Introduction
Breast cancer-associated gene 3 (BCA3), also known
as A kinase interacting protein 1 (Akip1) was first identified in
mRNA screens of breast and prostate cancer cell lines (1). BCA3 was found through a yeast
two-hybrid screen as a protein of unknown function that interacted
with the N-terminal 30 residues of PKAc (2). BCA3 was later characterized as an
important regulator of nuclear factor-κB (NF-κB) signaling by
interacting with p65 and protein kinase A (PKA) (3–5).
BCA3 is highly conserved among human, mouse and other species.
Multiple phosphorylation sites and functional domains, such as a
proline-rich domain (1), and SH2
and SH3 binding sites have been predicted based on its conserved
amino-acid sequence (6), which
suggests that BCA3 can interact with a wide variety of proteins.
Consistent with that prediction, BCA3 has been reported to interact
with several other proteins in addition to p65 and PKA (7–9).
The interaction of BCA3 with TAp73 has been reported to enhance the
sensitivity of cervical cancer cells to irradiation (7). BCA3 is a KyoT2 binding protein and
may compete with really interesting new gene 1 (RING1) in
regulating Notch signaling (8).
Moreover, BCA3 can interact with the mitochondrial localized
apoptosis inducing factor (AIF) and increase cardiomyocyte
tolerance to oxidant and ischemic stress (10). Some interactions cause
post-translational modifications of BCA3. Thus, interaction with
secretory protein with RING finger domain (SPRING) leads to the
ubiquitination of BCA3 (11).
Similarly, BCA3 is neddylated by its interaction with NEDD8 and
inhibits NF-κB signaling (9).
BCA3 was also identified as a Rac1-interacting
protein in a yeast 2-hybrid screen using an osteoclast cDNA library
as bait (12). Rac1 plays an
important role in colony stimulating factor 1 (CSF1)-induced
cytoskeleton remodeling in mature osteoclasts and the
overexpression of BCA3 in these cells attenuates CSF1-induced
cytoskeletal remodeling (13).
Both BCA3 and Rac1 have been reported to play a role in NF-κB
signaling (3,14,15). Thus, in this study, we sought to
determine whether their interaction also plays a role in NF-κB
signaling in vitro. Since it is unclear and controversial as
to whether BCA3 suppresses or promotes NF-κB signaling, we
selectively overexpressed BCA3 in osteoclasts and examined the
in vivo response of these animals to receptor activator of
nuclear factor-κB ligand (RANKL), given that NF-κB is required for
RANKL-induced osteoclastogenesis and bone resorption (16,17).
Materials and methods
Generation of BCA3 transgenic mice
To generate animals with the expression of BCA3
restricted to osteoclasts, the full-length cDNA for murine BCA3 was
inserted into exon 1 of the cathepsin K (Ctsk) gene in a C57BL/6
mouse genomic bacterial artificial chromosome (BAC) clone, which
was then used to create a transgenic animal model. The BAC clone
(RP23-422N18) encompassing the entire Ctsk locus was purchased from
the BACPAC Resources Center (https://bacpacresources.org). The cDNA for BCA3 was
cloned by RT-PCR as previously reported (12). The relevant portions of the
targeting vector used to introduce the BCA3 cDNA into the Ctsk
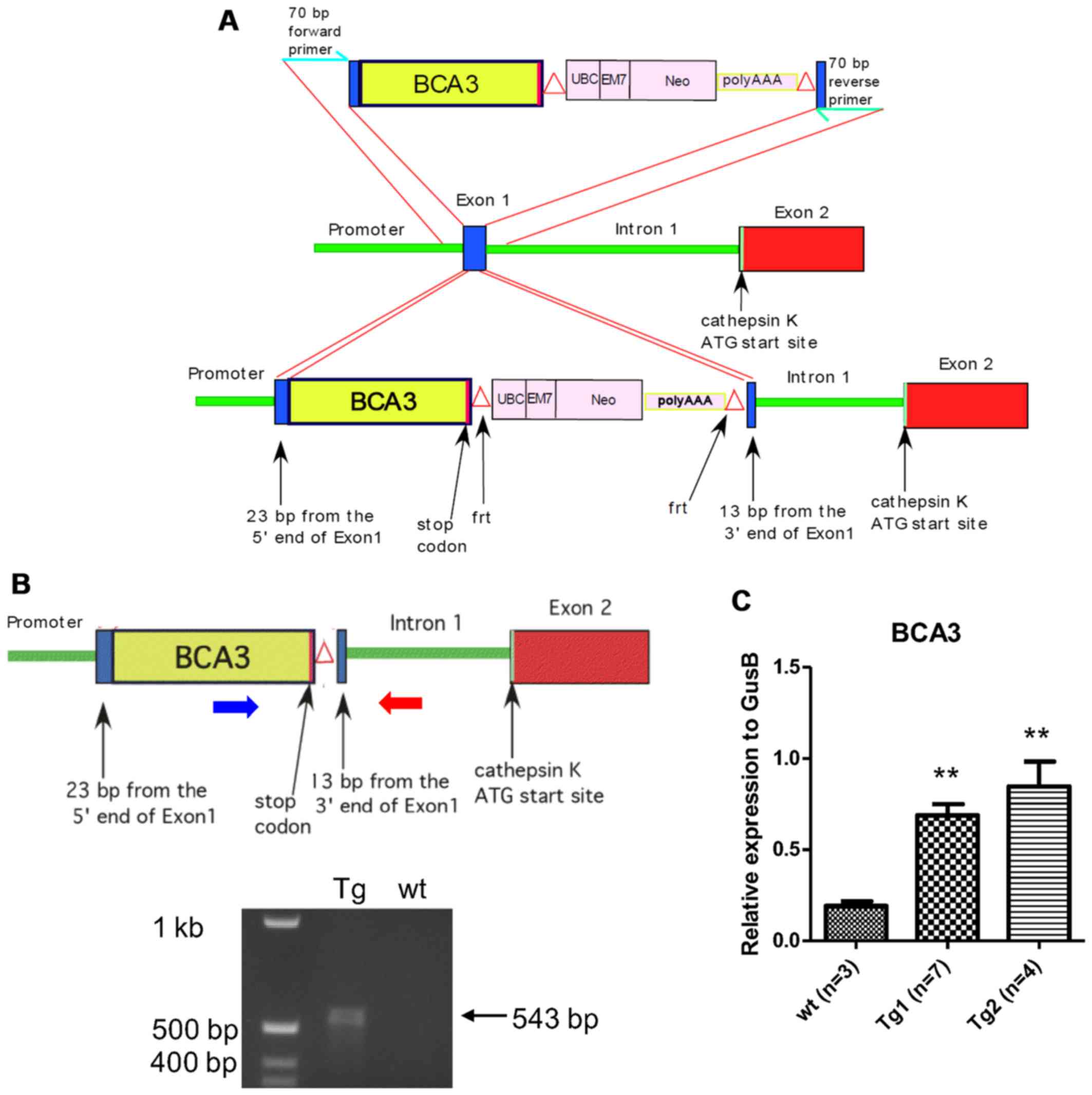
locus are shown in Fig. 1A.
Standard cloning techniques were used to assemble
the targeting vector that included 23 bp from the 5′ end of Ctsk
exon 1, the entire BCA3 coding sequence (including the endogenous
stop codon), a neomycin selection cassette flanked by frt sites
(18), and 13 base pairs from the
3′ end of exon 1. To ensure the efficient translation of BCA3 cDNA,
the 9 bp 5′ of the BCA3 cDNA initiation codon were derived from the
endogenous BCA3 Kozak sequence. Following assembly, the BCA3-NEO
segment was then PCR-amplified with oligonucleotide primers
representing 70 b of the Ctsk sequence immediately 5′ and 3′ of
exon 1, respectively, to provide flanking homology for targeted
homologous recombination into exon 1 of the Ctsk gene. This
construct was integrated into the Ctsk gene within the BAC clone by
homologous recombination using the Red recombination system of
bacteriophage λ as previously described (18–20). Selected clones were sequenced
across both Ctsk-insert junctions to confirm proper integration.
The Neo cassette was excised by inducing FLP recombinase F70L in
the DH10B bacterial host of the recombined BAC clone (21).
