Introduction
Periodontitis is a chronic-inflammatory disease with
a high incidence, which is initiated by microorganisms in the
dental biofilm and leads to the destruction of tooth-supporting
tissues and eventual tooth loss (1,2).
Periodontal ligament cells (PDLCs), as a type of stem-like cell,
play an important role in maintaining the dynamic stability and
function of the periodontium (3).
Human PDLCs not only function as support cells for the periodontal
tissues but also produce inflammatory mediators in response to
pathogens and proinflammatory stimuli; therefore, PDLCs are
important in the host immune response (3). The response of human PDLCs to
inflammatory stimuli is involved in the pathogenesis of chronic
periodontitis (4).
Periodontal pathogenic bacteria, such as
Porphyromanas gingivalis and Actinobacillus
actinomycetemcomitans, are likely to serve major roles in the
pathogenesis of periodontitis (5,6).
Lipopolysaccharide (LPS), a cell wall component of gram-negative
organisms, is also a potent inducer of the proinflammatory response
and initiates numerous host-mediated destructive processes
(4). Previous studies have shown
that when the human PDLCs are stimulated by LPS, the production
levels of the inflammatory cytokines cyclooxygenase-2 (COX-2),
interleukin-17 (IL)-17, tumor necrosis factor (TNF)-α and receptor
activator of nuclear factor-κB ligand (RANKL) are increased
(7). COX-2 is the rate-limiting
enzyme of prostaglandin E2 (PGE2) synthesis
(8). High levels of
PGE2 are detected in the gingiva and gingival crevicular
fluid of patients with periodontal disease (8), and PGE2 expression is
associated with bone resorption during the progression of
periodontal diseases (8–12). Smoking has been demonstrated to be
a risk factor for periodontitis; in tobacco-smoking patients with
periodontitis, nicotine, a major toxic substance in tobacco, has
been detected in the saliva and gingival crevicular fluid (13). Previous studies have shown that
nicotine inhibits the attachment and growth of human gingival
fibroblasts and human PDLCs, changes the periodontal tissue
microcirculation, and increases the absorption of alveolar bone
(13,14).
Following the discovery of RANKL and its decoy
receptor osteoprotegerin (OPG), it has been hypothesized that the
balance of the OPG/RANKL axis is critical in the regulation of
osteoclast differentiation and function (15). During the pathological process of
periodontitis, these inflammatory cytokines play an important role
in the destruction of the alveolar bone; thus, they are regarded as
targets for the suppression of inflammation in tobacco-smoking
patients with periodontitis.
Heme oxygenase-1 (HO-1), the most responsive of the
known induced enzymes, which can be induced to high levels within
hours by cytokines, hemoglobin, oxygen, hyperoxia, heat shock,
endotoxins, hydrogen peroxide, ultraviolet radiation, heavy metal
and nitric oxide (16), has
become a focus of medical research. Studies have demonstrated that
HO-1 has cell-protective functions (17,18). With heme as its substrate, HO-1
metabolizes and produces carbon monoxide (CO), biliverdin and
Fe2+ (19). CO has
been confirmed to serve a notable role in the biological processes
of many cells, including the inhibition of cell proliferation and
apoptosis, and the suppression of the immune inflammatory response
(20,21). CO-releasing molecules (CORMs) are
a new class of compounds, typically transition metal carbonyl
complexes, that are capable of liberating CO under the appropriate
conditions (22). Therefore,
CORMs may be of therapeutic interest due to their capacity to
modulate ongoing inflammatory reactions by delivering CO in a
controllable fashion (23). In
addition, CORMs have been widely used to increase understanding of
the biological function of CO (24,25).
Previously, the present research group found that
CORM-3 [tricarbonylchloro(glyconato)-ruthenium(II)], which is fully
water-soluble, and rapidly liberates CO when dissolved in
physiological solutions, increases HO-1 expression in vascular
endothelial cells (26). The
previous study also demonstrated that CORM-3 inhibits the
expression of adhesion molecules in human gingival fibroblasts
costimulated with TNF-α and IL-1β. In the present study, human
PDLCs were used as an in vitro model to investigate the
influence of CORM-3 on COX-2, PGE2, OPG and RANKL
expression in cells stimulated with LPS and nicotine. The possible
mechanism by which CORM-3 exerts this effect was also
investigated.
Materials and methods
Reagents
CORM-3 was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Dulbecco's modified Eagle's medium (DMEM) was
purchased from HyClone (GE Healthcare Life Sciences, Logan, UT,
USA); fetal bovine serum (FBS) was purchased from Biological
Industries (Kibbutz Beit-Haemek, Israel); 100X
Penicillin-Streptomycin Solution and LPS from Escherichia
coli were purchased from Beijing Solarbio Science and
Technology Co., Ltd. (Beijing, China); nicotine was purchased from
Cerilliant Corp. (Round Rock, TX, USA); antibodies against COX-2
(ab62331), OPG (ab73400), RANKL (ab9957) and HO-1 (ab13248) were
purchased from Abcam (Cambridge, UK); the antibody against
PGE2 (4a-2639R) was purchased from 4A Biotech Co., Ltd.
(Beijing, China); glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
antibody (10494-1-AP), α-tubulin antibody (11224-1-AP) and the
secondary antibodies horseradish peroxidase (HRP)-conjugated
affinipure goat anti-rabbit IgG (H+L) (SA00001-2) and
HRP-conjugated affinipure goat anti-mouse IgG (H+L) (SA00001-1)
were purchased from Proteintech Group, Inc. (Chicago, IL, USA); and
the primers for OPG, RANKL, PGE2, COX-2 and HO-1, and
the small interfering RNAs (siRNAs) were purchased from GenePharma
(Shanghai, China).
Cell culture
Human PDLCs were isolated with an explant culture
technique from normal periodontal ligament tissues obtained from
impacted wisdom teeth being removed or the teeth of patients
undergoing orthodontic treatment. Informed consent was obtained
from the patients prior to the surgery. The study was approved by
the Ethics Committee of the School of Dentistry, Shandong
University (Jinan, China). Briefly, the tissues were cut into
1-mm2 explants and placed in 25-cm2 culture
bottles (Corning Inc., Corning, NY, USA) containing 100 U/ml
penicillin G, 100 mg/ml streptomycin and 20% heat-inactivated FBS
at 37°C in a humidified atmosphere of 5% CO2 and 95%
air. After 5–7 days, the cells were detached with 0.025% trypsin
and 0.05% EDTA diluted with culture medium and then subcultured at
a ratio of 1:2, and the concentration of the FBS was changed to
10%; the other components in the medium remained the same. Cells
between the 4th and 6th passages were used in the subsequent
experiments.
Cell Counting kit-8 (CCK-8) assay
The toxicities of different concentrations of CORM-3
to the human PDLCs were assessed using a CCK-8 (WST-8) assay. The
PDLCs were seeded and cultured in 96-well plates (8,000
cells/well). The cells were divided into five groups and were
treated with CORM-3 at 0, 100, 200, 400 and 800 μM for 24 h.
After this, the CCK-8 reagent was added to every well, and the
cells were incubated in a 37°C incubator for ≥0.5 h in the dark.
The optical density (OD) values were read at 450 nm using a
microplate reader (SPECTROstar Nano; BMG Labtech, Ortenberg,
Germany). The data were collected every 0.5 h within 4 h of the
addition of CCK-8.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
PDLCs were grown in 6-well plates (200,000
cells/well) and incubated in fresh medium containing various
stimuli. For the first part of the experiment, the cells were
divided into four groups as follows: Control group; LPS + nicotine
group, in which the cells were incubated with LPS (1 μg/ml)
and nicotine (5 mM) for 24 h; CORM-3 + LPS + nicotine group, in
which the cells were pretreated with CORM-3 (400 μM) for 6 h
prior to incubation with LPS (1 μg/ml) and nicotine (5 mM);
and deactivated CORM-3 + LPS + nicotine group, in which the cells
were pretreated with deactivated CORM-3 (400 μM) for 6 h
prior to incubation with LPS and nicotine. Deactivated CORM-3 was
produced by dissolving CORM-3 in water to provide a 400 μM
aqueous solution, and putting the solution in a vacuum device for
24 h prior to use.
