Introduction
Chronic kidney disease (CKD) is a global health
burden that affects >10% of adults worldwide (1). Despite advances in therapeutic
strategies, CKD is considered an independent risk factor for
cardiovascular disease (2–4).
Furthermore, CKD is associated with high morbidity and mortality in
earlier stages (3). For patients
with stage 5 on CKD on dialysis, the mortality rate is 10–100 times
greater than that in an age-matched population with normal renal
function (3). In terms of
economics, CKD is associated with high healthcare cost and loss of
productivity (3,5). Therefore, optimal management of CKD
is necessary for improvement of global public health problems.
Tubulointerstitial fibrosis is a common final
feature of kidney disease, irrespective of glomerular, tubular and
capillary injury (6). The
fibrotic process is characterized by sustained inflammation,
including inflammatory cell infiltration and secretion of
cytokines, excessive extracellular matrix (ECM) accumulation and
imbalance of ECM degradation, leading to organ dysfunction
(7,8). Myofibroblasts are key mediators of
fibrosis that produce and secrete ECM following activation
(6). This fibrotic process is
mediated by transforming growth factor-β (TGF-β) and its downstream
signaling pathways, which have roles in cellular proliferation,
differentiation and growth (9).
Our previous study reported that tamoxifen, a selective estrogen
receptor (ER) modulator, has a protective effect on renal fibrosis
through suppression of TGF-β-induced renal fibroblast activation,
ECM deposition and inflammation via modulation of ERα-dependent
TGF-β/Smad signaling (10).
Therefore, identifying for a novel therapeutic target to reverse
renal fibrosis is a challenge for preventing and treating
progressive CKD.
Valproic acid (VPA) is a branched short-chain fatty
acid that is prescribed for treatment of seizure disorders, mood
disorders and migraine headaches (11). The mechanism of action is
associated with regulation of γ-amino butyrate neurotransmitter
activity. Previously, VPA has been reported to have anticancer
activity via regulation of cell differentiation and apoptosis by
inhibition of histone deacetylase (HDAC) activity, and is
considered a class I HDAC inhibitor (12,13). HDACs function by removing the
acetyl groups from acetylated histones and/or non-histone proteins
to regulate gene transcription and protein functions (14). HDACs are expressed in the
developing kidney, renal tubular cells and renal fibroblasts, and
have been reported to be involved in cellular proliferation,
survival, differentiation and immunological responses. Several
studies have investigated the protective effect of VPA in a renal
injury model, including diabetic nephropathy (15), adriamycin-induced glomerular
sclerosis model (16) and
sepsis-induced kidney injury (17). However, the effect of VPA on
unilateral ureteral obstruction (UUO)-induced fibroblast
activation, inflammation and ECM accumulation, and their mechanisms
remain elusive. Therefore, the current study aimed to investigate
whether inhibition of HDAC1 by VPA has a protective effect on
UUO-induced renal fibrosis via modulation of renal inflammation and
ECM gene transcription.
Materials and methods
Animal experiments
The animal experiment protocol was reviewed and
approved by the Institutional Animal Care and Use Committee of
Chonbuk National University (Jeonju, Korea). Male C57BL/6 mice
(n=60; 7 weeks old; weighing 20–23 g) were purchased from Orient
Bio, Inc. (Seoul, Korea) and maintained in a room under controlled
temperature (23±1°C), humidity, lighting (12 h light/12 h dark
cycle) and with free access to chow and water. For the experiment,
the mice were divided into four groups: i) Sham with vehicle
treatment; ii) UUO with vehicle treatment; iii) sham with VPA
treatment; and iv) UUO with VPA treatment (n=15 each group). VPA
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was dissolved in
PBS. PBS was used as the vehicle. VPA (300 mg/kg) was administered
by intraperitoneal injection once a day for 5 days prior to UUO
surgery and continued for 14 days after UUO surgery.
Renal fibrosis was induced by UUO operation as
described previously (10). In
brief, mice were anesthetized via intraperitoneal injection of
ketamine (100 mg/kg; Huons Co., Ltd., Seoul, Korea) and xylazine
(10 mg/kg; Bayer, Newbury, UK) and placed on temperature-controlled
operating table with body temperature maintained at 37°C. Following
midline incision in the abdomen, the right proximal ureteral was
exposed and ligated at two separated points using 3–0 black silk.
The sham operation was performed using the same method without
ligation of the ureter. At 2 weeks after UUO, the obstructed kidney
was harvested, prepared for histological examination, and stored at
−80°C for western blot analysis and cytokine assays.