The engineered Ctsk/BCA3 BAC clone was then
micro-injected into C57BL/6 X SJL F2 oocytes and transgenic animals
were generated using standard techniques. Briefly, to generate
zygotes for microinjection, 3-week old B6;SJLF1 females (JAX) were
superovulated by IP injection with 5IU pregnant mare's serum
gonadotropin and 46 h later, IP injection of 5IU human chorionic
gonadotropin. Subsequent to the administration of human chorionic
gonadotropin, the females were mated with B6;SJL F1 stud males
(JAX) between the age of 10 weeks to 12 months. Females with a
copulatory plug the following morning (0.5 dpc) were euthanized and
zygotes were collected from oviducts. The engineered Ctsk/BCA3 BAC
clone was microinjected at a concentration of 5 ng/µl in TE
into the pronucleus of approximately 225 zygotes, and injected
zygotes were transferred to the oviducts of pseudopregnant females
after injection, 25–30/mouse, via standard embryo transfer surgery.
At weaning, tail biopsies of pups resulting from microinjection
were collected and genotyped, and 2 founder mice containing the
transgene were identified. These founders were bred with 6–8 weeks
old wild-type C57Bl/6 mice to establish 2 independent lines. This
ensured that the transgenic animals remained heterozygous during
breeding. Resultant transgenic mice were identified by PCR
amplification of a unique Ctsk-BCA3 junction sequence. Transgenic
animals and their wild type littermates, used in all experiments
were studied at 12 weeks of age. For the bone density experiments,
10 transgenic animals (4 female, 6 male) and 16 wild type
littermate controls (6 female, 10 male) were examined. For the
RANKL infusion experiments, 10 transgenic animals (5 female, 5
male) and 9 wild type litter mate controls (5 female, 4 male) were
examined. The use of animals in this study was approved by the Yale
Animal Care and Use Committee.
Cells
The pZen murine fibroblast cell line (22) was a generous gift from Dr Lawrence
Rohrschneider. The MC3T3E1 (preosteoblasts), 293, NIH3T3
(fibroblasts) and HeLa (cervical cancer) cell lines were purchased
from the American Type Culture Collection (ATCC, Manassas, VA,
USA). The pZen and MC3T3E1 cells were grown in α-MEM
(Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal
bovine serum (FBS) (Atlanta Biologicals, Lawrenceville, GA, USA)
and 1% P/S (Gibco, Grand Island, NY, USA). The 293, HeLa and NIH3T3
cells were grown in Dulbecco's modified Eagle's medium (DMEM; high
glucose) supplemented with 10% FBS, 1% P/S and 10 mM sodium
pyruvate (all from Gibco).
RNA isolation and qPCR
Total bone RNA from BCA3 transgenic and wild-type
animals was isolated from the tibiae and femurs of mice by rapidly
isolating the long bones, removing soft tissue and immediately
placing them in liquid nitrogen. The bones were pulverized to a
powder in liquid nitrogen and RNA isolated using TRIzol®
(Invitrogen, Carlsbad, CA, USA) following the manufacturer's
recommended protocol. cDNA was synthesized using the iScript cDNA
synthesis kit (Bio-Rad, Hercules, CA, USA). TaqMan probes and
primer sets for mouse BCA3 (Mm00498591) and β-glucuronidase (GusB,
Mm00446953) were purchased from Applied Biosystems (Foster City,
CA, USA) and RT-PCR was performed using iQ Supermix (Bio-Rad). qPCR
was performed using a BioRad MyiQ2 detection system. All qPCR
reactions were performed in duplicate and cycling conditions were
95°C for 20 sec and 60°C for 1 min for 40 cycles. The relative
expression level of each transcript was determined using the
comparative CT method using GusB as an endogenous reference.
Cloning, DNA transfection and luciferase
assay
The NF-κB luciferase construct was a generous gift
from Sankar Ghosh (Chair of Microbiology and Immunology Albert
Einstein College of Medicine). Rac1 and different BCA3 constructs
were cloned by RT-PCR as previously described (12). We divided the full length BCA3
protein into 3 fragments: fragment 1, 2 and 3 which correspond to
amino-acid 1–75, 76–125, and 126–221 of full length BCA3 protein,
respectively. The Rac1 cDNA was subcloned into the pEGFPC1 vector
(Clontech, Mountain View, CA, USA) to generate a Rac1-EGFP fusion
protein. The cells were plated one day prior to transfection in
24-well plates at a density of 4–8×104/well. Rac1,
full-length BCA3 or truncated BCA3 constructs, or empty pcDNA4
vector were co-transfected with the NF-κB luciferase construct
using the Extreme Gene HP reagent (Roche, Indianapolis, IN, USA) at
a ratio of 1:3. Twenty-four hours after transfection, the cells
were stimulated with or without tumor necrosis factor-α (TNF-α) (10
ng/ml) in culture medium containing 1% FCS for a further 24 h
before harvesting for luciferase assay. Luciferase assays were
performed using the dual luciferase reporter assay system with
pGL4.73 vector (both from Promega, Madison, WI, USA) as the
internal control. Data are presented as the fold increase over
cells transfected with empty pGL3 basic vector.
Staining and confocal microscopy
The 293 cells were plated on glass chamber slides
(BD Falcon, Bedford, MA, USA) and transfected with pEGFP-Rac1
vector and His tagged BCA3 vector using the Extreme Gene HP reagent
as described above. Cells were cultured for 48 h after transfection
and fixed with 3.7% formaldehyde (Thermo Scientific, Rockford, IL,
USA). Cells were then stained with Alexa 555 conjugated anti-His
tag antibody (Upstate, Lake Placid, NY, USA) for BCA3. The slides
were imaged using a Zeiss LSM 710 Duo confocal microscope (Carl
Zeiss Microimaging, Thornwood, NY, USA).
Immunoprecipitation and immunoblots
Anti-Rac1 antibody (Cat no. 05-389; Millipore,
Billerica, MA, USA) was cross-linked to protein A agarose beads
(Calbiochem, Darmstadt, Germany) using dimethyl pimelimidate
(Sigma-Aldrich) prior to immunoprecipitation. The 293 cells were
transfected with full-length His-tagged BCA3 or truncated
His-tagged BCA3 constructs as described above. The cells were
cultured for 48 h after transfection and whole cell lysates
prepared using HTNG lysis buffer (50 mM HEPES, 150 mM NaCl, 1%
Triton X-100, 10% glycerol, 1.5 mM MgCl2 and 1 mM EGTA).
Equal amounts of whole cell lysates were incubated with protein A
agarose coupled Rac1 antibody at 4°C overnight. Beads were washed 3
times in HTNG lysis buffer and bound proteins eluted using 1X
Laemmli sample buffer (Bio-Rad) with heating to 95°C. Equal volumes
of eluted protein, and amounts of whole cell lysates equivalent to
the input into the immunoprecipitation experiments, were subjected
to sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto nitrocellulose paper (Trans-Blot
Transfer Medium; Bio-Rad). After SDS-PAGE and trans-blotting, the
nitrocellulose membrane was probed with an anti-His antibody (Cat
no. 05-949; Millipore). The blots were developed using HRP
conjugated secondary antibodies (Cat no. W4021; Promega) followed
by enhanced chemiluminescence detection (ECL detection kit;
Amersham Inc., Piscataway, NJ, USA).
RANKL injection and minipump
infusion
Recombinant murine soluble RANKL was generously
provided by Amgen, Inc. (Thousand Oaks, CA, USA). The in
vivo hypercalcemia assay using high-dose RANKL injections was
performed as previously described by Lacey et al (23). Briefly, transgenic or wild-type
animals were administered 1.5 mg/kg of RANKL subcutaneously every
12 h for 7 doses at which point the serum calcium concentration was
measured. To assess RANKL-induced bone loss, the cytokine was
infused for 2 weeks using Alzet osmotic minipumps (Durect,
Cupertino, CA, USA). RANKL at a constant dose of 0.4 mg/kg/day or
phosphate-buffered saline (PBS) was administered for 14 days using
minipumps that deliver 0.25 µl/h. The pumps were
equilibrated in 0.9% NaCl overnight at 37°C and then implanted into
an interscapular subcutaneous pocket in either control or BCA3
transgenic mice using Isothesia® (Butler Animal Health
Supply, Dublin, OH, USA) for anesthesia. After 14 days of infusion,
the animals were sacrificed and serum collected to measure type I
collagen carboxyterminal telopeptide (CTX). Bones were harvested
for densitometric analysis by microCT.
