Introduction
Acute pancreatitis (AP) is one of the most
catastrophic upper abdominal disorders (1,2).
AP is characterized by auto-digestion of the pancreas following
intra-acinar zymogen activation and the release of pancreatic
activated enzymes, which causes acinar cell injury, systemic
inflammatory response syndrome and even persistent multiple organ
failure (3-5). Approximately 20% of cases of AP are
associated with multi-organ dysfunction and local complications,
and patients with persistent organ failure within the first few
days are at an increased risk of succumbing to the disease, with a
mortality rate approaching 30% (2). In addition, the development of
infected necrosis among patients with severe acute pancreatitis
(SAP) is associated with extremely high mortality (6-8).
The renin-angiotensin (Ang) system (RAS) is
important in the maintenance of cardiovascular homeostasis, fluid
and salt balance, and has been implicated to play a role in
diabetes, chronic renal disease and hepatic fibrosis (9–11).
Increased activity of the arm of the RAS comprising Ang-converting
enzyme (ACE), AngII and AngII receptor 1 (AT1), namely the
ACE-AngII-AT1 axis, may aggravate the development of pancreatitis
(12-14). However, ACE2, Ang-(1-7) and the
Ang-(1-7) receptor Mas constitute another arm of the RAS, the
ACE2-Ang-(1-7)-Mas axis, which may counteract the ACE-AngII-AT1
axis (15,16). ACE2 cleaves AngII into the
vasodilatory peptide Ang-(1-7), which has certain functions
opposing those of AngII; AngII promotes vasoconstriction, cell
proliferation, pro-thrombotic activity and inflammation, whereas
Ang-(1-7) has vasodilatory, anti-proliferative, anti-thrombotic and
anti-inflammatory functions (17–19). Thus, Ang-(1-7) is considered an
important active product in the pathophysiology of numerous
diseases, such as heart failure, diabetes, disuse-induced skeletal
muscle atrophy and microcirculation (20-24). The manufacture and use of the
Ang-(1-7)-specific receptor (Mas) antagonist, D-Ala-7-Ang-(1-7),
also known as A779, has allowed further analysis of the Mas
receptor for Ang-(1-7). Oruc et al (25) demonstrated that the activation of
the RAS is closely associated with AP. In addition, previous
studies conducted by the present research group have shown that SAP
is associated with upregulation of the ACE2-Ang-(1-7)-Mas axis and
promotes increased circulating levels of Ang-(1-7) (26-28). However, whether the
ACE2-Ang-(1-7)-Mas axis serves a protective role in the
pathogenesis of pancreatitis, and the signaling pathway through
which the ACE2-Ang-(1-7)-Mas axis protects pancreatic cells from
inflammation remain unknown. The p38-mitogen-activated protein
kinase (p38 MAPK), a member of the MAPK family, is considered to be
an important kinase in stress signalling (29). The p38 MAPK signaling transduction
pathway plays an essential role in the regulation of a number of
cellular processes, including inflammation, cell cycling, cell
differentiation, cell growth and cell death (30,31). MAPK activation initiates the
downstream induction of nuclear factor-κB (NF-κB), which is an
essential regulator of the expression of numerous genes involved in
the function and development of the immune system and in
inflammatory responses (32-34). There is also evidence to indicate
that p38 MAPK is involved in the activation of pro-inflammatory
nuclear transcription factors such as NF-κB in isolated pancreatic
acinar cells (35); p38 MAPK has
been demonstrated to regulate NF-κB pathway activation in AR42J
cells (36). These findings
indicate that the p38 MAPK/NF-κB signaling pathway potentially
serves a role in the pathogenesis of AP. Thus, the aforementioned
studies suggest the possibility that the ACE2-Ang-(1-7)-Mas axis
protects pancreatic acinar cells from damage via the p38 MAPK/NF-κB
signaling pathway.
The present study was conducted to investigate the
hypothesis that the ACE2-Ang-(1-7)-Mas axis protects pancreatic
cells from damage through the p38 MAPK/NF-κB signaling pathway. The
aim of the study was to examine the effect of the
ACE2-Ang-(1-7)-Mas axis on caerulein (CAE)-stimulated MPC-83 cells,
and to identify whether this axis contributes to the pathogenesis
of pancreatitis through the p38 MAPK/NF-κB pathway in acinar
cells.
Materials and methods
Cell culture and treatments
MPC-83 mouse pancreatic acinar cancer cells (Cancer
Institute and Hospital, Chinese Academy of Medical Sciences,
Beijing, China) were cultured in RPMI-1640 medium (HyClone; GE
Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA), 100 U/ml penicillin and 100 mg/ml streptomycin. The cells
were incubated at 37°C in a humidified atmosphere containing 5%
CO2. All experiments were carried out 24 h after the
cells were seeded.
The cells were stimulated with 10 nmol/l (M) CAE
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for various time
periods to create a model of AP (37). CAE is a functional analog of
cholecystokinin that causes exocrine secretion and an inflammatory
response in pancreatic cells (38). Generally, 10 nM CAE is used to
induce the inflammation of pancreatic acinar cells as a model of AP
(39). The cells were divided
into six groups: Control, CAE (AP), CAE + Ang-(1-7), CAE + A779,
SB203580 and DX600 groups. The control group comprised normally
growing MPC-83 cells without stimulation. In the CAE group, MPC-83
cells were stimulated with 10 nM CAE for 2, 6, 12, 24 or 48 h. In
the CAE + Ang-(1-7), CAE + A779, SB203580 and DX600 groups, a 24 h
time period for stimulation with CAE was chosen on the basis of
preliminary experiments (data not shown). Our preliminary
experiments showed that the inflammation-related cytokines of the
pancreatitis were most obvious at the 24 h time-point. Prior to
stimulation with CAE for 24 h, the cells were mock pretreated or
pretreated with Ang-(1-7) (1×10−7, 1×10−6 or
1×10−5 M; Sigma-Aldrich; Merck KGaA), Ang-(1-7)
antagonist A779 (1×10−7, 1×10−6 or
1×10−5 M; Sigma-Aldrich; Merck KGaA), 1×10−5
M p38 MAPK inhibitor SB203580 (Beyotime Institute of Biotechnology,
Shanghai, China) or 1×10−6 M ACE2 inhibitor DX600
(Anaspec Inc., Fremont, CA, USA), respectively, for 30 min. Cells
from each group were harvested following the 24 h stimulation with
CAE. Cells were then seeded onto glass coverslips in 6-well plates
at 1×105 cells/well for immunocytochemical analysis in
triplicates.
Immunofluorescence assay of ACE2, Mas
receptor and p38 MAPK
In brief, MPC-83 cells were incubated in 4%
paraformaldehyde for 40 min at 37°C. Following this fixation step,
the cells were blocked with 1% bovine serum albumin (Sigma-Aldrich;
Merck KGaA) for 30 min at 37°C. The cells were then incubated with
rabbit anti-mouse anti-ACE2 (1:200; ab59351; Abcam, Cambridge, UK)
and anti-Mas receptor (1:100; AAR-013; Alomone Labs, Jerusalem,
Israel) antibodies at 4°C overnight. The primary antibodies were
then incubated for 40 min at 37°C with goat anti-rabbit fluorescein
isothiocyanate (FITC) green fluorescent probes (1:100;
bs-0295M-FITC; BIOSS, Beijing, China) as the secondary antibody.
Cell nuclei were stained with DAPI (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) for 5 min at 37°C. Immunofluorescence was analyzed
by using a fluorescence microscope. Cells incubated with normal
rabbit serum (Sigma-Aldrich; Merck KGaA) instead of a primary
antibody served as negative control.
Western blotting of ACE2, Mas receptor,
p38 MAPK, phosphorylated (p)-p38 MAPK and NF-κB
Cells were washed three times with cold
phosphate-buffered saline (PBS), followed by lysis on ice with
lysis buffer (BIOSS) for 30 min. The total protein concentration
was determined using a BCA Protein αssay kit (Pierce; Thermo Fisher
Scientific, Inc.). Subsequently, 25 µg protein-containing lysate
sample was separated using 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred onto
polyvinylidene difluoride membranes, which were then blocked by
incubation with 5% non-fat dry milk at 37°C for 2 h. The blocked
membranes were incubated with rabbit anti-mouse ACE2 monoclonal
antibody (1:400; ab59351), mouse β-actin monoclonal antibody
(1:500; ab3280) (both from Abcam, Cambridge, UK), rabbit anti-mouse
Mas receptor polyclonal antibody (1:200; AAR-013; Alomone Labs) or
rabbit anti-mouse p38 MAPK (1:800), p-p38 MAPK or NF-κB p65
monoclonal antibody (all 1:800; cat. nos. 9212, 4511 and 8242,
respectively; Cell Signaling Technology, Inc., Danvers, MA, USA) at
4°C overnight. Subsequent to washing three times with TBST, the
membranes were incubated for 1 h at room temperature with
horseradish peroxidase (HRP)-conjugated goat anti-rabbit (1:8,000;
sc-2004) or goat anti-mouse (1:8,000; sc-2031) secondary antibodies
(both from Santa Cruz Biotechnology, Inc.).