For the second part of the experiment, the cells
were divided into four groups as follows: Control group; scrambled
siRNA control group, in which the cells were transiently
transfected with scrambled siRNA; HO-1 siRNA + LPS + nicotine
group, in which the cells were transiently transfected with HO-1
siRNA prior to incubation with LPS (1 μg/ml) and nicotine (5
mM); and HO-1 siRNA + CORM-3 + LPS + nicotine group, in which the
cells transiently transfected with HO-1 siRNA were pretreated with
CORM-3 (400 μM) for 6 h prior to incubation with LPS and
nicotine.
Total RNA was extracted from the cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc., waltham, MA,
USA) according to the manufacturer's protocol. The RNA
concentration was assessed using a UV spectrophotometer (GeneQuant
pro DNA/RNA Calculator; GE Healthcare Life Sciences). The cDNA was
amplified at 42°C by combining RNA, gDNA eraser, gDNA eraser buffer
and RNase-free dH2O, with PrimeScript RT enzyme mix,
PrimeScript buffer, RT primer mix and RNase free dH2O,
using RNA PCR kit (AMV) Ver.3.0 (Takara Bio, Inc., Otsu, Japan)
according to the manufacturer's protocol. qPCR was performed using
SYBR Premix Ex Taq (Takara Bio., Inc.) with a Biometra thermocycler
(TGradient; Biometra GmbH, Göttingen, Germany). The reaction
conditions for the qPCR were 45 cycles of denaturation at 95.8°C
for 30 sec, annealing at 55–60.8°C for 30 sec, and extension at
72.8°C for 1 min. The primer sequences for differentiation markers
were as follows: PGE2 forward, 5′-CGATGCTCATGCTCTTCGC-3′
and reverse, 5′-GGGAGACTGCATAGATGACAGG-3′; COX-2 forward,
5′-CTGGCGCTCAGCCATACAG-3′ and reverse,
5′-CGCACTTATACTGGTCAAATCCC-3′; RANKL forward,
5′-TCCATGTTCGTGGCCCTC-3′ and reverse, 5′-GCAGTGAGTGCCATCTTCTG-3′;
OPG forward, 5′-GTGTGCGAATGCAAGGAAGG-3′ and reverse,
5′-CCACTCCAAATCCAGGAGGG-3′; HO-1 forward, 5′-AGGCCAAGACTGCGTTCCT-3′
and reverse, 5′-AACTGTCGCCACCAGAAAGCTGAG-3′; GAPDH forward,
5′-GCACCGTCAAGGCT GAGAAC-3′ and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′.
The 2−ΔΔCq method was used for quantification (27).
Western blot analysis
PDLCs were grown in 6-well plates and incubated in
fresh medium containing various stimuli; the groups were
established as described above for the first and second part of the
experiment. The cells in the 6-well plates from each set of
experiments were harvested and washed three times in cold
phosphate-buffered saline. The cells were then solubilized in
ice-cold radioimmunoprecipitation assay lysis buffer (Solarbio,
Beijing, China), and after 30 min on ice, the lysates were
clarified by centrifugation with the speed of 12,000 × g at 4°C for
5 min. The protein samples (20 μg), which were quantified
using a BCA kit (Solarbio), were mixed with a quarter-volume of 5X
SDS-PAGE loading buffer, boiled for 5 min to denature, and then
separated by 10% SDS-polyacrylamide electrophoresis (Gel
Preparation kit; Boster Biological Technology, Pleasanton, CA,
USA). Following electrophoresis, the proteins were transferred to
polyvinylidene fluoride membranes (0.45 μm) via
electrophoretic transfer. The membranes were blocked in 5% dry milk
(1 h), rinsed and incubated with antibodies (COX-2, 1:1,000; OPG, 1
μg/ml; RANKL, 1 μg/ml; PGE2, 1:1,000;
HO-1, 4 μg/ml; GAPDH, 1:2,000; α-tubulin, 1:2,000) in
Tris-buffered saline (TBS) overnight at 4°C. The primary antibodies
were then removed by washing the membranes three times in TBS. The
primary antibodies were labelled via incubation with 0.1 mg/ml
HRP-labeled secondary antibodies for 1 h. Following three washes in
TBS, the bands were visualised using a western blot
chemiluminescence reagent (EMD Millipore, Billerica, MA, USA) and
ChampChem™ automated chemiluminescence system (Sage Creation
Science Co., Ltd., Beijing, China), and then imaged using Lane 1D
4.0 gel imaging analysis software (Sage Creation Science Co.,
Ltd.).
Transient transfection
Human PDLCs were transfected with HO-1 siRNA as
follows. The siRNA target sequences for HO-1 were: forward,
5′-GGGUCCUUACAUUCAGCUUTT-3′ and reverse,
5′-AAGCUGAGUGUAAGGACCCTT-3′. Four groups were established as
defined above for the second part of the experiment. In brief, the
cells in 6-well plates (150,000 cells/well) were transfected with 5
μl siRNA for 6 h using Lipofectamine® 3000
Transfection reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The cells were cultured
in Opti-MEM® Reduced Serum Medium (Gibco; Thermo Fisher
Scientific, Inc.) and transiently transfected with the scrambled
siRNA or HO-1 siRNA, followed by treatment with LPS and nicotine in
the presence or absence of CORM-3.
Statistical analysis
Differences among the groups were analyzed using
one-way analysis of variance, and comparisons between groups were
performed using Tukey's test. All values are expressed as the mean
and standard deviation, and P<0.05 was considered to indicate a
statistically significant difference. The statistical analysis was
conducted using SPSS version 22.0 software (IBM Corp., Armonk, NY,
USA).
Results
Cytotoxicity of CORM-3 to human
periodontal ligament cells
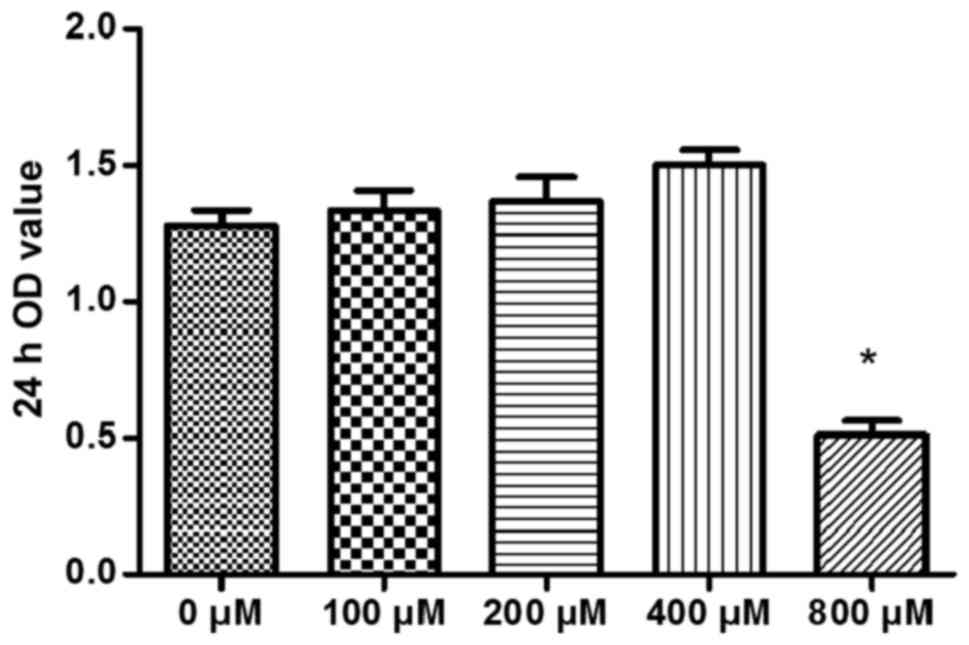
To evaluate the cytotoxicity of CORM-3 to human
PDLCs, the PDLCs were treated with CORM-3 at concentrations of 0,
100, 200, 400 and 800 μM for 24 h in 96-well plates (8,000
cells/well). After 24 h, the OD values were determined (Fig. 1). CORM-3 at 800 μM was
significantly cytotoxic to human PDLCs, but 400 μM of CORM-3
exhibited no significant cytotoxicity to the cells. The cells
treated with CORM-3 at 400 μM exhibited a certain degree of
cell proliferation, but this finding was not statistically
significant (Fig. 1). Similar
results were obtained in three independent experiments. Therefore,
a CORM-3 concentration of 400 μM was selected for use in
subsequent experiments.
