Introduction
Acute kidney injury (AKI) is a critical care
syndrome in clinical practice, which is characterized by a rapid
decline in renal function, and is responsible for high morbidity
and mortality rates of patients in intensive care units (1). The clinical symptoms of AKI are
caused by diverse components, including sepsis-induced infection
(2), liver transplantation
(3), cardiac surgery (4) and contrast media (5). Lipopolysaccharide (LPS) is an
endotoxin produced following infection by Gram negative bacteria,
and remains the most common trigger for AKI (6). In addition, LPS-induced AKI often
develops in patients with decreased immune-competence, resulting in
severe renal failure and exacerbated mortality rates (7). It has been suggested the onset of
LPS-induced AKI is associated with an elevation of inflammatory
cytokines (8) and transcription
factors, for example nuclear factor (NF)-κB, which consequentially
induce pro-inflammatory mediators, including tumor necrosis
factor-α (TNF-α), interleukin (IL)-1 and IL-6, which coordinate the
elimination of pathogens and infected cells (9). Although the pathogenic factors of
AKI are clear, the complex biological and molecular mechanisms
underlying AKI remain to be fully elucidated.
MicroRNAs (miRNAs; ~22 nt) are important in gene
regulation and are involved in diverse biological processes,
including cell growth, migration, apoptosis and differentiation, by
binding to the 3′-untranslated regions of mRNAs (10). In addition to feedback regulation
at the level of transcription, miRNAs have been identified to serve
in post-transcriptional negative feedback loops, which modulate
inflammatory signaling (11).
miR-146a was previously found to be transcriptionally induced by
NF-κB in response to the activation of innate immune signaling in
monocytes (12) and inhibit
activation of the NF-κB pathway (13). In addition, the miR-146a-induced
suppression of NF-κB-driven monocyte/macrophage activation and
atherosclerosis are regulated by the expression of cellular
apolipoprotein E (14). A defect
in the NF-κB/miR-146a negative feedback loop may be involved in the
pathogenesis of diabetic neuropathy (15). These previous findings demonstrate
that miR-146a is involved in a negative feedback loop to control
the NF-κB pathway. However, the function of the miR-146a/NF-κB
pathway in AKI remains to be fully elucidated. Therefore, the
identification of miR-146a/NF-κB, and the clarification of its
potential mechanisms, is critical for developing efficient
therapies against AKI.
Long non-coding RNAs (lncRNAs; ~200 nt) are a class
of RNAs, which do not encode proteins (16). Accumulating evidence shows that
lncRNAs are widely expressed in various human somatic tissues and
are involved in diverse cellular events, including epigenetic
regulation, gene transcription, mRNA processing and gene
translation (17). Compared with
the well-reported dysregulated miRNAs, which are involved in
various diseases, only a small number of lncRNAs have been
investigated in relation to their functions in kidney cancer,
including PRINS (18), CRNDE
(19) and H19 (20), and even fewer have been recognized
in the process of AKI. In the field of nephropathy,
metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was
revealed to be upregulated in clear cell renal cell carcinoma
(ccRCC) and acted as a novel molecule involved in the progression
of ccRCC (21). However, whether
MALAT1 was associated with the malignant progression of AKI remains
to be elucidated and warrants further investigations to examine the
underlying mechanisms.
The present study aimed to determine the expression
and function of MALAT1, miR-146a and NF-κB pathway factors in AKI
tissues and cells. The present study also investigated the
interactions among the three regulators in the regulation of AKI
malignant behavior and the potential molecular pathways
involved.
Materials and methods
Animals
Male Sprague-Dawley rats (n=30; age, 6 weeks;
weight, 22–25 g) were obtained from the Laboratory Animal Center of
Zhejiang University (Hangzhou, China) and were maintained in a
standard laboratory in an air-conditioned room at 18–26°C with free
access to food and water. Artificial lighting was provided on a
12/12-h light/dark cycle. All experimental protocols were performed
according to the Guide for the Care and Use of Laboratory Animals
and were approved by the Ethics Review Board for Animal Studies of
the School of Medicine, Zhejiang University.
In vivo AKI animal model
Following adaptation to the laboratory environment
for 1 week, the rats were selected for establishment of the AKI
model. The animals were randomly divided into two groups
(n=15/group), comprising a control and LPS-treated AKI group. The
animals in the AKI model groups were administered an
intraperitoneal injection of LPS (10 mg/kg) 6 h prior to sacrifice
by cervical dislocation following the inhalation of
1,1,1-trichloroethane (TCE) (22,23). Blood samples were obtained and
centrifuged at 3,600 × g for 10 min at 4°C to obtain serum. The
blood samples were then stored at −20°C and kidney samples were
stored at −80°C for further analysis.
Cell culture
The NRK-52E normal rat kidney epithelial cell line
and HK-2 human kidney tubular epithelial cell line were purchased
from the Institute of Biochemistry and Cell Biology of the Chinese
Academy of Sciences (Shanghai, China). The cells were maintained in
Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc.) and
antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin) in a
humidified atmosphere of 5% CO2 and 95% O2 at
37°C.
In vivo LPS treatment
The LPS was prepared in ultrapure water and used to
produce the working dilution at a concentration of 0.5 µg/ml
(24) in serum-free DMEM for
direct application to cell cultures (25). The NRK-52E and HK-2 cells were
plated in 96-well plates at a density of 5×104 cells/ml
per well for 24 h, following which they were challenged with LPS
and then incubated for another 24 h. Cell viability was assessed
using an MTT assay.
Cell transfection
The plasmid for the overexpression of MALAT1 was
constructed by inserting the amplified MALAT1 cDNA using a MiRcute
miRNA first-strand cDNA synthesis kit (Tiangen Biotech Co., Ltd.,
Beijing, China) according to the manufacturer's instructions into
the pCDNA3.1 vector (Invitrogen; Thermo Fisher Scientific, Inc.),
resulting in pcDNA MALAT1. Small interfering RNA (si)-MALAT1 targe
ting MALAT1 and negative control siRNA were purchased from Qiagen
GmbH (Hilden, Germany). The miR-146a mimic/negative control mimic
and the miR-146a inhibitor/negative control inhibitor were obtained
from Guangzhou RiboBio Co., Ltd. (Guangzhou, China). For cell
transfection, the cells were incubated with si-MALAT1/pcDNA MALAT1,
miR-146a mimic/miR-146a inhibitor or their corresponding controls
using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions, and the cells
were harvested 48 h later for subsequent experiments.
The specific primers used were as follows: human
si-MALAT1 sense, GCAAAUGAAAGCUACCAAUTT and antisense,
AUUGGUAGCUUUCAUUUGCTT; rat si-MALAT1 sense, GCAGUUUAGGAGAUUGUAATT
and antisense, UUACAAUCUCCUAAACUGCTT; negative control siRNA sense,
UUCUCCGAACGUGUCACGUTT and antisense, ACGUGACACGUUCGGAGAATT.
Western blot analysis
Total protein was extracted from the kidneys and
cultured cells, and the concentrations were estimated by collecting
equal number of cells and homogenizing using RIPA lysis buffer. The
protein concentrations of the samples were determined using a BCA
protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and standards. The proteins (50 µg) were denatured with
sodium dodecyl sulfate (SDS) sample buffer, separated by 10% SDS
polyacrylamide gel electrophoresis and then transferred onto PVDF
membranes (EMD Millipore, Billerica, MA, USA). Following blocking
with 5% skim milk for 2 h at room temperature, the membranes were
incubated with primary antibodies including phosphorylated
inhibitor of NF-κB (p-IκBα; cat. no. 2859, 1:1,000), IκBα (cat. no.