Enhanced chemiluminescence HRP substrate (EMD
Millipore, Billerica, MA, USA) was used to detect the
immune-reactive bands and densitometric analysis of the bands was
performed using Image Lab software 3.0 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Analysis of p38 MAPK, NF-κB, tumor
necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-8 and IL-10
mRNA expression using reverse transcription-quantitative polymerase
chain reaction (RT-qPCR)
MPC-83 cells were collected and total RNA was
extracted using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.)
in accordance with the manufacturer's protocol. Total RNA (2 µg)
was subjected to first-strand cDNA synthesis using random primers,
M-MLV reverse transcriptase and RNase inhibitor provided in a
Revert Aid First Strand cDNA synthesis kit (Fermentas; Thermo
Fisher Scientific, Inc., Pittsburgh, PA, USA). The resultant cDNA
was subjected to qPCR using specific primers as follows: p38 MAPK
forward, 5′-GAGCTGTTGACCGGAAGAAC-3′ and reverse, 5′-GGC
TTGGCATCCTGTTAATG-3′; NF-κB forward, 5′-CCCGAC TTGTTTGGGTGAT-3′ and
reverse, 5′-TCCGTCTCCAGG AGGTTAA-3′; TNF-α forward,
5′-GGTGCCTATGTCTCA GCCTCTT-3′ and reverse, 5′-GCACCTCCACTTGGTGG
TTT-3′; IL-6 forward, 5′-AGTTGCCTTCTTGGGACTGA-3′ and reverse,
5′-TCCACGATTTCCTAGAGAAC-3′; IL-8 forward,
5′-TGAGCTGCGCTGTCAGTGCCT-3′ and reverse,
5′-AGAAGCCAGCGTTCACCAGA-3′; IL-10 forward,
5′-ATTTGAATTCCCTGGGTGAGAAG-3′ and reverse,
5′-CACAGGGGAGAAATCGATGACA-3′; and β-actin forward,
5′-CATCCGTAAAGACCTCTATGCCAAC-3′ and reverse,
5′-ATGGAGCCACCGATCCACA-3′. qPCR was performed with a Power
SYBR-Green PCR Master mix using an ABI 7500 instrument (both from
Applied Biosystems; Thermo Fisher Scientific, Inc.). The qPCR
reaction was conducted at 95°C for 5 min, 95°C for 15 sec, and 60°C
for 1 min for 40 cycles. Data analysis was performed using the
2−ΔΔCq method described by Livak and Schmittgen
(40).
Statistical analysis
All data are presented as mean ± standard deviation
unless otherwise specified. All experiments were repeated at least
three times independently. Results were analyzed using SPSS 13.0
(SPSS, Inc., Chicago, IL, USA), and analysis of variance followed
by post hoc analysis using Newman-Keuls tests was used to compare
differences among groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
Localization of ACE2, Mas receptor and
p38 MAPK in MPC-83 cells
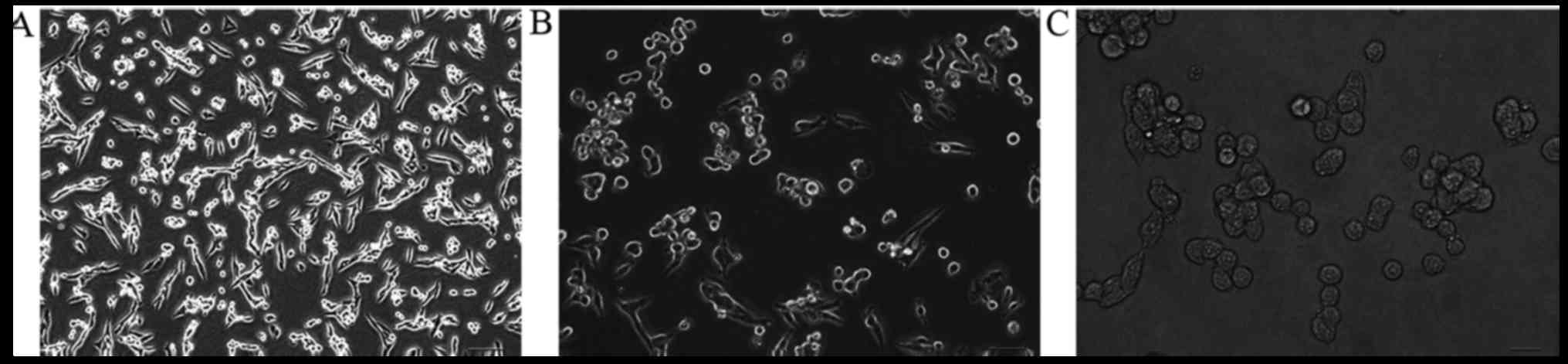
Untreated MPC-83 cells initially cultured in fresh
RPMI-1640 media adhered to the walls of the tissue culture flasks
at 24 h after incubation. The cells exhibited a long fusiform
morphology at a magnification of ×100, and the cells appeared to
grow well (Fig. 1).
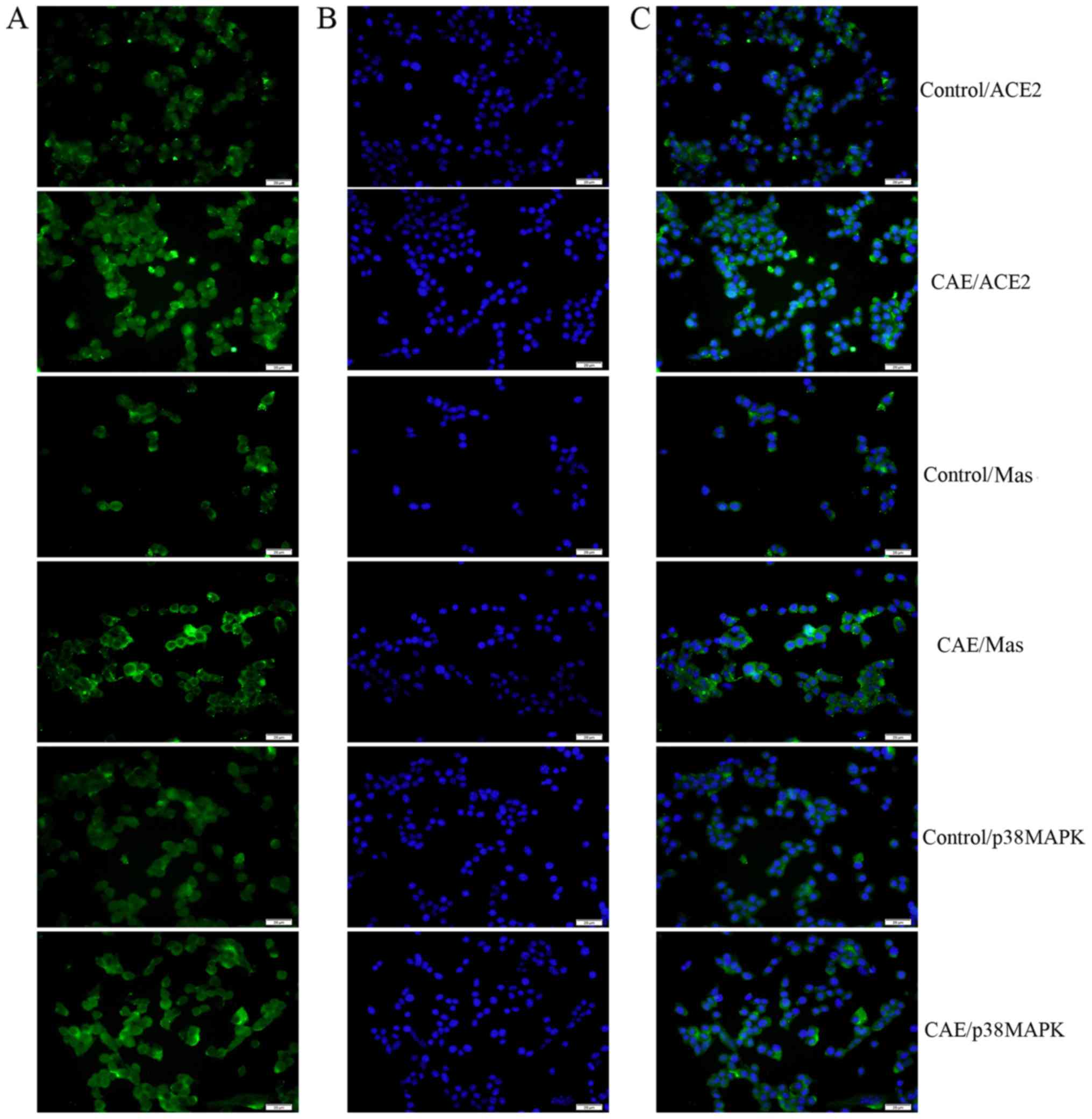
Immunofluorescence assays demonstrated that ACE2 and p38 MAPK were
present mainly in the cytoplasm, while the Mas receptor was
observed mainly in the cell membrane. Furthermore, the expression
of ACE2, Mas receptor and p38 MAPK immunofluorescence levels were
upregulated in the AP model MPC-83 cells than the control (Fig. 2).
ACE2, Mas receptor, p38 MAPK, p-p38 MAPK
and NF-κB protein levels are upregulated in AP model MPC-83
cells
Western blot analysis revealed that ACE2 protein
levels in the MPC-83 cells were significantly increased (P<0.05)
by stimulation with CAE for 2-48 h compared with those in the
unstimulated control group. Following exposure to CAE for 24 h,
ACE2 expression increased to 1.16±0.04 which was 3-fold greater
(P<0.05) than that in the control group (0.37±0.01) (Fig. 3A and B). Mas receptor protein
expression peaked following 12 h of AP induction at 1.22±0.10,
which was significantly greater compared with that in the control
group (0.52±0.07, P<0.05) (Fig. 3C
and D). ACE2 and Mas receptor protein levels were upregulated
in the AP model, suggesting that the ACE2-Ang-(1-7)-Mas axis was
involved in the pathological process in these cells.
