherosclerosis in patients with type 2
diabetes. Exp Clin Endocrinol Diabetes. 118:75–80. 2010. View Article : Google Scholar
|
|
4
|
Iacobellis G, Iorio M, Napoli N, Cotesta
D, Zinnamosca L, Marinelli C, Petramala L, Minisola S, D'Erasmo E
and Letizia C: Relation of adiponectin, visfatin and bone mineral
density in patients with metabolic syndrome. J Endocrinol Invest.
34:e12–e15. 2011. View Article : Google Scholar
|
|
5
|
Unlütürk U, Harmanci A, Yildiz BO and
Bayraktar M: Dynamics of Nampt/visfatin and high molecular weight
adiponectin in response to oral glucose load in obese and lean
women. Clin Endocrinol (Oxf). 72:469–474. 2010. View Article : Google Scholar
|
|
6
|
de Luis DA, Aller R, Gonzalez Sagrado M,
Conde R, Izaola O and de la Fuente B: Serum visfatin levels and
metabolic syndrome criteria in obese female subjects. Diabetes
Metab Res Rev. 29:576–581. 2013.PubMed/NCBI
|
|
7
|
Chen S, Sun L, Gao H, Ren L, Liu N and
Song G: Visfatin and oxidative stress influence endothelial
progenitor cells in obese populations. Endocr Res. 40:83–87. 2015.
View Article : Google Scholar
|
|
8
|
Boini KM, Zhang C, Xia M, Han WQ, Brimson
C, Poklis JL and Li PL: Visfatin-induced lipid raft redox signaling
platforms and dysfunction in glomerular endothelial cells. Biochim
Biophys Acta. 1801:1294–1304. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiao K, Zou WH, Yang Z, Rehman Zia ur,
Ansari AR, Yuan HR, Zhou Y, Cui L, Peng KM and Song H: The role of
visfatin on the regulation of inflammation and apoptosis in the
spleen of LPS-treated rats. Cell Tissue Res. 359:605–618. 2015.
View Article : Google Scholar
|
|
10
|
van der Veer E, Nong Z, O'Neil C, Urquhart
B, Freeman D and Pickering JG: Pre-B-cell colony-enhancing factor
regulates NAD+-dependent protein deacetylase activity
and promotes vascular smooth muscle cell maturation. Circ Res.
97:25–34. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moschen AR, Kaser A, Enrich B, Mosheimer
B, Theurl M, Niederegger H and Tilg H: Visfatin, an adipocytokine
with proinflammatory and immunomodulating properties. J Immunol.
178:1748–1758. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vallejo S, Romacho T, Angulo J, Villalobos
LA, Cercas E, Leivas A, Bermejo E, Carraro R, Sánchez-Ferrer CF and
Peiró C: Visfatin impairs endothelium-dependent relaxation in rat
and human mesenteric microvessels through nicotinamide
phosphoribosyltransferase activity. PLoS One. 6:e272992011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Urbich C and Dimmeler S: Endothelial
progenitor cells: Characterization and role in vascular biology.
Circ Res. 95:343–353. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmidt-Lucke C, Rössig L, Fichtlscherer
S, Vasa M, Britten M, Kämper U, Dimmeler S and Zeiher AM: Reduced
number of circulating endothelial progenitor cells predicts future
cardiovascular events: Proof of concept for the clinical importance
of endogenous vascular repair. Circulation. 111:2981–2987. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Werner N, Kosiol S, Schiegl T, Ahlers P,
Walenta K, Link A, Böhm M and Nickenig G: Circulating endothelial
progenitor cells and cardiovascular outcomes. N Engl J Med.
353:999–1007. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Denny MF, Thacker S, Mehta H, Somers EC,
Dodick T, Barrat FJ, McCune WJ and Kaplan MJ: Interferon-alpha
promotes abnormal vasculogenesis in lupus: A potential pathway for
premature atherosclerosis. Blood. 110:2907–2915. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fujii H, Li SH, Szmitko PE, Fedak PW and
Verma S: C-reactive protein alters antioxidant defenses and
promotes apoptosis in endothelial progenitor cells. Arterioscler
Thromb Vasc Biol. 26:2476–2482. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tie G, Yan J, Yang Y, Park BD, Messina JA,
Raffai RL, Nowicki PT and Messina LM: Oxidized low-density
lipoprotein induces apoptosis in endothelial progenitor cells by
inactivating the phosphoinositide 3-kinase/Akt pathway. J Vasc Res.