9247, 1:1,000), B-cell lymphoma 2 (Bcl-2; cat. no. 3498, 1:1,000),
Bcl-2-associated X protein (Bax; cat. no. 2772, 1:1,000), and
caspase-3 (cat. no. 9662, 1:1,000) (all from Cell Signaling
Technology, Boston, MA, USA), np65 (cat. no. ab83063, 1:1,000;
Abcam, Cambridge, MA, USA), lamin B (cat. no. 13435, 1:1,000) and
β-actin (cat. no. 8457, 1:1,000) (both from Cell Signaling
Technology) overnight at 4°C. Following the addition of anti-rabbit
horseradish peroxidase-conjugated IgG secondary antibodies (cat.
no. ab6721, 1:2000; Abcam) for 2 h at room temperature. The protein
bands on the membranes were detected using an enhanced
chemiluminescence system (UVP, Inc., Upland, CA, USA). The
intensity values of the bands were adjusted relative to the
expression of the internal reference (β-actin and lamin B).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA from the tissues and cells was extracted
using TRIzol (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. The mRNA and miRNA were polyadenylated
using a poly-A polymerase-based First-Strand Synthesis kit (Takara
Biotechnology Co., Ltd., Shanghai, China) and reverse transcription
of the total mRNA was performed using a PrimeScript RT reagent kit
according to the manufacturer's instructions. The reactions were
incubated in a 96-well plate at 95°C for 10 min, followed by 40
cycles of 95°C for 15 sec, 60°C for 30 sec and 72°C for 30 sec. The
resulting amplification were quantified on an ABI 7500HT system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) using
SYBR-Green I. U6 or GAPDH were used as endogenous controls.
Relative fold expression levels were calculated with the
comparative quantification cycle (2−ΔΔCq) method
(26).
The primer sequences were listed as follows:
miRNA-146a forward, 5′-ACACTCCAGCTGGGTGAGAACTGAATTCCA-3′ and
reverse, 5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAACCCATG-3′; and
MALAT1 forward, 5′-GCGACGAGTTGTGCTGCTATCT-3′ and reverse,
5′-ACACTGCTCTGGGTCTGCTTTT-3′.
Assessment of biochemical parameters
Serum levels of blood urea nitrogen (BUN; mmol/l)
and creatinine (Cr; µmol/l) were detected using specific
kits according to the manufacturer's instructions. The serum
expression levels of TNF-α and IL-6 (ng/l) were determined using
commercial ELISA kits according to the manufacturer's instructions.
All assays were performed in triplicate.
Cell viability assay
Cell proliferation was measured using an MTT assay.
The cells were seeded in a 96-well plate and cultured in normal
medium. Following transfection, the cells were incubated in 0.1
mg/ml MTT at 37°C for 4 h and lysed in dimethyl sulfoxide at room
temperature for 10 min. The absorbance in each well was measured on
a microplate reader at a wavelength of 490 nm (Bio-Rad
Laboratories, Inc.).
RNA immunoprecipitation (RIP)
LncBase Predicted v.2 bioinformatics tools
(http://carolina.imis.athena-innovation.gr/) was used
to predict the potential interaction of lncRNA MALAT1 and miR-146a.
The cells were co-transfected with a pcDNA-MS2/pcDNA-MALAT1-MS2
vector and miR-146a mimic or non-targeting miRNA mimic. At 48 h
post-transfection, the cells were washed and lysed in
radioimmunoprecipitation buffer containing 10% proteinase inhibitor
cocktail and 1 mM phenylmethylsulfonyl fluoride. A fraction of the
whole cell lysate was used for RNA isolation, and the remaining
lysate was subjected to IP using an antibody against immunoglobulin
G (cat. no. 5873, 1:20; Cell Signaling Technology) for 18 h at 4°C.
The RNA from the whole cell lysates and the RIP fractions were
extracted with TRIzol according to the manufacturer's instructions.
The relative mRNA expression levels of MALAT1 and miR-146a were
determined using RT-qPCR analysis, as described above. The relative
mRNA enrichment in the RIP fractions was computed based on the
ratio of relative mRNA levels in the RIP fractions and the relative
mRNA levels in the whole cell lysates (input).
Statistical analysis
All data are expressed as the mean ± standard
deviation and were analyzed using SPSS 16.0 software for Windows
(SPSS, Inc., Chicago, IL, USA). An independent sample t-test was
used to determine the statistical differences between two groups.
Multiple group comparisons were tested using one-way analysis of
variance (ANOVA). Pearson's correlation analysis was used to assess
the correlation between MALAT1 and miR-146a. P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of MALAT1 and miR-146a are
inversely correlated in the LPS-induced AKI model
To examine aberrant gene expression in response to
kidney injury, a rat AKI model was successfully established. Serum
and infarcted kidney tissues were isolated for subsequent analysis
using RT-qPCR and western blot analyses. The results showed a
significant several-fold increase in serum level of BUN in the AKI
group, compared with that in the normal control (Fig. 1A), in line with markedly elevated
Cr production (Fig. 1B).
Concomitantly, the ELISA results revealed that LPS significantly
enhanced the expression of serum inflammatory factors IL-6 and
TNF-α (Fig. 1C and D). In
addition, the RT-qPCR analysis indicated that the relative
expression level of miR-146a was decreased following LPS treatment,
compared with that in the control group (Fig. 1E), whereas the expression of
MALAT1 was upregulated in the kidneys stimulated by LPS (0.5
µg/ml; Fig. 1F). To
further confirm the role of MALAT1 in the AKI rats, the potential
correlation between MALAT1 and miR-146a was examined using
Pearson's correlation analysis. The expression of MALAT1 negatively
correlated with the expression of miR-146a (P=0.0219; r=−0.7084;
Fig. 1G). The results of the
western blot analysis for the protein expression levels of p-IκBα,
np65 and apoptosis-associated regulators showed that the production
of p-IκBα, np65, cleaved (c)-caspase-3 and Bax was increased in the
model group, compared with the control group, which was in contrast
to the level of Bcl-2 in the kidney tissues (Fig. 1H). These results indicated that
the NF-κB np65-mediated pathways affected the LPS-induced
upregulation of MALAT1 and downregulation of miR-146a.
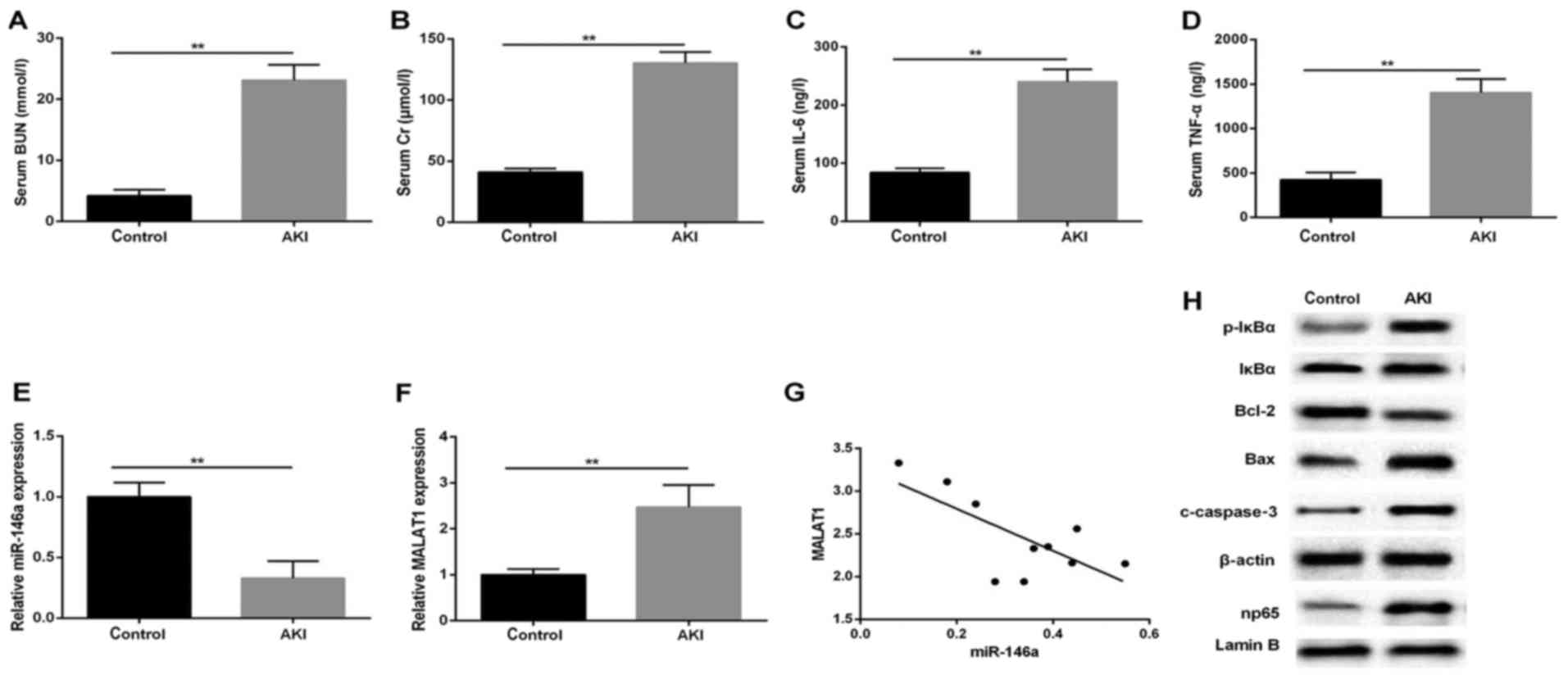 | Figure 1MALAT1 expression negatively
correlates with the expression of miR-146a in a LPS-induced AKI rat
model. Serum levels of (A) BUN (mmol/l), (B) Cr (µmol/l),
(C) IL-6 and (D) TNF-α (ng/l). Reverse transcription-quantitative
polymerase chain reaction analysis of the expression of (E)
miR-146a and (F) MALAT1 in rat AKI tissues (n=15 for each group);
U6 and GAPDH were used as internal controls, respectively. (G)
Correlation analysis between MALAT1 and miR-146a (P=0.0219;
r=0.7084). (H) Western blot analysis of p-IκBα, np65, c-caspase-3,
Bax and Bcl-2 in tissues. β-actin was used as an internal control.