CORM-3 suppresses the nicotine- and
LPS-induced inflammatory response via the release of CO
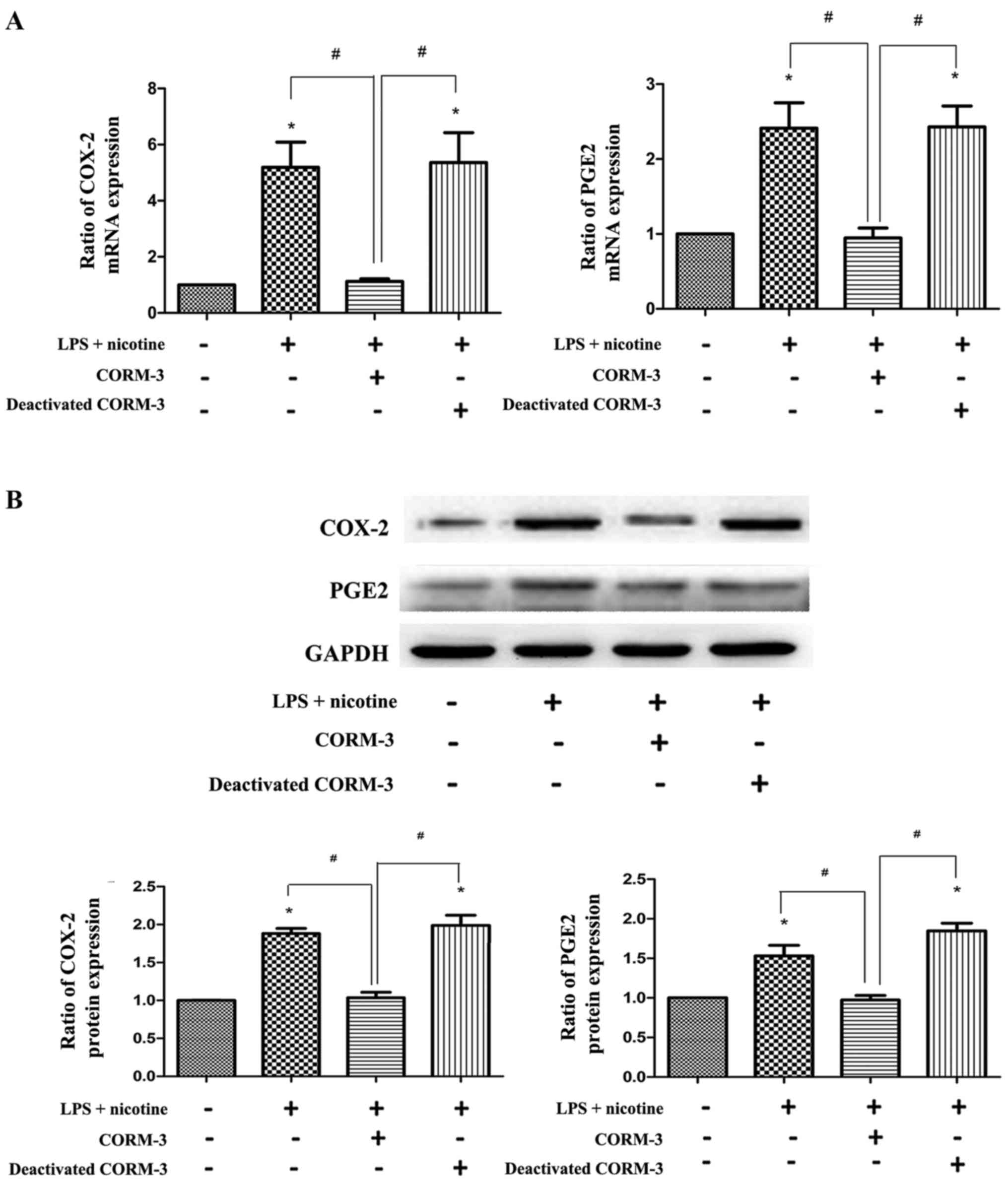
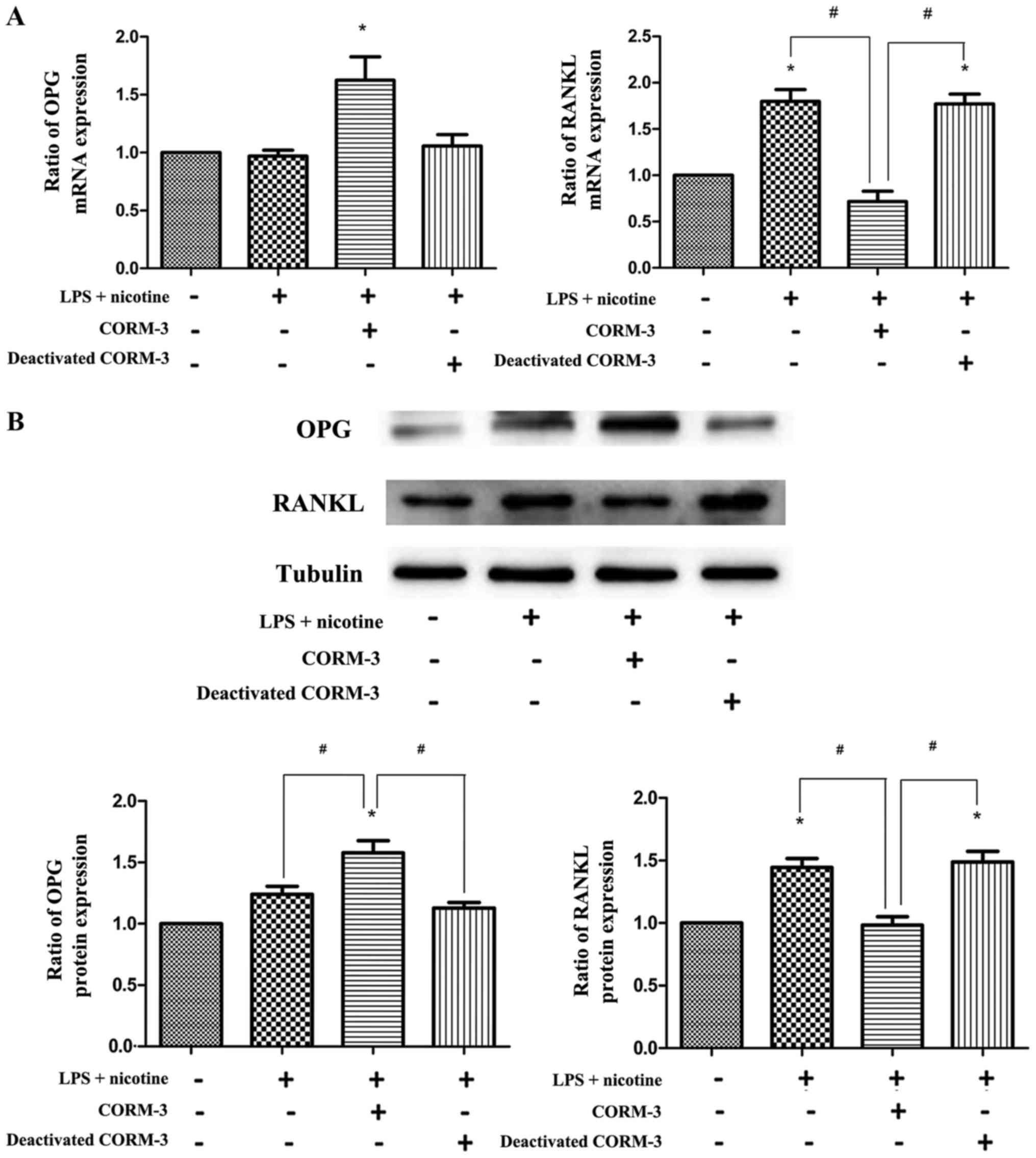
To determine the effects of CORM-3 on the
inflammatory response and osteoclastogenesis in vitro, the
expression of COX-2, PGE2, OPG and RANKL induced in
human PDLCs by LPS (1 μg/ml) and nicotine (5 mM) with or
without CORM-3 (400 μM) pretreatment was examined.
