Introduction
Reduced glutathione (GSH) and its related enzymes
constitute the key antioxidant system in the body for counteraction
of oxidative stress injury (1).
Among these enzymes, glutathione peroxidase (GPx) and its eight GPx
isozymes (GPx1-8) that have been identified to date in mammals
(2) can detoxify reactive oxygen
species (ROS), including hydrogen peroxide and free radicals, in
the presence of GSH. GPx3, which accounts for more than 97% of all
plasma selenium in mice (3), has
been shown to be upregulated in alcohol-induced hepatic injury to
protect the liver from oxidative stress (4). According to a study on GPx gender
differences, both GPx activities and GPx3 concentrations in serum
are higher in females than in males (5).
Acetaminophen (APAP) is a widely available
antipyretic and analgesic drug; however, its overdose can inflict
severe damage on the liver by formation of its reactive metabolite,
N-acetyl-p-benzoquinone imine (NAPQI). NAPQI can
usually be detoxified by GSH, but its excess causes depletion of
GSH, leading to production of ROS, and hence liver injury (6). It is noteworthy that males are more
susceptible to APAP-induced liver injury than females, suggesting
the resistance of females to APAP hepatotoxicity (7–12).
This difference between genders is likely to be due to
gender-dependent activities of GPx in mouse liver (13), although the involvement of gender
differences in the levels and/or activities of other
antioxidant-related enzymes, such as glutathione-S-transferase π
(7) and glutamate-cysteine ligase
(8), cannot be ignored. Indeed,
animals overexpressing plasma GPx have strong resistance to
APAP-induced hepatotoxicity (13). We have recently shown in mice
that, among isozymes of GPx, the degree of APAP-induced
hepatotoxicity depends on the mRNA expression levels of GPx3 in the
blood (14). This finding
therefore raises the question as to whether the gender difference
in the GPx3 level contributes to the difference in APAP-induced
hepatotoxicity between genders. In association with the resistance
to APAP in females, 17β-estradiol, a major female hormone,
specifically attenuates acute hepatic damage and decreases
mortality in APAP-overdosed male mice (15). Expression of the GPx3 gene is
sensitive to circulating estrogens in skeletal muscle (16). A second question, therefore, is
whether the hepatoprotective action of 17β-estradiol on APAP
toxicity is mediated by increased synthesis of GPx3.
To answer these questions, the present study was
carried out to determine the role of the GPx3 protein in
APAP-induced hepatotoxicity in vivo and in vitro. In
in vivo experiments in mice, we examined whether the degree
of the GPx3 level accounts for the gender differences in
APAP-induced hepatic injury, and whether GPx3 mediates the
17β-estradiol reduction in APAP-induced hepatotoxicity. In in
vitro experiments, to better clarify the role of cellular GPx3
in APAP-induced injury, we evaluated NAPQI-induced cellular injury
when cellular GPx3 expression was altered by either transfected
GPx3 siRNA or a transfected GPx3 expression vector.
Materials and methods
Animals and chemicals
Four-week-old adult male or female ddY mice (20–25
g) were obtained from Japan SLC Inc. (Hamamatsu, Japan). The
animals were maintained on a 12-h light/dark cycle in a
temperature- and humidity-controlled room. The experiments were
conducted in accordance with the standards established by the
Japanese Pharmacological Society and were approved by the Tohoku
Medical and Pharmaceutical University of Institutional Animal Care
and Use Committee (experimental no. 16014). The animals were
allowed free access to laboratory pellet chow (CE-2; Clea Japan,
Inc., Tokyo, Japan) and water before the experiments. APAP was
purchased from Junsei Chemical Co., Ltd. (Nagano, Japan).
17β-estradiol was obtained from Sigma Chemical Co. (St. Louis, MO,
USA). NAPQI was purchased from Toronto Research Chemicals Inc.
(Toronto, Canada). All other reagents, unless stated, were of the
highest grade available and were supplied by either Sigma or Wako
Pure Chemical Industries, Ltd. (Osaka, Japan).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
assay
The expression levels of mRNA were quantified using
RT-qPCR according to our previously described methods (14). The animals were acclimated for 5–7
days prior to a pre-dose blood draw. Blood was drawn from the tail
vein of each mouse into tubes containing 1 mM EDTA as an
anticoagulant. Total RNA was isolated using the ISOGEN reagent
(Nippon Gene Co., Ltd., Tokyo, Japan), and RNA concentrations were
determined using the NanoDrop 1000 (Thermo Fisher Scientific,
Waltham, MA, USA). Total RNA from each sample (0.1 µg) was reverse
transcribed into single-stranded cDNA using the ReverTra Ace kit
(Toyobo Co., Ltd., Osaka, Japan). Aliquots of the resulting cDNA
preparations were then subjected to qPCR analysis using the KOD
SYBR® qPCR Mix (Toyobo Co., Ltd.). A CFX
Connect™ Real-Time PCR system (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) was used to determine mRNA expression
levels of the GPx3 gene (GenBank accession no. NM_008161.3). The
mRNA level was normalized against that of the GAPDH-encoding locus
(GenBank accession no. NM_001289726). The sequences of the primer
pairs used were obtained from the Takara Perfect Real Time Primers
(Takara Bio, Shiga, Japan). The results of all assays were checked
against melting curves in order to confirm the presence of single
PCR products. At least two independent experiments were conducted
and samples were assessed in (at least) triplicate in each
experiment.
APAP hepatotoxicity and plasma
concentration
Mice were orally administered (p.o.) APAP (500 mg/kg
in 10 ml/kg saline) at 18:00 h, and blood was collected 18 h later
for determination of the serum activity of alanine aminotransferase
(ALT) and aspartate aminotransferase (AST) using a colorimetric kit
(Wako, Tokyo, Japan) as described in our previous reports (17,18). Male mice were pretreated with
17β-estradiol at a dose of 0.2 mg/kg that was administered by
intraperitoneal (i.p.) injection 4 h before treatment with APAP.