Histological examination
The kidneys were fixed via immersion in 4%
paraformaldehyde at 4°C for 24 h, dehydrated by washing in a series
of increasing ethanol concentrations (70, 95 and 100%) for 1 h, and
then embedded in paraffin. The block was cut into 5-μm
sections and stained with 0.5% periodic acid-Schiff stain (PAS) for
15 min and Masson's trichrome (MTC) using biebrich scarlet-acid
fuchsin solution and phosphomolybdic-phosphotungstic acid solution
for 15 min. Immunohistochemical and immunofluorescence staining was
performed as described previously (10). For immunohistochemical staining,
the following primary antibodies were used: goat anti-mouse type I
collagen (1310-01; 1:100; SouthernBiotech, Birmingham, AL, USA),
hamster anti-mouse intercellular adhesion molecule-1 (ICAM-1;
553249; 1:100; BD Biosciences, San Jose, CA, USA) or rabbit
anti-mouse monocyte chemoattractant protein-1 (MCP-1; 70R50662;
1:100; Fitzgerald Industries International, Acton, MA, USA). For
immunofluorescence staining, rabbit anti-fibroblast specific
protein-1 (FSP-1) antibody (ab93283; 1:100; Abcam, Cambridge, UK),
anti-α-smooth muscle actin (α-SMA; BD Biosciences), rat anti-mouse
Ki-67 (14-5698-82; 1:200; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) or rat anti-mouse F4/80 (14-4801-82; 1:200; eBioscience,
San Diego, CA, USA) were used as primary antibodies and then FITC-
or Cy3-labeled as secondary antibodies (Chemicon; Merck & Co.,
Inc., Whitehouse Station, NJ, USA). The nuclear staining was
performed using 300 nM DAPI solution for 3 min (Molecular Probes;
Thermo Fisher Scientific, Inc.). For morphometric analysis, two
observers unaware of the origins of samples used a Zeiss Z1
microscope or Zeiss LSM 510 confocal microscope (Carl Zeiss AG,
Oberkochen, Germany) to evaluate all slides. The tubular injury was
scored at six levels on the basis of the percentage of tubular
dilatation, epithelial desquamation and loss of brush border in 10
randomly chosen, non-overlapping fields at a magnification of ×200
under a light microscope: 0, none; 0.5, <10%; 1, 10 to 25%; 2,
25 to 50%; 3, 50 to 75%; and 4, >75%. The fibrotic areas and
areas positive for type I collagen, ICAM-1 and MCP-1 were measured
in 10 randomly chosen, non-overlapping fields at a magnification of
×200 using ImageJ software (http://rsb.info.nih.gov/ij). The number of Ki-67 and
α-SMA double-positive myofibroblasts and F4/80-positive macrophages
was counted at a magnification of ×400.
Picrosirius red stain
For evaluation of the collagen deposition after
ureteral obstruction, paraffin-embedded tissue sections were
stained with Picrosirius red (10). The Picrosirius red-positive areas
were measured in 10 randomly chosen, non-overlapping fields at a
magnification of ×200 using ImageJ software.
Western blotting
Western blot analysis was performed as described
previously (10). Primary
antibodies α-SMA (A2547; mouse) and vimentin (V6630; mouse)
(1:1000; Sigma-Aldrich; Merck KGaA), ICAM-1 (sc-1511; goat;
1:1,000), fibronectin (sc-6953; goat; 1:500), and Smad7 (sc-11392;
rabbit; 1:1,000) (Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
type I collagen (1310-01; goat; 1:1,000; SouthernBiotech),
phospho-Smad2 (3101; rabbit), phospho-Smad3 (9520; rabbit),
acetyl-histone H3 (14932; rabbit), and histone H3 (9715; rabbit)
(1:1,000; Cell Signaling Technology Inc., Danvers, MA, USA), and
Smad2/3 (07-408; rabbit; 1:1,000; EMD Millipore, Billerica, MA,
USA) were used. GAPDH (AP0063; rabbit; 1:2,000; Bioworld
Technology, Inc., St. Louis Park, MN, USA) was used as an internal
control. All signals were analyzed by densitometric scanning
(LAS-3000; FujiFilm, Tokyo, Japan).
Measurement of renal MCP-1 and TGF-β1
levels
The MCP-1 (MJE00) and TGF-β1 (MB100B) levels were
determined by ELISA kits (R&D Systems, Inc., Minneapolis, MN,
USA) according to the manufacturer's instructions.
Cell culture experiment
In vitro experiments were performed using rat
renal fibroblast cell line (NRK-49F; American Type Culture
Collection, Manassas, VA, USA). NRK-49F cells were cultured in
Dulbecco's modified Eagle's medium with 4 mM L-glutamine adjusted
to contain 1.5 g/l sodium bicarbonate and 4.5 g/l glucose
supplemented with 5% (vol/vol) heat-inactivated fetal bovine serum
and antibiotics (100 U/ml penicillin G and 100 μg/ml
streptomycin) at 37°C with 5% CO2 in 95% air. To
investigate the effect of VPA on myofibroblast activation and ECM
protein expression, subconfluent NRK-49F cells were incubated with
VPA (0.1, 1 and 2.5 mM) for 30 min and then stimulated with TGF-β1
(2 ng/ml; Sigma-Aldrich; Merck KGaA) for 24 h. To examine the
effects of VPA on HDAC1-mediated ECM protein expression, HDAC1
small interfering RNA (siRNA; 25 pmol; sc-270070; Santa Cruz
Biotechnology, Inc.) or non-target control siRNA (25 pmol;
sc-37007; Santa Cruz Biotechnology, Inc.) were tranfected using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) in subconfluent NRK49F cells. Following knockdown
of HDAC1 for 24 h, cells were incubated with VPA (2.5 mM) for 30
min and then stimulated with TGF-β1 (2 ng/ml) for 24 h.