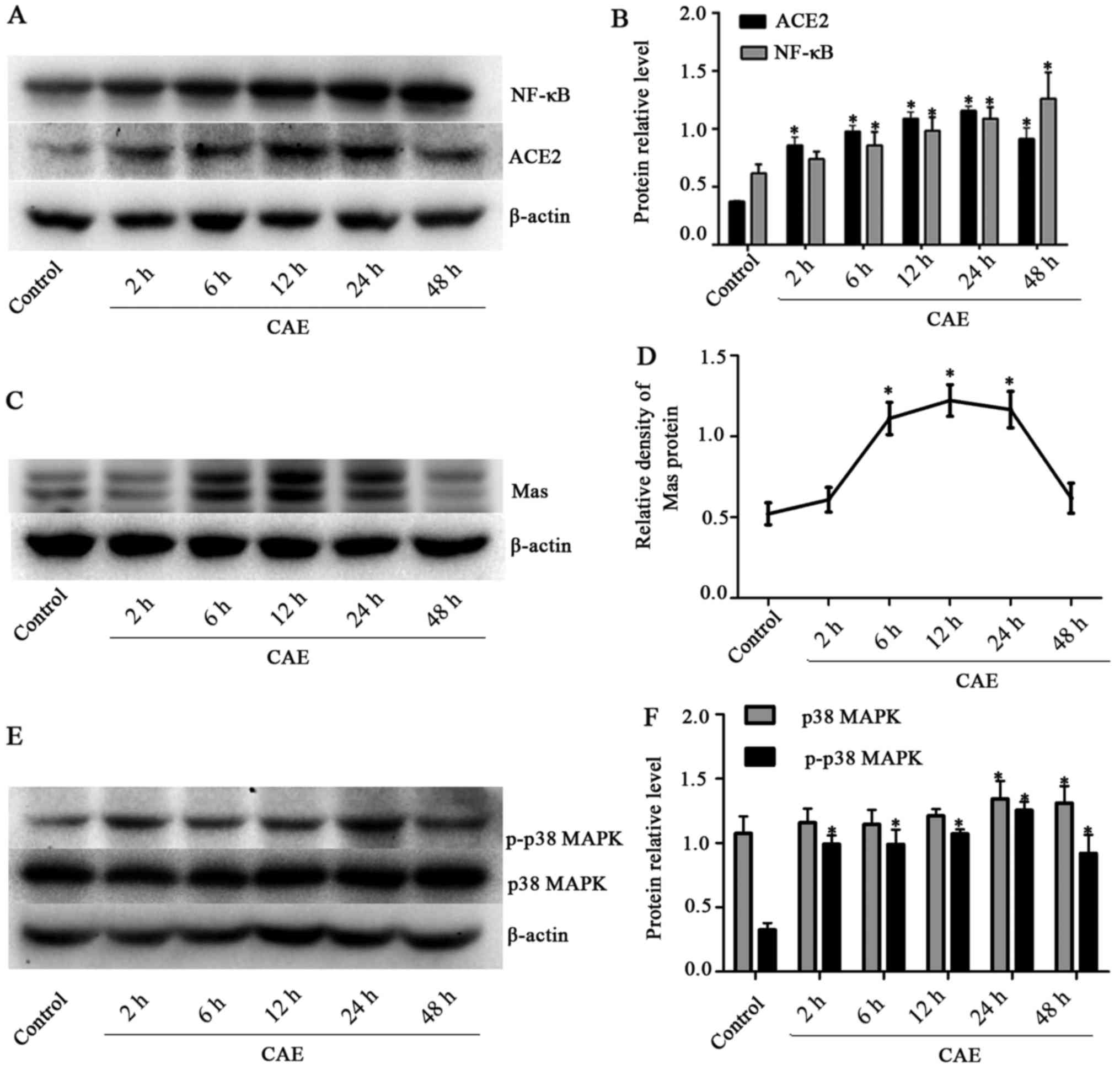 | Figure 3Time course of ACE2, Mas, p38 MAPK,
p-p38 MAPK and NF-κB protein levels in cultured MPC-83 cells
stimulated with CAE for various time periods. (A) Western blotting
of ACE2 and NF-κB, and (B) quantified ACE2 and NF-κB protein
expression levels. (C) Western blotting of Mas and (D) quantified
Mas protein levels. (E) Western blotting of p38 MAPK and p-p38 MAPK
and (F) quantified p38 MAPK and p-p38 MAPK protein levels. β-actin
was measured in the same gel as an internal standard, the optical
density of each band was quantified, and results are presented as
the ratio of the target protein to β-actin. *P<0.05
vs. the control group. ACE2, angiotensin-converting enzyme 2; Mas,
receptor for angiotensin-(1-7); p38 MAPK, p38 mitogen-activated
protein kinase; p-, phosphorylated; NF-κB, nuclear factor-κB; CAE,
caerulein. |
p38 MAPK total proteins contain p-p38 MAPK and
unphosphorylated p38 MAPK. In the western blot analysis, p38 MAPK
total protein levels in the AP model cells were significantly
greater (P<0.05) compared with those in the control group
(1.07±0.13) following stimulation with CAE for 24 h (1.34±0.14) and
48 h (1.31±0.13). The p-p38 MAPK levels were significantly higher
in the CAE groups compared with the control group (0.32±0.05) for
stimulation periods between 2 h (0.99±0.07) and 48 h (0.92±0.14).
In addition, the level of p-p38 MAPK peaked (1.46±0.10) at 24 h and
then started to decrease (Fig. 3E and
F). NF-κB expression underwent a dynamic change during AP,
demonstrating a significant elevation. The levels of NF-κB were
upregulated (P<0.05) ~2-fold following 48 h of exposure to CAE
(1.26±0.20) compared with the control group (0.62±0.07) (Fig. 3A and B). These results demonstrate
that ACE2, Mas receptor protein levels and the p38 MAPK and NF-κB
pathway were upregulated during AP.
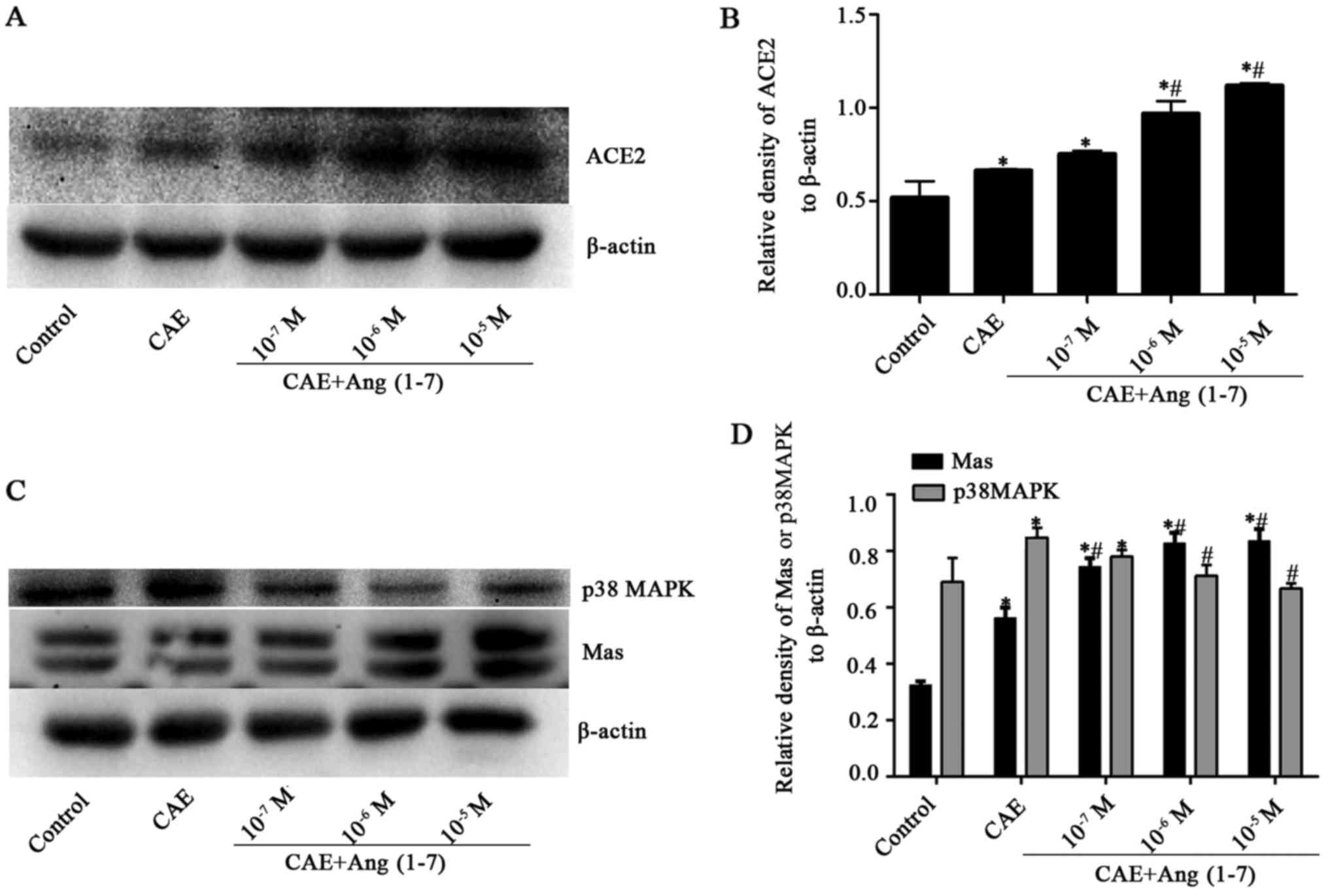
Ang-(1-7) upregulates ACE2 and Mas
receptor protein but downregulates p38 MAPK, p-p38 MAPK and NF-κB
in AP model MPC-83 cells
Whether Ang-(1-7), a component of the
ACE2-Ang-(1-7)-Mas axis, promotes the expression of the axis was
investigated using western blot analysis. The results revealed that
when AP model MPC-83 cells were treated with different
concentrations (1×10−7, 1×10−6 and
1×10−5 M) of Ang-(1-7), the ACE2 and Mas receptor
protein levels were significantly increased compared with those in
the CAE group, with the exception that ACE2 induction by
1×10−7 M Ang-(1-7) was not statistically significant
compared with the CAE group. ACE2 and Mas receptor protein
expression levels increased from those CAE group as the Ang-(1-7)
concentration increased to 10−5 M Ang-(1-7) [from
0.75±0.02 to 1.12±0.01 and from 0.56±0.04 to 0.84±0.04,
respectively (P<0.05) (Fig.
4)].