The plasma concentration of APAP was measured using a modification
of the method of Hori et al (19). Briefly, an aliquot (100 µl) of
plasma was diluted with an equal volume of 0.2 M
Na2HPO4 and 0.1 M citric acid buffer (pH
3.0), and the mixture then was combined with 300 µl of ethyl
acetate. The resulting samples were centrifuged at 3,000 × g for 5
min and the upper layer was retained and the lower layer was
discarded. After the removal of solvents by evaporation under
nitrogen gas, the sample was dissolved in 100 µl of the mobile
phase (methanol and 1 N acetic acid at a 70:30 ratio, v/v), and
injected into a high-performance liquid chromatography system
consisting of an Alliance 2695 Separations Module (Waters, Milford,
MA, USA). The flow rate was kept constant at a rate of 0.4 ml/min,
and peaks were monitored at a wavelength of 254 nm for 10 min. The
concentration of APAP was calculated using an APAP standard
curve.
GPx activity
Mouse plasma GPx was measured using a Glutathione
Peroxidase Assay kit (catalog no. 703102; Cayman Chemical Co., Ann
Arbor, MI, USA). The plasma samples were collected from a cut of
the tail vein on the day before APAP treatment. The GPx level in
each plasma sample (20 µl) was analyzed according to the protocol
supplied by the manufacturer of the kit.
Cell culture
The Huh-7 human liver cancer cell line and the K562
human erythroleukemia cell line were supplied by the Cell Resource
Center for Biomedical Research, Tohoku University (Sendai, Japan).
The cells were maintained in RPMI-1640 medium supplemented with 10%
fetal bovine serum, 100 U/ml penicillin G, and 100 µg/ml
streptomycin at 37°C in a humidified 5% CO2-95% air
incubator under standard conditions. The cells were counted,
excluding cells stained with 0.2% Trypan blue. To maintain
exponential growth, cells were seeded at a density of
5×104 cells/ml and were passaged every 3–4 days. Cells
were cultured in 2 ml aliquots in 35-mm dishes for other
assays.
Cell survival assay
Cellular survival was assessed using the
water-soluble tetrazolium WST-1 (sodium 5-(2,4-disulfophe
nyl)-2-(4-iodophenyl)-3-(4-nitrophenyl)-2H tetrazolium inner salt)
assay, which detects metabolically competent cells with an intact
mitochondrial electron transport chain (20). Briefly, 1×104 cells
were seeded into 96-well plates and cultured overnight. The cells
were incubated with NAPQI for the indicated times, and medium
containing the WST-1 solution (0.5 mM WST-1 and 0.02 mM
1-methoxy-5-methylphenazinium methylsulfate; 1-PMS) was added to
each well. The cells were incubated for 60 min at 37°C, and
absorption at a wavelength of 438 nm (ref. 620 nm) was measured
using a SH-1200 Microplate Reader® (Corona, Hitachinaka,
Japan). Control cells were treated with 0.1% DMSO. Cell viability
was calculated using the following formula: Absorbance in the
treated sample/absorbance in the control ×100 (%).
GPx3 knockdown
siRNA-GPx3 (siGPx3) and siRNA-control [non-targeting
siRNA; negative control (Neg)] were transfected into Huh-7 or K562
cells using HyperFect transfection reagent (Qiagen, Inc., Valencia,
CA, USA) according to the protocol supplied by the manufacturer. A
non-targeting siRNA was used as a control for the
non-sequence-specific effects of the transfected siRNAs. The siRNAs
used were siGPx3, a Silencer® Select Pre-designed siRNA
Product (ID no. s6109; Ambion, Austin, TX, USA), and negative
control siRNA from AllStars Neg. Control siRNA (ID no. AM4611;
Qiagen, Inc.). Briefly, 5×104 cells containing each
siRNA (final concentration, 10 nM) and the HyperFect reagent were
incubated for 24 h for assessment of GPx3 expression or cytotoxic
effects induced by NAPQI.
GPx3 overexpression
The cells were transfected with a GFP-tagged ORF
clone of GPx3 cloned into a pCMV6-AC-GFP vector (OriGene
Technologies, Inc., Rockville, MD, USA). Plasmid DNA was
transfected into Huh-7 or K562 cells using ViaFect™ Transfection
Reagent (Promega Corp., Madison, WI, USA) and the Neon™
Transfection System (Invitrogen Life Technologies, Carlsbad, CA,
USA), respectively, according to the instructions provided by the
manufacturer. The presence of the transfected vector in the cells
was confirmed by fluorescence microscopic observation (488 nm, GFP
fluorescence) as previously described (21).
Western blot analysis
The cells were washed with phosphate buffered saline
(PBS) and lysed in CelLytic M® (Sigma-Aldrich, St.
Louis, MO, USA) to collect a total cell lysate according to the
manufacturer's instructions. Protein concentration was measured
using the BCA™ protein assay kit (Thermo Fisher
Scientific, Inc., Rockford, IL, USA) according to the instructions
provided by the manufacturer. Following electrophoreses of protein
samples (30 µg) on a 10% SDS-polyacrylamide gel, the protein
was transferred to a polyvinylidene difluoride membrane. The
membrane was blocked with Blocking One® (Nacalai Tesque,
Inc., Kyoto, Japan) for 1 h and then incubated with a primary
antibody overnight at 4°C. Antibodies against human GPx3 (mouse
monoclonal; ab27325; Abcam, Cambridge, MA, USA) and against β-actin
as the loading control (rabbit polyclonal; #4967; Cell Signaling
Technology, Inc., Danvers, MA, USA) were used. The membrane was
then washed with wash buffer (PBS containing 0.05% Tween-20) and
incubated with horseradish peroxidase-linked secondary antibody
[anti-mouse IgG (#7076) or anti-rabbit IgG (#7074); Cell Signaling
Technology, Inc.] for 1 h. After another wash with wash buffer,
protein signals were analyzed by enhanced chemiluminescence with
the Pierce® Western Blotting substrate (Thermo Fisher
Scientific, Inc.).
Statistical analysis
Statistical analysis was performed with two-way
analysis of variance, followed by the Bonferroni test to compare
among multiple groups. Data are expressed as means ± standard error
of the mean (SEM). P<0.05 was considered to indicate a
statistically significant difference. Statistical analyses were
performed using Ekuseru-Toukei 2012 software (Social Survey
Research Information Co., Ltd., Tokyo, Japan).