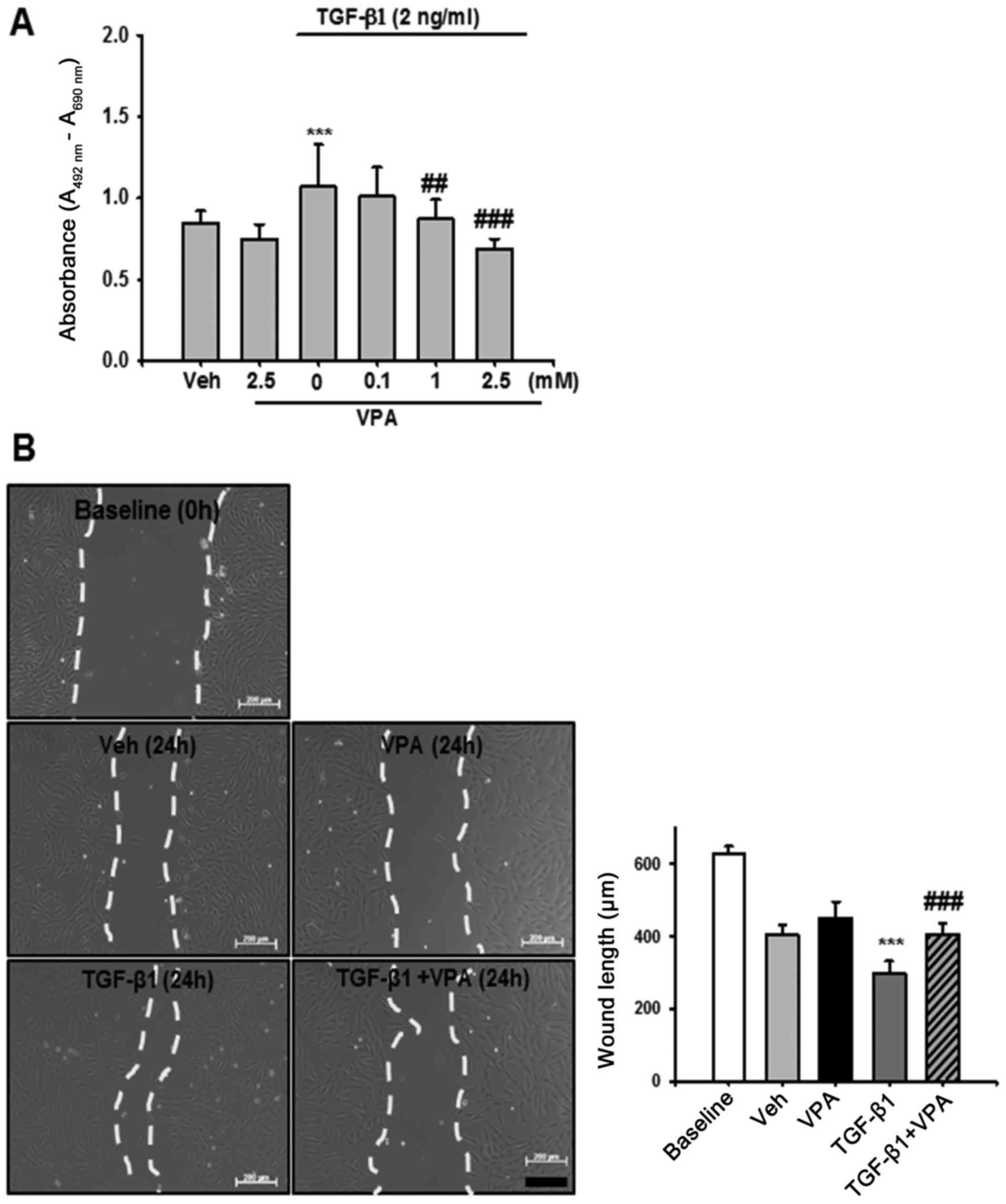
Cell proliferation assay
After 24 h treatment with VPA (0.1, 1 and 2.5 mM)
and TGF-β1 (2 ng/ml), proliferation of NRK-49F cells was determined
by a colorimetric assay (Cell Proliferation kit II; Roche
Diagnostics GmbH, Mannheim, Germany) according to the
manufacturer's protocol. All experimental values were determined
from triplicate wells.
Wound healing assay
Subconfluent NRK-49F cells were cultured in 6-well
dishes. Prior to treatment with VPA and TGF-β1, dishes were
scratched using a sterile 200 μl pipette tip, causing three
separate wounds. The cells were incubated with VPA (2.5 mM) for 30
min and then stimulated with TGF-β1 (2 ng/ml) for 24 h. Wound
lengths were measured using the ImageJ program. At 0 h after
scratching, this wound length was used as the control.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assay was performed using the acetyl-histone H3
immunoprecipitation kit (17-245; EMD Millipore) according to the
manufacturer's protocol. In brief, subconfluent NRK-49F cells were
incubated with VPA (2.5 mM) for 30 min and then stimulated with
TGF-β1 (2 ng/ml) for 1 h. For cross-linking histones to DNA, cells
were treated with 1% formaldehyde for 10 min at 37°C. After washing
and harvesting the cells, genomic DNA fragments were obtained by
sonicating cell lysates. The cross-linked histone-DNA complexes
were immunoprecipitated using antibody against acetyl-histone H3
and normal rabbit IgG as a negative control. Following
precipitation with salmon sperm DNA/protein A agarose slurry, the
histone-DNA complexes were eluted and reversed by heating at 65°C
for 4 h. The DNA was recovered by phenol/chloroform extraction. The
inputs consisted of 5% chromatin before immunoprecipitation. The
ChIP-enriched DNA and input DNA were analyzed by quantitative real
time polymerase chain reaction (qRT-PCR) using the following
primers: Fibronectin 1 (Fn1) promoter forward,
5′-CGTACCCTGGAAAGTC-3′ and reverse, 5′-CTAAGCCTACCTAACACCGA-3′;
type I collagen α1 (Col1α1) promoter forward,
5′-GCAGACTCTTCTAGCCGCTG-3′ and reverse, 5′-CTATGTCGGCAGACAGGCTC-3′.
qRT-PCR was performed using a SYBR-Green PCR Master Mix (Applied
Biosystems, Carlsbad, CA, USA) on a rotor-gene Q 2plex system
(Qiagen, Hilden, Germany) to measure ChIP-enriched DNA and input
DNA expression. The PCR program was as follows: 2 min at 50°C, 10
min at 95°C, then 95°C for 15 sec, and 60°C for 1 min for 40
cycles. The relative expression of specific promotor sequences in
immunoprecipitated DNA was determined using the ΔΔCt method. The
relative enrichment of promoter DNA was normalized to the
input.