Bone density measurements
In vivo bone density measurements were
performed by dual-energy X-ray absorptiometry using a PIXImus
densitometer (Lunar, Madison, WI) as previously described (24). Anesthetized mice (ketamine at 100
mg/kg body wt and xylazine at 10 mg/kg body wt given i.p.) were
placed in the prone position and scans performed with a
1.270-mm-diameter collimator, 0.762-mm line spacing, 0.380-mm point
resolution, and an acquisition time of 5 min. The spine window is a
rectangle spanning a length of the spine from T1 to the beginning
of the sacrum. The femur window encompasses the entire right femur
of each mouse. The coefficient of variation for total body bone
mineral density (BMD) is 1.5%. Microcomputed tomography was
performed as previously described (25). Briefly, femurs were stripped of
soft tissue and stored in 70% EtOH at 4°C. Specimens were analyzed
in 70% EtOH by cone beam microfocus X-ray computed tomography using
a Scanco µCT-35 instrument (Scanco, Brutissellen,
Switzerland). Images were acquired at 55 kVp, with an integration
time of 500 msec and an isometric voxel size of 6 mm. Segmentation
of bone from marrow and soft tissue was performed in conjunction
with a constrained Gaussian filter (support = 1; 3X3X3 voxel
window; σ= 0.8) to reduce noise, applying density thresholds of 250
and 420 for the trabecular and cortical compartments of the femur,
respectively. Volumetric regions for trabecular analysis were
selected within the endosteal borders of the distal femoral
metaphysis to include the secondary spongiosa located 1 mm from the
growth plate and extending 1 mm proximally. Cortical morphometry
was quantified and averaged volumetrically through 233 serial
cross-sections (1.4 mm) centered on the diaphyseal midpoint between
proximal and distal growth plates. Both 2- and 3-D µCT data
included bone volume to total volume fraction (BV/TV), and
trabecular number (Tb.N), thickness (Tb.Th), spacing (Tb.Sp) and
connectivity density (Conn.D). Cortical thickness, averaged for
both cortices (Ct.Th), was also quantified.
Biochemical measurements
Serum type I collagen carboxy-terminal telopeptide
(CTX) was measured by EIA (Ratlaps EIA kit; Immunodiagnostic
Systems Inc., Scottsdale, AZ, USA). Serum calcium was measured in
the Yale New Haven Hospital Consolidated Laboratory (Department of
Laboratory Medicine) using a multichannel autoanalyzer (Roche DPP
Autoanalyzer).
Statistical analysis
An unpaired t-test or one-way ANOVA with Bonferroni
post-hoc testing was used as indicated in the figure legends.
Two-way ANOVA was used with Bonferroni post-hoc test to assess the
change of CTX and bone mass data by µCT in the low-dose
RANKL infusion experiment. Two-way ANOVA was also applied to the
analysis of the effect of BCA3 on NF-κB signaling in different cell
types (Fig. 6). Data presented
are the means ± SEM. The error bars in figures reflect SEM. A value
of p<0.05 was considered to indicate a statistically significant
difference.
Results
BCA3-Rac1 interaction promotes NF-κB
signaling in 293 cells
BCA3 is thought to affect NF-κB signaling by
shuttling PKA to the nucleus and enhancing the phosphorylation of
p65 (4,5). Rac1 has also been reported to
activate the NF-κB pathway although the details of that molecular
mechanism remain unclear (26,27). Since, as noted, BCA3 can bind
Rac1, experiments were performed to determine the impact of this
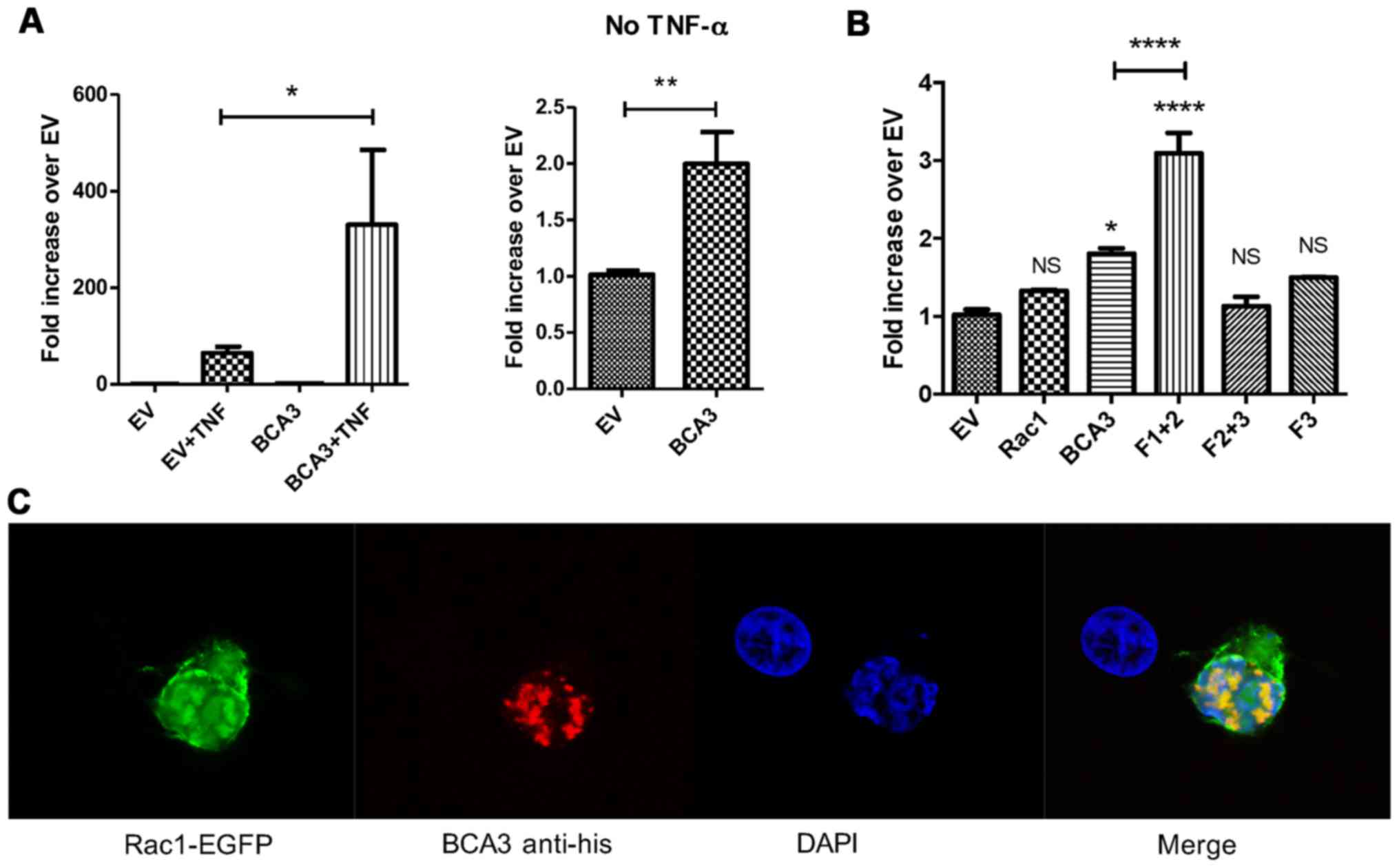
interaction on NF-κB signaling. The overexpression of BCA3 alone in
293 cells activated NF-κB signaling (Fig. 2A, left panel). The expression of
BCA3 also robustly enhanced TNF-α-induced NF-κB signaling compared
to the empty vector (EV) control (Fig. 2A, right panel). These data are
consistent with those of published studies demonstrating that BCA3
augments NF-κB signaling in 293 cells (2–4).
We have previously defined the region of BCA3 responsible for
interacting with Rac1 by using three deletion constructs: fragment
1 (aa 1–75), fragment 2 (aa 76–125) and fragment 3 (aa 126–221)
(12). Fragment 2 was identified
as a putative Rac1 binding site. In the present study, the
overexpression of BCA3 fragment 1+2 alone activated NF-κB
signaling, while fragment 2+3 or fragment 3 alone showed no
activity (Fig. 2B). As previously
reported, we were unable to individually express fragment 1 or 2
(12). The augmentation of NF-κB
signaling by fragment 1+2 was even stronger than the wild-type full
length BCA3. These results suggest that the amino terminal 125
amino acids of BCA3 contain an NF-κB activating sequence, while
fragment 3 may have the function to prevent BCA3 being
over-activated. The overexpression of Rac1 did not have a
significant effect on NF-κB activity (Fig. 2B).
To investigate BCA3 Rac1 interactions in 293 cells,
the cells were transfected with EGFP-tagged Rac1 and His-tagged
BCA3. Confocal microscopy showed BCA3 staining to be primarily
nuclear in a speckled pattern, while Rac1 was found both in the
cytoplasm and nucleus (Fig. 2C).