Results
Female mice are resistant to APAP
hepatotoxicity and show a higher GPx3 activity
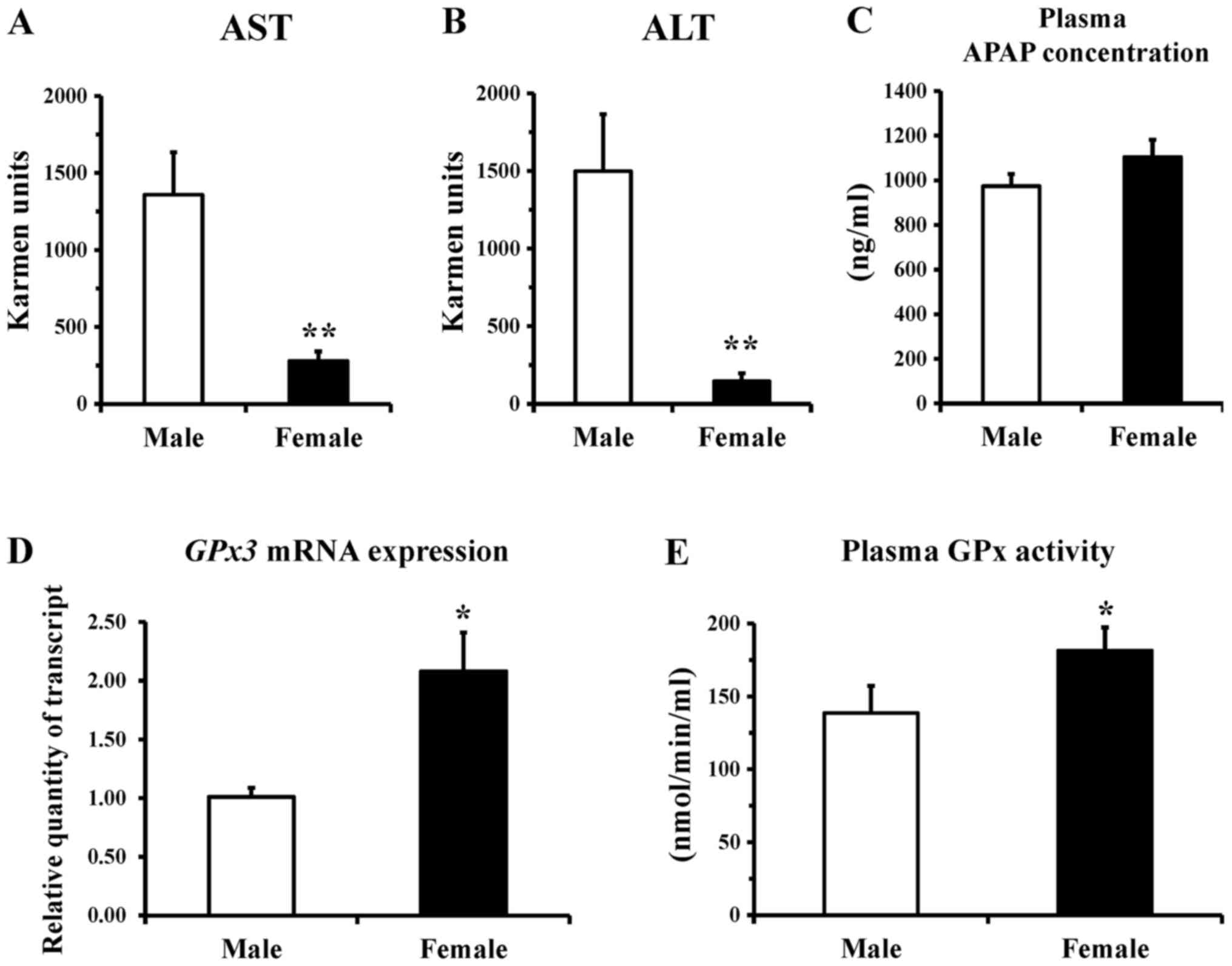
There was no significant difference in serum
activities of AST and ALT, which are markers of liver injury,
between male and female mice before APAP treatment. These values
were 135±28 KU and 48±8 KU, respectively, in male mice, and 104±18
KU and 38±11 KU, respectively, in female mice. Compared to these
basal values, the serum activities of AST and ALT after oral
administration of APAP were increased 10- and 31-fold, respectively
in male mice, whereas they were only increased 3- and 4-fold,
respectively in female mice (Fig. 1A
and B). These results suggested that APAP inflicted damage to
the liver of the mice (ddY mice), and that sensitivity to APAP
hepatotoxicity was much lower in females than in males. It was
noted, however, that the plasma APAP concentration after the
treatment was not significantly different between the male and
female mice (Fig. 1C). Female
mice also showed a slight but significantly higher activity of
plasma GPx (Fig. 1E), and a
2-fold higher expression of Gpx3 mRNA (Fig. 1D), relative to the male mice,
suggesting higher activity of this antioxidant-related enzyme in
females.