**P<0.01 vs. control group. All data are expressed as
the mean ± standard deviation of at least three independent
experiments. MALAT1, metastasis-associated lung adenocarcinoma
transcript 1; LPS, lipopolysaccharide; AKI, acute kidney injury;
miR, microRNA; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α;
BUN, blood urea nitrogen; Cr, creatinine; p-IκBα, phosphorylated
inhibitor of nuclear factor-κB; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein. |
MALAT1 silencing protects NRK-52E kidney
cells from LPS challenge
To determine the abnormal factors expressed in
LPS-injured kidney epithelial cells, NRK-52E and HK-2 cells were
used for investigation of the mechanism. The NRK-52E cells were
subjected to LPS treatment, simulating the LPS-induced AKI model
in vitro. Cell viability (Fig.
2A) was detected, and changes in the expression of MALAT1,
miR-146a and np65, and the production of TNF-α in the supernatant
were analyzed. Similar results were observed in the LPS-treated
NRK-52E renal tubular epithelial cells, including increased
production of TNF-α, IL-6 and MALAT1 (Fig. 2B–D). By contrast, cell viability
was inhibited and the expression of miR-146a was markedly
downregulated in the LPS treatment group, compared with that in the
control cells (Fig. 2E).
Simultaneously, the production of p-IκBα and np65 at the protein
level were assessed using western blot analysis. As shown in
Fig. 2F, the levels of NF-κB
signaling pathway-associated molecules p-IκBα and np65 were higher
in the NRK-52E cells induced by LPS, compared with those in the
controls. However, the above-mentioned results were reversed by
si-MALAT1 in the NRK-52E cells. These results indicated that MALAT1
may be important in LPS-induced AKI, involve the LPS-induced
production of TNF-α via NF-κB np65-mediated pathways.
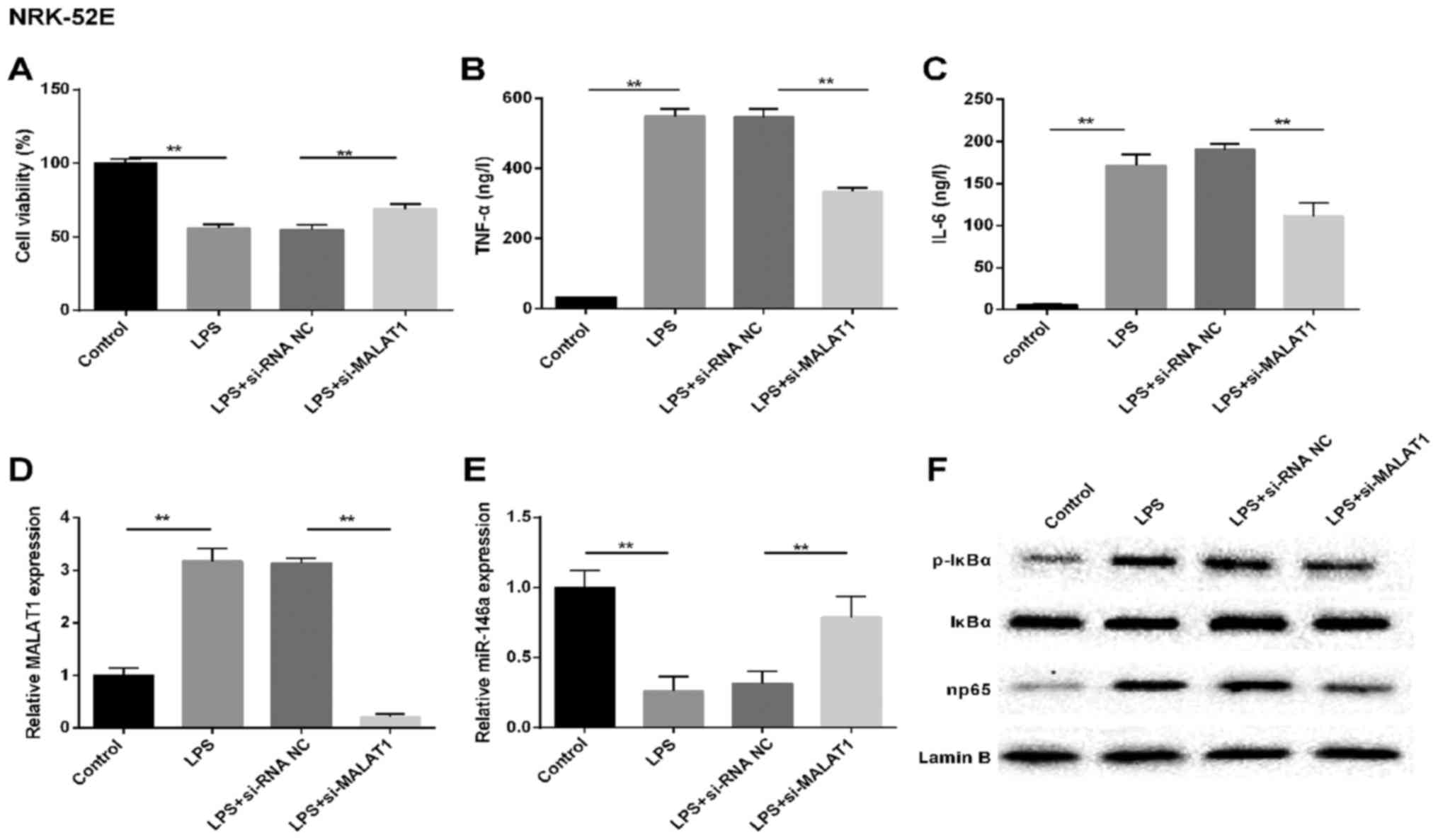 | Figure 2MALAT1 silencing protects NRK-52E
renal tubular epithelial cells from LPS challenge. In vitro,
cells were randomly divided into four groups: Control, LPS,
LPS+si-RNA NC and LPS+si-MALAT1. (A) Cell viability was assessed
using the MTT method. Expression levels of (B) TNF-α and (C) IL-6
(ng/l). Reverse transcription-quantitative polymerase chain
reaction analysis of the expression of (D) MALAT1 and (E) miR-146a
in cells. U6 and GAPDH were used as internal controls,
respectively. (F) Western blot analysis of protein levels of NF-κB
regulatory factors in NRK-52E cells. Lamin B was used as an