BCA3 and Rac1 fluorescence exhibited significant nuclear
co-localization consistent with a molecular interaction (Fig. 2C). To further confirm a BCA3-Rac1
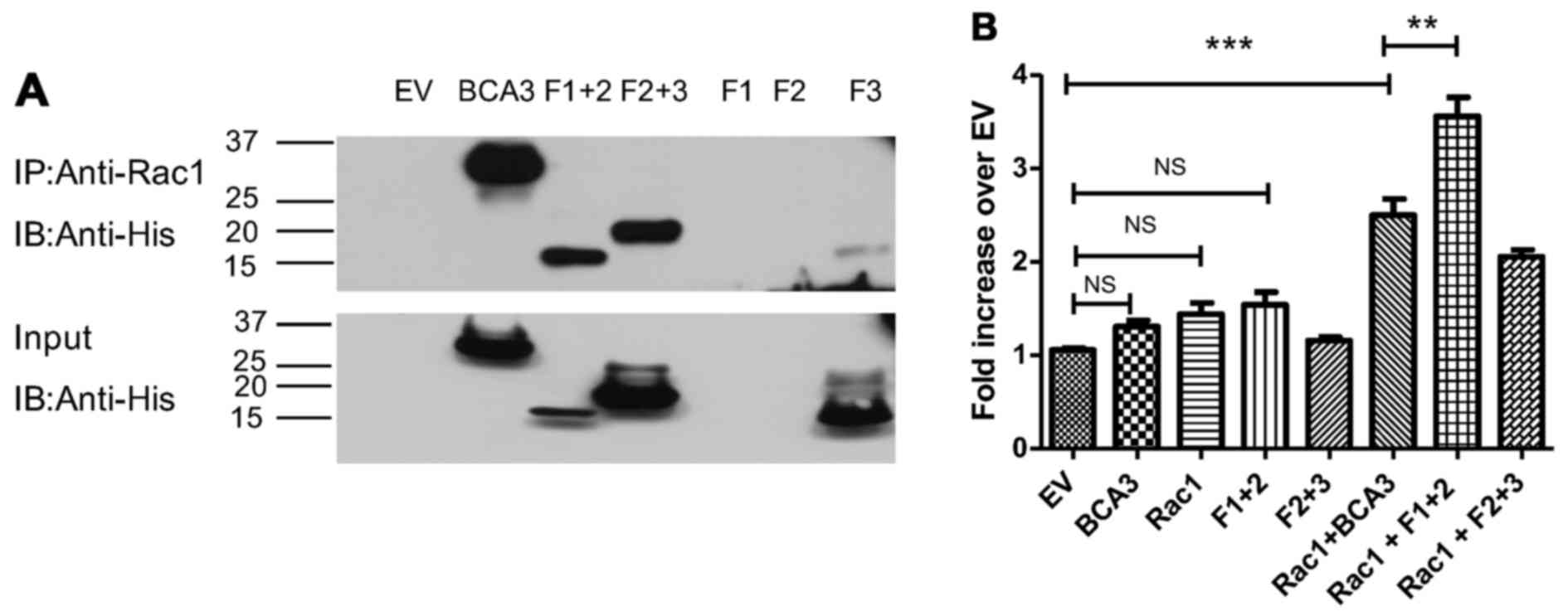
interaction, His-tagged BCA3 or His-tagged BCA3 fragments were
transiently transfected into 293 cells, and lysates were prepared
and immunoprecipitated with an antibody to Rac1. The Rac
immunoprecipitates were examined by immunoblotting with an anti-His
antibody. As shown in Fig. 3A,
Rac1 co-precipitated full-length BCA3, fragment 1+2 and 2+3. By
contrast, very little of fragment 3 co-precipitated with Rac1
(Fig. 3A). As noted, fragments 1
and 2 could not be individually expressed (lanes 5 and 6).
When adjusted for the level of input (Fig. 3A, bottom panel), fragment 1+2
appeared to have Rac1 binding ability similar to full-length BCA3
and to have more activity than fragment 2+3. Since fragment 1+2 was
also more effective at activating NF-κB than fragment 2+3, this
raised the possibility that the interaction with Rac1 may augment
the effect of BCA3 on NF-κB signaling. To examine this hypothesis,
BCA3 and its fragments were co-transfected with Rac1 into 293 cells
with the NF-κB reporter construct and luciferase assays performed
(Fig. 3B). As these experiments
required 3 simultaneous transfections, lesser amounts of DNA were
used for each construct. When only 2 vectors were being analyzed
(e.g., NF-κB reporter and full-length BCA3) an empty vector was
also transfected to ensure that equivalent amounts of DNA were used
in every experimental condition so that variation in the amount of
DNA introduced into the cell would not confound the results. For
this reason, although full-length BCA3 and fragment 1+2 both
increased NF-κB activity when introduced into the cells alone
(Fig. 2A and B), in the
experiments summarized in Fig. 3,
these increases did not reach statistical significance.
As shown above, Rac1 alone did not stimulate NF-κB
signaling (Fig. 3B). However, the
expression of both Rac1 and full-length BCA3 led to the synergistic
activation of NF-κB. The expression of both Rac1 and fragment 1+2
was even more effective at activating NF-κB signaling, while Rac1
plus fragment 2+3 was less effective than Rac1 plus either
full-length BCA3 or fragment 1+2.
Osteoclast-specific overexpression of
BCA3 in mice
The role and importance of BCA3 as well as that of
the Rac1/BCA3 interaction in regulating NF-κB signaling remain to
be fully defined. Our previous study suggested that BCA3 may have
an important function in osteoclasts (12). BCA3 has been reported to play an
important role in NF-κB signaling, which is required for
RANKL-dependent osteoclast formation and activation (28). We were unable to find a cell model
of osteoclasts in which to test the interaction of BCA3 and NF-κB
in vitro. Despite studies to the contrary (29), we have not been able to
demonstrate cytokine-responsive NF-κB signaling in RAW 264.7 cells,
which are a commonly used model of preosteoclasts. Primary murine
preosteoclasts are extremely resistant to transfection even using
lentiviral infection. Although we have successfully introduced one
construct into primary pre-osteoclast cultures (30), given the relatively low efficiency
of even this method, the introduction of multiple constructs
required to study NF-κB signaling as was done for the cells lines
just described, is not feasible in preosteoclasts. Therefore in an
effort to clarify the function of BCA3 in osteoclasts in
vivo, studies were conducted in transgenic mice selectively
overexpressing BCA3 in osteoclasts. Since RANKL is required for
normal osteoclasts function and is the final common effector of
bone resorption in vivo, we hypothesized that if NF-κB
signaling was augmented by BCA3 in osteoclasts, the bone-resorbing
effects of RANKL would be enhanced.
A full-length BCA3 cDNA was introduced into exon 1
of the Ctsk gene contained in a BAC DNA clone as described above
and summarized in Fig. 1A and B.
Two transgenic lines were created and the presence of the transgene
was confirmed by PCR genotyping. To confirm the overexpression of
BCA3 in bone, RNA was extracted from tibiae and femurs from two
different transgenic lines and their wild-type siblings and
analyzed by qPCR. There was a significant increase in BCA3
transcript expression in both transgenic lines (Fig. 1C). Since line 2 showed a higher
level of BCA3 transcript expression this line was used in
subsequent experiments. Since Ctsk expression is restricted to late
preosteoclasts and mature osteoclasts in bone, the overexpression
of BCA3 in the whole bone tissue indicates a very robust
overexpression of BCA3 in osteoclasts since these cells make up
only a small fraction of the cellular material in bone.