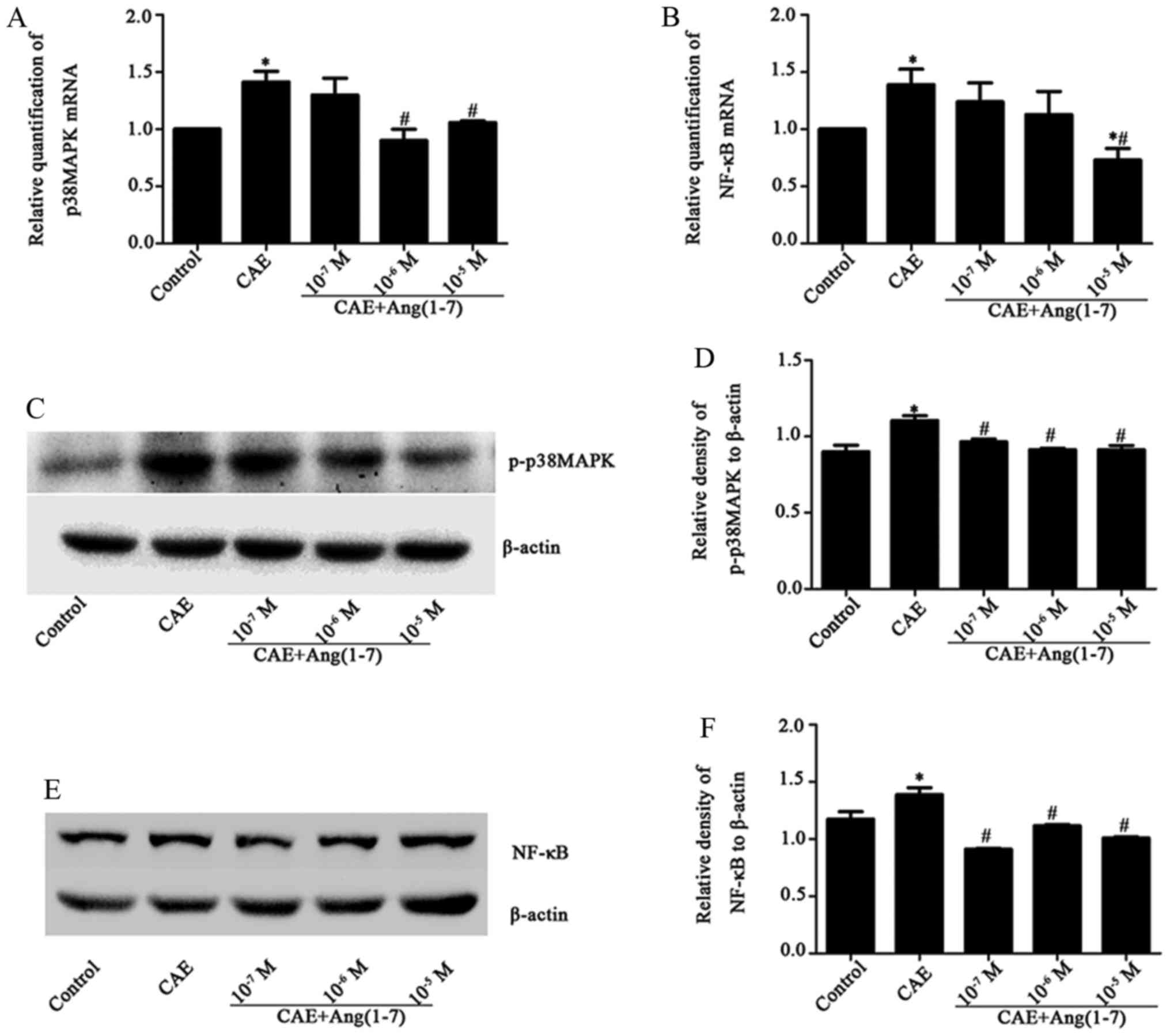
The stimulation of MPC-83 cells with CAE caused a
significant increase in p38 MAPK mRNA levels in the CAE group
(1.41±0.23) compared with the control group (Fig. 5A). Similarly, NF-κB mRNA levels
increased following stimulation with CAE (1.39±0.33, P<0.050
(Fig. 5B). Conversely, following
treatment with Ang-(1-7) at various concentrations
(1×10−7, 1×10−6 and 1×10−5 M), an
attenuated inflammatory response occurred, as evidenced by
decreased p38 MAPK (Fig. 5A) and
NF-κB (Fig. 5B) mRNA levels
compared with those in the CAE group. In addition, p38 MAPK and
p-p38 MAPK protein levels in the AP model MPC-83 cells were
inhibited by Ang-(1-7) in a dose-dependent manner [for p38 MAPK
1×10−6 and 1×10−5 M, P<0.05) (Fig. 4C and D); for p-p38 MAPK
1×10−7, 1×10−6 and 1×10−5 M,
P<0.05) (Fig. 5C and D)].
Similarly, NF-κB protein levels were significantly decreased
(P<0.05) by all three concentrations of Ang-(1-7) compared with
those in the CAE group (Fig. 5E and
F). Thus, in the MPC-83 cell model of AP, Ang-(1-7)
downregulated the p38 MAPK/NF-κB signaling pathway. These results
indicate that Ang-(1-7) promoted the expression of the
ACE2-Ang-(1-7)-Mas axis and downregulated the p38 MAPK/NF-κB
signaling pathway in MPC-83 cells.
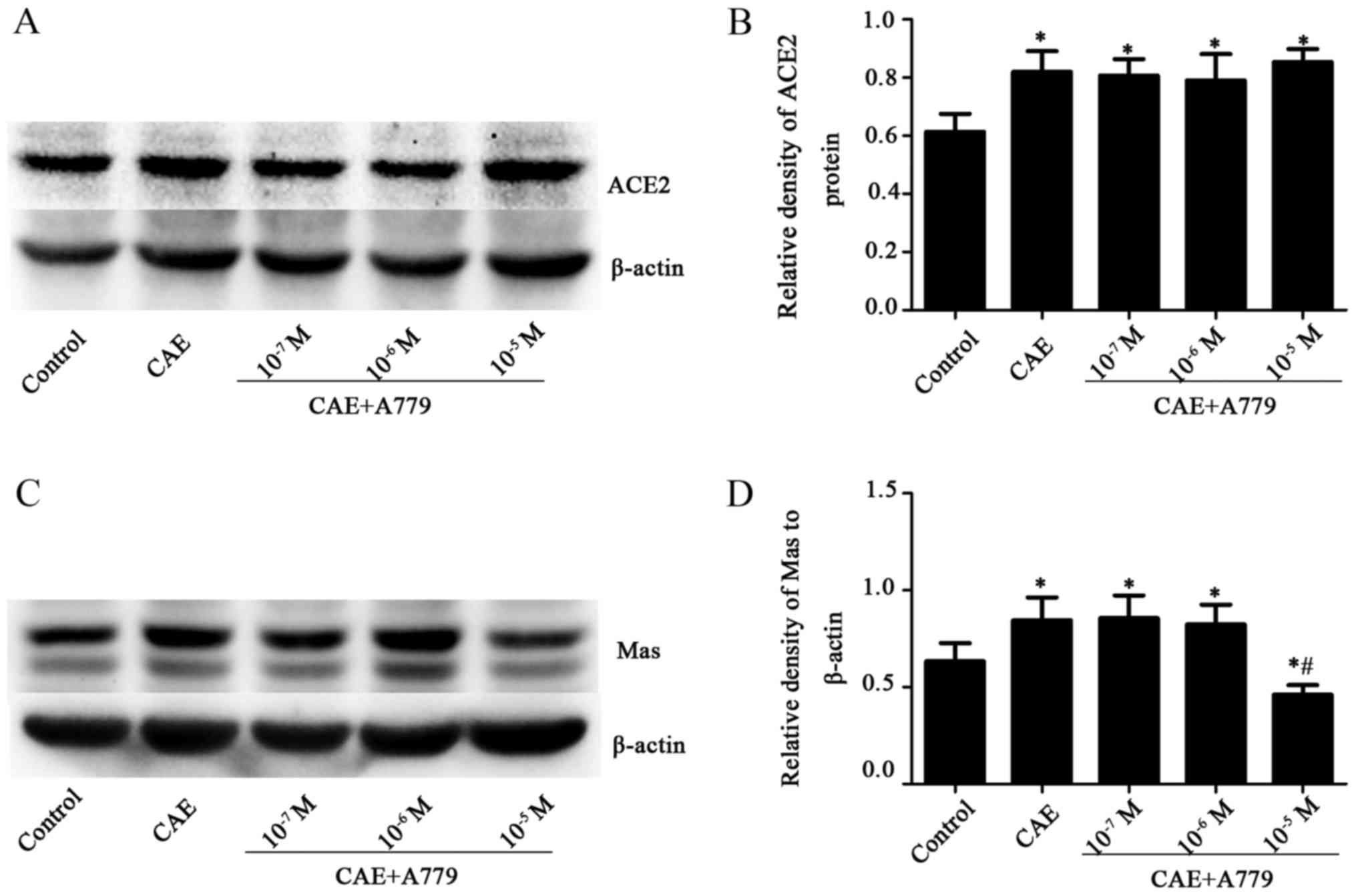
Ang-(1-7) receptor antagonist A779
downregulates Mas without altering ACE2, and upregulates p38 MAPK,
p-p38 MAPK and NF-κB levels in AP model MPC-83 cells
ACE2 levels were increased significantly (P<0.05)
in the CAE and CAE + A779 groups compared with the control group.
However, no significant difference was identified between the
groups treated with CAE and CAE + A779 (1×10−7,
1×10−6 and 1×10−5 M) (Fig. 6A and B). The stimulation of MPC-83
cells with CAE caused a significant increase in Mas receptor levels
in the CAE group (0.84±0.12) compared with the control group
(0.63±0.09, P<0.050) (Fig. 6C and
D). In the 1×10−5 M A779 + CAE group, the Mas
receptor expression (0.46±0.05) was significantly decreased
(P<0.05) compared with those in the control and CAE groups
(Fig. 6C and D).