17β-estradiol attenuates APAP-induced
hepatotoxicity and increases GPx3 in male mice
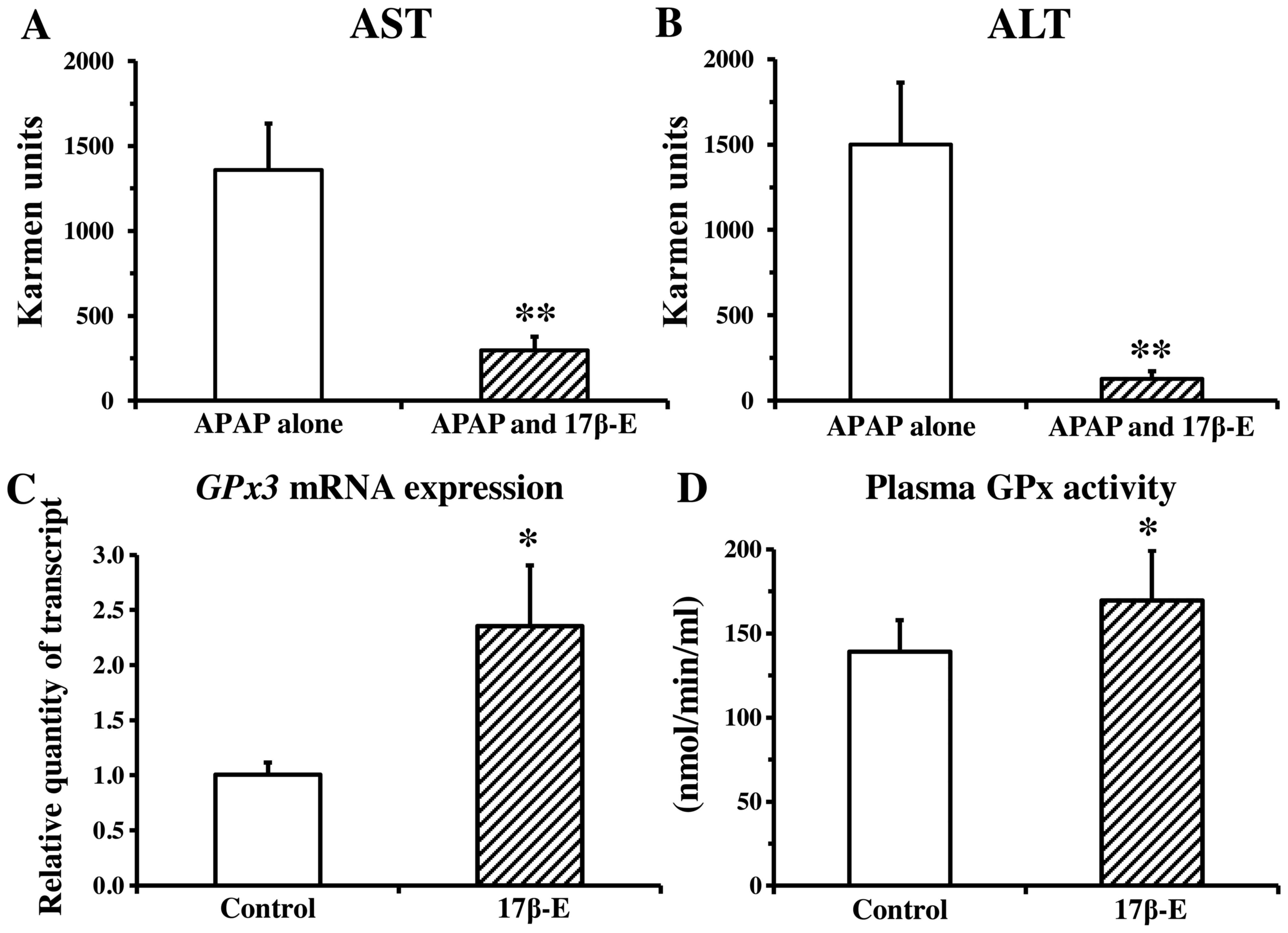
We examined the effect of 17β-estradiol on
APAP-induced hepatotoxicity in male mice. In a preliminary
experiment, pretreatment with 17β-estradiol did not influence
plasma APAP concentrations (data not shown). As shown in Fig. 2A and B, serum aminotransferase
activities in the group treated with a combination of 17β-estradiol
and APAP were much lower than those in the group treated with APAP
alone, suggesting a protective action of 17β-estradiol against
APAP-induced hepatotoxicity. The blood level of Gpx3 mRNA
expression was approximately 2-fold higher (Fig. 2C) and the activities of GPx in
plasma were marginally significantly elevated (Fig. 2D) in the 17β-estradiol-treated
group compared with the control group, suggesting that
17β-estradiol enhances the activities of the antioxidant-related
enzyme in males.
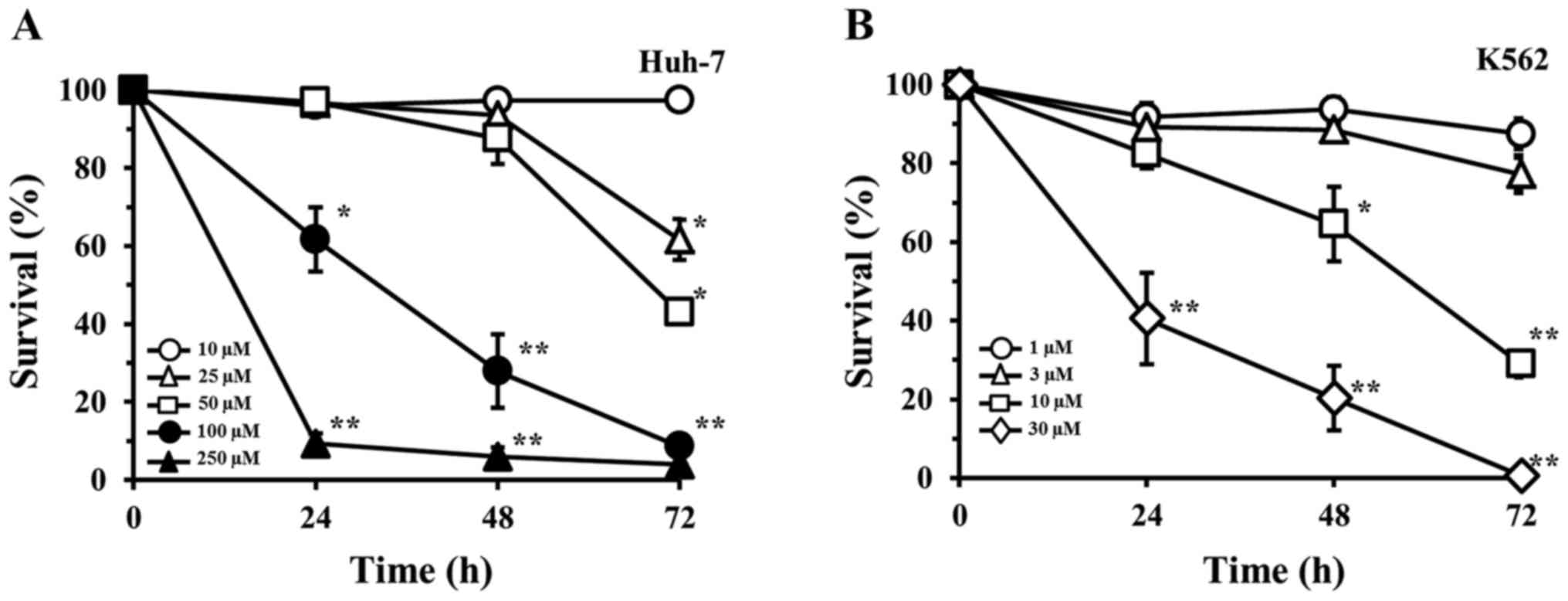
NAPQI-induced reduction in cell survival
is affected by cellular GPx3 expression
To determine the effects of GPx3 on APAP-induced
toxicity at the cellular level, in vitro experiments were
performed with heterogeneous cultured human cell lines (the human
hepatoma Huh-7 cells and the human erythroleukemia K562 cells). The
extent of cell injury in response to various concentrations of
NAPQI, as judged by analysis of cell survival, is shown in Fig. 3A and B. NAPQI reduced the survival
of both Huh-7 and K562 cell lines in a concentration- and
time-dependent manner. The amount of NAPQI required to inhibit cell
survival was higher in the Huh-7 cells than this amount in the K562
cells; e.g., the 50% inhibitory concentrations (IC50) at
72 h of incubation were calculated as 35.5 and 5.4 µM,
respectively, indicating that the NAPQI sensitivity was 6.6-fold
lower in Huh-7 cells than that noted in the K562 cells.