47:519–530. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun Y, Chen S, Song G, Ren L, Wei L, Liu
N, Zhang D and Lv X: Effect of visfatin on the function of
endothelial progenitor cells in high-fat-fed obese rats and
investigation of its mechanism of action. Int J Mol Med.
30:622–628. 2012.PubMed/NCBI
|
|
20
|
Bakogiannis C, Tousoulis D, Androulakis E,
Briasoulis A, Papageorgiou N, Vogiatzi G, Kampoli AM, Charakida M,
Siasos G, Latsios G, et al: Circulating endothelial progenitor
cells as biomarkers for prediction of cardiovascular outcomes. Curr
Med Chem. 19:2597–2604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peng J, Liu B, Ma QL and Luo XJ:
Dysfunctional endothelial progenitor cells in cardiovascular
diseases: Role of NADPH oxidase. J Cardiovasc Pharmacol. 65:80–87.
2015. View Article : Google Scholar
|
|
22
|
Joseph B, Marchetti P, Formstecher P,
Kroemer G, Lewensohn R and Zhivotovsky B: Mitochondrial dysfunction
is an essential step for killing of non-small cell lung carcinomas
resistant to conventional treatment. Oncogene. 21:65–77. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Salvesen GS and Dixit VM: Caspases:
Intracellular signaling by proteolysis. Cell. 91:443–446. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Korsmeyer SJ: BCL-2 gene family and the
regulation of programmed cell death. Cancer Res. 59(Suppl 7):
1693s–1700s. 1999.PubMed/NCBI
|
|
26
|
Youle RJ and Strasser A: The BCL-2 protein
family: Opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar
|
|
27
|
Laudes M, Oberhauser F, Schulte DM, Freude
S, Bilkovski R, Mauer J, Rappl G, Abken H, Hahn M, Schulz O, et al:
Visfatin/PBEF/Nampt and resistin expressions in circulating blood
monocytes are differentially related to obesity and type 2 diabetes
in humans. Horm Metab Res. 42:268–273. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Y, Ingram DA, Murphy MP, Saadatzadeh
MR, Mead LE, Prater DN and Rehman J: Release of proinflammatory
mediators and expression of proinflammatory adhesion molecules by
endothelial progenitor cells. Am J Physiol Heart Circ Physiol.
296:H1675–H1682. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Adamis AP and Berman AJ: Immunological
mechanisms in the pathogenesis of diabetic retinopathy. Semin
Immunopathol. 30:65–84. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Satofuka S, Ichihara A, Nagai N, Yamashiro
K, Koto T, Shinoda H, Noda K, Ozawa Y, Inoue M, Tsubota K, et al:
Suppression of ocular inflammation in endotoxin-induced uveitis by
inhibiting nonproteolytic activation of prorenin. Invest Ophthalmol
Vis Sci. 47:2686–2692. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Murugeswari P, Shukla D, Rajendran A, Kim
R, Namperumalsamy P and Muthukkaruppan V: Proinflammatory cytokines
and angiogenic and anti-angiogenic factors in vitreous of patients
with proliferative diabetic retinopathy and eales' disease. Retina.
28:817–824. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tang JN, Shen DL, Liu CL, Wang XF, Zhang
L, Xuan XX, Cui LL and Zhang JY: Plasma levels of C1q/TNF-related
protein 1 and interleukin 6 in patients with acute coronary
syndrome or stable angina pectoris. Am J Med Sci. 349:130–136.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kitaba S, Murota H, Terao M, Azukizawa H,
Terabe F, Shima Y, Fujimoto M, Tanaka T, Naka T, Kishimoto T, et
al: Blockade of interleukin-6 receptor alleviates disease in mouse
model of scleroderma. Am J Pathol. 180:165–176. 2012. View Article : Google Scholar
|
|
34
|
Okiyama N, Sugihara T, Iwakura Y, Yokozeki
H, Miyasaka N and Kohsaka H: Therapeutic effects of interleukin-6
blockade in a murine model of polymyositis that does not require
interleukin-17A. Arthritis Rheum. 60:2505–2512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kitagawa K, Matsumoto M, Sasaki T,
Hashimoto H, Kuwabara K, Ohtsuki T and Hori M: Involvement of
ICAM-1 in the progression of atherosclerosis in APOE-knockout mice.