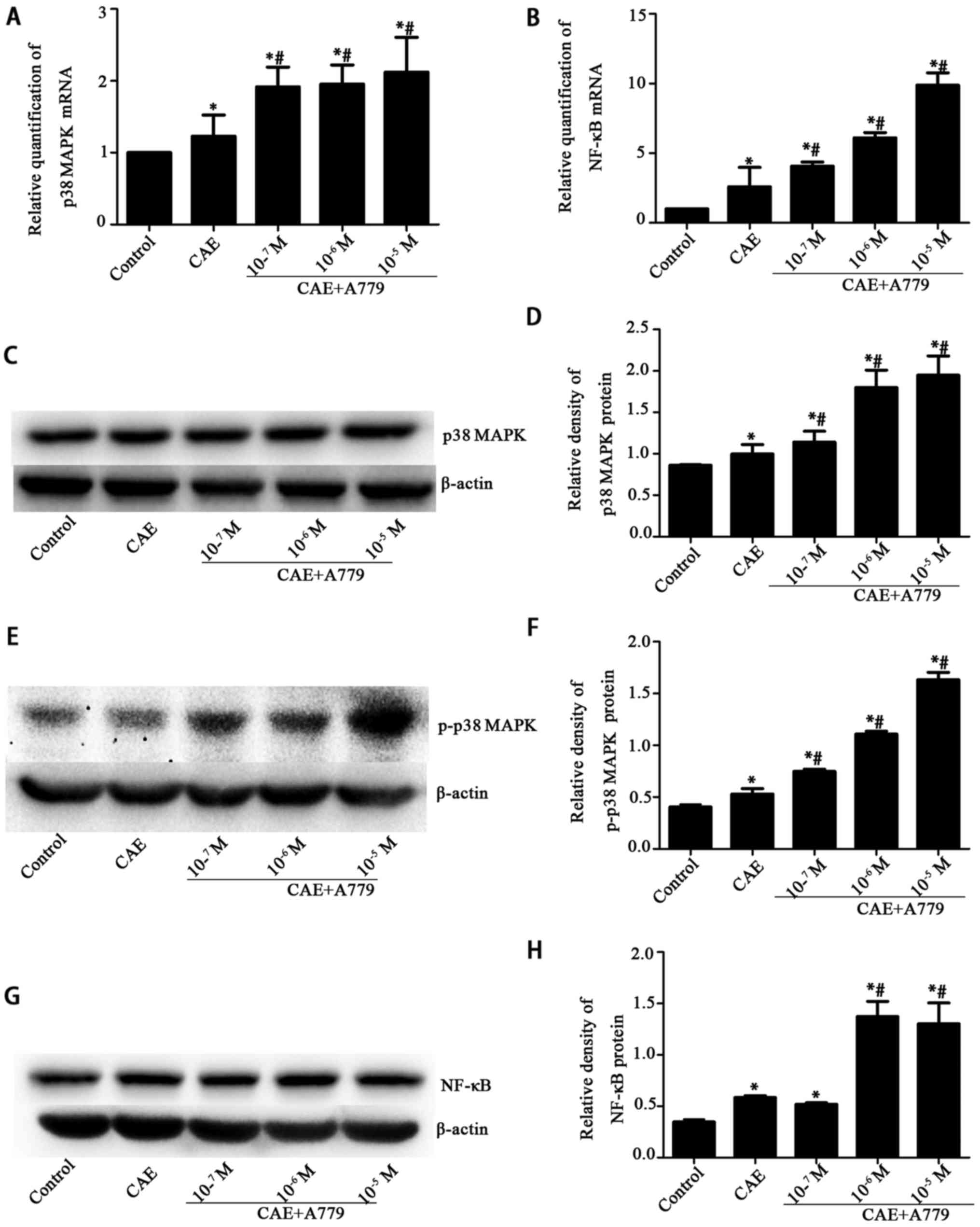
p38 MAPK and NF-κB mRNA levels were significantly
increased following AP induction (to 1.23±0.29 and 2.60±1.39,
respectively; P<0.05 vs. the control group), and further
increased by A779 treatment at different concentrations
(1×10−7, 1×10−6 or 1×10−5 M) in a
dose-dependent manner (Fig. 7A and
B). The changes in p38 and p-p38 MAPK protein levels were
comparable with the changes in p38 MAPK and NF-κB mRNA levels
(Fig. 7C–F). The NF-κB protein
levels were significantly increased (P<0.05) following treatment
with 1×10−6 or 1×10−5 M A779 in comparison
with those in the CAE group (Fig. 7G
and H).
These results indicate that A779 downregulated the
Mas receptor, which downregulated the normal processes associated
with the ACE2-Ang-(1-7)-Mas axis, and upregulated p38 MAPK, p-p38
MAPK and NF-κB expression in the MPC-83 cell model of AP. The
results further illustrate the association between the
ACE2-Ang-(1-7)-Mas axis and the p38 MAPK/NF-κB pathway. However,
ACE2 expression was not significantly changed by any of the tested
concentrations of A779.
SB203580 downregulates ACE2, Mas
receptor, p-p38 MAPK and NF-κB; conversely, DX600 upregulates p-p38
MAPK and NF-κB in AP model MPC-83 cells
Western blotting (Fig.
8) revealed that following treatment with SB203580, an
attenuated response to stimulation with CAE occurred, as evidenced
by significantly decreased levels of ACE2 in the SB203580 group
compared with the CAE group (0.16±0.07 vs 0.28±0.03) (Fig. 8A and B), Mas receptor (0.14±0.01
vs. 0.21±0.02) (Fig. 8A and D),
p-p38 MAPK (0.40±0.12 vs. 0.47±0.01) (Fig. 8C and E) and NF-κB (0.66±0.04 vs.
0.76±0.05) (Fig. 8C and F)
compared with those in the CAE group, but no significant effect on
p38 MAPK (0.66±0.00 vs. 0.65±0.03) (Fig. 8C and E). By contrast, in the DX600
group, p-p38 MAPK (Fig. 8E) and
NF-κB (Fig. 8F) protein levels
were significantly increased (P<0.05) compared with those in the
CAE group, whereas the protein levels of Mas receptor (Fig. 8A), p38 MAPK (Fig. 8E) underwent no significant
changes.
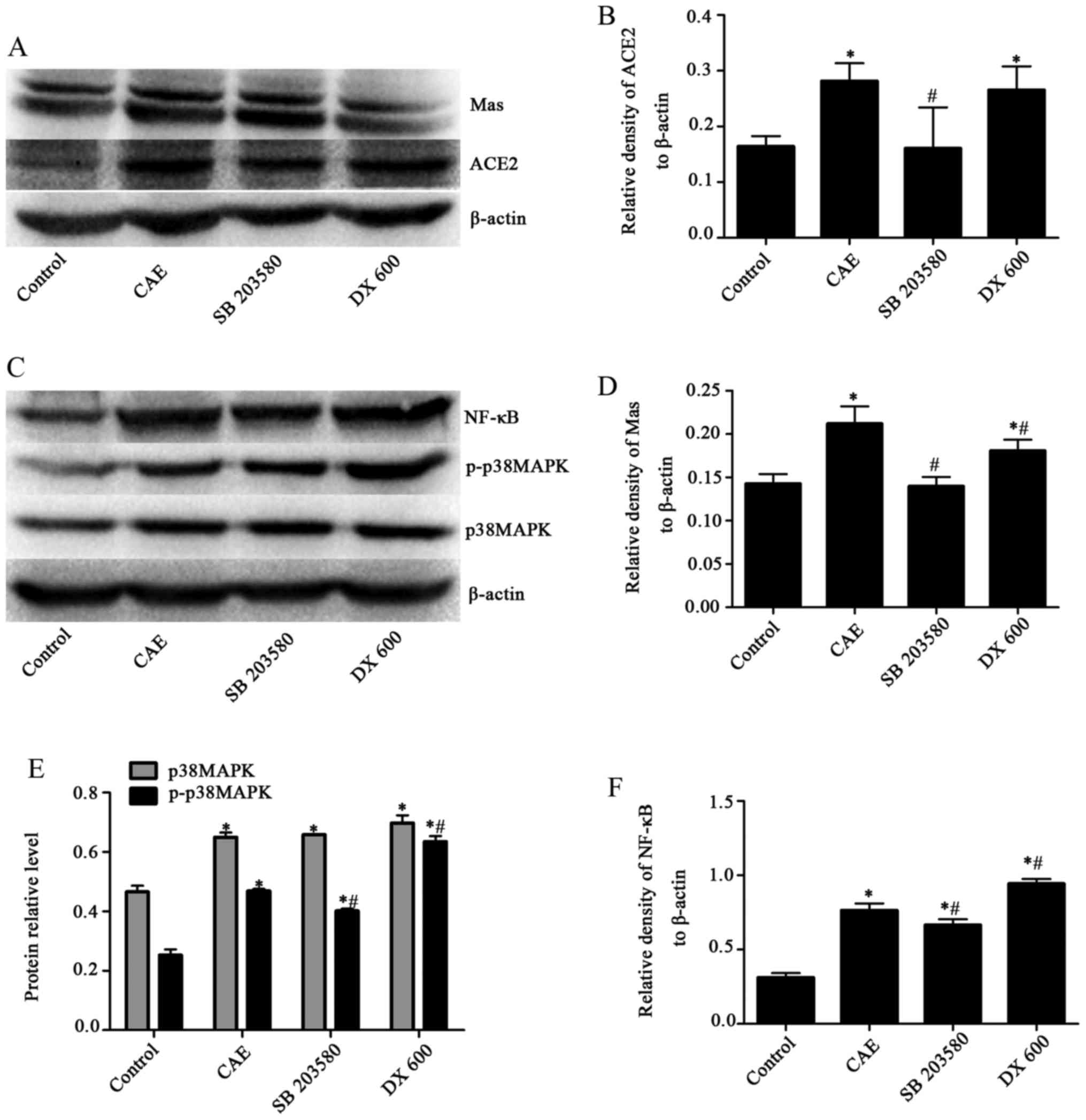 | Figure 8Effects of SB203580 and DX600 on
ACE2, Mas receptor, p38 MAPK, p-p38 MAPK and NF-κB protein levels
in the control, CAE, SB203580 and DX600 groups of MPC-83 cells as
determined by western blotting. (A) Western blotting of ACE2 and
Mas receptor. (B) Quantified expression levels of ACE2. (C) Western
blotting of p38 MAPK, p-p38 MAPK and NF-κB. Quantified expression
levels of (D) Mas, (E) p38 MAPK, p-p38 MAPK and (F) NF-κB proteins.