Statistical analysis
Data are expressed as mean ± standard deviation.
Multiple comparisons were examined for significant differences
using analysis of variance, followed by individual comparison with
the Tukey post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
VPA decreases UUO-induced renal tubular
injury and fibrosis
VPA has been reported as a class I HDAC inhibitor,
therefore the effect of VPA on histone H3 acetylation (H3Ac)
following UUO surgery. In sham-operated kidneys, VPA treatment
increased H3Ac at lysine 9 and 14 compared with the vehicle-treated
group. UUO kidneys from VPA-treated mice exhibited an increase in
H3Ac compared to vehicle-treated UUO mice. These data suggest that
VPA treatment significantly increases H3Ac in both sham and UUO
kidneys (Fig. 1A).
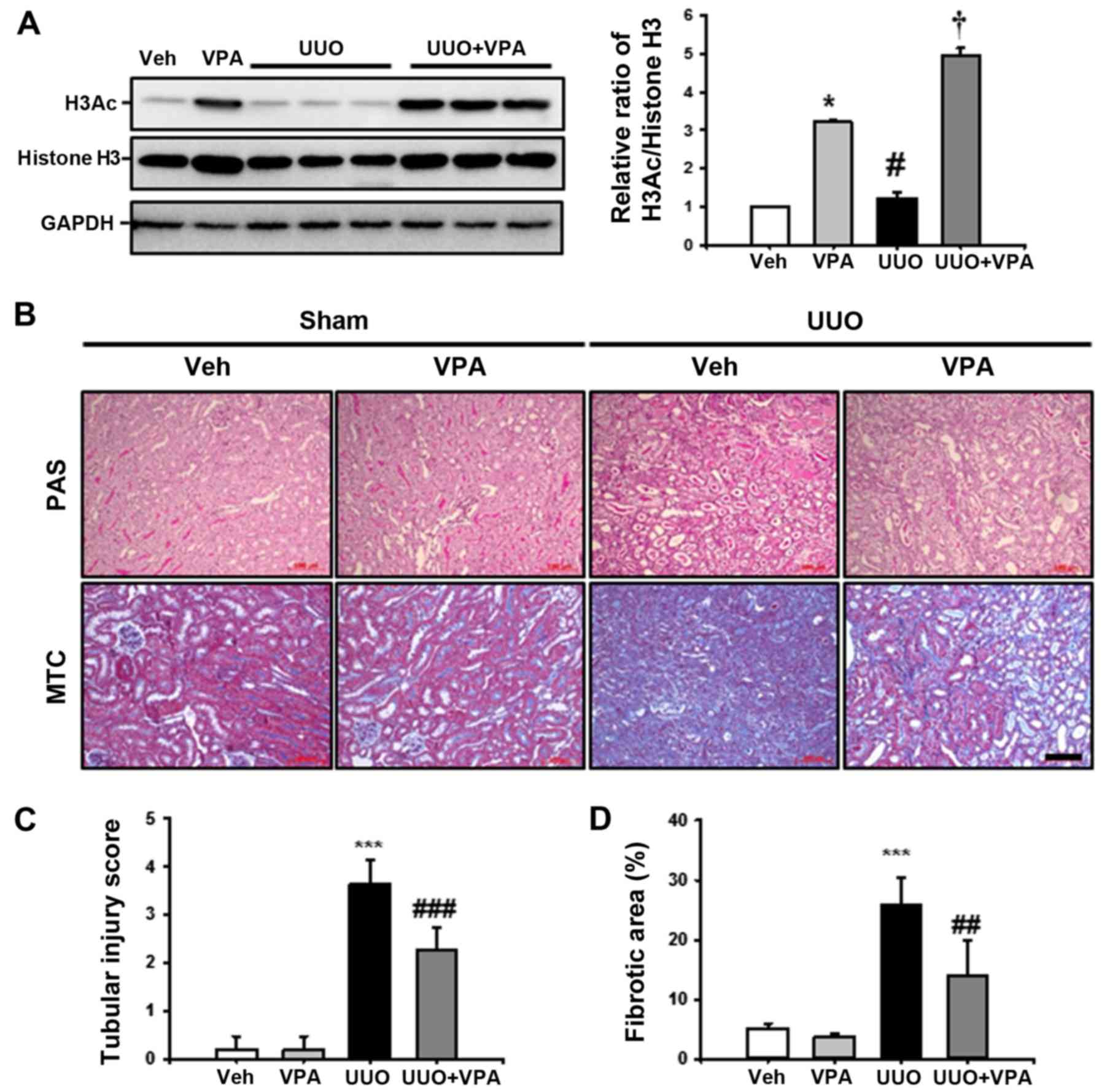 | Figure 1Effect of VPA on UUO-induced renal
tubular injury and fibrosis. (A) Representative western blot
analysis of H3Ac at lysine 9 and 14 from sham- and UUO-operated
mice treated with Veh or VPA. Data from densitometric analyses are
presented as the relative ratio of each protein to histone H3. The
relative ratio measured in the kidneys from sham-operated mice
treated with Veh is arbitrarily presented as 1. Data are expressed
as the mean ± SD of 3 independent experiments.