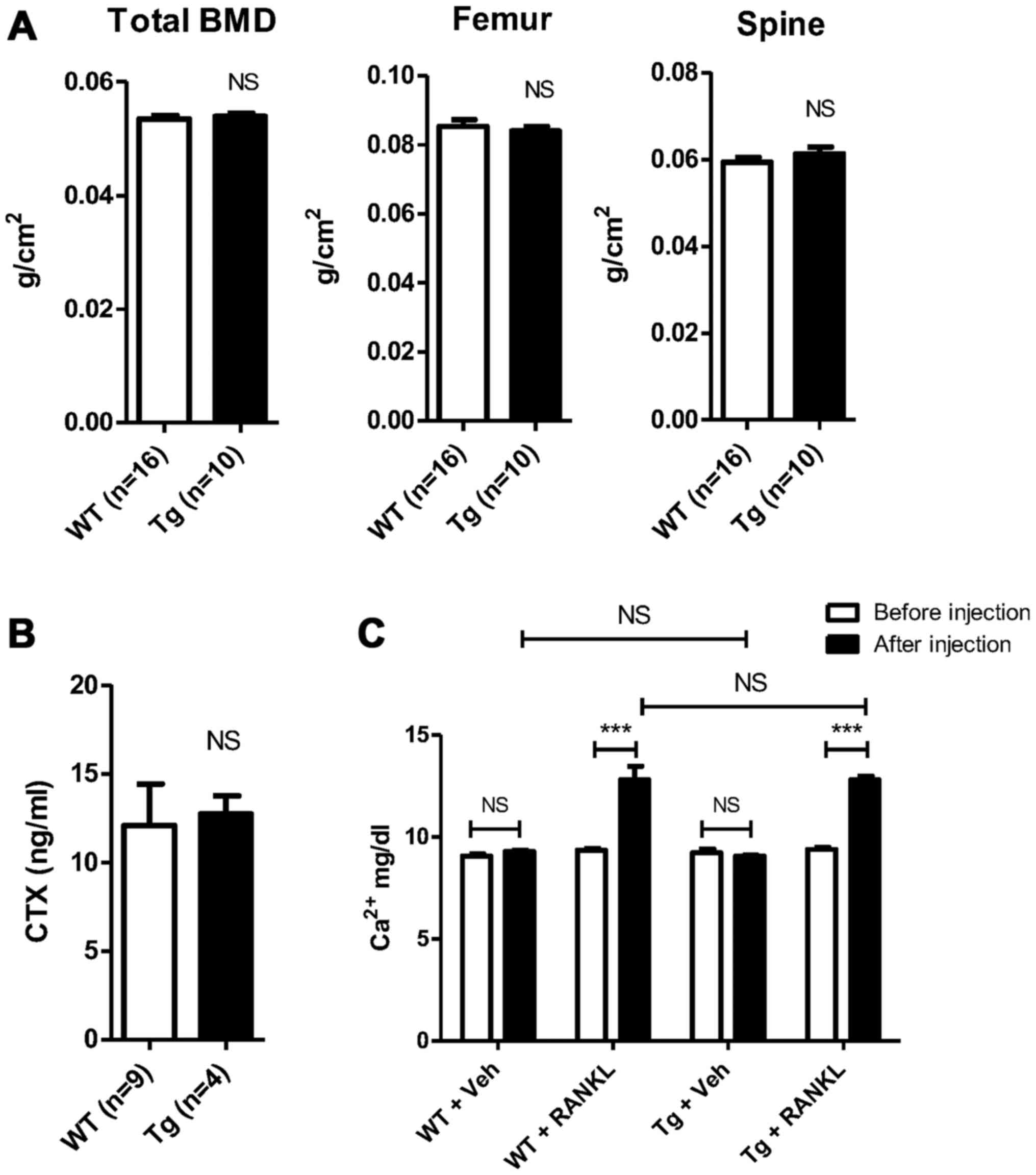
BCA3 transgenic mice have normal bone
density and serum levels of CTX
Bone density was measured by DXA in 12-week old
transgenic mice and littermate controls. There were no statistical
differences in total body BMD (0.0539±0.0005 vs. 0.0535±0.0006
g/cm2, Tg vs. Wt, n=10 and 16 in Tg and Wt groups,
respectively, p=NS) ; femur BMD (0.0840±0.0013 vs. 0.0853±0.0020
g/cm2, Tg vs. Wt, n=10 and 16 in Tg and Wt groups,
respectively, p=NS) or spinal BMD (0.0612±0.0016 vs. 0.0594±0.0011
g/cm2, Tg vs. Wt, n=10 and 16 in Tg and Wt groups,
respectively, p=NS) (Fig. 4A).
Micro CT analyses revealed no statistical differences in the
trabecular bone density in either the femur or spine (data not
shown). Consistent with these data, serum CTX values were similar
in transgenic and control animals, suggesting no change in
osteoclast resorptive activity (12.72±1.020 vs. 12.10±2.344 ng/ml,
Tg vs. Wt, n=4 and 9 in Tg and Wt groups respectively, p=NS)
(Fig. 4B).
Transgenic line 1 also showed normal bone density
both by DXA and µCT analysis (data not shown) and was not
further studied since, as noted, the expression level of BCA3 was
lower than in transgenic line 2.
BCA3 overexpression does not alter the in
vivo resorptive response to RANKL
Age and gender-matched BCA3 transgenic mice and
their wild-type siblings were treated with high-dose RANKL (1.5
mg/kg) or an equivalent volume of PBS administered subcutaneously
every 12 h for a total of 7 doses. Serum calcium was measured
before the first dose and after the final dose. There was no change
in serum calcium levels with PBS treatment in either group (Tg,
9.2±0.2 vs. 9.1±0.1 mg/dl, baseline vs. PBS, n=3, p=NS; Wt, 9.1±0.1
vs. 9.3±0.1 mg/dl, baseline vs. PBS, n=3, p=NS) (Fig. 4C). As expected, RANKL treatment
caused significant hypercalcemia in both groups compared to either
baseline or to the vehicle treatment values in the animals
receiving PBS (Tg, 9.4±0.1 vs. 12.8±0.2 mg/dl, baseline vs. RANKL,
n=4, p<0.0001; Wt, 9.4±0.1 vs. 12.8±0.7 mg/dl, baseline vs.
RANKL, n=4, p<0.0001) (Fig.
4C). However, there was no difference in the magnitude of
hypercalcemia induced based on genotype. The serum calcium values
following RANKL treatment did not differ between the 2 groups
(12.8±0.2 vs. 12.8±0.7 mg/dl, Tg vs. Wt, n=4 in both groups,
p=NS).
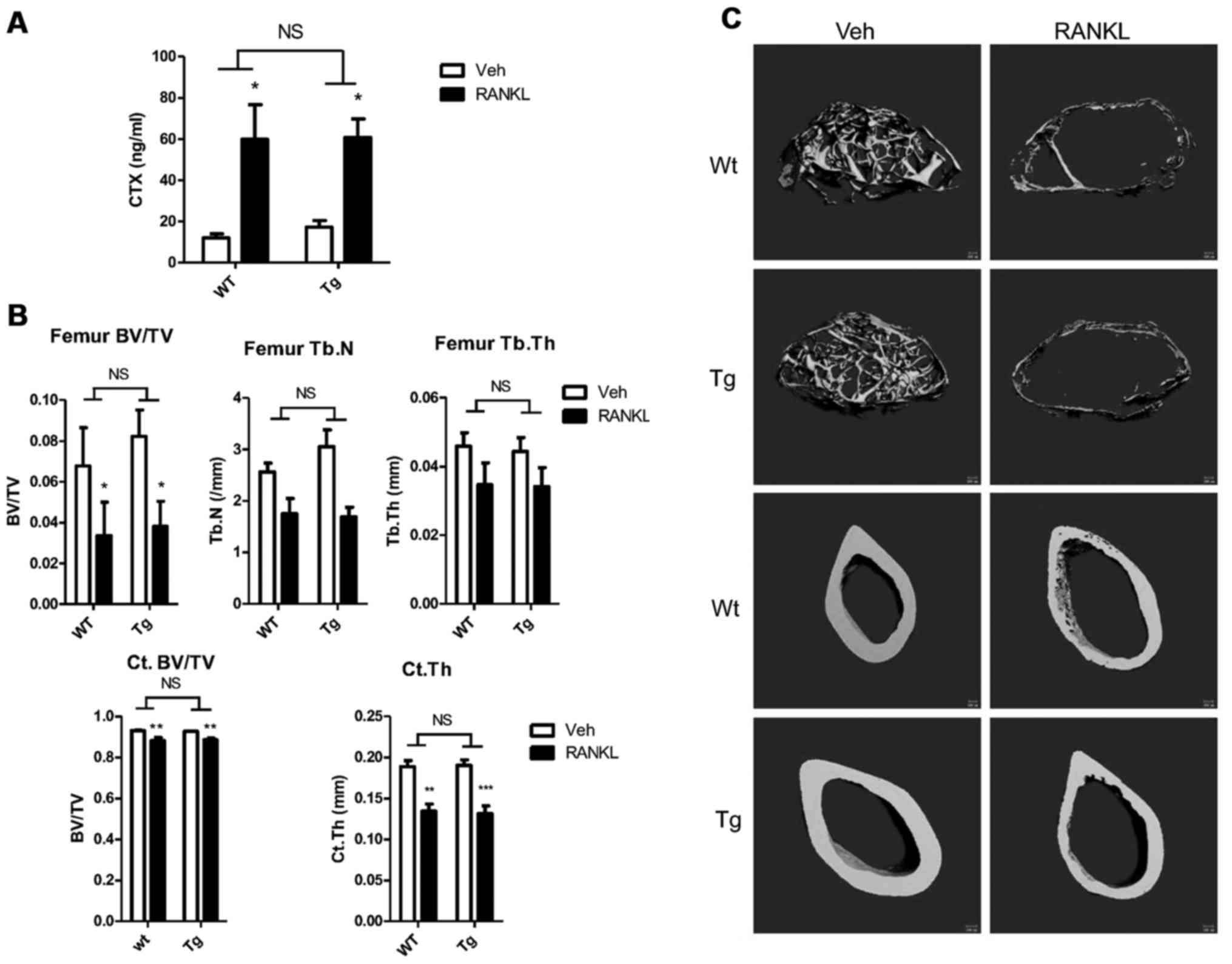
The low-dose infusion of RANKL leads to measurable
bone loss in mice (31). Thus, to
determine whether a lower dose and longer exposure to RANKL may
reveal subtle differences in the osteoclastic response of wild-type
and transgenic mice, both groups were administered RANKL (0.4
mg/kg/day) or PBS via an Alzet minipump for 14 days. After 14 days,
the mice were sacrificed and femurs and serum were obtained for μCT
and CTX analyses.