internal control. **P<0.01 between groups. All data
are expressed as the mean ± standard deviation of at least three
independent experiments. MALAT1, metastasis-associated lung
adenocarcinoma transcript 1; miR, microRNA; LPS,
lipopolysaccharide; si, small interfering RNA; NC, negative
control; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α;
p-IκBα, phosphorylated inhibitor of nuclear factor-κB. |
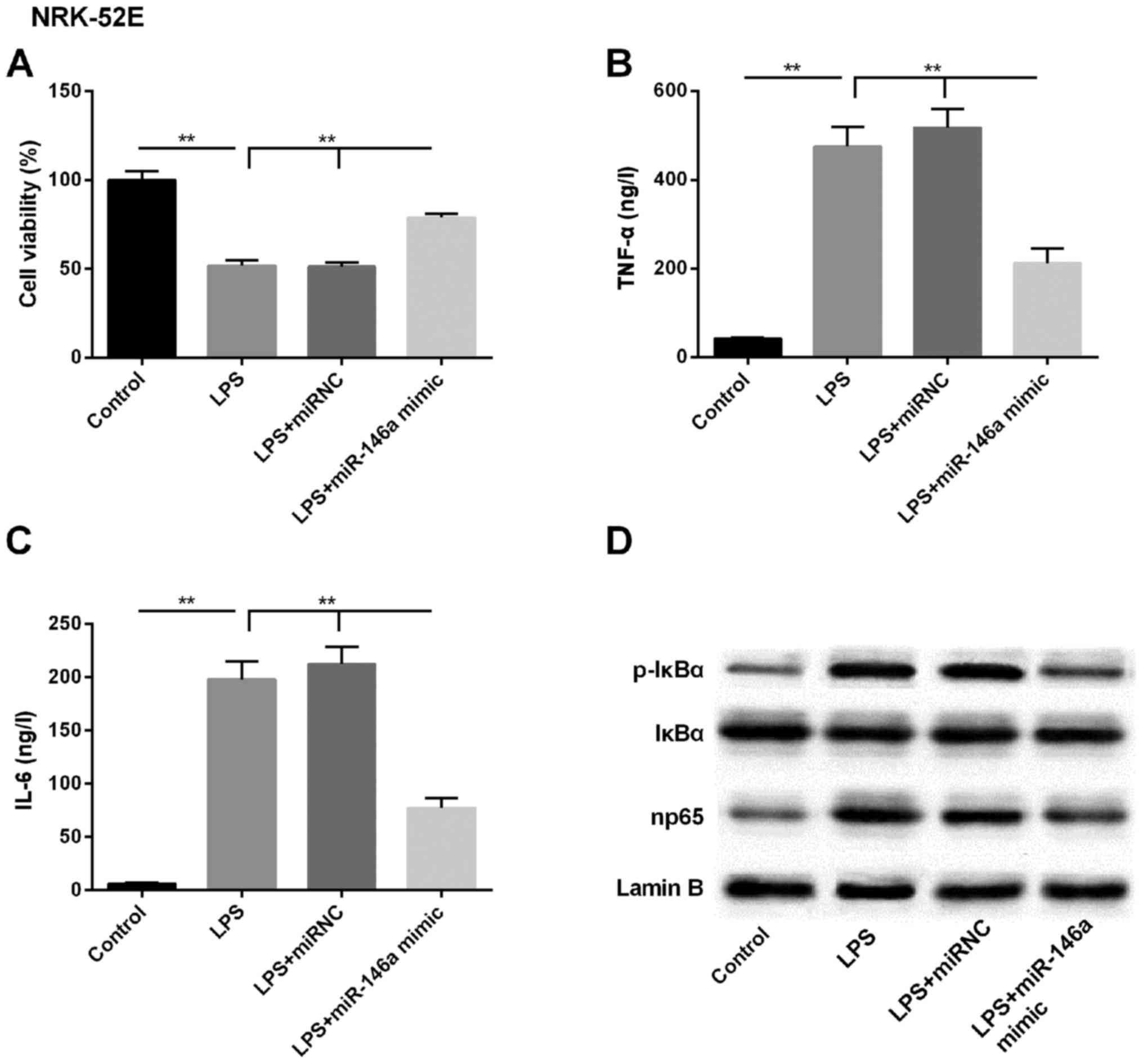
miR-146a attenuates LPS-induced
expression of NF-κB pathway-associated molecules
As the expression of miR-146a was downregulated
following LPS treatment, the overexpression of miR-146a was
enforced by transfecting the miR-146a mimic into NRK-52E cells. As
shown in Fig. 3A, the LPS-induced
downregulation in cell viability was reversed by the miR-146a
mimic, to close to the baseline level. Further analysis of the
levels of pro-inflammatory cytokines in the supernatant showed that
the LPS-induced production of TNF-α and IL-6 were significantly
attenuated in the miR-146a-overexpressing kidney epithelial cells
(Fig. 3B and C). Subsequently,
the expression of NF-κB pathway-associated regulators (p-IκBα and
np65) were examined using western blot analysis, and it was found
that the basal levels of these proteins were suppressed in the
cells transfected with miR-146a mimic, compared with those in the
LPS, and LPS+miR-NC groups (Fig.
3D). Therefore, the miR-146a-mediated expression of NF-κB and
np65 pathway members may be the underlying LPS-induced mechanism
during the pathogenesis of AKI.
Protective effect of MALAT1-silencing on
LPS-treated NRK-52E cells is reversed by miR-146a inhibitor
To further detect the effect of MALAT1 and miR-146a
on inflammatory response-induced kidney injury, the NRK-52E cells
treated with LPS were transfected with si-MALAT1 or siRNA NC,
miR-146a inhibitor or miR-NC. As shown in Fig. 4A, the LPS-induced increase of cell
viability was markedly attenuated in the miR-146a-inhibited NRK-52E
cells. The expression levels of TNF-α and IL-6 were markedly
upregulated in cells co-transfected with miR-146a inhibitor and
si-MALAT1, compared with those in the corresponding control group
(Fig. 4B and C). The western blot
analysis showed that the levels of p-IκBα and np65 were promoted by
the inhibition of miR-146a, compared with that in the control
(Fig. 4D). Therefore, the
cytoprotective effect induced by si-MALAT1 was undermined by
miR-146a-inhibition.