*P<0.05 vs. Veh; #P<0.05 vs. VPA;
†P<0.05 vs. UUO. (B) Representative PAS and
MTC-stained sections of kidneys from sham- and UUO-operated mice
treated with Veh or VPA. Scale bar, 100 μm. Bar graphs show
the semi-quantitative scoring of (C) tubular injury stained by PAS
and (D) the area fractions (%) of tubulointerstitial fibrosis
stained by MTC in the sham and UUO-operated kidneys. Randomly
chosen, non-overlapping fields (n=10) at a magnification of ×200
were quantified (n=15/each group). Data are expressed as the mean ±
SD of 3 independent experiments. ***P<0.001 vs. Veh
or VPA; ##P<0.01 vs. UUO; ###P<0.001
vs. UUO. Veh, vehicle; VPA, valproic acid; Sham, sham-operated
mice; UUO, unilateral ureteral obstruction-operated mice; H3Ac,
acetylated histone 3; PAS, periodic acid-Schiff stain; MTC,
Masson's trichrome; SD, standard deviation. |
To investigate the effect of VPA on UUO-induced
renal tubulointerstitial fibrosis, kidney sections were examined
following PAS and MTC staining. After 2 weeks of ureteral
obstruction, UUO kidneys from vehicle-treated mice exhibited an
increase in tubular dilatation, inflammatory cells and
fibroblast-like cell infiltration, and tubulointerstitial fibrosis
compared with sham-operated kidneys treated with vehicle or VPA.
The VPA-treated UUO kidneys exhibited relatively preserved normal
tubular structures and significantly reduced tubular injury,
tubulointerstitial inflammation and fibrosis (Fig. 1B–D). These data suggest that VPA
ameliorates UUO-induced tubular injury and fibrosis in
vivo.
VPA reduces UUO-induced renal
interstitial fibroblast activation and proliferation
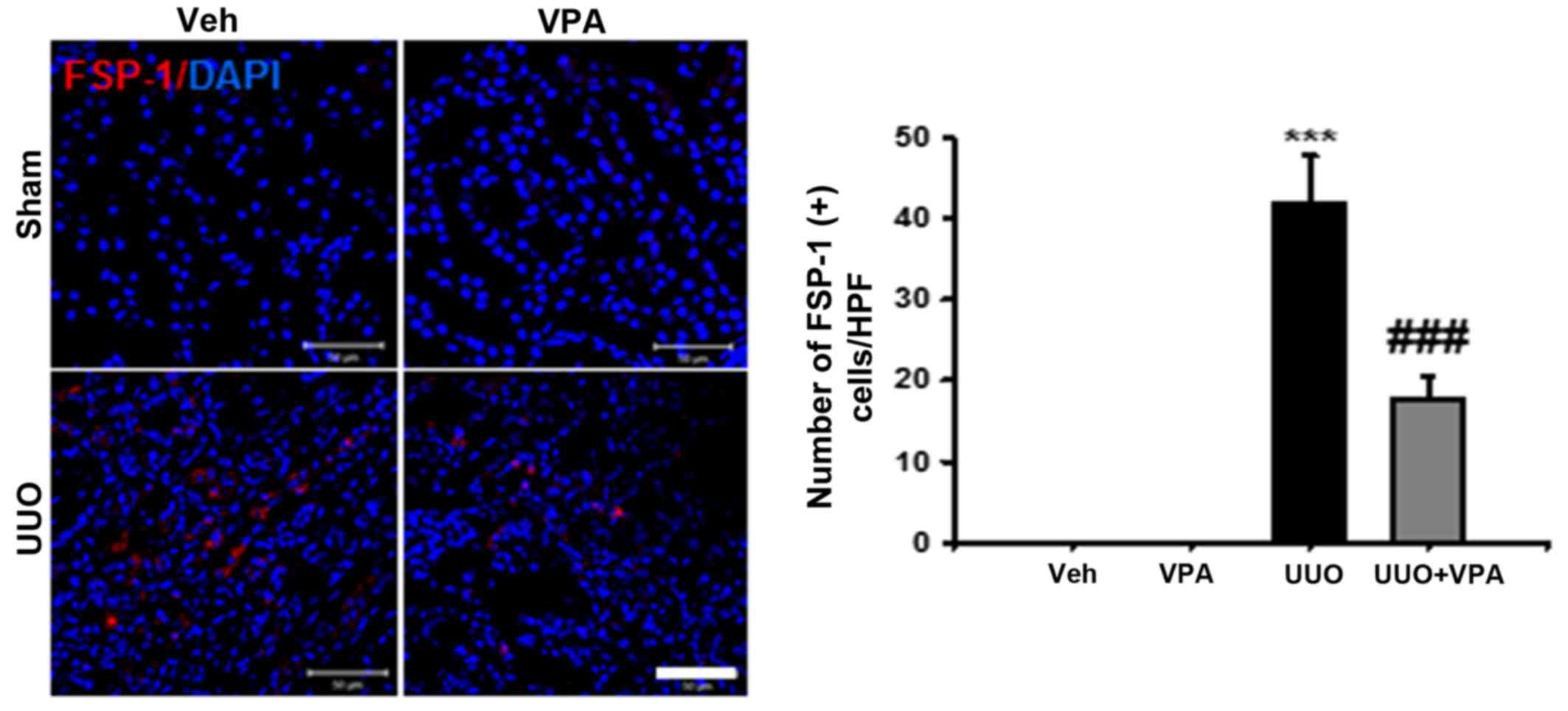
Renal interstitial fibroblast activation is one
mechanism of renal fibrogenesis (18). Therefore, we evaluated renal
interstitial fibroblast expression after ureteral obstruction using
anti-FSP-1 antibody. At 2 weeks after ureteral obstruction, the
number of FSP-1-positive fibroblasts was increased in the
tubulointerstitial area compared with sham-operated kidneys. VPA
treatment significantly decreased the number of FSP-1-positive
fibroblasts in UUO kidneys (Fig.