RANKL caused significant and equivalent increases in
serum CTX levels in both groups compared to PBS treatment (Tg,
17.3±3.2 vs. 60.8±9.0 ng/ml, n=4 vs. 6, PBS vs. RANKL respectively,
p<0.05; Wt, 12.0±2.0 vs. 59.8±16.9 ng/ml, n=4 vs. 5, PBS vs.
RANKL, respectively, p<0.05) (Fig.
5A). Although the fold increase in serum CTX levels was
slightly greater in the Wt animals (3.5- vs. 5.0-fold, Tg vs. Wt),
there was no difference in the mean serum CTX levels following
RANKL infusion based on genotype (60.8±9.0 vs. 59.8±16.9 ng/ml, n=6
vs. 5, Tg vs. Wt, respectively, p=NS).
RANKL administration induced significant bone loss
in both wild-type and transgenic mice compared to the PBS-infused
animals. As shown in Fig. 5B
(upper panel) 2 weeks of RANKL infusion resulted in a 53.6% decline
in trabecular bone mass in the femur of transgenic animals and a
comparable 50.5% decline in the controls. There was no difference
in the mean trabecular bone volume following RANKL infusion based
on genotype (3.82±1.22 vs. 3.36±1.6%, n=6 vs. n=5, Tg vs. Wt,
respectively, p=NS). Trabecular number was slightly higher in the
PBS-infused BCA transgenic animals than in the controls infused
with saline (3.05±0.36 vs. 2.56±0.17/mm, n=3 vs. 4; Tg vs. Wt,
p=NS); however trabecular number was similar in both groups
following RANKL administration (1.69±0.19 vs. 1.75±0.30/mm, Tg vs.
Wt, p=NS). The percentage decrease in trabecular thickness in the
RANKL-infused compared to the PBS-infused animals (23.1 vs. 24.4%,
Tg vs. Wt) and the final values for the parameter (0.034±0.006 vs.
0.035±0.006 mm, n=6 vs. 5, Tg vs. Wt, respectively, p=NS) did not
differ between the 2 groups. Cortical bone volume was similar in
the 2 groups following saline infusion and declined by the same
extent with RANKL treatment (4.5 vs. 5.0%, Tg vs. Wt) such that the
values after RANKL infusion did not differ (88.61±0.92 vs.
88.30±1.31%, n=6 vs. 4, Tg vs. Wt, respectively, p=NS) (Fig. 5B, lower panel). Similarly,
cortical thickness did not differ in either the saline-treated
(0.190±0.007 vs. 0.189±0.007 mm, n=4 vs. 4, Tg vs. Wt,
respectively, p=NS) or RANKL-treated (0.131±0.009 vs. 0.134±0.009
mm, n=6 vs. 4, Tg vs. Wt, respectively, p=NS) groups based on
genotype. As with all other parameters, RANKL induced a significant
decline in this parameter that was equivalent in both groups of
animals (Fig. 5B, lower panel).
Fig. 5C shows representative
images of cortical and trabecular bone from saline and
RANKL-infused animals. In both RANKL-treated groups, marked loss of
trabecular bone as well as cortical thinning are evident.
Effect of BCA3 on NF-κB signaling is
cell-type dependent
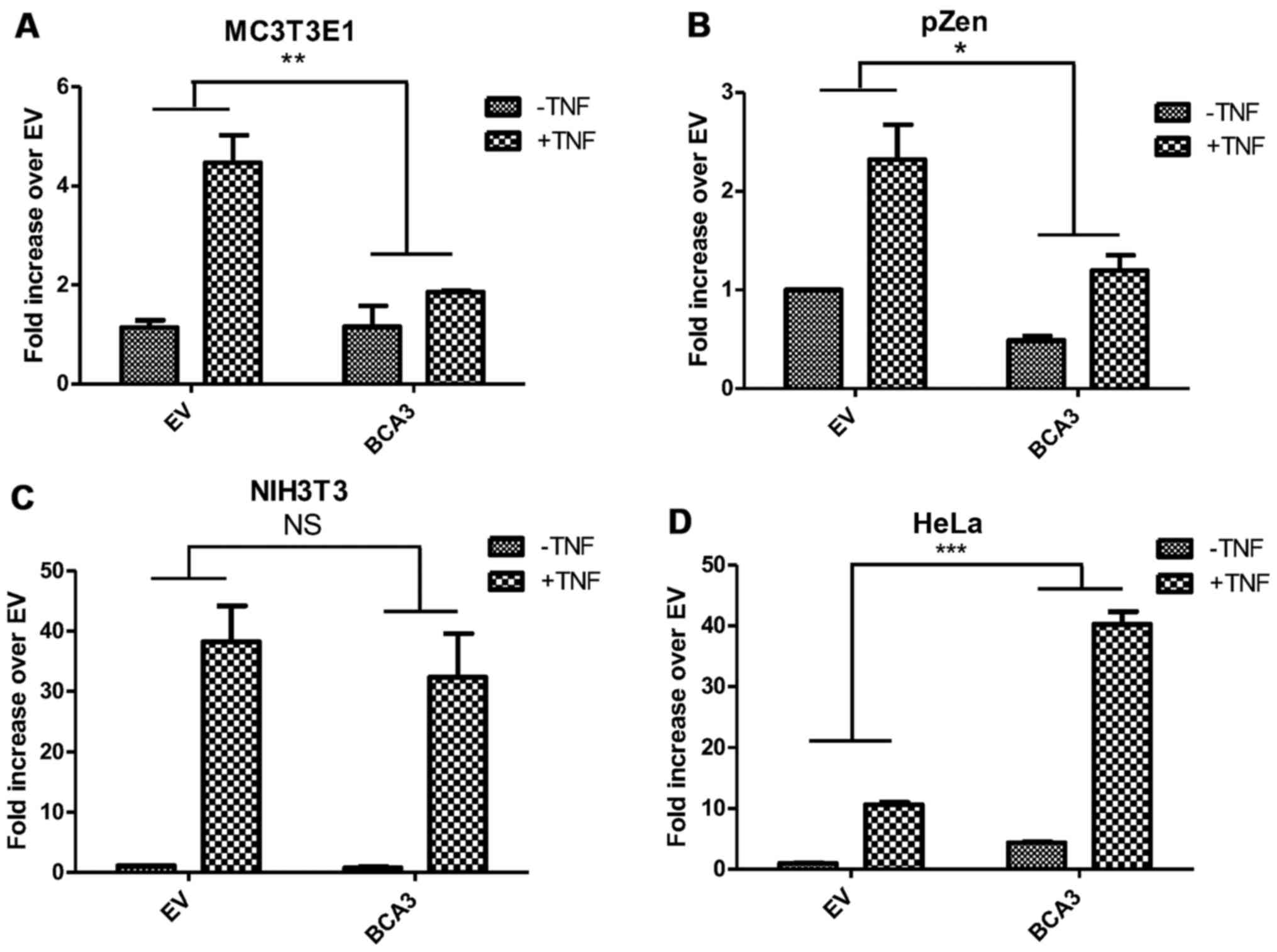
Although BCA3 augments NF-κB signaling in 293 cells,
it has also been reported to inhibit the NF-κB pathway in HeLa
cells (9). Thus, to investigate
the impact of BCA3 on NF-κB signaling in cells other than the 293
line, experiments were conducted in HeLa cells, pZen cells (a mouse
embryonic fibroblast line overexpressing the CSF1 receptor),
MC3T3E1 cells (a murine osteoblast-like cell line) and NIH3T3
cells. The pZen cells differ from NIH3T3 cells in that they are
transformed and have tumorigenic potential (22). In pZen cells, BCA3 inhibited NF-κB
signaling both in the absence and presence of TNF-α treatment
(Fig. 6B). The results in MC3T3E1
cells were similar to those in pZen cells with an even greater
inhibition of TNF-α-induced NF-κB signaling (Fig. 6A). However, BCA3 had no effect on
NF-κB signaling in NIH3T3 cells (Fig.