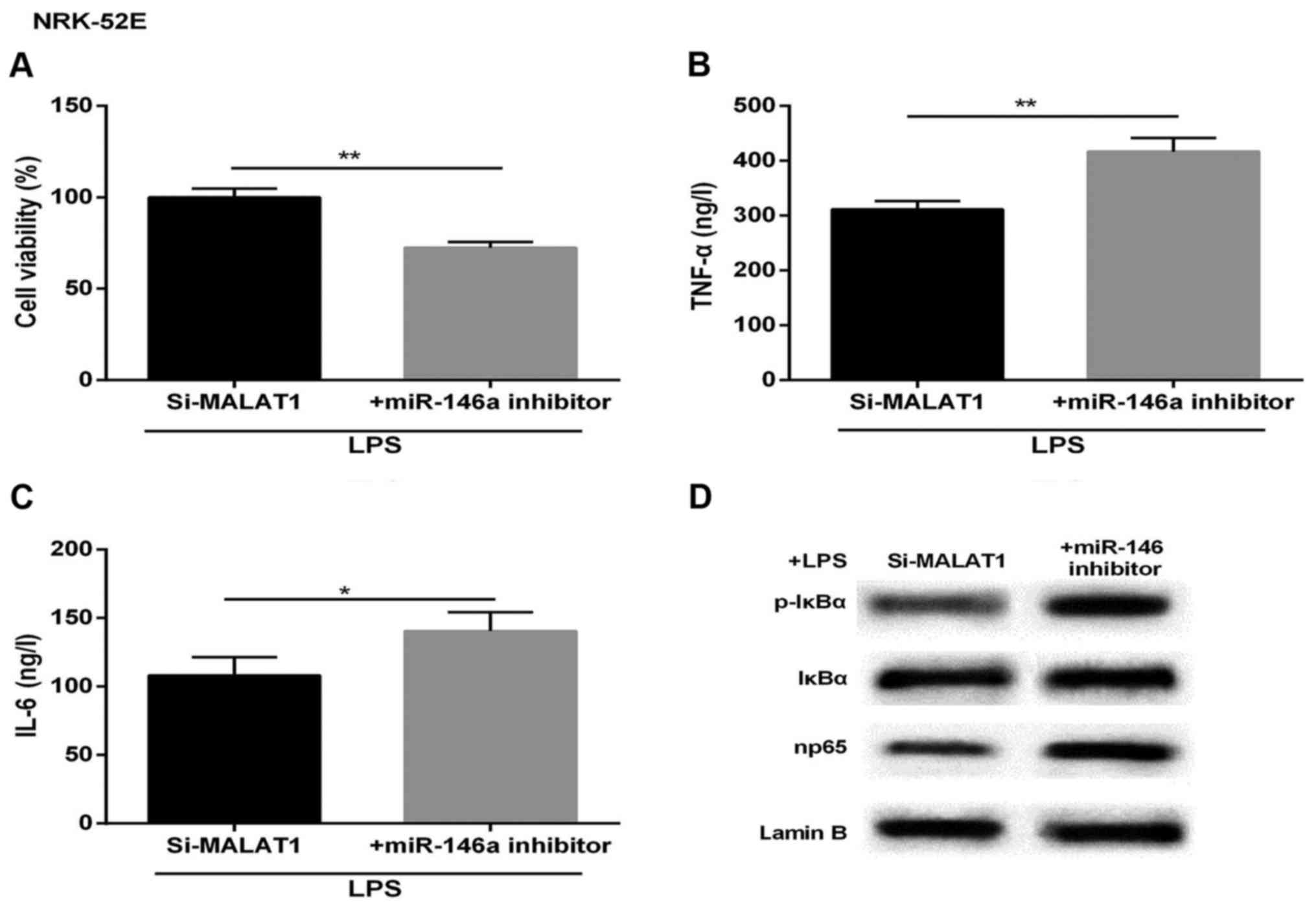 | Figure 4Cytoprotective effects of si-MALAT1 on
LPS-treated acute kidney injury is reversed by the miR-146a
inhibitor. Following LPS treatment, the cells were divided into
si-MALAT1 and si-MALAT1+miR-146a inhibitor groups. (A) Cell
viability. Expression of (B) TNF-α, (C) IL-6 (ng/l) and (D) NF-κB
regulatory proteins in NRK-52E cells. Lamin B was used as an
internal control. *P<0.05 and **P<0.01
between groups. All data are expressed as the mean ± standard
deviation of at least three independent experiments. MALAT1,
metastasis-associated lung adenocarcinoma transcript 1; miR,
microRNA; LPS, lipopolysaccharide; si, small interfering RNA; IL-6,
interleukin-6; TNF-α, tumor necrosis factor-α; NF-κB, nuclear
factor-κB; p-IκBα, phosphorylated inhibitor of NF-κB. |
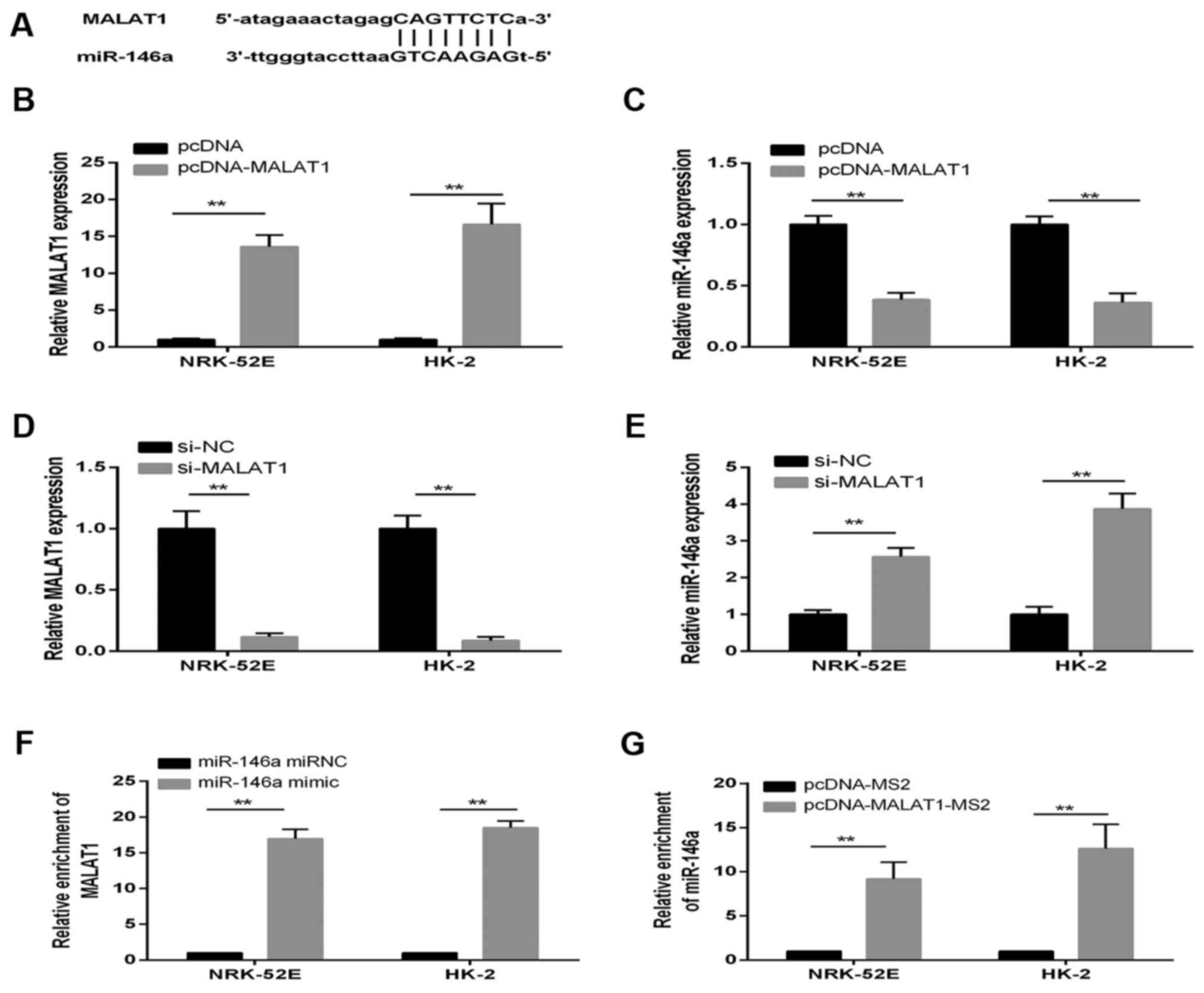
MALAT1 is a direct target of
miR-146a
There is increasing evidence that lncRNAs may be
competing endogenous RNAs or molecular sponges in downregulating
the expression and biological functions of miRNAs. Using a
bioinformatics database (starBase v2.0), the present study
determined that MALAT1 harbored one putative binding site for
miR-146a (Fig. 5A). To confirm
the hypothesis that miR-146a directly binds to MALAT1, the
expression of miR-146a in pcDNA-MALAT1 cells was measured using
RT-qPCR analysis. The expression of MALAT1 was confirmed to be
significantly upregulated in NRK-52E and HK-2 cells transfected
with pcDNA-MALAT1; whereas the expression of miR-146a was decreased
in the pcDNA-MALAT1 group (Fig. 5B
and C). By contrast, the expression of MALAT1 and miR-146a
showed the opposite results when the cells were treated with
si-MALAT1, compared with those in the si-NC control group (Fig. 5D and E).