Results are presented as the ratio of the target protein to
β-actin. *P<0.05 vs. the control group;
#P<0.05 vs. the CAE group. Mas, receptor for
angiotensin-(1–7); p38 MAPK, p38 mitogen-activated protein kinase;
p-, phosphorylated; NF-κB, nuclear factor-κB; CAE, caerulein. |
Ang-(1-7) downregulates the mRNA levels
of inflammatory factors IL-6, TNF-α and IL-8, and upregulates IL-10
mRNA levels in AP MPC-83 cells
The effects of Ang-(1-7) on inflammatory factors
were evaluated using the Ang-(1-7) receptor antagonist A779.
Following treatment with 1×10−7, 1×10−6 or
1×10−5 M Ang-(1-7), the IL-6 mRNA levels were decreased
significantly (P<0.05) compared with those in the CAE group
(Fig. 9A). In addition, TNF-α and
IL-8 mRNA levels were significantly reduced by all concentrations
of AngII compared with those in the CAE group; in the
1×10−5 M Ang-(1-7) group, the TNF-α levels were reduced
from 0.83±0.11 and 0.68±0.27 and the IL-8 levels were reduced from
0.64±0.03 to 0.43±0.29 (both P<0.05) (Fig. 9B and C). Furthermore, following
treatment with 1×10−6 or 1×10−5 M Ang-(1-7),
the IL-10 mRNA levels were increased significantly (P<0.05)
(Fig. 9D) compared with those in
the CAE group.
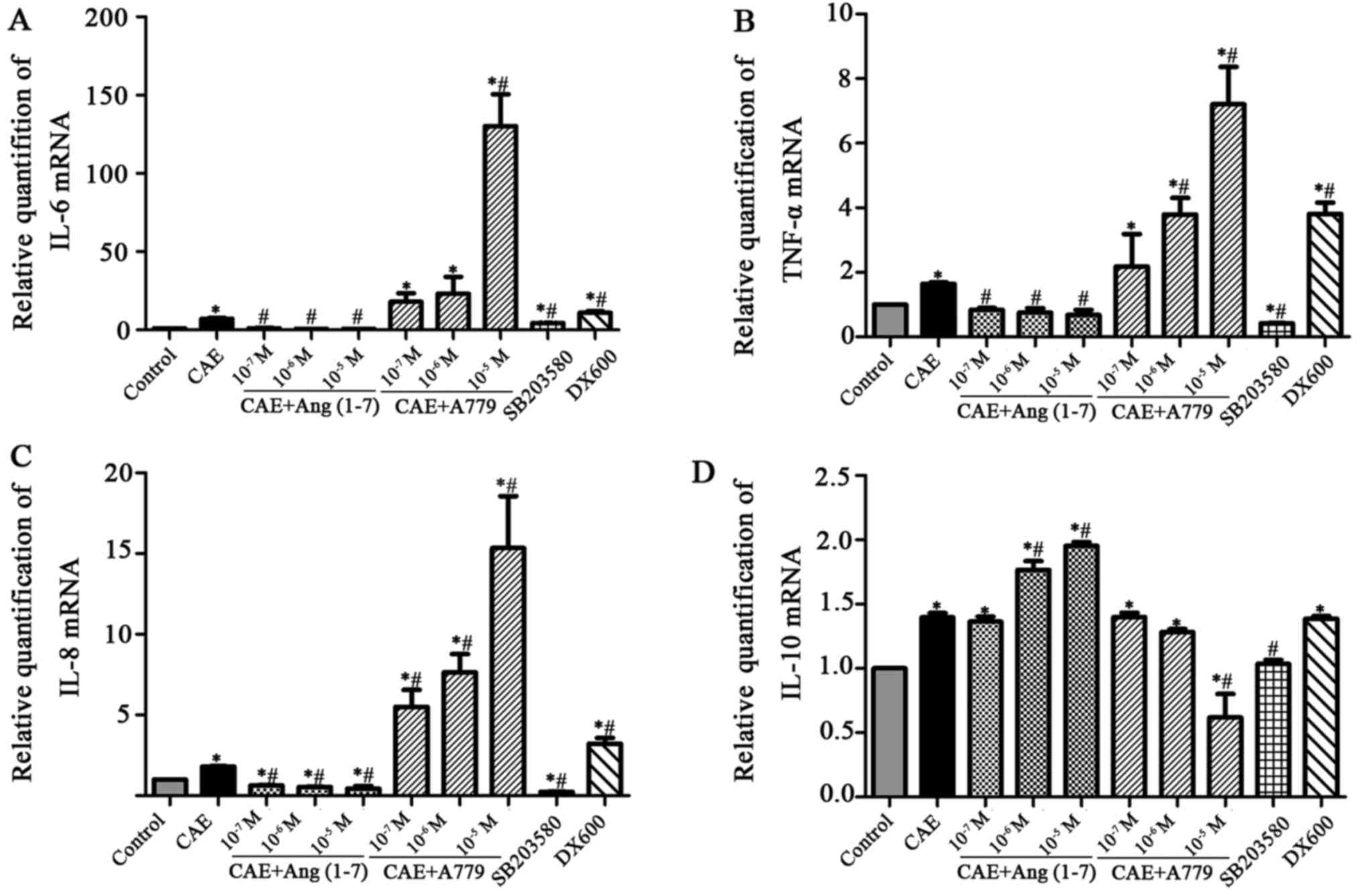 | Figure 9Effects of Ang-(1-7), A779, SB203580
and DX600 on IL-6, TNF-α, IL-8 and IL-10 mRNA levels in MPC-83
cells as determined by reverse transcription-quantitative
polymerase chain reaction. Relative quantification of (A) IL-6, (B)
TNF-α, (C) IL-8 and (D) IL-10 mRNA expression in the control, CAE,
CAE + Ang-(1-7), CAE + A779, SB203580 and DX600 groups.
*P<0.05 vs. the control group; #P<0.05
vs. the CAE group. Ang, angiotensin; A779, Ang-(1-7) antagonist;
SB203580, p38 MAPK inhibitor; DX600, ACE2 inhibitor; ACE2,
angiotensin-converting enzyme 2; IL, interleukin; TNF, tumor
necrosis factor; CAE, caerulein. |
Following stimulation with A779 (1×10−5
M), the IL-6 level of the cells was significantly increased
(130.26±35.25) compared with that in the CAE control group
(P<0.05) (Fig. 9A). Similarly,
the TNF-α levels in the 1×10−6 and 1×10−5 M
A779 groups (1.54±0.09 and 7.21±2.01) was significantly increased
compared with that in the CAE group (Fig. 9B). The expression of IL-8 mRNA was
increased in a significant and concentration-dependent manner by
A779 (from 5.50±1.82 in the 1×10−7 group to 15.34±5.57
in the 1×10−6 group) compared with that in the CAE group
(Fig. 9C). In addition, following
treatment with A779 (1×10−5 M), the IL-10 level was
significantly decreased compared with that in the CAE group
(Fig. 9D).
IL-6, TNF-α, IL-8 and IL-10 mRNA levels were
downregulated significantly (P<0.05) in the SB203580 group
compared with the CAE group. By contrast, IL-6, TNF-α and IL-8 mRNA
levels were upregulated significantly (P<0.05) in the DX600
group compared with the CAE group. However, there was no
significant difference in IL-10 mRNA levels between the CAE and
DX600 groups (1.40±0.06 vs. 1.39±0.04, respectively; P>0.05).
These results indicate that Ang-(1-7) downregulates the
inflammatory response in MPC-83 cells, and that A779 functions in
opposition to Ang-(1-7).
Discussion
The present study demonstrates that ACE2 and Mas
receptor protein levels are significantly elevated in the MPC-83
cell model of AP, suggesting that the ACE2-Ang-(1-7)-Mas axis is
important in protecting acinar cells from inflammation. This was in
accordance with previous findings in animal models of pancreatitis,
in which the inhibition of the ACE-AngII-AT1 axis significantly
decreased pancreatic injury (25,41). Notably, ACE2 congeners have been
confirmed to act as RAS antagonists, and the catalytic efficiency
of ACE2 in the hydrolysis of AngII to Ang-(1-7) has been
demonstrated to be >400-fold greater than that for the
hydrolysis of AngI to Ang-(1-9) in the local RAS of the heart and
kidneys (42). Previous studies
have shown that the binding of Ang-(1-7) to the Mas receptor
induces numerous effects, including protection of the vascular
endothelium, vasodilation, protection of renal tubular cells and
diuresis (43,44), possibly by inhibiting the
proliferation and inflammatory reactions occurring in response to
hypertensive challenge by AngII. Pancreatic acinar cells synthesize
and release cytokines and chemokines, resulting in the recruitment
of inflammatory cells, including neutrophils, lymphocytes and
macrophages when under oxidative stress or exposed to infection
(45). The recruitment and
activation of various inflammatory cells lead to further acinar
cell injury and cause an elevation of various pro-inflammatory
mediators, including TNF-α and IL-6, as well as anti-inflammatory
factors such as IL-10 (46,47). The present study demonstrates that
exogenous Ang-(1-7) increased ACE2 and Mas receptor expression, but
decreased IL-6 and TNF-α mRNA expression in CAE-treated pancreatic
acinar cells; further evidence was provided by the application of
the Mas receptor antagonist A779. The results suggest that the
ACE2-Ang-(1-7)-Mas axis inhibited the production of inflammatory
factors and protected MPC-83 cells from damage. However, to the
best of our knowledge, the specific signaling pathway through which
the ACE2-Ang-(1-7)-Mas axis protects acinar cells from inflammatory
injury remains unknown.