6C). In contrast to the findings in these 2 cell lines, BCA3
significantly increased NF-κB signaling in HeLa cells (Fig. 6D).
Discussion
BCA3 was first characterized as a proline-rich
protein overexpressed in breast and prostate cancer cells.
Bioinformatics studies have identified multiple functional sites in
its coding sequence, including a PDZ domain, a proline-rich domain,
multiple SH2 domains and phosphorylation sites. Although the
functions of BCA3 are largely unknown, its multiple predicted
functional sites suggest that it may be an adaptor protein in
signaling pathways. Consistent with this notion BCA3 has been
reported to interact with several proteins including TAp73, Rac1,
PKA, p65 and mitochondrial AIF.
The role of BCA3 in NF-κB signaling is an area of
present interest. The BCA3-p65 interaction has been reported to
facilitate the nuclear retention and phosphorylation of p65,
thereby enhancing NF-κB signaling (3). Moreover the level of BCA3 expression
modulates PKA-dependent p65 phosphorylation in HEK293 and MCF7
cells (4). The present study
demonstrated that BCA3 robustly increased NF-κB activity when
overexpressed in 293 cells and synergized with TNF-α in activating
NF-κB-dependent transcription. In addition, c-terminally truncated
BCA3 was a more potent agonist than the full-length protein,
suggesting that the last 96 amino acids of BCA3 may have inhibitory
activity. Consistent with our data that fragment 1+2 most
effectively activates NF-κB signaling, p65 has been reported to
interact with a region of BCA3 encoded by exon 4 (5), which is contained in fragment 2.
However the carboxyterminal exons 5 and 6 of BCA3 reportedly
interact with PKA (5). We found
that fragment 1+2, which lacks exons 5 and 6, effectively activated
NF-κB signaling while fragment 2+3, which contains exons 5 and 6 of
BCA3 did not.
We have previously characterized BCA3 as a
Rac1-interacting protein in osteoclasts. In the present study we
extended this finding to 293 cells. In these cells the interaction
occurred primarily in the nucleus, while in osteoclasts it was
particularly prominent in the perinuclear region (12). In 293 cells, Rac1 and BCA3
synergized in promoting NF-κB activity. Rho family small GTPases
such as Rac1 are able to activate NF-κB signaling although the
mechanism is still unclear (26,32,33). Unlike Rac2 and Rac3, Rac1 has a
strong polybasic region, which contains a nuclear localization
signal (34–36). Geranylgeranyl modification of the
carboxyterminus of Rac1, which binds to RhoGDI, results in
cytoplasmic localization of Rac1. However, it has been shown that
Rac1 accumulates in the nucleus during the G2 phase of the cell
cycle and promotes cell division (34). In the present study, the
overexpression of wild-type Rac1 in 293 cells, had no effect on
NF-κB signaling, since 293 cells express minimal amounts of BCA3
(10). The co-transfection of
BCA3 with Rac1 greatly enhanced NF-κB activity and it is possible
that BCA3 functioned as a molecular shuttle to bring Rac1 into the
nucleus. How the Rac1/BCA3 interaction results in synergistic
activation of NF-κB signaling is currently unclear.
This study also confirmed that fragment 2 of BCA3
was responsible for its interaction with Rac1 since
co-immunoprecipitation experiments demonstrated that fragments 1+2
and 2+3, but not fragment 3 alone were pulled down with Rac1.
Fragment 1+2 showed more robust interaction with Rac1 than did
fragment 2+3 or full-length BCA3 suggesting an inhibitory effect of
fragment 3. Consistent with this notion and with our hypothesis
that fragment 2 contains the binding site for Rac1, it was fragment
1+2 that best synergized with Rac1 in activating NF-κB
signaling.
NF-κB signaling is required for osteoclast
differentiation, activation and survival (28,37,38). The RANK signaling cascade entrains
the NF-κB complex and is critical to the resorptive effects of
RANKL (28). RANKL potently
activates NF-κB signaling in mature osteoclasts (28). Since BCA3 modulates NF-κB
signaling, we hypothesized that it would impact the actions of
RANKL by altering RANKL-dependent NF-κB signaling in preosteoclasts
and mature osteoclasts. However, restricted overexpression of BCA3
in osteoclasts did not alter the skeletal phenotype of transgenic
animals. To determine whether stressing the skeletons of these
animals with exogenous RANKL would uncover a functionally
significant change in RANKL activity, transgenic mice were tested
for RANKL tolerance using two treatment strategies. We found that
overexpression of BCA3 in osteoclasts did not alter the response to
either short-term high-dose or longer-term lower dose RANKL
administration. These data suggest that BCA3 is not a major
regulator of NF-κB signaling in mature osteoclasts. Since BCA3
expression in our transgenic mice was restricted in its expression
to mature osteoclasts and perhaps late preosteoclasts, our data do
not preclude the possibility that BCA3 plays an important role in
NF-κB-dependent signaling in early osteoclast differentiation.
The role of BCA3 in NF-κB signaling remains
controversial. Although studies using 293 cells and breast cancer
cell lines have indicated that BCA3 augments NF-κB signaling, it
also has been reported to suppress NF-κB signaling in HeLa cells
(9). By contrast, another group
(3) reported that BCA3 enhanced
the nuclear retention and phosphorylation of p65 in HeLa cells,
consistent with enhanced of NF-κB signaling. In this study, the
effects of BCA3 on NF-κB signaling were cell-type specific. We
found that overexpression of BCA3 could augment, attenuate or have
no effect on NF-κB signaling depending on the cellular context.
These data suggest that the effect of BCA3 on NF-κB signaling may
depend on its interacting partners, which may be cell
type-specific. The significance of the BCA3/NF-κB interaction in
vivo remains to be determined.
Acknowledgments
This study was supported by NIH grants nos. DE12459
and DK045228 and by a P30 Core Center Award (AR46032), which
supports the Yale Core Center for Musculoskeletal Disorders. This
study was also supported by the Science and Technology Commission
of Shanghai Municipality grant no. 14pj1407200 and the National
Natural Science Foundation of China grant no. 81400855.
Glossary
Abbreviations
Abbreviations:
|
NF-κB
|
nuclear factor-κB
|
|
BCA3
|
breast cancer-associated gene 3
|
|
PKA
|
protein kinase A
|
|
CSF1
|
colony stimulating factor 1
|
|
RANK
|
receptor activator of nuclear
factor-κB
|
|
RANKL
|
receptor activator of nuclear factor
κB ligand
|
|
BAC
|
bacterial artificial chromosome
|
|
Ctsk
|
cathepsin K
|
|
BMD
|
bone mineral density
|
|
EV
|
empty vector
|
|
CTX
|
type I collagen carboxyterminal
telopeptide
|
|
BV
|
bone volume
|
|
TV
|
tissue volume
|
|
Tb.N
|
trabecular number
|
|
Tb.Th
|
trabecular thickness
|
|
Ct.Th
|
cortical thickness
|
References
|
1
|
Kitching R, Li H, Wong MJ, Kanaganayakam
S, Kahn H and Seth A: Characterization of a novel human breast
cancer associated gene (BCA3) encoding an alternatively spliced
proline-rich protein. Biochim Biophys Acta. 1625:116–121. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sastri M, Barraclough DM, Carmichael PT
and Taylor SS: A-kinase-interacting protein localizes protein
kinase A in the nucleus. Proc Natl Acad Sci USA. 102:349–354. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gao N, Asamitsu K, Hibi Y, Ueno T and
Okamoto T: AKIP1 enhances NF-kappaB-dependent gene expression by
promoting the nuclear retention and phosphorylation of p65. J Biol
Chem. 283:7834–7843. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gao N, Hibi Y, Cueno M, Asamitsu K and
Okamoto T: A-kinase-interacting protein 1 (AKIP1) acts as a
molecular determinant of PKA in NF-kappaB signaling. J Biol Chem.
285:28097–28104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
King CC, Sastri M, Chang P, Pennypacker J
and Taylor SS: The rate of NF-κB nuclear translocation is regulated
by PKA and A kinase interacting protein 1. PLoS One. 6:e187132011.
View Article : Google Scholar
|
|
6
|
Leon DA and Cànaves JM: In silico study of
breast cancer associated gene 3 using LION Target Engine and other
tools. Biotechniques. 35:1222–1226. 12281230–1231. 2003.PubMed/NCBI
|
|
7
|
Leung TH and Ngan HY: Interaction of TAp73
and breast cancer-associated gene 3 enhances the sensitivity of
cervical cancer cells in response to irradiation-induced apoptosis.