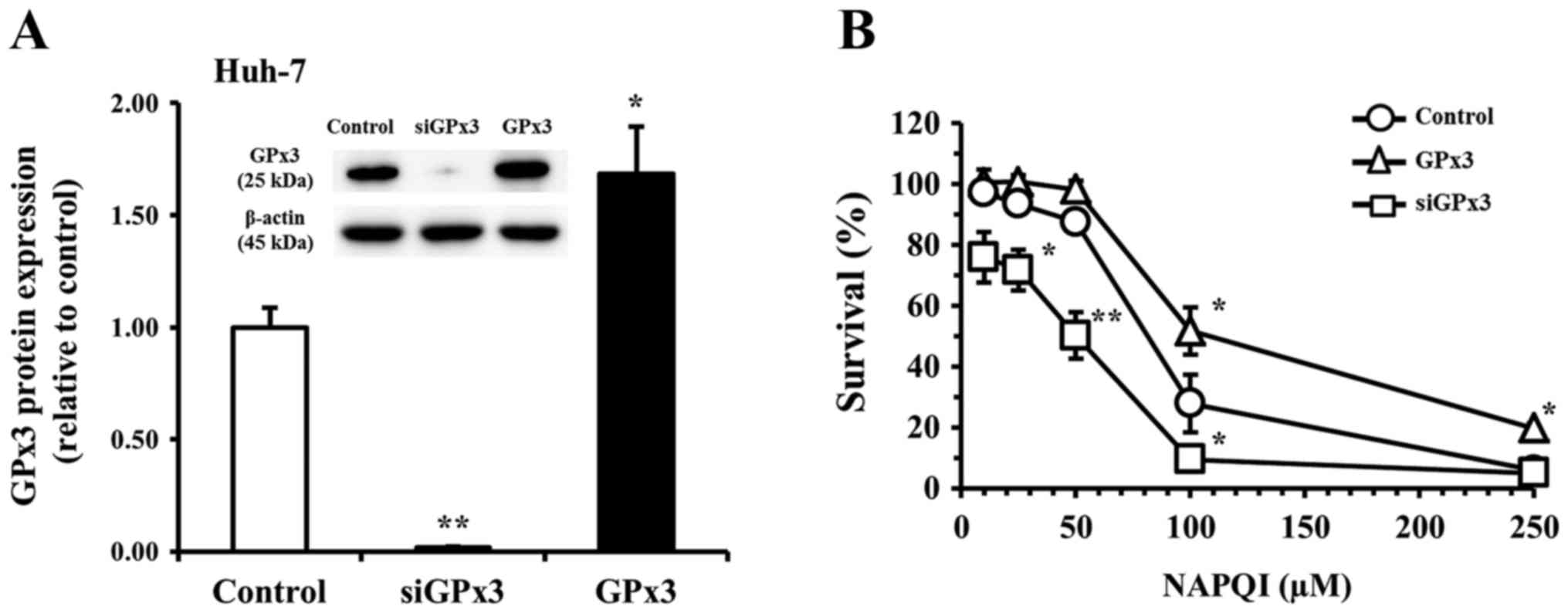
We next examined the potential role of GPx3 in these
effects of NAPQI by analysis of the effect of changes in cellular
GPx3 expression brought about by transfection of siGPx3 or a GPx3
expression vector on the NAPQI-induced reduction of cell survival
in the Huh-7 (Fig. 4) or K562
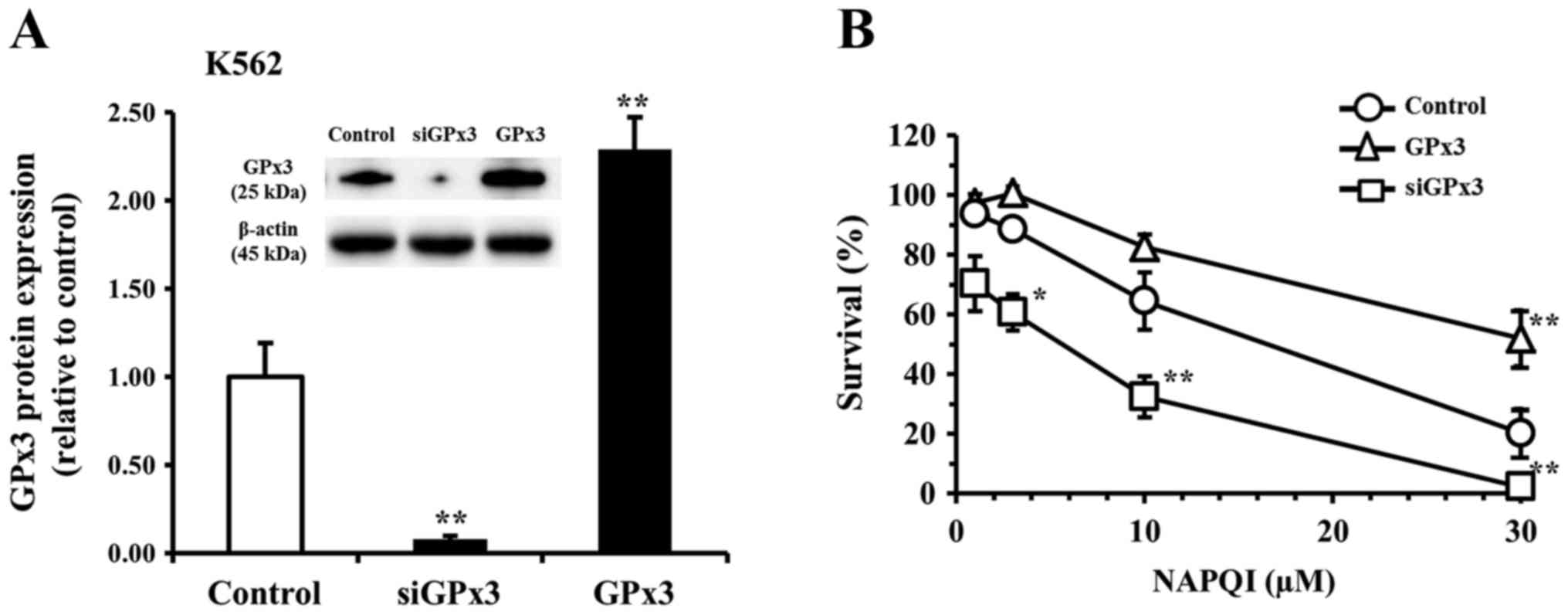
(Fig. 5) cell line. After
transfection of Huh-7 and K562 cells with siGPx3 for 24 h, the
expression level of GPx3 was barely detectable in either cell line
compared to the control (non-transfected) group. The knockdown
efficacy of GPx3 mRNA was estimated to be >99% by RT-qPCR. The
expression levels of GPx3 in the GPx3 vector-transfected group were
1.7-fold and 2.3-fold higher in the Huh-7 and K562 cells,
respectively, than that in the respective non-transfected control
group (Figs. 4A and 5A). In the non-transfected control Huh-7
group, incubation (for 48 h) with NAPQI reduced cell survival to
97, 94, 88, 28, and 6% of the control at a concentration of 10, 25,
50, 100, and 250 µM, respectively (Fig. 4B). In the non-transfected control
K562 group, incubation (for 48 h) with NAPQI reduced cell survival
to 94, 89, 65, and 20% of the control at a concentration of 1, 3,
10, and 30 µM of NAPQI, respectively (Fig. 5B). The reduction in cell survival
by NAPQI was potentiated in each siGPx3-transfected group, but was
inhibited in each GPx3-transfected group (GPx3 overexpression
group) vs. the respective non-treated control in both cell lines
(Figs. 4B and 5B). In a preliminary experiment, neither
transfection with control siRNA nor with vector alone affected cell
survival of either cell line at incubation times of 72 h or
shorter. These results suggested that GPx3 has a protective role
against NAPQI-induced cellular injury in both Huh-7 and K562
cells.
Discussion
The present study demonstrated that APAP caused
liver injury in vivo, as assessed by increases in serum AST
and ALT levels in ddY mice, and that females were less sensitive to
APAP hepatotoxicity than males (Fig.
1). Resistance of females to APAP-induced hepatotoxicity has
also been observed in another mouse species, C57BL/6 mice (7,9,10).
These findings suggest that APAP induces gender-dependent but
species-independent liver injury in mice. It is well established
that there is a gender difference in the activities of liver
metabolic enzymes, which are involved in metabolism and excretion
of drugs. However, the gender difference in APAP-induced
hepatotoxicity is not likely to be due to a difference in