Co-treatment with LPS and nicotine resulted in increased COX-2,
PGE2 (Fig. 2) and
RANKL (Fig. 3) expression.
However, with CORM-3 pretreatment, an inhibitory effect on these
three inflammatory cytokines was observed, and the expression of
OPG exhibited a significant increase (Fig. 3). By contrast, deactivated CORM-3,
which is not able to release CO, did not attenuate LPS- and
nicotine-induced inflammatory cytokine expression in the human
PDLCs, which demonstrated that the effect of CORM-3 is mediated by
CO. These results were verified by the evaluation of mRNA (Figs. 2A and 3A) and protein (Figs. 2B and 3B) expression. The expression levels of
COX-2, PGE2 and RANKL in the CORM-3 + LPS + nicotine
group were decreased compared with those in the LPS + nicotine
group and the deactivated CORM-3 + LPS + nicotine group. The
expression levels of OPG were increased in the CORM-3 + LPS +
nicotine group in comparison with those in the LPS + nicotine group
and deactivated CORM-3 + LPS + nicotine group.
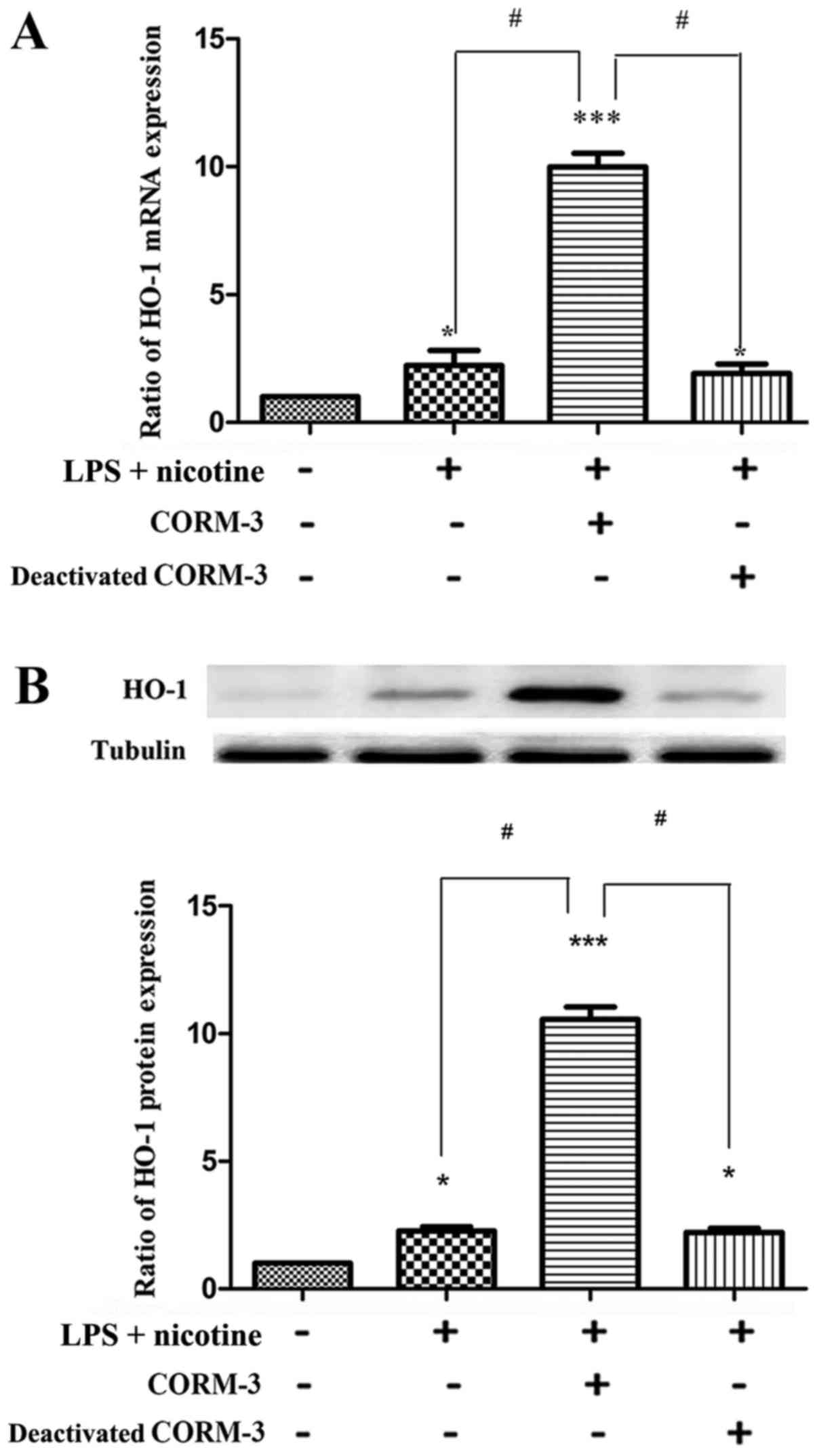
CORM-3 promotes HO-1 expression in HPDLCs
incubated with or without LPS and nicotine via the release of
CO
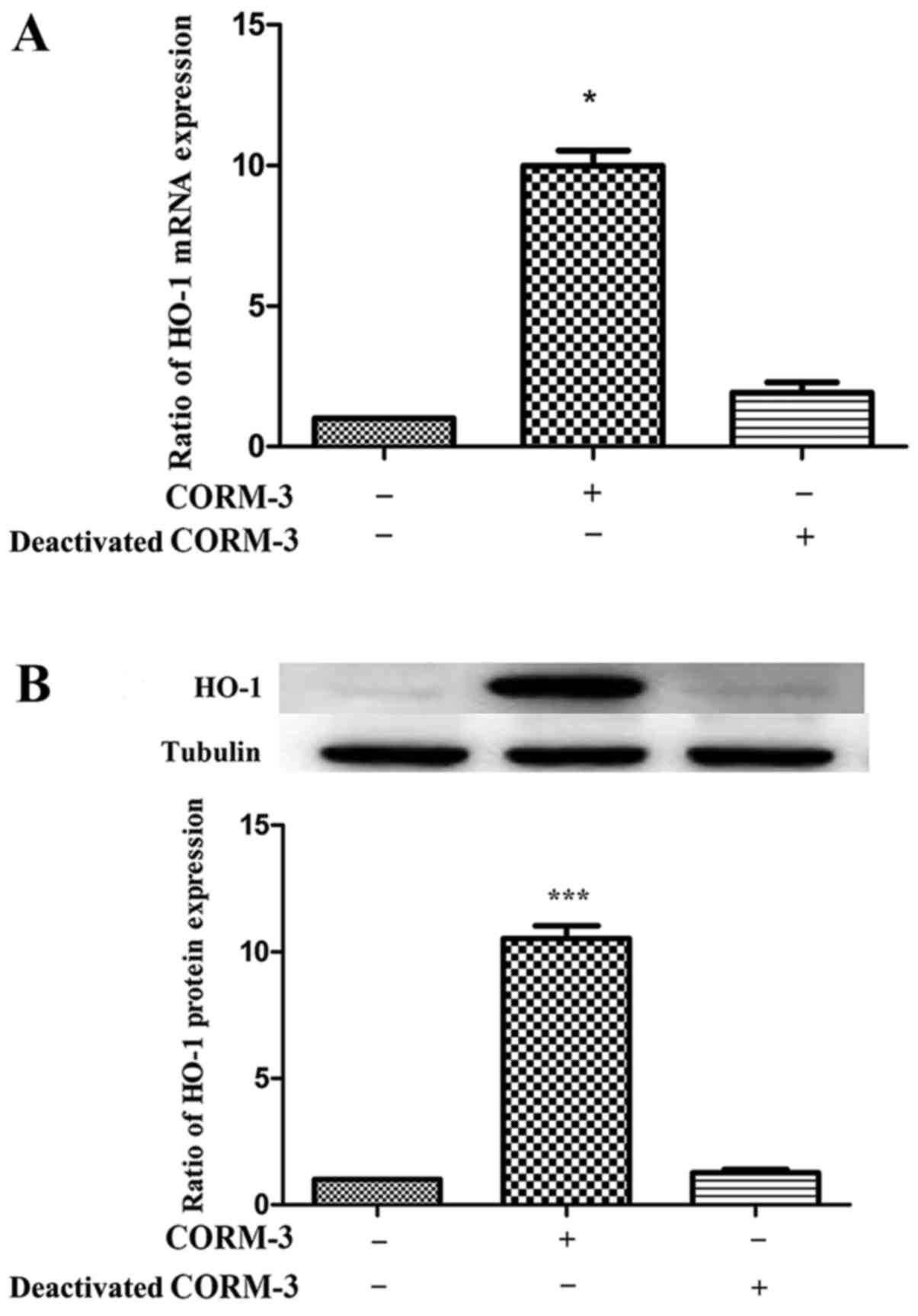
First, human PDLCs were examined for HO-1 expression
induced by CORM-3 and it was found that HO-1 expression was
upregulated significantly compared with that in the control group,
as shown in Fig. 4. In addition,
deactivated CORM-3 did not exhibit an inductive effect on HO-1
expression, which indicated that the effect of CORM-3 was mediated
by CO. These results were confirmed for HO-1 mRNA (Fig. 4A) and protein expression (Fig. 4B). Following this, human PDLCs
were examined for HO-1 expression induced by LPS and nicotine, with
or without CORM-3 pretreatment. As shown in Fig. 5, nicotine- and LPS-induced HO-1
expression levels were comparable to those in the control cultures.