In 2004, Ren et al (48) demonstrated that p38 MAPK activity
in the pancreas was significantly higher than the basal activity 24
h after the induction of SAP, and that the p38 MAPK signal
transduction pathway served an important role in the pathogenesis
of SAP. In addition, a study has observed that the pancreatic
expression of NF-κB increases in rats with acute necrotizing
pancreatitis (49). However, the
mechanism by which the p38 MAPK and NF-κB signaling pathways are
regulated remains unclear. In the present in vitro study,
p38 MAPK activation was observed in association with the
inflammatory response, and p38 MAPK protein levels increased in an
approximately time-dependent manner during the AP process,
indicating that CAE acts as an environmental stressor and activates
the p38 MAPK signaling pathway. In general cells, mitogen- and
stress-activated kinase 1/2, which is a downstream substrate of p38
MAPK, directly phosphorylates and activates transcription factors
including NF-κB isoform p65 and histone H3 (49). The involvement of the
transcription factor NF-κB in AP has been demonstrated in
pancreatic acinar cells, where it induces the release of numerous
pro-inflammatory cytokines, including IL-6 and TNF-α (50). In the present study, NF-κB protein
levels were observed to increase in a time-dependent manner in the
pancreatic acinar cells following stimulation with CAE, indicating
that CAE induced an NF-κB-mediated inflammatory response. This is
consistent with a previous study of AP in which CAE induced the
expression of inhibitor of κB kinase (IKK)α, phosphorylated IKKα
and NF-κB p65, and NF-κB p50 (51). However, the pharmacological
inhibition of MAPK during the onset of AP has resulted in mixed
outcomes and the role of the p38 MAPK in AP pathogenesis remains
controversial. A number of previous studies (52-54) have suggested an adverse effect of
NF-κB activation on pancreatitis-associated injury; however, one
study (55) proposed a protective
role via the induction a self-defending genetic program prior to
the onset of pancreatic cellular injury. The present results
suggest that MAPK and NF-κB may have harmful functions during the
course of AP.
In the present study, the results show that
SC203580, a selective inhibitor of p38 MAPK, did not downregulate
p38 MAPK expression, whereas, Ang-(1-7) and SB203580 downregulated
p-p38 MAPK and NF-κB protein expression, and inflammatory factor
(IL-6, TNF-α and IL-8) mRNA expression in AP MPC-83 cells. These
results were supported by application of A779 and the ACE2
inhibitor DX600, which exhibited opposite effects. Those results
are consistent with previous results from a study of ACE2 knock-out
and ACE2 transgenic animals (51). Furthermore, in the present study,
Ang-(1-7) upregulated the mRNA level of the anti-inflammatory
factor IL-10, while A779 reduced it. These results suggest that the
ACE2-Ang-(1-7)-Mas axis contributes to the progression of AP though
the p38 MAPK/NF-κB signaling pathway.
The p38 MAPK pathway positively regulates the
activity of NF-κB, and p38 MAPK has been shown to affect the
activity of IKK and p65 (56).
Furthermore, it has been demonstrated that p38 activity is required
to enhance the accessibility of the cryptic NF-κB binding sites
contained in histone H3 phosphorylated promoters, indicating that
p38-dependent H3 phosphorylation may mark promoters for increased
NF-κB recruitment (57). In the
present study, p38 MAPK, p-p38 MAPK and NF-κB protein levels
increased in parallel, indicating that p38 MAPK plays a significant
role in the activation of NF-κB signaling in pancreatic cells.
Moreover, the present study also found that the expression of ACE2,
Mas, NF-κB and pro-inflammatory cytokines was downregulated
following the inhibition of p38 MAPK signaling in MPC-83 cells.
Accordingly, it may be concluded that the functions of p38 MAPK are
mediated by NF-κB signaling in the inflammatory response of
pancreatitis, and that p38 MAPK is a key factor in the activation
of the ACE2-Ang-(1-7)-Mas axis.
In conclusion, the present study indicates that the
ACE2-Ang-(1-7)-Mas axis protects MPC-83 cells from damage via
inhibition of the p38 MAPK/NF-κB pathway. Considering the vital
role of the ACE2-Ang-(1-7)-Mas axis in the pathogenesis of
pancreatitis, substantial efforts to develop clinical strategies to
counter the regulatory axis expression or its activity are
recommended, for example, by upregulation of the expression of ACE2
and/or increasing the tissue levels of Ang-(1-7). Understanding the
specific interaction of ACE2-Ang-(1-7)-Mas axis components with the
p38 MAPK/NF-κB pathway may help in the development of therapeutic
agents useful for ameliorating the inflammation reaction and injury
of organs in patients with pancreatitis.
Acknowledgments
The present study was supported by the Beijing
Natural Science Foundation of 2014 year (grant no. 7142044) and the
National Natural Science Foundation of China of 2016 year (grant
no. 81571933). The study was also supported by the National Natural
Science Foundation of China (grant no. 81441060).
References
|
1
|
Akay C, Yaman H, Oztosun M, Cakir E,
Yildirim AO, Eyi YE, Agilli M, Akgul EO, Aydin I, Kaldirim U, et
al: The protective effects of taurine on experimental acute
pancreatitis in a rat model. Hum Exp Toxicol. 32:522–529. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pan Z, Feng L, Long H, Wang H, Feng J and
Chen F: Effects of local pancreatic renin-angiotensin system on the
microcirculation of rat with severe acute pancreatitis. Korean J
Physiol Pharmacol. 19:299–307. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Singh P and Garg PK: Pathophysiological
mechanisms in acute pancreatitis: Current understanding. Indian J
Gastroenterol. 35:153–166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singh VK, Wu BU, Bollen TL, Repas K,
Maurer R, Mortele KJ and Banks PA: Early systemic inflammatory
response syndrome is associated with severe acute pancreatitis.
Clin Gastroenterol Hepatol. 7:1247–1251. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mofidi R, Duff MD, Wigmore SJ, Madhavan
KK, Garden OJ and Parks RW: Association between early systemic
inflammatory response, severity of multiorgan dysfunction and death
in acute pancreatitis. Br J Surg. 93:738–744. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu M, Shi L, Chen M, Chen S and Zou X:
Effects of c-Jun N-terminal kinase signaling pathway on severe
acute pancreatitis-associated lung injury. Pancreas. 41:358–366.
2012. View Article : Google Scholar
|
|
7
|
van Santvoort HC, Bakker OJ, Bollen TL,
Besselink MG, Ahmed Ali U, Schrijver AM, Boermeester MA, van Goor
H, Dejong CH, van Eijck CH, et al Dutch Pancreatitis Study Group: A
conservative and minimally invasive approach to necrotizing
pancreatitis improves outcome. Gastroenterology. 141:1254–1263.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Petrov MS, Shanbhag S, Chakraborty M,
Phillips AR and Windsor JA: Organ failure and infection of
pancreatic necrosis as determinants of mortality in patients with
acute pancreatitis. Gastroenterology. 139:813–820. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gul R, Ramdas M, Mandavia CH, Sowers JR
and Pulakat L: RAS-mediated adaptive mechanisms in cardiovascular
tissues: Confounding factors of RAS blockade therapy and
alternative approaches. Cardiorenal Med. 2:268–280. 2012.
View Article : Google Scholar
|
|
10
|
Lenos MG and Tsaniklidou SM: Comment on
'Angiotensin-converting-enzyme 2 inhibits liver fibrosis in mice'.
Hepatology. 51:7182010. View Article : Google Scholar
|
|
11
|
Verma A, Shan Z, Lei B, Yuan L, Liu X,
Nakagawa T, Grant MB, Lewin AS, Hauswirth WW, Raizada MK, et al:
ACE2 and Ang-(1-7) confer protection against development of
diabetic retinopathy. Mol Ther. 20:28–36. 2012. View Article : Google Scholar :
|
|
12
|
Zhang XP, Li ZJ and Zhang J: Inflammatory
mediators and microcirculatory disturbance in acute pancreatitis.
Hepatobiliary Pancreat Dis Int. 8:351–357. 2009.PubMed/NCBI
|
|
13
|
Ibiş M, Yüksel O, Yilmaz G, Köklü S,
Yilmaz FM, Ertuğrul I, Uçar E and Altiparmak ME: Serum angiotensin
converting enzyme levels in pancreatic diseases.
Hepatogastroenterology. 55:1814–1817. 2008.
|
|
14
|
Ulmasov B, Xu Z, Talkad V, Oshima K and
Neuschwander-Tetri BA: Angiotensin II signaling through the AT1a
and AT1b receptors does not have a role in the development of
cerulein-induced chronic pancreatitis in the mouse. Am J Physiol
Gastrointest Liver Physiol. 299:G70–G80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ahmadian E, Pennefather PS, Eftekhari A,
Heidari R and Eghbal MA: Role of renin-angiotensin system in liver
diseases: An outline on the potential therapeutic points of
intervention. Expert Rev Gastroenterol Hepatol. 10:1279–1288. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guo YJ, Li WH, Wu R, Xie Q and Cui LQ:
ACE2 overexpression inhibits angiotensin II-induced monocyte
chemoattractant protein-1 expression in macrophages. Arch Med Res.
39:149–154. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Verdecchia P, Gentile G, Angeli F and
Reboldi G: Beyond blood pressure: Evidence for cardiovascular,
cerebrovascular, and renal protective effects of renin-angiotensin
system blockers. Ther Adv Cardiovasc Dis. 6:81–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fraga-Silva RA, Da Silva DG, Montecucco F,
Mach F, Stergiopulos N, da Silva RF and Santos RA: The
angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas receptor
axis: A potential target for treating thrombotic diseases. Thromb
Haemost. 108:1089–1096. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Santos RA, Ferreira AJ, Verano-Braga T and
Bader M: Angiotensin-converting enzyme 2, angiotensin-(1-7) and
Mas: New players of the renin-angiotensin system. J Endocrinol.