metabolism and excretion of this drug, since the concentrations of
free APAP in bile, urine, or serum were not reported to be
different between male and female mice (7). We also confirmed that there was no
difference in the plasma concentration of APAP between genders. It
is unlikely, therefore, that the resistance to APAP-induced
hepatotoxicity in females is due to the difference in APAP
pharmacokinetics between genders.
It is well established that APAP hepatotoxicity is
initiated by the formation of NAPQI, which causes depletion of GSH,
leading to production of ROS, and hence hepatic injury. Some
previous reports have indicated that the activity of
glutamate-cysteine ligase, a rate-limiting enzyme in GSH synthesis,
is higher in female mice than in male mice, and therefore early
recovery of hepatic GSH may confer resistance to APAP-induced liver
injury. These findings suggest that gender differences in APAP
hepatotoxicity can be attributed to the activity of
glutamate-cysteine ligase (8–11).
Other reports have shown that GSH amount or GSH-related enzymatic
activity in mouse liver is involved in the gender difference in
APAP hepatotoxicity (7, 8). The analysis of blood samples in the
present study demonstrated that both blood GPx3 mRNA expression and
plasma GPx activity (i.e., GPx3 activity) were higher in female
mice than in male mice. Since GPx3 can protect some tissues
including liver from oxidative stress by ROS, the resistance to
APAP toxicity observed in female mice is thought to be due, at
least in part, to higher GPx3 activity. These data suggested that
endogenous estrogens may play a role in the resistance to APAP
toxicity and the increased activity of GPx that was observed in
female mice. In an in vivo experiment, we found that
treatment of male mice with 17β-estradiol, which is one of the most
active estrogens, markedly inhibited the APAP-induced increases in
serum AST and ALT, and increased both Gpx3 mRNA expression and
plasma GPx activity vs. the non-treated control mice. These data
(Fig. 2) were comparable to those
in female mice (Fig. 1). These
findings strongly suggested that higher activity of GPx3
contributes to the beneficial action of 17β-estradiol and to the
resistance of females against APAP hepatotoxicity. Another study
demonstrated that 17β-estradiol is capable of attenuating APAP
toxicity without a change in normal GSH levels in male liver
(15). Thus, 17β-estradiol can
protect the liver from APAP-induced toxicity, and the antioxidant
activity of estrogen is attributed to its effect on powerful
antioxidant enzymes in the whole body.