An RIP assay was performed to determine whether
MALAT1 and miR-146a were in the expected RNA-induced silencing
complex (RISC). The RNA levels were examined using RT-qPCR
analysis, which revealed significant relative enrichment of mRNA
levels of MALAT1 and miR-146a, compared with those in the control
group, respectively (Fig. 5F and
G). Therefore, MALAT1 inhibition was confirmed to restore the
expression of miR-146a in a RISC-dependent manner, and there was a
negative linear correlation between MALAT1 and miR-146a.
MALAT1 regulates the NF-κB pathway via
affecting the expression of miR-146a
The mechanism underlying the effect of MALAT1 in AKI
mediated by the miR-146a/NF-κB signaling pathway was assessed in
NRK-52E rat renal tubular epithelial cells, and HK-2 human renal
tubular epithelial cells were used to confirm the findings. In
cells co-treated with LPS and si-MALAT1, cell survival rate
increased and the secretion of inflammatory factors decreased,
compared with the LPS-only induction group. However, this effect
was eliminated by miR-146a inhibitor (Fig. 6A–C). The change in the expression
of miR-146a in the supernatant was analyzed accordingly, and
similar results were observed in the treated groups (Fig. 6D). In addition, western blot
analysis of the expression of p-IκBα and np65 showed that these two
proteins were increased in the LPS+si-MALAT1+miR-146a inhibitor
group, compared with those in cells transfected with LPS+si-MALAT1
(Fig. 6E). These results
indicated that MALAT1 regulated kidney injury through the
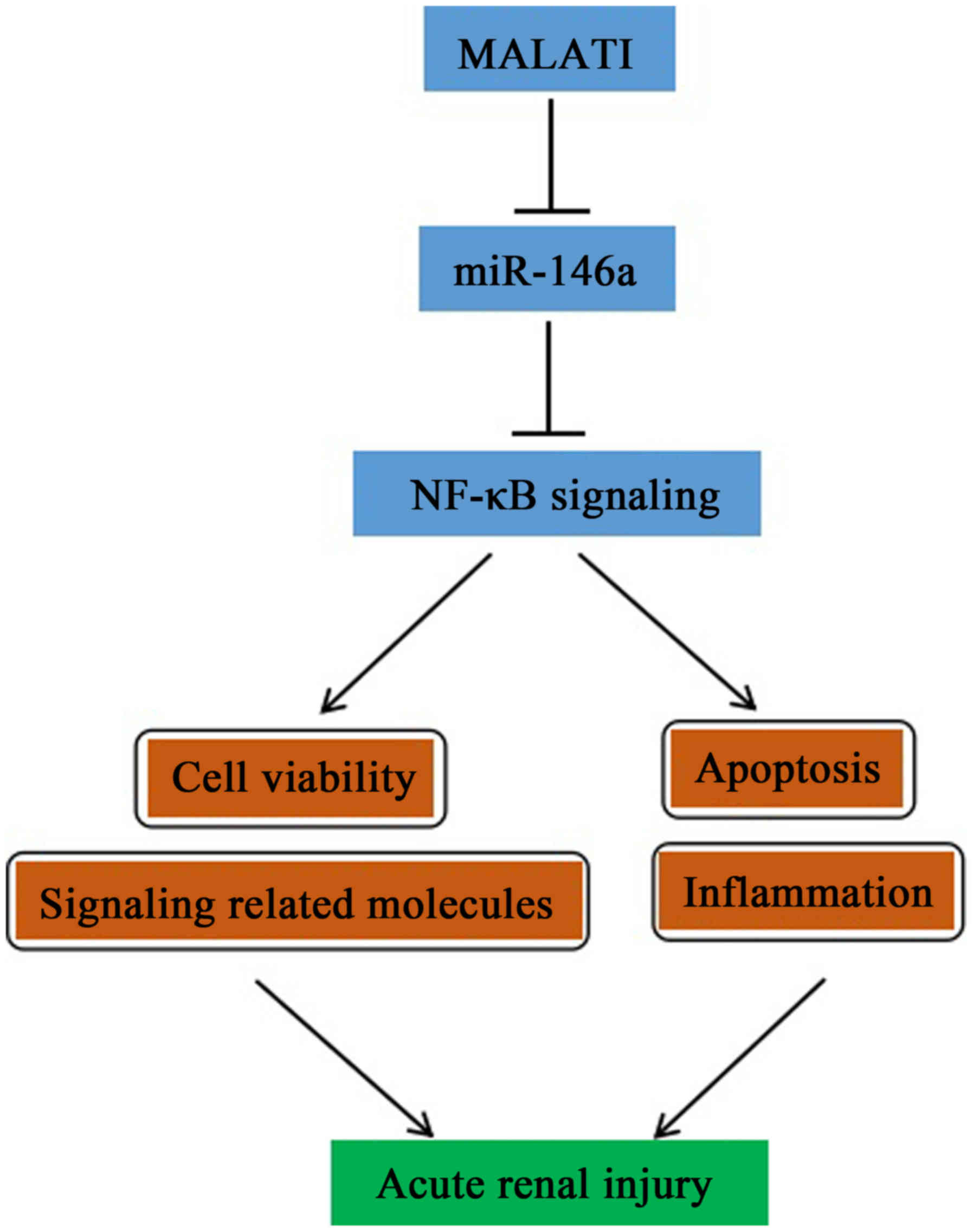
miR-146a/NF-κB signaling pathway in HK-2 cells. A diagram of the
mechanism is shown in Fig. 7.