2).
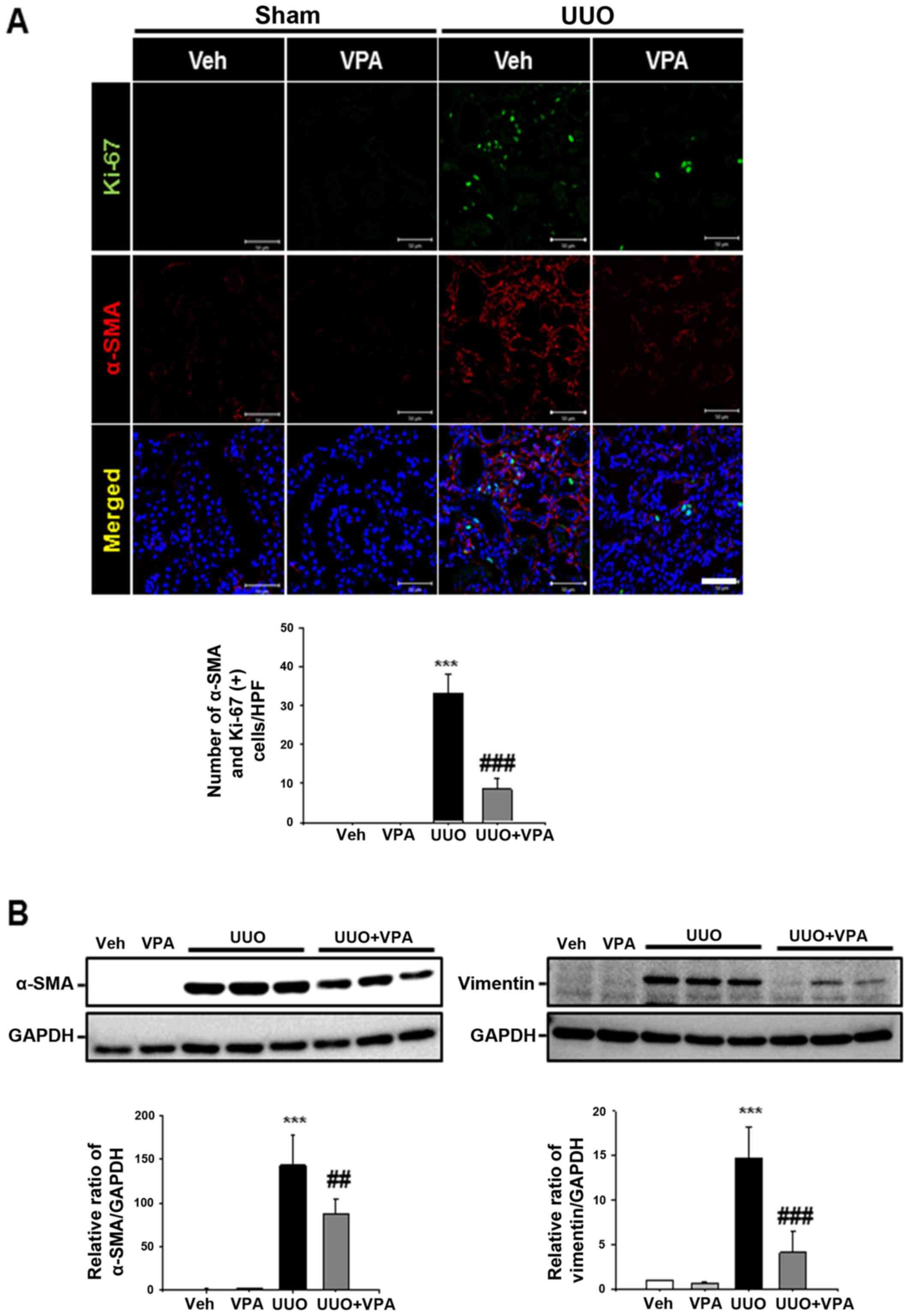
To address the effect of VPA on UUO-induced
myofibroblast activation and proliferation, double-positive α-SMA
and Ki-67 myofibroblast expression was evaluated following ureteral
obstruction. The number of α-SMA- and Ki-67-double positive
myofibroblasts was increased in the tubulointerstitial area
compared with sham-operated kidneys. VPA treatment mitigated the
myofibroblast proliferation and infiltration in UUO kidneys
(Fig. 3A). Protein expression of
α-SMA and vimentin was significantly increased in UUO kidneys from
vehicle-treated mice compared with sham-operated kidneys. UUO
kidneys from VPA-treated mice exhibited decreased α-SMA and
vimentin expression of ~39.8 and 67.5%, respectively, compared with
the UUO kidneys from vehicle-treated mice (Fig. 3B). These data suggest that VPA
treatment mitigates UUO-induced renal myofibroblast activation and
proliferation.