Atherosclerosis. 160:305–310. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bielinski SJ, Pankow JS, Li N, Hsu FC,
Adar SD, Jenny NS, Bowden DW, Wasserman BA and Arnett D: ICAM1 and
VCAM1 polymorphisms, coronary artery calcium, and circulating
levels of soluble ICAM-1: the multi-ethnic study of atherosclerosis
(MESA). Atherosclerosis. 201:339–344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tanaka T, Narazaki M and Kishimoto T: IL-6
in inflammation, immunity, and disease. Cold Spring Harb Perspect
Biol. 6:a0162952014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH,
Koo TH, Jang HO, Yun I, Kim KW, Kwon YG, et al: Visfatin enhances
ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB
activation in endothelial cells. Biochim Biophys Acta.
1783:886–895. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wan R, Liu Y, Li L, Zhu C, Jin L and Li S:
Urocortin increased endothelial ICAM1 by cPLA2-dependent NF-kappaB
and PKA pathways in HUVECs. J Mol Endocrinol. 52:43–53. 2013.
View Article : Google Scholar
|
|
40
|
Matsuda A, Yang WL, Jacob A, Aziz M,
Matsuo S, Matsutani T, Uchida E and Wang P: FK866, a visfatin
inhibitor, protects against acute lung injury after intestinal
ischemia-reperfusion in mice via NF-κB pathway. Ann Surg.
259:1007–1017. 2014. View Article : Google Scholar
|
|
41
|
Patel ST, Mistry T, Brown JE, Digby JE,
Adya R, Desai KM and Randeva HS: A novel role for the adipokine
visfatin/pre-B cell colony-enhancing factor 1 in prostate
carcinogenesis. Peptides. 31:51–57. 2010. View Article : Google Scholar
|
|
42
|
Yang H, Yang T, Baur JA, Perez E, Matsui
T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A,
et al: Nutrient-sensitive mitochondrial NAD+ levels
dictate cell survival. Cell. 130:1095–1107. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hasmann M and Schemainda I: FK866, a
highly specific noncompetitive inhibitor of nicotinamide
phosphoribosyltransferase, represents a novel mechanism for
induction of tumor cell apoptosis. Cancer Res. 63:7436–7442.
2003.PubMed/NCBI
|
|
44
|
Wosikowski K, Mattern K, Schemainda I,
Hasmann M, Rattel B and Löser R: WK175, a novel antitumor agent,
decreases the intracellular nicotinamide adenine dinucleotide
concentration and induces the apoptotic cascade in human leukemia
cells. Cancer Res. 62:1057–1062. 2002.PubMed/NCBI
|
|
45
|
Crabtree HG: Observations on the
carbohydrate metabolism of tumours. Biochem J. 23:536–545. 1929.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kroemer G and Pouyssegur J: Tumor cell
metabolism: Cancer's Achilles' heel. Cancer Cell. 13:472–482. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tan BK, Chen J, Digby JE, Keay SD, Kennedy
CR and Randeva HS: Increased visfatin messenger ribonucleic acid
and protein levels in adipose tissue and adipocytes in women with
polycystic ovary syndrome: Parallel increase in plasma visfatin. J
Clin Endocrinol Metab. 91:5022–5028. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kocelak P, Olszanecka-Glinianowicz M,
Owczarek A, Bożentowicz-Wikarek M, Brzozowska A, Mossakowska M,
Zdrojewski T, Grodzicki T, Więcek A and Chudek J: Plasma
visfatin/nicotinamide phosphoribosyltransferase levels in
hypertensive elderly - results from the PolSenior substudy. J Am
Soc Hypertens. 9:1–8. 2015. View Article : Google Scholar
|
|
49
|
Rongvaux A, Galli M, Denanglaire S, Van
Gool F, Drèze PL, Szpirer C, Bureau F, Andris F and Leo O:
Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing
factor/visfatin is required for lymphocyte development and cellular
resistance to genotoxic stress. J Immunol. 4685–4695. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Borradaile NM and Pickering JG:
Nicotinamide phosphoribosyltransferase imparts human endothelial
cells with extended replicative lifespan and enhanced angiogenic
capacity in a high glucose environment. Aging Cell. 8:100–112.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Adya R, Tan BK, Punn A, Chen J and Randeva
HS: Visfatin induces human endothelial VEGF and MMP-2/9 production
via MAPK and I3K/Akt signalling pathways: Novel insights into
visfatin-induced angiogenesis. Cardiovasc Res. 78:356–365. 2008.
View Article : Google Scholar
|