216:R1–R17. 2013. View Article : Google Scholar
|
|
20
|
Patel VB, Zhong JC, Grant MB and Oudit GY:
Role of the ACE2/angiotensin 1-7 axis of the renin-angiotensin
system in heart failure. Circ Res. 118:1313–1326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Liu J, Luo JY, Tian XY, Cheang
WS, Xu J, Lau CW, Wang L, Wong WT, Wong CM, et al: Upregulation of
Angiotensin- (1-7)-mediated signaling preserves endothelial
function through reducing oxidative stress in diabetes. Antioxid
Redox Signal. 23:880–892. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Márquez-Miranda V, Abrigo J, Rivera JC,
Araya-Durán I, Aravena J, Simon F, Pacheco N, González-Nilo FD and
Cabello- Verrugio C: The complex of PAMAM-OH dendrimer with
Angiotensin-(1-7) prevented the disuse-induced skeletal muscle
atrophy in mice. Int J Nanomedicine. 12:1985–1999. 2017. View Article : Google Scholar
|
|
23
|
Durand MJ, Zinkevich NS, Riedel M,
Gutterman DD, Nasci VL, Salato VK, Hijjawi JB, Reuben CF, North PE
and Beyer AM: Vascular actions of angiotensin 1-7 in the human
microcirculation: Novel role for telomerase. Arterioscler Thromb
Vasc Biol. 36:1254–1262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bihl JC, Zhang C, Zhao Y, Xiao X, Ma X and
Chen Y, Chen S, Zhao B and Chen Y: Angiotensin-(1-7) counteracts
the effects of Ang II on vascular smooth muscle cells, vascular
remodeling and hemorrhagic stroke: Role of the NFкB inflammatory
pathway. Vascul Pharmacol. 73:115–123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oruc N, Ozutemiz O, Nart D, Yuce G, Celik
HA and Ilter T: Inhibition of renin-angiotensin system in
experimental acute pancreatitis in rats: A new therapeutic target?
Exp Toxicol Pathol. 62:353–360. 2010. View Article : Google Scholar
|
|
26
|
Wang Y, Wang J, Liu R, Qi H, Wen Y, Sun F
and Yin C: Severe acute pancreatitis is associated with
upregulation of the ACE2-angiotensin-(1-7)-Mas axis and promotes
increased circulating angiotensin-(1-7). Pancreatology. 12:451–457.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu R, Qi H, Wang J, Wang Y, Cui L, Wen Y
and Yin C: Angiotensin-converting enzyme (ACE and ACE2) imbalance
correlates with the severity of cerulein-induced acute pancreatitis
in mice. Exp Physiol. 99:651–663. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu R, Qi H, Wang J, Wang Y, Cui L, Wen Y
and Yin C: Ulinastatin activates the renin-angiotensin system to
ameliorate the pathophysiology of severe acute pancreatitis. J
Gastroenterol Hepatol. 29:1328–1337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hemi R, Yochananov Y, Barhod E,
Kasher-Meron M, Karasik A, Tirosh A and Kanety H: p38
mitogen-activated protein kinase-dependent transactivation of ErbB
receptor family: A novel common mechanism for stress-induced IRS-1
serine phosphorylation and insulin resistance. Diabetes.
60:1134–1145. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao SC, Yin HB, Liu HX and Sui YH:
Research progress on MAPK signal pathway in the pathogenesis of
osteoarthritis. Zhongguo Gu Shang. 27:441–444. 2014.In Chinese.
PubMed/NCBI
|
|
31
|
Ono K and Han J: The p38 signal
transduction pathway: Activation and function. Cell Signal.
12:1–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu H, Zhu X, Lin R, Li Z and Chen L:
Suppressive effects of Gua Lou Gui Zhi decoction on MCAO-induced NO
and PGE2 production are dependent on the MAPK and NF-κB signaling
pathways. Mol Med Rep. 14:5141–5147. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moreno JA, Sullivan KA, Carbone DL,
Hanneman WH and Tjalkens RB: Manganese potentiates nuclear
factor-kappaB-dependent expression of nitric oxide synthase 2 in
astrocytes by activating soluble guanylate cyclase and
extracellular responsive kinase signaling pathways. J Neurosci Res.
86:2028–2038. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Carter AB and Hunninghake GW: A
constitutive active MEK→ERK pathway negatively regulates NF-kappa
B-dependent gene expression by modulating TATA-binding protein
phosphorylation. J Biol Chem. 275:27858–27864. 2000.PubMed/NCBI
|
|
35
|
Williard DE, Twait E, Yuan Z, Carter AB
and Samuel I: Nuclear factor kappa B-dependent gene transcription
in cholecystokinin-and tumor necrosis factor-alpha-stimulated
isolated acinar cells is regulated by p38 mitogen-activated protein
kinase. Am J Surg. 200:283–290. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Twait E, Williard DE and Samuel I:
Dominant negative p38 mitogen-activated protein kinase expression
inhibits NF-kappaB activation in AR42J cells. Pancreatology.
10:119–128. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu JH, Lim JW and Kim H: Altered gene
expression in cerulein-stimulated pancreatic acinar cells:
Pathologic mechanism of acute pancreatitis. Korean J Physiol
Pharmacol. 13:409–416. 2009. View Article : Google Scholar
|
|
38
|
Rolny P, Arlebäck A, Funch-Jensen P, Kruse
A, Ravnsbaeck J and Järnerot G: Paradoxical response of sphincter
of Oddi to intravenous injection of cholecystokinin or ceruletide.
Manometric findings and results of treatment in biliary dyskinesia.
Gut. 27:1507–1511. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cui L, Liu R, Li C, Yu X, Liu X, Hou F,
Chi C, Yin C and Wang C: Angiotensin (1-7) attenuates caerulein
induced pancreatic acinar cell apoptosis. Mol Med Rep.
16:3455–3460. 2017.PubMed/NCBI
|
|
40
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
41
|
Tsang SW, Ip SP and Leung PS: Prophylactic
and therapeutic treatments with AT 1 and AT 2 receptor antagonists
and their effects on changes in the severity of pancreatitis. Int J
Biochem Cell Biol. 36:330–339. 2004. View Article : Google Scholar
|
|
42
|
Vickers C, Hales P, Kaushik V, Dick L,
Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, et al:
Hydrolysis of biological peptides by human angiotensin-converting
enzyme-related carboxypeptidase. J Biol Chem. 277:14838–14843.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ren T, He H, Yu X, Fan J, Tan J and Liu J:
Angiotensin-(1-7) inhibits hypoxia-induced renal tubular
epithelial-to-mesenchymal transition in rats. Xi Bao Yu Fen Zi Mian
Yi Xue Za Zhi. 29:593–596. 2013.In Chinese. PubMed/NCBI
|
|
44
|
Su Z, Zimpelmann J and Burns KD:
Angiotensin-(1-7) inhibits angiotensin II-stimulated
phosphorylation of MAP kinases in proximal tubular cells. Kidney
Int. 69:2212–2218. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pérez S, Pereda J, Sabater L and Sastre J:
Redox signaling in acute pancreatitis. Redox Biol. 5:1–14. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee WS, Shin JS, Jang DS and Lee KT:
Cnidilide, an alkylphthalide isolated from the roots of Cnidium
officinale, suppresses LPS-induced NO, PGE2, IL-1β, IL-6 and TNF-α
production by AP-1 and NF-κB inactivation in RAW 264.7 macrophages.
Int Immunopharmacol. 40:146–155. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Makhija R and Kingsnorth AN: Cytokine
storm in acute pancreatitis. J Hepatobiliary Pancreat Surg.
9:401–410. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ren HB, Li ZS, Xu GM, Tu ZX, Shi XG, Jia
YT and Gong YF: Dynamic changes of mitogen-activated protein kinase
signal transduction in rats with severe acute pancreatitis. Chin J
Dig Dis. 5:123–125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xu M, Wang KN, Wu K and Wang XP:
Pyrrolidine dithiocarbamate inhibits nuclear factor κB and
Toll-like receptor 4 expression in rats with acute necrotizing
pancreatitis. Gut Liver. 9:411–416. 2015. View Article : Google Scholar
|
|
50
|
Vaquero E, Gukovsky I, Zaninovic V,
Gukovskaya AS and Pandol SJ: Localized pancreatic NF-kappaB
activation and inflammatory response in taurocholate-induced
pancreatitis. Am J Physiol Gastrointest Liver Physiol.
280:G1197–G1208. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bettaieb A, Xi Y, Hosein E, Coggins N,
Bachaalany S, Wiede F, Perez S, Griffey SM, Sastre J, Tiganis T, et
al: Pancreatic T cell protein-tyrosine phosphatase deficiency
ameliorates cerulein- induced acute pancreatitis. Cell Commun
Signal. 12:132014. View Article : Google Scholar
|
|
52
|
Li G, Wu X, Yang L, He Y, Liu Y, Jin X and
Yuan H: [Corrigendum] TLR4-mediated NF-κB signaling pathway
mediates HMGB1-induced pancreatic injury in mice with severe acute
pancreatitis. Int J Mol Med. 38:13132016. View Article : Google Scholar
|
|
53
|
Shi Q, Liao KS, Zhao KL, Wang WX, Zuo T,
Deng WH, Chen C, Yu J, Guo WY, He XB, et al: Hydrogen-rich saline
attenuates acute renal injury in sodium taurocholate-induced severe
acute pancreatitis by inhibiting ROS and NF-κB pathway. Mediators
Inflamm. 2015:6850432015. View Article : Google Scholar
|
|
54
|
Yang X, Jin H, Liu K, Gu Q and Xu X: A
novel peptide derived from human pancreatitis-associated protein
inhibits inflammation in vivo and in vitro and blocks NF-kappa B
signaling pathway. PLoS One. 6:e291552011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Steinle AU, Weidenbach H, Wagner M, Adler
G and Schmid RM: NF-kappaB/Rel activation in cerulein pancreatitis.
Gastroenterology. 116:420–430. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kefaloyianni E, Gaitanaki C and Beis I:
ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved
in NF-kappaB transactivation during oxidative stress in skeletal
myoblasts. Cell Signal. 18:2238–2251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Saccani S, Pantano S and Natoli G:
p38-Dependent marking of inflammatory genes for increased NF-kappa
B recruitment. Nat Immunol. 3:69–75. 2002. View Article : Google Scholar
|