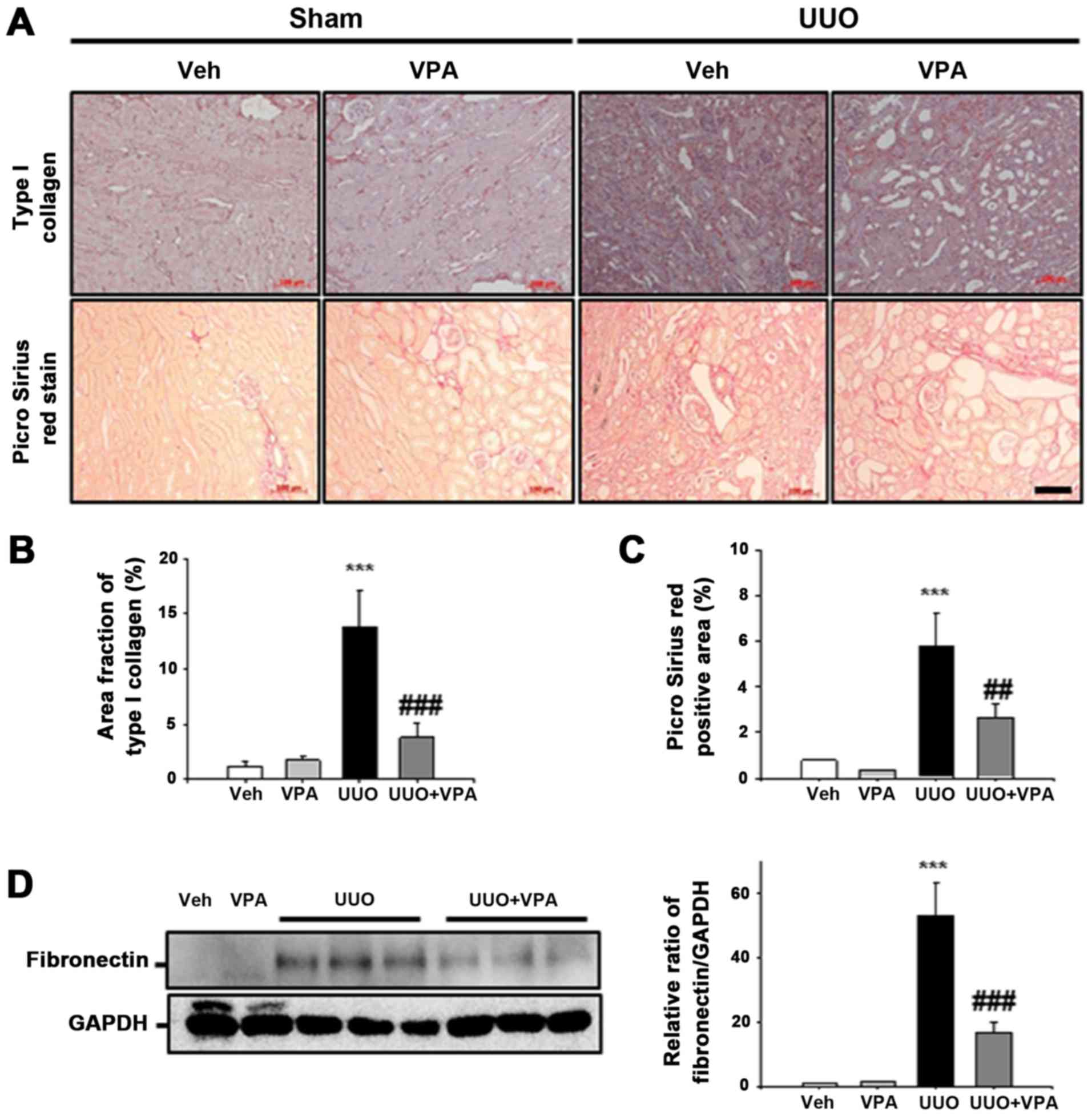
VPA decreases UUO-induced fibronectin and
type I collagen expression
ECM deposition is an important process during renal
fibrogenesis (18). Whether VPA
regulates ECM deposition following ureteral obstruction was
evaluated in the current study. In immunohistochemistry analysis,
UUO kidneys exhibited an increase in type I collagen expression in
the tubulointerstitial area compared with sham-operated kidneys.
Collagen fibril deposition was also evaluated by Picrosirius red
stain. Deposition of collagen fibrils was significantly increased
in the tubulointerstitial areas of UUO kidneys from vehicle-treated
mice. VPA treatment significantly decreased the UUO-induced
increase in type I collagen expression. UUO kidneys from
VPA-treated mice exhibited a decrease in Picrosirius red-positive
areas of ~55.1% compared with UUO kidneys from vehicle-treated mice
(Fig. 4A–C). In western blot
analysis, fibronectin expression following ureteral obstruction was
significantly increased compared to that in sham-operated kidneys.
However, VPA treatment significantly decreased the UUO-induced
increase of fibronectin expression by ~68.4% (Fig. 4D). These data suggest that VPA
ameliorates the UUO-induced increase in ECM deposition.
VPA ameliorates UUO-induced renal
tubulointerstitial inflammation
Inflammation promotes progressive renal fibrosis
(19). Therefore, the effects of
VPA on macrophage infiltration, cell adhesion molecules and
chemoattractant factor expression were examined following ureteral
obstruction. The number of F4/80-positive macrophages was
significantly increased in the tubulointerstitial areas in the UUO
kidney compared with sham-operated kidneys (Fig. 5A). VPA treatment significantly
reduced F4/80-positive macrophage infiltration in UUO kidneys
compared with vehicle-treated UUO kidneys.