Furthermore, CORM-3 pretreatment significantly promoted the mRNA
and protein expression of HO-1 in human PDLCs compared with the
respective levels in the nicotine + LPS group (Fig. 5). It may be concluded that CORM-3
promotes HO-1 expression and, notably, its expression was elevated
compared with that in human PDLCs only co-treated with LPS and
nicotine. It was also observed that deactivated CORM-3, which is
not able to release CO, did not have an inductive effect on
HO-1.
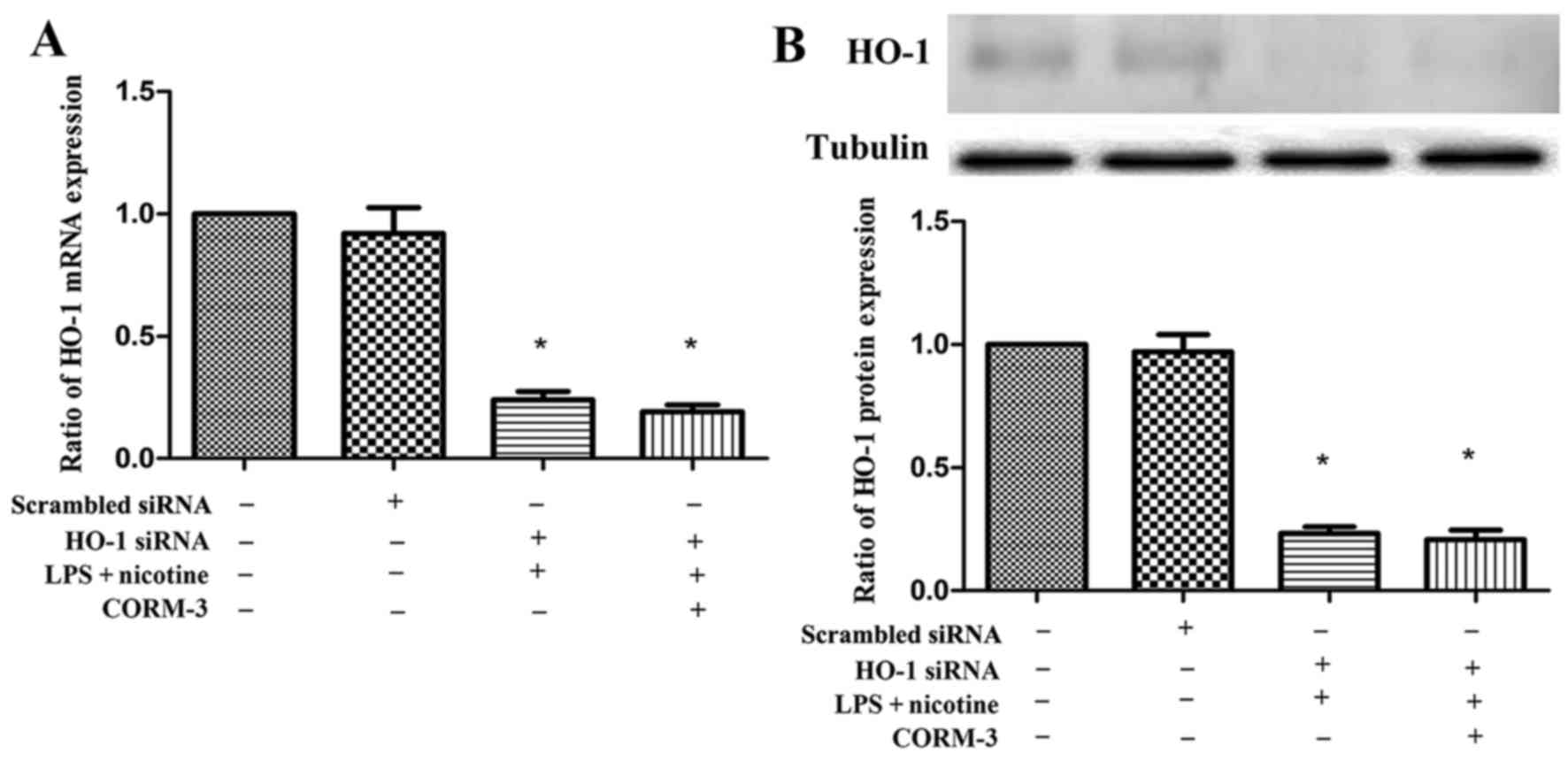
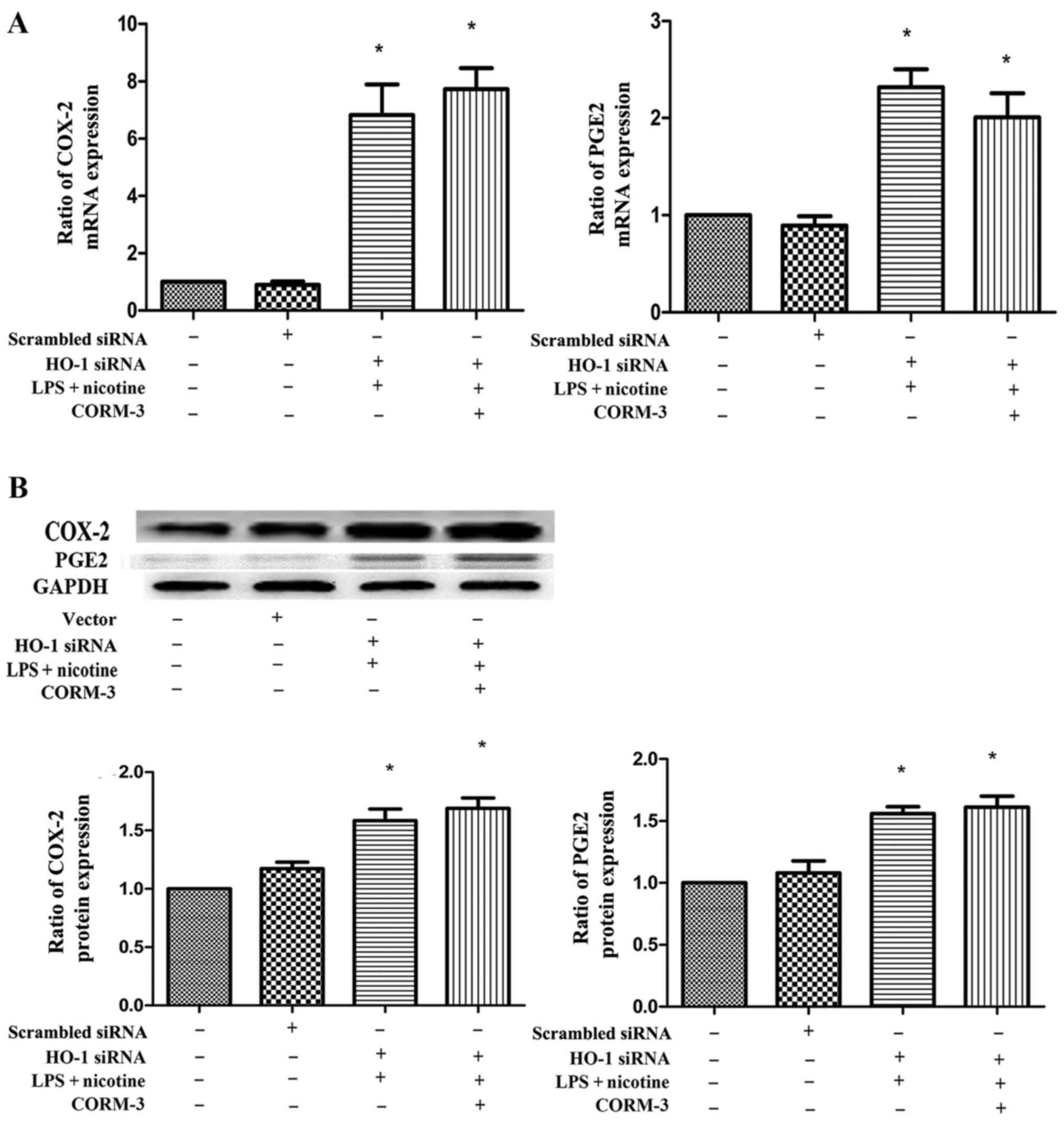
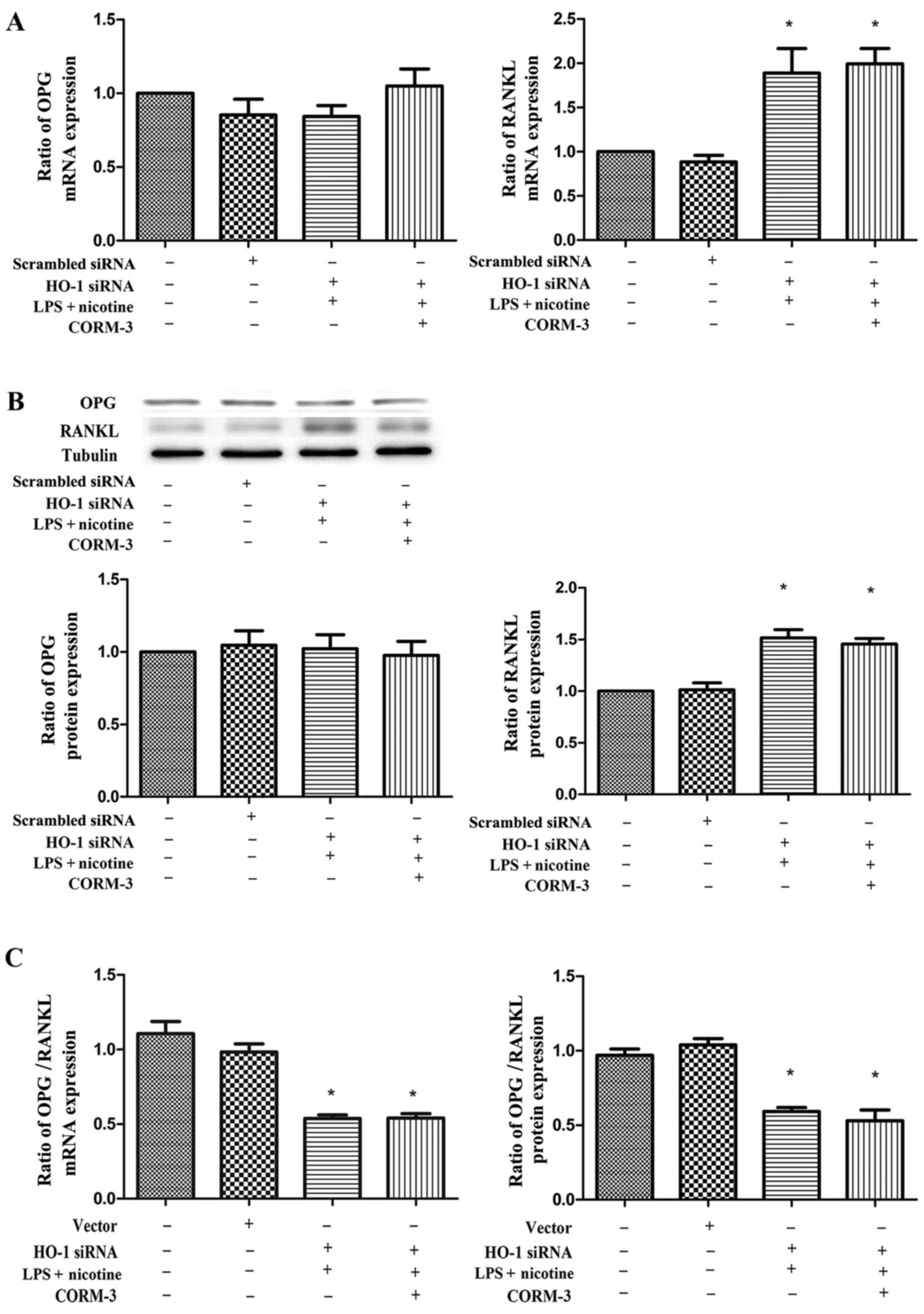
Effect of silencing HO-1 on the LPS- and
nicotine-induced inflammatory response with or without CORM-3
pretreatment
To examine whether HO-1 expression is involved in
the nicotine- and LPS-mediated induction of COX-2, PGE2,
OPG and RANKL production, human PDLCs were transfected with HO-1
siRNA and then subjected to 24 h exposure to LPS and nicotine with
or without CORM-3 pretreatment for 6 h. The mRNA and protein
expression levels of HO-1 following transfection with HO-1 siRNA
were significantly suppressed (Fig.
6). As shown in Figs. 7 and
8, HO-1 siRNA transfection
abolished the anti-inflammatory effects of CORM-3, and the COX-2,
PGE2 and RANKL expression levels were not downregulated
by CORM-3. These results were verified by mRNA (Figs. 7A and 8A) and protein (Figs. 7B and 8B) expression profiles. The expression
levels of COX-2, PGE2 and RANKL in the HO-1 siRNA +
CORM-3 + LPS + nicotine group were increased in comparison with
those in the control groups, and were not statistically different
in comparison with those in the HO-1 siRNA + LPS + nicotine group.
Following transient transfection with HO-1 siRNA, CORM-3 exhibited
no effect on OPG expression (Fig.
8). However, it was observed that the ratio of OPG/RANKL in the
HO-1 siRNA + CORM-3 + LPS + nicotine group was decreased in
comparison with that in the control groups and was not
statistically different in comparison with that in HO-1 siRNA + LPS
+ nicotine group (Fig. 8C).