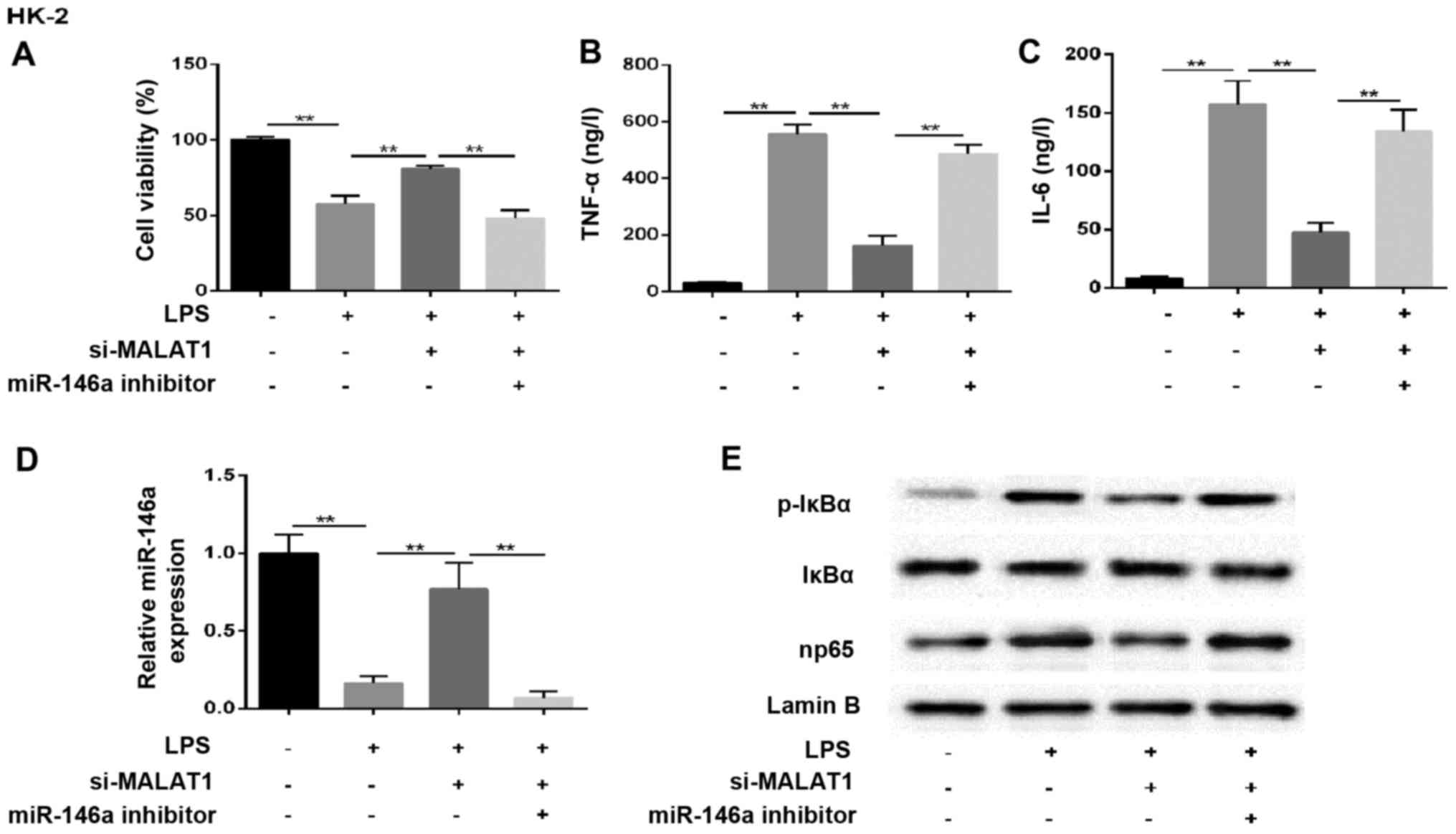 | Figure 6MALAT1 regulates the NF-κB pathway by
affecting the expression of miR-146a. HK-2 cells were randomly
divided into four groups: control, LPS, LPS+si-MALAT1 and
LPS+si-MALAT1+miR-146a inhibitor. (A) Cell viability. Expression of
(B) TNF-α and (C) IL-6 (ng/l). (D) Reverse
transcription-quantitative polymerase chain reaction analysis of
miR-146a. U6 was used as the internal control. (E) Expression of
NF-κB regulatory proteins in HK-2 cells. Lamin B was used as an
internal control. **P<0.01 between groups. All data
are expressed as the mean ± standard deviation of at least three
independent experiments. MALAT1, metastasis-associated lung
adenocarcinoma transcript 1; miR, microRNA; LPS,
lipopolysaccharide; si, small interfering RNA; IL-6, interleukin-6;
TNF-α, tumor necrosis factor-α; NF-κB, nuclear factor-κB; p-IκBα,
phosphorylated inhibitor of NF-κB. |
Discussion
In the present study, it was demonstrated that
MALAT1 was upregulated in damaged tissues and renal tubular
epithelial cells. The inhibition of MALAT1 impaired the malignant
behavior of AKI and attenuated the expression of np65. By contrast,
the expression of miR-146a was downregulated in renal tissue
samples and cell lines. The restoration of miR-146a suppressed the
cell inflammatory response, promoted cell proliferation and reduced
the expression of np65. miR-146a was also found to bind to MALAT1
in a sequence-specific manner, and there was reciprocal repression
between miR-146a and MALAT1, possibly induced by the RISC. NF-κB
was identified as an important downstream regulatory factor of
miR-146a and was involved in the malignant progression of AKI.
NF-κB was also confirmed to promote the adverse effects of AKI by
enhancing the expression of cell inflammatory factors and promoting
apoptosis.
AKI is a complex disorder for which currently there
is no accepted definition; it is typically characterized by kidney
function detriment, resulting in profound effects on patient health
(27). Several factors can cause
AKI, including predominantly acute hypoxia, trauma, and toxic or
septic insults (28). Notably,
LPS also induces the pathogenesis of AKI and leads to kidney
failure and multi-organ damage; however, the pathogenesis of human
AKI is complex and only partially elucidated (29). Therefore, it is important to
examine the potential mechanism and identify effective therapeutic
strategies for LPS-induced AKI.
miRNAs are vital in several biological processes
through mRNA degradation or translational suppression (30). miR-146a is widely expressed in
kidney diseases, and its urinary excretion is specifically
associated with the development of interstitial lesions and
correlated with inflammatory cell infiltration (31). Cheng et al demonstrated
that the anti-inflammatory activity of miR-146a is mediated by the
suppression of pro-inflammatory transcription factor NF-κB
(11). In the present study, it
was found that the levels of miR-146a were markedly decreased in
vitro, accompanied by increased protein levels of np65.
Therefore, miR-146a may be a potential target in the process of
LPS-induced AKI.
There is substantial evidence that lncRNAs are
aberrantly expressed in various diseases, and are important
regulators of physiological and pathological responses. Previous
studies have indicated that they lncRNAs act as miRNA sponges or
miRNA inhibitors (antagomirs), which interact with miRNAs and
regulate the expression of miRNA target genes (32). For example, the overexpression of
miR21 partly abrogated the cancer susceptibility 2-mediated
inhibition of human renal cell carcinoma (RCC) 786-O and A498 cell
proliferation and migration (33). The expression of HOTAIR was found
to be inversely correlated with that of miR-141 in RCC, in which
miR-141 bound to HOTAIR in a sequence-specific manner, and
suppressed the expression and functions of HOTAIR, including
proliferation and invasion (34).
Cardiac hypertrophy-related factor was found to directly bind to
miR-489 and regulate the expression of myeloid differentiation
primary response 88 in cardiac hypertrophy (35). The present study investigated
whether MALAT1 had similar effects on the levels of miR-146a in
LPS-induced AKI. MALAT1 is present at high levels in several human
cell types and is conserved across several mammalian species,
indicating its functional importance (36). In the present study, MALAT1 was
significantly increased in tissues and cell lines, and the
inhibitory effect of MALAT1 on miR-146a levels was observed in
NRK-52E and HK-2 renal tubular epithelial cells. The downregulation
of MALAT1 mitigated cytokine secretion and the immune response,
increased cell viability and increased the expression of miR-146a.
These data indicated that MALAT1 may be directly or indirectly
involved in inflammation and the progression of AKI. It was also
confirmed that the inhibition of miR-146a reversed the promoting
effects of si-MALAT1 on np65 and p-IκBα. The knockdown of MALAT1
promoted the functions of miR-146a. These results revealed that
MALAT1 was involved in LPS-induced AKI through regulating the
expression and function of miR-146a. However, the exact mechanism
by which MALAT1 regulates miR-146a remains to be elucidated and
warrants further investigation.
In conclusion, based on the results of the present
study, a simplified pathway to describe the possible involvement of
MALAT1/miR-146a/NF-κB signaling in LPS-induced AKI was
demonstrated. The findings not only provide novel insight for
clarifying the complex molecular mechanisms of specific miRNAs and
lncRNAs in LPS-induced AKI, but also assist in facilitating the
development of miRNA- and lncRNA-directed therapeutic strategies
for kidney injury.
Acknowledgments
This study was supported by the Hangzhou Health
Science and Technology Plan Project (grant no. 2017A71).
References
|
1
|
Segev G, Langston C, Takada K, Kass PH and
Cowgill LD: Validation of a clinical scoring system for outcome
prediction in dogs with acute kidney injury managed by
hemodialysis. J Vet Intern Med. 30:803–807. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Angeli P, Tonon M, Pilutti C, Morando F
and Piano S: Sepsis-induced acute kidney injury in patients with
cirrhosis. Hepatol Int. 10:115–123. 2016. View Article : Google Scholar
|
|
3
|
Linkermann A: Nonapoptotic cell death in
acute kidney injury and transplantation. Kidney Int. 89:46–57.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pistolesi V, Di Napoli A, Fiaccadori E,
Zeppilli L, Polistena F, Sacco MI, Regolisti G, Tritapepe L,
Pierucci A and Morabito S: Severe acute kidney injury following
cardiac surgery: short-term outcomes in patients undergoing
continuous renal replacement therapy (CRRT). J Nephrol. 29:229–239.