It should be noted, however, that estrogens exert
antioxidant actions through a variety of mechanisms; they both
reduce the production of ROS and upregulate a number of cellular
antioxidative defense molecules (22–24). The antioxidant potency of
estrogens, which is shared with other phenolic agents, is thought
to be due to the presence of a benzene ring that scavenges hydroxyl
radicals (25). On the other
hand, the antioxidant effect of estrogens on rat hepatocytes is not
dependent on the chemical structure of the estrogens per se,
but rather on their hormonal effects (26). Within the cell, estrogens bind to
their membrane-bound receptors, activate the MAP kinase-NFκB
pathway, and increase transcription of antioxidant enzymes,
especially GPx and superoxide dismutase (SOD) 2 (27). In addition, the activity of
certain antioxidant enzymes including GPx is increased in
lymphocytes treated with estrogen and its receptor agonists
(28). These findings in
biochemical and immune fields support the effect of estrogen to
enhance the activity of antioxidant enzymes. Thus, the antioxidant
effect of estrogens is likely to be due to both direct and indirect
protective activities against ROS, although the detailed mechanisms
remain unclear.
Intracellular overexpression of GPx can detoxify the
final products of oxidative stress and is more efficient in
protecting cells in vitro against ROS than SOD or catalase
(29). To clarify the role of GPx
in cell damage induced by NAPQI in vitro, we investigated
whether NAPQI-induced cell death is altered by a change in cellular
GPx3 expression brought about by transfection of either siGPx3 or a
GPx3 expression vector into Huh-7 or K562 cells (Figs. 4 and 5). These experiments showed that
NAPQI-induced cell death was reduced by increased GPx3 expression
and was enhanced by decreased GPx3 expression, suggesting a crucial
role for intracellular GPx in preventing the oxidative stress
induced by NAPQI. The role of GPx in APAP-induced hepatotoxicity
could be further clarified by analysis using knock-out mice of GPx.
However, we did not perform this analysis, as there is no available
knock-out in mouse strain (ddY) used in the present study. There is
also previous evidence that shows that the antioxidant action of
GPx contributes to inhibition of tumor progression and recurrence.
The expression of GPx3 is downregulated within tumor tissues in
several types of cancers (30–33). In addition, lower amounts of
plasma GPx3 is correlated with tumor progression and recurrence in
hepatocellular carcinoma (HCC) patients, and overexpression of GPx3
or administration of recombinant GPx3 inhibits the proliferation
and invasiveness of HCC cells (34). In our experiments, there was no
significant change in cell growth by transfection of siGPx3 or a
GPx3 expression vector during incubation for 72 h after
transfection compared to the non-transfected or control-transfected
cells. However, longer incubation could possibly change the growth
of these transfected cells.
In conclusion, GPx3 is an important factor for
inhibition of APAP-induced hepatotoxicity both in vivo and
in vitro. To our knowledge, this is the first report to show
a hepatoprotective role of cellular GPx3, although there have been
many reports of the attenuation of APAP-induced liver injury by
antioxidant enzymes. Nevertheless, more studies are required to
determine why GPx3 transcription is regulated by estrogen or other
factors and to investigate the mechanisms of resistance to APAP
toxicity.
Acknowledgments
This study was supported in part by a Grant-in-Aid
for Scientific Research (C) (KAKENHI 25460220) from the Japan
Society for the Promotion of Science, and by a Matching Fund
Subsidy for Private Universities from the Ministry of Education,
Culture, Sports, Science and Technology of Japan.
References
|
1
|
Deneke SM and Fanburg BL: Regulation of
cellular glutathione. Am J Physiol. 257:L163–L173. 1989.PubMed/NCBI
|
|
2
|
Brigelius-Flohé R and Maiorino M:
Glutathione peroxidases. Biochim Biophys Acta. 1830:3289–3303.
2013. View Article : Google Scholar
|
|
3
|
Olson GE, Whitin JC, Hill KE, Winfrey VP,
Motley AK, Austin LM, Deal J, Cohen HJ and Burk RF: Extracellular
glutathione peroxidase (Gpx3) binds specifically to basement
membranes of mouse renal cortex tubule cells. Am J Physiol Renal
Physiol. 298:F1244–F1253. 2010. View Article : Google Scholar :
|
|
4
|
Li YG, Ji DF, Zhong S, Shi LG, Hu GY and
Chen S: Saponins from Panax japonicus protect against
alcohol-induced hepatic injury in mice by upregulating the
expression of GPX3, SOD1 and SOD3. Alcohol Alcohol. 45:320–331.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rush JW and Sandiford SD: Plasma
glutathione peroxidase in healthy young adults: Influence of gender
and physical activity. Clin Biochem. 36:345–351. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nelson SD: Molecular mechanisms of the
hepatotoxicity caused by acetaminophen. Semin Liver Dis.
10:267–278. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dai G, He L, Chou N and Wan YJ:
Acetaminophen metabolism does not contribute to gender difference
in its hepatotoxicity in mouse. Toxicol Sci. 92:33–41. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sheng Y, Liang Q, Deng Z, Ji L and Wang Z:
Acetaminophen induced gender-dependent liver injury and the
involvement of GCL and GPx. Drug Discov Ther. 7:78–83.
2013.PubMed/NCBI
|
|
9
|
Botta D, Shi S, White CC, Dabrowski MJ,
Keener CL, Srinouanprachanh SL, Farin FM, Ware CB, Ladiges WC,
Pierce RH, et al: Acetaminophen-induced liver injury is attenuated
in male glutamate-cysteine ligase transgenic mice. J Biol Chem.
281:28865–28875. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McConnachie LA, Mohar I, Hudson FN, Ware
CB, Ladiges WC, Fernandez C, Chatterton-Kirchmeier S, White CC,
Pierce RH and Kavanagh TJ: Glutamate cysteine ligase modifier
subunit deficiency and gender as determinants of
acetaminophen-induced hepatotoxicity in mice. Toxicol Sci.
99:628–636. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Masubuchi Y, Nakayama J and Watanabe Y:
Sex difference in susceptibility to acetaminophen hepatotoxicity is
reversed by buthionine sulfoximine. Toxicology. 287:54–60. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Du K, Williams CD, McGill MR and Jaeschke
H: Lower susceptibility of female mice to acetaminophen
hepatotoxicity: Role of mitochondrial glutathione, oxidant stress
and c-jun N-terminal kinase. Toxicol Appl Pharmacol. 281:58–66.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mirochnitchenko O, Weisbrot-Lefkowitz M,
Reuhl K, Chen L, Yang C and Inouye M: Acetaminophen toxicity.