Discussion
Previous studies have reported on the nicotine- and
LPS-mediated induction of proinflammatory cytokines, including
COX-2, PGE2 and RANKL, in human PDLCs (28–32). In addition, CORM-3 has been
demonstrated to modulate inflammatory processes in a variety of
in vivo and in vitro models (33,34). However, to the best of our
knowledge, the inhibitory effect of CORM-3 on the induction of
proinflammatory cytokines by LPS and nicotine in human PDLCs has
not been demonstrated. In the present study, the effect of CORM-3
on the LPS and nicotine-stimulated inflammatory response and the
role of HO-1 in the process was investigated.
Human PDLCs were selected for use in the study as
they are the group of cells that are most highly affected by LPS
and nicotine in periodontitis; they also play the most important
role in the regeneration of periodontal attachment (3). The host inflammatory response to
smoking and dental plaque are considered major causative factors in
the local tissue destruction observed in periodontitis (13,14,19); thus, the present study was
conducted using nicotine- and LPS-treated human PDLCs with
pretreatment with or without CORM-3.
The results of the study suggested that when human
PDLCs were exposed to LPS and nicotine, COX-2 and PGE2
production increased. The results also demonstrated that LPS and
nicotine upregulated RANKL expression and downregulated OPG
expression in human PDLCs. Furthermore, it was found that
pretreatment with CORM-3 effectively reduced LPS- and
nicotine-induced COX-2, PGE2 and RANKL levels and
promoted OPG levels in human PDLCs. By contrast, deactivated
CORM-3, which is not able to release CO, did not attenuate LPS- and
nicotine-induced inflammatory cytokine expression in human PDLCs.
Thus, it may be hypothesized that CORM-3exertsanti-inflammatory and
anti-osteoclastogenic effects via the release of CO.
Among the many molecules involved in the
inflammatory process, COX-2 is highly expressed during periodontal
disease and is responsible for the production of PGE2,
which is involved in inflammation (35,36). Studies have reported that
PGE2 mediates bone resorption through the activation of
osteoclasts and RANKL in vitro (10) and in vivo (37). RANKL is essential in
osteoclastogenesis, and PGE2 is considered a major
target for periodontal therapy since it is inducible and present in
the cells involved in periodontal disease (37,38). These inflammatory molecules
activate osteoclasts and induce bone resorption as a result of an
exacerbated host response (39).
Previous studies have reported that HO-1 induction
by proinflammatory cytokines, nitric oxide, mechanical stress and
hydrogen peroxide may exert cytoprotective and anti-inflammatory
effects in human pulp and PDLCs via the production of CO with heme
as its substrate (40–43). In addition, HO-1 serves critical
roles in the regulation of cell growth and differentiation and in
the control of cellular responses to cytokines and other stresses
(44). The results of the present
study demonstrated that LPS and nicotine induced HO-1 expression in
human PDLCs, and CORM-3 induced HO-1 expression more strongly than
the combination of LPS and nicotine.
The present authors hypothesized that HO-1 induction
by CORM-3 in human PDLCs may be responsible for the
anti-inflammatory effects of CORM-3 against the inflammation
induced by LPS and nicotine. One finding of the present study was
that CORM-3 inhibited the inflammatory effects induced by the
combination of LPS and nicotine. The inhibitory effect of CORM-3
may have been mediated via the release of CO, since the deactivated
CORM-3 did not suppress the inflammatory response to LPS and
nicotine stimulation. In addition, following HO-1 siRNA
transfection, the anti-inflammatory effects of CORM-3 as a
downregulator of COX-2 and PGE2 expression and RANKL
production vanished and the ratio of OPG/RANKL decreased
concurrently. These findings indicate that the anti-inflammatory
effects of CORM-3 are dependent upon HO-1 overexpression in human
PDLCs. Based on these findings, it is proposed that CORM-3, as a
controllable extrinsic molecule, represents a novel preventive or
therapeutic agent for periodontitis. A number of studies have
postulated putative mechanisms by which HO-1 exerts its
anti-inflammatory effect (17,18); however, these mechanisms require
further elucidation. In the future, the present research group
plans to study these cell-signalling pathways and mechanisms
further.
In conclusion, the results of the present study
demonstrate for the first time that anti-inflammatory and
anti-osteoclastogenic effects of CORM-3 proceed via HO-1-dependent
pathways in periodontal disease models, which are mediated through
the expression of OPG/RANKL and COX-2/PGE2. Thus, it is
suggested that CORM-3 may be of potentially useful therapeutic
value for the treatment of periodontitis resulting from dental
plaque and smoking.
Acknowledgments
This study was supported by the Shandong Province
Natural Science (grant no. ZR2015HM019), the Jinan College and
University Science and Technology Innovation Program (grant no.
201401259) and the Special Funds for Education and Awards of
Shandong Province (grant no. 2014-94).
References
|
1
|
Listgarten MA: Bacteria and periodontitis.
J Can Dent Assoc. 62:12–13. 1996.PubMed/NCBI
|
|
2
|
Sanz M and van Winkelhoff AJ; Working
Group 1 of Seventh European Workshop on Periodontology: Periodontal
infections: Understanding the complexity - consensus of the Seventh
European Workshop on Periodontology. J Clin Periodontol. 38(Suppl
11): 3–6. 2011. View Article : Google Scholar
|
|
3
|
Socransky SS and Haffajee AD: The
bacterial etiology of destructive periodontal disease: Current
concepts. J Periodontol. 63(Suppl 4): 322–331. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yamaji Y, Kubota T, Sasaguri K, Sato S,
Suzuki Y, Kumada H and Umemoto T: Inflammatory cytokine gene
expression in human periodontal ligament fibroblasts stimulated
with bacterial lipopolysaccharides. Infect Immun. 63:3576–3581.
1995.PubMed/NCBI
|
|
5
|
Holt SC, Kesavalu L, Walker S and Genco
CA: Virulence factors of Porphyromonas gingivalis. Periodontol
2000. 20:168–238. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Slots J and Listgarten MA: Bacteroides
gingivalis, Bacteroides intermedius and Actinobacillus
actinomycetemcomitans in human periodontal diseases. J Clin
Periodontol. 15:85–93. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Graves DT and Cochran D: The contribution
of interleukin-1 and tumor necrosis factor to periodontal tissue
destruction. J Periodontol. 74:391–401. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Offenbacher S, Heasman PA and Collins JG:
Modulation of host PGE2 secretion as a determinant of
periodontal disease expression. J Periodontol. 64(Suppl 5):
432–444. 1993.PubMed/NCBI
|
|
9
|
Fukushima H, Jimi E, Okamoto F, Motokawa W
and Okabe K: IL-1-induced receptor activator of NF-kappa B ligand
in human periodontal ligament cells involves ERK-dependent
PGE2 production. Bone. 36:267–275. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shimizu N, Ozawa Y, Yamaguchi M, Goseki T,
Ohzeki K and Abiko Y: Induction of COX-2 expression by mechanical
tension force in human periodontal ligament cells. J Periodontol.
69:670–677. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Offenbacher S, Odle BM, Gray RC and Van
Dyke TE: Crevicular fluid prostaglandin E levels as a measure of
the periodontal disease status of adult and juvenile periodontitis
patients. J Periodontal Res. 19:1–13. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Offenbacher S, Odle BM and Van Dyke TE:
The use of crevicular fluid prostaglandin E2 levels as a
predictor of periodontal attachment loss. J Periodontal Res.