2016. View Article : Google Scholar
|
|
5
|
Chalikias G, Drosos I and Tziakas DN:
Prevention of contrast-induced acute kidney injury: an update.
Cardiovasc Drugs Ther. 30:515–524. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu D, Chen M, Ren X, Ren X and Wu Y:
Leonurine ameliorates LPS-induced acute kidney injury via
suppressing ROS-mediated NF-κB signaling pathway. Fitoterapia.
97:148–155. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kawara M, Matsunaga R, Yamamoto Y, Yoneda
G, Fujino R, Nishi K, Jono H and Saito H: Nephropreventive effect
of shikonin on murine LPS-induced septic acute kidney injury via
Nrf2 activation with antioxidative responses. J Clin Exp Nephrol.
1:192016. View Article : Google Scholar
|
|
8
|
Heemskerk S, Masereeuw R, Russel FG and
Pickkers P: Selective iNOS inhibition for the treatment of
sepsis-induced acute kidney injury. Nat Rev Nephrol. 5:629–640.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao G, Su Z, Song D, Mao Y and Mao X: The
long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced
inflammatory response through its interaction with NF-κB. FEBS
Lett. 590:2884–2895. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Frixa T, Donzelli S and Blandino G:
Oncogenic microRNAs: key players in malignant transformation.
Cancers (Basel). 7:2466–2485. 2015. View Article : Google Scholar
|
|
11
|
Cheng HS, Sivachandran N, Lau A, Boudreau
E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI and Fish JE:
MicroRNA-146 represses endothelial activation by inhibiting
pro-inflammatory pathways. EMBO Mol Med. 5:1017–1034. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taganov KD, Boldin MP, Chang KJ and
Baltimore D: NF-kappaB-dependent induction of microRNA miR-146, an
inhibitor targeted to signaling proteins of innate immune
responses. Proc Natl Acad Sci USA. 103:12481–12486. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao JL, Rao DS, Boldin MP, Taganov KD,
O'Connell RM and Baltimore D: NF-kappaB dysregulation in
microRNA-146a-deficient mice drives the development of myeloid
malignancies. Proc Natl Acad Sci USA. 108:9184–9189. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li K, Ching D, Luk FS and Raffai R:
Abstract 155: Micro-RNA-146a suppression of NF-κB-driven
monocyte/macrophage activation and atherosclerosis is regulated by
cellular ApoE expression. Arterioscler Thromb Vasc Biol.
35:A1552015.
|
|
15
|
Yousefzadeh N, Alipour MR and Soufi FG:
Deregulation of NF-κB-miR-146a negative feedback loop may be
involved in the pathogenesis of diabetic neuropathy. J Physiol
Biochem. 71:51–58. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang YA, Chen X, You ZH, Huang DS and
Chan KC: ILNCSIM: improved lncRNA functional similarity calculation
model. Oncotarget. 7:25902–25914. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gong W, Zheng J, Liu X, Ma J, Liu Y and
Xue Y: Knockdown of NEAT1 restrained the malignant progression of
glioma stem cells by activating microRNA let-7e. Oncotarget.
7:62208–62223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu TM, Palanisamy K, Sun KT, Day YJ, Shu
KH, Wang IK, Shyu WC, Chen P, Chen YL and Li CY: RANTES mediates
kidney ischemia reperfusion injury through a possible role of
HIF-1α and lncRNA PRINS. Sci Rep. 6:184242016. View Article : Google Scholar
|
|
19
|
Shao K, Shi T, Yang Y, Wang X, Xu D and
Zhou P: Highly expressed lncRNA CRNDE promotes cell proliferation
through Wnt/β-catenin signaling in renal cell carcinoma. Tumour
Biol. Oct 6–2016.Epub ahead of print. View Article : Google Scholar
|
|
20
|
Wang L, Cai Y, Zhao X, Jia X, Zhang J, Liu
J, Zhen H, Wang T, Tang X, Liu Y, et al: Down-regulated long
non-coding RNA H19 inhibits carcinogenesis of renal cell carcinoma.
Neoplasma. 62:412–418. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang HM, Yang FQ, Chen SJ, Che J and
Zheng JH: Upregulation of long non-coding RNA MALAT1 correlates
with tumor progression and poor prognosis in clear cell renal cell
carcinoma. Tumour Biol. 36:2947–2955. 2015. View Article : Google Scholar
|
|
22
|
Nair AR, Masson GS, Ebenezer PJ, Del Piero
F and Francis J: Role of TLR4 in lipopolysaccharide-induced acute
kidney injury: protection by blueberry. Free Radic Biol Med.
71:16–25. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shin S, Kim Y, Jeong S, Hong S, Kim I, Lee
W and Choi S: The therapeutic effect of human adult stem cells
derived from adipose tissue in endotoxemic rat model. Int J Med
Sci. 10:8–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qi M, Yin L, Xu L, Tao X, Qi Y, Han X,
Wang C, Xu Y, Sun H, Liu K, et al: Dioscin alleviates
lipopolysaccharide-induced inflammatory kidney injury via the
microRNA let-7i/TLR4/MyD88 signaling pathway. Pharmacol Res.
111:509–522. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hei Z, Zhang A, Wei J, Gan X, Wang Y, Luo
G and Li X: Lipopolysaccharide effects on the proliferation of
NRK52E cells via alternations in gap-junction function. J Trauma
Acute Care Surg. 73:67–72. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−ΔΔC(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
27
|
Schrier RW, Wang W, Poole B and Mitra A:
Acute renal failure: definitions, diagnosis, pathogenesis, and
therapy. J Clin Invest. 114:5–14. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bihorac A, Baslanti TO, Cuenca AG, Hobson
CE, Ang D, Efron PA, Maier RV, Moore FA and Moldawer LL: Acute
kidney injury is associated with early cytokine changes after
trauma. J Trauma Acute Care Surg. 74:1005–1013. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heyman SN, Khamaisi M, Rosen S and
Rosenberger C: In vivo models of acute kidney injury. Drug Discov
Today Dis Models. 7:51–56. 2010. View Article : Google Scholar
|
|
30
|
Kurozumi A, Goto Y, Matsushita R, Fukumoto
I, Kato M, Nishikawa R, Sakamoto S, Enokida H, Nakagawa M, Ichikawa
T, et al: Tumor-suppressive microRNA-223 inhibits cancer cell
migration and invasion by targeting ITGA3/ITGB1 signaling in
prostate cancer. Cancer Sci. 107:84–94. 2016. View Article : Google Scholar
|
|
31
|
Ichii O, Otsuka S, Sasaki N, Namiki Y,
Hashimoto Y and Kon Y: Altered expression of microRNA miR-146a
correlates with the development of chronic renal inflammation.
Kidney Int. 81:280–292. 2012. View Article : Google Scholar
|
|
32
|
Yoon JH, Abdelmohsen K and Gorospe M:
Functional interactions among microRNAs and long noncoding RNAs.
Semin Cell Dev Biol. 34:9–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cao Y, Xu R, Xu X, Zhou Y, Cui L and He X:
Downregulation of lncRNA CASC2 by microRNA-21 increases the
proliferation and migration of renal cell carcinoma cells. Mol Med
Rep. 14:1019–1025. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chiyomaru T, Fukuhara S, Saini S, Majid S,
Deng G, Shahryary V, Chang I, Tanaka Y, Enokida H, Nakagawa M, et
al: Long noncoding RNA HOTAIR is targeted and regulated by
microRNA-141 in renal carcinoma cells. J Biol Chem.
289:12550–12565. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu Q, Han L, Yan W, Ji X, Han R, Yang J,
Yuan J and Ni C: miR-489 inhibits silica-induced pulmonary fibrosis
by targeting MyD88 and Smad3 and is negatively regulated by lncRNA
CHRF. Sci Rep. 6:309212016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gutschner T, Hämmerle M and Diederichs S:
MALAT1 - a paradigm for long noncoding RNA function in cancer. J
Mol Med (Berl). 91:791–801. 2013. View Article : Google Scholar
|