Opposite effects of two forms of glutathione peroxidase. J Biol
Chem. 274:10349–10355. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kanno S, Tomizawa A and Yomogida S:
Detecting mRNA Predictors of acetaminophen-induced hepatotoxicity
in mouse blood using quantitative real-time PCR. Biol Pharm Bull.
39:440–445. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chandrasekaran VR, Periasamy S, Liu LL and
Liu MY: 17β-Estradiol protects against
acetaminophen-overdose-induced acute oxidative hepatic damage and
increases the survival rate in mice. Steroids. 76:118–124. 2011.
View Article : Google Scholar
|
|
16
|
Baltgalvis KA, Greising SM, Warren GL and
Lowe DA: Estrogen regulates estrogen receptors and antioxidant gene
expression in mouse skeletal muscle. PLoS One. 5:e101642010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kanno S, Ishikawa M, Takayanagi M,
Takayanagi Y and Sasaki K: Potentiation of acetaminophen
hepatotoxicity and mortality by doxapram in mice. Biol Pharm Bull.
21:934–937. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kanno S, Tomizawa A, Hiura T, Osanai Y,
Kakuta M, Kitajima Y, Koiwai K, Ohtake T, Ujibe M and Ishikawa M:
Melatonin protects on toxicity by acetaminophen but not on
pharmacological effects in mice. Biol Pharm Bull. 29:472–476. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hori Y, Iwasaki Y, Kuroki Y, Komiyayama Y,
Nakatani H and Namera A: Practical analysis of toxic substances
useful for clinical toxicology–4–Acetaminophen. Chudoku Kenkyu.
15:385–390. 2002.
|
|
20
|
Berridge MV, Herst PM and Tan AS:
Tetrazolium dyes as tools in cell biology: New insights into their
cellular reduction. Biotechnol Annu Rev. 11:127–152. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kanno S, Kurauchi K, Tomizawa A, Yomogida
S and Ishikawa M: Pifithrin-alpha has a p53-independent
cytoprotective effect on docosahexaenoic acid-induced cytotoxicity
in human hepatocellular carcinoma HepG2 cells. Toxicol Lett.
232:393–402. 2015. View Article : Google Scholar
|
|
22
|
Huh K, Shin US, Choi JW and Lee SI: Effect
of sex hormones on lipid peroxidation in rat liver. Arch Pharm Res.
17:109–114. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gómez-Zubeldia MA, Hernandez R, Viguera J,
Arbues JJ, Aparicio A and Millán JC: Effect of bilateral
ovariectomy and ovarian steroid hormones on the antioxidant systems
and plasma malondialdehyde levels in Wistar rats. Endocr Res.
26:97–107. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kumar S, Lata K, Mukhopadhyay S and
Mukherjee TK: Role of estrogen receptors in pro-oxidative and
anti-oxidative actions of estrogens: A perspective. Biochim Biophys
Acta. 1800:1127–1135. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sugioka K, Shimosegawa Y and Nakano M:
Estrogens as natural antioxidants of membrane phospholipid
peroxidation. FEBS Lett. 210:37–39. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ruiz-Larrea MB, Leal AM, Martín C,
Martínez R and Lacort M: Antioxidant action of estrogens in rat
hepatocytes. Rev Esp Fisiol. 53:225–229. 1997.PubMed/NCBI
|
|
27
|
Borrás C, Gambini J, Gómez-Cabrera MC,
Sastre J, Pallardó FV, Mann GE and Viña J: 17beta-oestradiol
up-regulates longevity- related, antioxidant enzyme expression via
the ERK1 and ERK2[MAPK]/NFkappaB cascade. Aging Cell. 4:113–118.
2005. View Article : Google Scholar
|
|
28
|
Priyanka HP, Krishnan HC, Singh RV, Hima L
and Thyagarajan S: Estrogen modulates in vitro T cell responses in
a concentration- and receptor-dependent manner: Effects on
intracellular molecular targets and antioxidant enzymes. Mol
Immunol. 56:328–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Michiels C, Raes M, Toussaint O and
Remacle J: Importance of Se-glutathione peroxidase, catalase, and
Cu/Zn-SOD for cell survival against oxidative stress. Free Radic
Biol Med. 17:235–248. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Barrett CW, Ning W, Chen X, Smith JJ,
Washington MK, Hill KE, Coburn LA, Peek RM, Chaturvedi R, Wilson
KT, et al: Tumor suppressor function of the plasma glutathione
peroxidase gpx3 in colitis-associated carcinoma. Cancer Res.
73:1245–1255. 2013. View Article : Google Scholar :
|
|
31
|
He Y, Wang Y, Li P, Zhu S, Wang J and
Zhang S: Identification of GPX3 epigenetically silenced by CpG
methylation in human esophageal squamous cell carcinoma. Dig Dis
Sci. 56:681–688. 2011. View Article : Google Scholar
|
|
32
|
Peng DF, Razvi M, Chen H, Washington K,
Roessner A, Schneider-Stock R and El-Rifai W: DNA hypermethylation
regulates the expression of members of the Mu-class glutathione
S-transferases and glutathione peroxidases in Barrett's
adenocarcinoma. Gut. 58:5–15. 2009. View Article : Google Scholar
|
|
33
|
Murawaki Y, Tsuchiya H, Kanbe T, Harada K,
Yashima K, Nozaka K, Tanida O, Kohno M, Mukoyama T, Nishimuki E, et
al: Aberrant expression of selenoproteins in the progression of
colorectal cancer. Cancer Lett. 259:218–230. 2008. View Article : Google Scholar
|
|
34
|
Qi X, Ng KT, Lian QZ, Liu XB, Li CX, Geng
W, Ling CC, Ma YY, Yeung WH, Tu WW, et al: Clinical significance
and therapeutic value of glutathione peroxidase 3 (GPx3) in
hepatocellular carcinoma. Oncotarget. 5:11103–11120. 2014.
View Article : Google Scholar : PubMed/NCBI
|