21:101–112. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McGuire JR, McQuade MJ, Rossmann JA,
Garnick JJ, Sutherland DE, Scheidt MJ and Van Dyke TE: Cotinine in
saliva and gingival crevicular fluid of smokers with periodontal
disease. J Periodontol. 60:176–181. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tanur E, McQuade MJ, McPherson JC,
Al-Hashimi IH and Rivera-Hidalgo F: Effects of nicotine on the
strength of attachment of gingival fibroblasts to glass and
non-diseased human root surfaces. J Periodontol. 71:717–722. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krishnan V and Davidovitch Z: Cellular,
molecular, and tissue-level reactions to orthodontic force. Am J
Orthod Dentofacial Orthop. 129:469.e1–469.e32. 2006. View Article : Google Scholar
|
|
16
|
Panahian N, Yoshiura M and Maines MD:
Overexpression of heme oxygenase-1 is neuroprotective in a model of
permanent middle cerebral artery occlusion in transgenic mice. J
Neurochem. 72:1187–1203. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Llesuy SF and Tomaro ML: Heme oxygenase
and oxidative stress. Evidence of involvement of bilirubin as
physiological protector against oxidative damage. Biochim Biophys
Acta. 1223:9–14. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Poss KD and Tonegawa S: Heme oxygenase 1
is required for mammalian iron reutilization. Proc Natl Acad Sci
USA. 94:10919–10924. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pi SH, Jeong GS, Oh HW, Kim YS, Pae HO,
Chung HT, Lee SK and Kim EC: Heme oxygenase-1 mediates nicotine-
and lipopolysaccharide-induced expression of cyclooxygenase-2 and
inducible nitric oxide synthase in human periodontal ligament
cells. J Periodontal Res. 45:177–183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Otterbein LE, Bach FH, Alam J, Soares M,
Tao Lu H, Wysk M, Davis RJ, Flavell RA and Choi AM: Carbon monoxide
has anti-inflammatory effects involving the mitogen-activated
protein kinase pathway. Nat Med. 6:422–428. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sethi JM, Otterbein LE and Choi AM:
Differential modulation by exogenous carbon monoxide of TNF-alpha
stimulated mitogen-activated protein kinases in rat pulmonary
artery endothelial cells. Antioxid Redox Signal. 4:241–248. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Motterlini R, Clark JE, Foresti R,
Sarathchandra P, Mann BE and Green CJ: Carbon monoxide-releasing
molecules: Characterization of biochemical and vascular activities.
Circ Res. 90:E17–E24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alcaraz MJ, Guillen MI, Ferrandiz ML,
Megías J and Motterlini R: Carbon monoxide-releasing molecules: A
pharmacological expedient to counteract inflammation. Curr Pharm
Des. 14:465–472. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Foresti R, Hammad J, Clark JE, Johnson TR,
Mann BE, Friebe A, Green CJ and Motterlini R: Vasoactive properties
of CORM-3, a novel water-soluble carbon monoxide-releasing
molecule. Br J Pharmacol. 142:453–460. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Motterlini R: Carbon monoxide-releasing
molecules (CO-RMs): Vasodilatory, anti-ischaemic and
anti-inflammatory activities. Biochem Soc Trans. 35:1142–1146.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song H, Bergstrasser C, Rafat N, Höger S,
Schmidt M, Endres N, Goebeler M, Hillebrands JL, Brigelius-Flohé R,
Banning A, et al: The carbon monoxide releasing molecule (CORM-3)
inhibits expression of vascular cell adhesion molecule-1 and
E-selectin independently of haem oxygenase-1 expression. Br J
Pharmacol. 157:769–780. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Hong JY, Bae WJ, Yi JK, Kim GT and Kim EC:
Anti-inflammatory and anti-osteoclastogenic effects of zinc finger
protein A20 over-expression in human periodontal ligament cells. J
Periodontal Res. 51:529–539. 2016. View Article : Google Scholar
|
|
29
|
Noguchi K, Shitashige M, Yanai M, Morita
I, Nishihara T, Murota S and Ishikawa I: Prostaglandin production
via induction of cyclooxygenase-2 by human gingival fibroblasts
stimulated with lipopolysaccharides. Inflammation. 20:555–568.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gutiérrez-Venegas G, Maldonado-Frı'as S,
Ontiveros-Granados A and Kawasaki Cárdenas P: Role of p38 in nitric
oxide synthase and cyclooxygenase expression, and nitric oxide and
PGE2 synthesis in human gingival fibroblasts stimulated
with lipopolysaccharides. Life Sci. 77:60–73. 2005. View Article : Google Scholar
|
|
31
|
Wada N, Maeda H, Yoshimine Y and Akamine
A: Lipopolysaccharide stimulates expression of osteoprotegerin and
receptor activator of NF-kappa B ligand in periodontal ligament
fibroblasts through the induction of interleukin-1 beta and tumor
necrosis factor-alpha. Bone. 35:629–635. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chang YC, Tsai CH, Yang SH, Liu CM and
Chou MY: Induction of cyclooxygenase-2 mRNA and protein expression
in human gingival fibroblasts stimulated with nicotine. J
Periodontal Res. 38:496–501. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Urquhart P, Rosignoli G, Cooper D,
Motterlini R and Perretti M: Carbon monoxide-releasing molecules
modulate leukocyte-endothelial interactions under flow. J Pharmacol
Exp Ther. 321:656–662. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ferrándiz ML, Maicas N, Garcia-Arnandis I,
Terencio MC, Motterlini R, Devesa I, Joosten LA, van den Berg WB
and Alcaraz MJ: Treatment with a CO-releasing molecule (CORM-3)
reduces joint inflammation and erosion in murine collagen-induced
arthritis. Ann Rheum Dis. 67:1211–1217. 2008. View Article : Google Scholar
|
|
35
|
Bae WJ, Shin MR, Kang SK, Zhang-Jun, Kim
JY, Lee SC and Kim EC: HIF-2 inhibition suppresses inflammatory
responses and osteoclastic differentiation in human periodontal
ligament cells. J Cell Biochem. 116:1241–1255. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim YS, Shin SI, Kang KL, Chung JH, Herr
Y, Bae WJ and Kim EC: Nicotine and lipopolysaccharide stimulate the
production of MMPs and prostaglandin E2 by
hypoxia-inducible factor-1α up-regulation in human periodontal
ligament cells. J Periodontal Res. 47:719–728. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Saito S, Ngan P, Rosol T, Saito M, Shimizu
H, Shinjo N, Shanfeld J and Davidovitch Z: Involvement of PGE
synthesis in the effect of intermittent pressure and interleukin-1
beta on bone resorption. J Dent Res. 70:27–33. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Garlet GP, Cardoso CR, Silva TA, Ferreira
BR, Avila-Campos MJ, Cunha FQ and Silva JS: Cytokine pattern
determines the progression of experimental periodontal disease
induced by Actinobacillus actinomycetemcomitans through the
modulation of MMPs, RANKL, and their physiological inhibitors. Oral
Microbiol Immunol. 21:12–20. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yasuda H, Shima N, Nakagawa N, Yamaguchi
K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A,
et al: Osteoclast differentiation factor is a ligand for
osteoprotegerin/osteoclastogenesis-inhibitory factor and is
identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 95:3597–3602.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee TS and Chau LY: Heme oxygenase-1
mediates the anti-inflammatory effect of interleukin-10 in mice.
Nat Med. 8:240–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Min KS, Kwon YY, Lee HJ, Lee SK, Kang KH,
Lee SK and Kim EC: Effects of proinflammatory cytokines on the
expression of mineralization markers and heme oxygenase-1 in human
pulp cells. J Endod. 32:39–43. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Min KS, Hwang YH, Ju HJ, Chang HS, Kang
KH, Pi SH, Lee SK, Lee SK and Kim EC: Heme oxygenase-1 mediates
cytoprotection against nitric oxide-induced cytotoxicity via the
cGMP pathway in human pulp cells. Oral Surg Oral Med Oral Pathol
Oral Radiol Endod. 102:803–808. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pi SH, Kim SC, Kim HT, Lee HJ, Lee SK and
Kim EC: Defense mechanism of heme oxygenase-1 against cytotoxic and
receptor activator of nuclear factor-kappaB ligand inducing effects
of hydrogen peroxide in human periodontal ligament cells. J
Periodontal Res. 42:331–339. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee SK, Park DY, Lee HJ, Lee J, Choi MK,
Jeon BH, Jun CD, Lee SK and Kim EC: Functional interaction between
nitric oxide-induced iron homeostasis and heme oxygenase-1 in
immortalized and malignant oral keratinocytes. Cancer Lett.
249:283–293. 2007. View Article : Google Scholar
|