Cancer Res. 70:6486–6496. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Qin HY and Han H: Effect of KyoT2 binding
protein KBP1 on RBP-J-mediated transcriptional activity. Xi Bao Yu
Fen Zi Mian Yi Xue Za Zhi. 20:544–547. 2004.In Chinese. PubMed/NCBI
|
|
9
|
Gao F, Cheng J, Shi T and Yeh ET:
Neddylation of a breast cancer-associated protein recruits a class
III histone deacetylase that represses NFkappaB-dependent
transcription. Nat Cell Biol. 8:1171–1177. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sastri M, Haushalter KJ, Panneerselvam M,
Chang P, Fridolfsson H, Finley JC, Ng D, Schilling JM, Miyanohara
A, Day ME, et al: A kinase interacting protein (AKIP1) is a key
regulator of cardiac stress. Proc Natl Acad Sci USA. 110:E387–E396.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hashimoto M, Murata E and Aoki T:
Secretory protein with RING finger domain (SPRING) specific to
Trypanosoma cruzi is directed, as a ubiquitin ligase related
protein, to the nucleus of host cells. Cell Microbiol. 12:19–30.
2010. View Article : Google Scholar
|
|
12
|
Yu KP, Itokawa T, Zhu ML, Syam S, Seth A
and Insogna K: Breast cancer-associated gene 3 (BCA3) is a novel
Rac1-interacting protein. J Bone Miner Res. 22:628–637. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakai H, Chen Y, Itokawa T, Yu KP, Zhu ML
and Insogna K: Activated c-Fms recruits Vav and Rac during
CSF-1-induced cytoskeletal remodeling and spreading in osteoclasts.
Bone. 39:1290–1301. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Diebold I, Djordjevic T, Hess J and
Görlach A: Rac-1 promotes pulmonary artery smooth muscle cell
proliferation by upregulation of plasminogen activator inhibitor-1:
role of NFkappaB-dependent hypoxia-inducible factor-1alpha
transcription. Thromb Haemost. 100:1021–1028. 2008.
|
|
15
|
Williams LM, Lali F, Willetts K, Balague
C, Godessart N, Brennan F, Feldmann M and Foxwell BM: Rac mediates
TNF-induced cytokine production via modulation of NF-kappaB. Mol
Immunol. 45:2446–2454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu M, Qi X, Moreno JL, Farber DL and
Keegan AD: NF-κB signaling participates in both RANKL- and
IL-4-induced macrophage fusion: Receptor cross-talk leads to
alterations in NF-κB pathways. J Immunol. 187:1797–1806. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamashita T, Yao Z, Li F, Zhang Q, Badell
IR, Schwarz EM, Takeshita S, Wagner EF, Noda M, Matsuo K, et al:
NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB
ligand (RANKL) and tumor necrosis factor-induced osteoclast
precursor differentiation by activating c-Fos and NFATc1. J Biol
Chem. 282:18245–18253. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Valenzuela DM, Murphy AJ, Frendewey D,
Gale NW, Economides AN, Auerbach W, Poueymirou WT, Adams NC, Rojas
J, Yasenchak J, et al: High-throughput engineering of the mouse
genome coupled with high-resolution expression analysis. Nat
Biotechnol. 21:652–659. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Copeland NG, Jenkins NA and Court DL:
Recombineering: A powerful new tool for mouse functional genomics.
Nat Rev Genet. 2:769–779. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu P, Jenkins NA and Copeland NG: A
highly efficient recombineering-based method for generating
conditional knockout mutations. Genome Res. 13:476–484. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sarov M, Schneider S, Pozniakovski A,
Roguev A, Ernst S, Zhang Y, Hyman AA and Stewart AF: A
recombineering pipeline for functional genomics applied to
Caenorhabditis elegans. Nat Methods. 3:839–844. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rohrschneider LR, Rothwell VM and Nicola
NA: Transformation of murine fibroblasts by a retrovirus encoding
the murine c-fms proto-oncogene. Oncogene. 4:1015–1022.
1989.PubMed/NCBI
|
|
23
|
Lacey DL, Timms E, Tan HL, Kelley MJ,
Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S,
et al: Osteoprotegerin ligand is a cytokine that regulates
osteoclast differentiation and activation. Cell. 93:165–176. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yao GQ, Wu JJ, Troiano N, Zhu ML, Xiao XY
and Insogna K: Selective deletion of the membrane-bound colony
stimulating factor 1 isoform leads to high bone mass but does not
protect against estrogen-deficiency bone loss. J Bone Miner Metab.
30:408–418. 2012. View Article : Google Scholar
|
|
25
|
Kawano T, Zhu M, Troiano N, Horowitz M,
Bian J, Gundberg C, Kolodziejczak K and Insogna K: LIM kinase 1
deficient mice have reduced bone mass. Bone. 52:70–82. 2013.
View Article : Google Scholar :
|
|
26
|
Perona R, Montaner S, Saniger L,
Sánchez-Pérez I, Bravo R and Lacal JC: Activation of the nuclear
factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev.
11:463–475. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Montaner S, Perona R, Saniger L and Lacal
JC: Multiple signalling pathways lead to the activation of the
nuclear factor kappaB by the Rho family of GTPases. J Biol Chem.
273:12779–12785. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miyazaki T, Katagiri H, Kanegae Y,
Takayanagi H, Sawada Y, Yamamoto A, Pando MP, Asano T, Verma IM,
Oda H, et al: Reciprocal role of ERK and NF-kappaB pathways in
survival and activation of osteoclasts. J Cell Biol. 148:333–342.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ogasawara T1, Katagiri M, Yamamoto A,
Hoshi K, Takato T, Nakamura K, Tanaka S, Okayama H and Kawaguchi H:
Osteoclast differentiation by RANKL requires NF-kappaB-mediated
down-regulation of cyclin-dependent kinase 6 (Cdk6). J Bone Miner
Res. 19:1128–1136. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yao C, Yao GQ, Sun BH, Zhang C, Tommasini
SM and Insogna K: The transcription factor T-box3 regulates colony
stimulating factor 1-dependent Jun dimerization protein 2
expression and plays an important role in osteoclastogenesis. J
Biol Chem. 289:6775–6790. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lloyd SA, Yuan YY, Kostenuik PJ, Ominsky
MS, Lau AG, Morony S, Stolina M, Asuncion FJ and Bateman TA:
Soluble RANKL induces high bone turnover and decreases bone volume,
density, and strength in mice. Calcif Tissue Int. 82:361–372. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Frost JA, Swantek JL, Stippec S, Yin MJ,
Gaynor R and Cobb MH: Stimulation of NFkappa B activity by multiple
signaling pathways requires PAK1. J Biol Chem. 275:19693–19699.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boyer L, Travaglione S, Falzano L,
Gauthier NC, Popoff MR, Lemichez E, Fiorentini C and Fabbri A: Rac
GTPase instructs nuclear factor-kappaB activation by conveying the
SCF complex and IkBalpha to the ruffling membranes. Mol Biol Cell.
15:1124–1133. 2004. View Article : Google Scholar :
|
|
34
|
Michaelson D, Abidi W, Guardavaccaro D,
Zhou M, Ahearn I, Pagano M and Philips MR: Rac1 accumulates in the
nucleus during the G2 phase of the cell cycle and promotes cell
division. J Cell Biol. 181:485–496. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
van Hennik PB, ten Klooster JP, Halstead
JR, Voermans C, Anthony EC, Divecha N and Hordijk PL: The
C-terminal domain of Rac1 contains two motifs that control
targeting and signaling specificity. J Biol Chem. 278:39166–39175.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sandrock K, Bielek H, Schradi K, Schmidt G
and Klugbauer N: The nuclear import of the small GTPase Rac1 is
mediated by the direct interaction with karyopherin alpha2.
Traffic. 11:198–209. 2010. View Article : Google Scholar
|
|
37
|
Vaira S, Alhawagri M, Anwisye I, Kitaura
H, Faccio R and Novack DV: RelA/p65 promotes osteoclast
differentiation by blocking a RANKL-induced apoptotic JNK pathway
in mice. J Clin Invest. 118:2088–2097. 2008.PubMed/NCBI
|
|
38
|
Ikeda F, Matsubara T, Tsurukai T, Hata K,
Nishimura R and Yoneda T: JNK/c-Jun signaling mediates an
anti-apoptotic effect of RANKL in osteoclasts. J Bone Miner Res.
23:907–914. 2008. View Article : Google Scholar : PubMed/NCBI
|