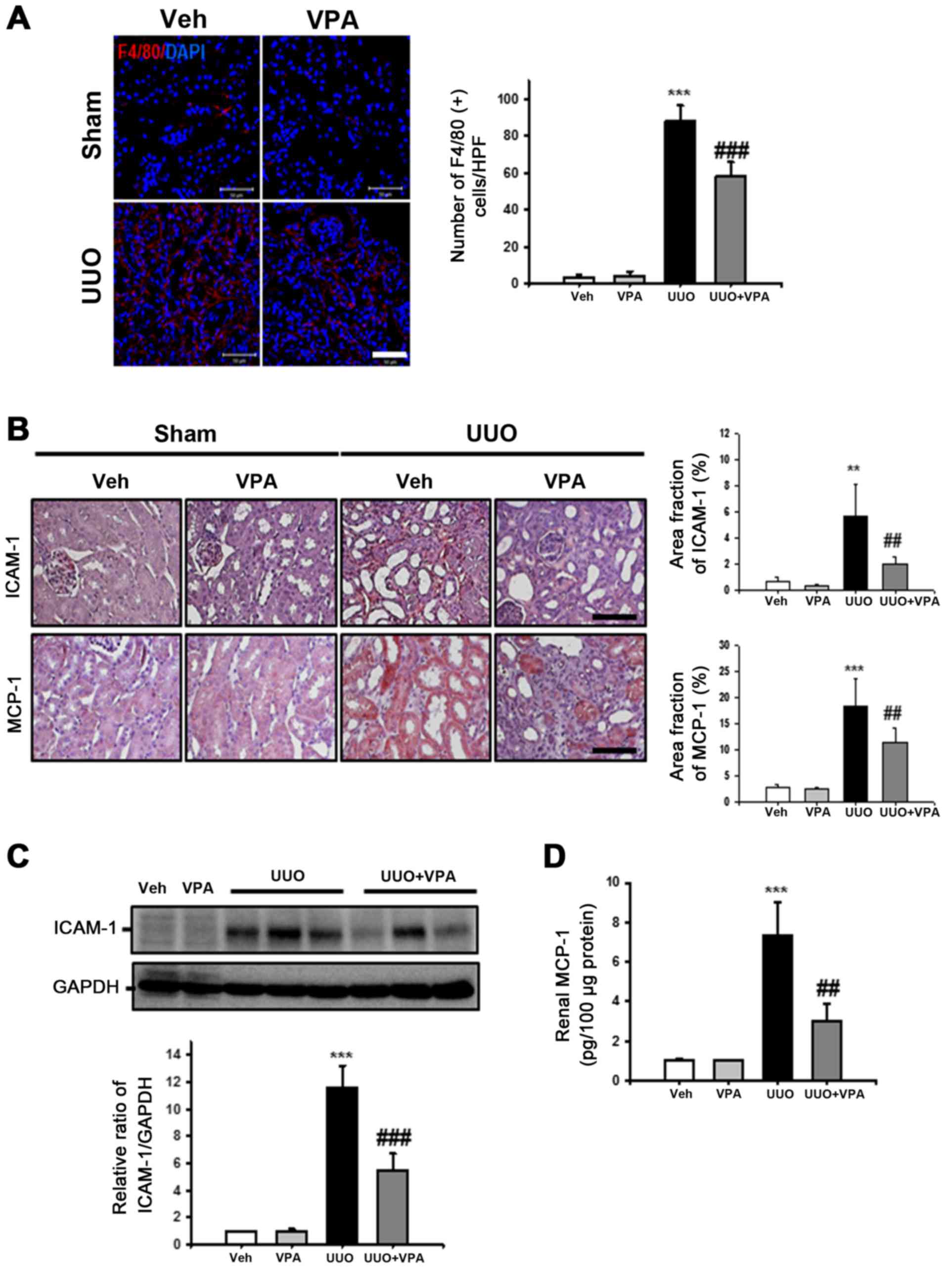 | Figure 5Effect of VPA on UUO-induced renal
inflammation. (A) Representative immunofluorescence staining of
F4/80-positive macrophages from kidneys of sham- and UUO-operated
mice treated with Veh or VPA. The nucleus was stained by DAPI. The
bar graph presents the number of F4/80-positive macrophages from 10
randomly chosen, non-overlapping fields at a magnification of ×400
(n=15/each groups). Bar, 50 μm. Data are expressed as the
mean ± SD. (B) Representative immunohistochemistry of ICAM-1 and
MCP-1 staining from kidneys of sham- and UUO-operated mice treated
with Veh or VPA. Bar, 100 μm. The bar graph shows the area
fractions (%) of ICAM-1 and MCP-1 from ten randomly chosen,
non-overlapping fields at magnification of ×200 (n=15/each groups).
Data are expressed as the mean ± SD. (C) Representative western
blot analysis of ICAM-1 from sham- and UUO-operated mice treated
with Veh or VPA. Data from densitometric analyses are presented as
the relative ratio of each protein to GAPDH. The relative ratio
measured in the kidneys from sham-operated mice treated with Veh is
arbitrarily presented as 1. Data are expressed as mean ± SD of 3
independent experiments. (D) Renal MCP-1 levels from sham- and
UUO-operated mice treated with Veh or VPA were measured by ELISA.
The levels were normalized to 100 μg of kidney protein. Data
are expressed as the mean ± SD of 3 independent experiments.
***P<0.001 vs. Veh or VPA; **P<0.01 vs.
Veh or VPA; ##P<0.01 vs. UUO;
###P<0.001 vs. UUO. Sham, sham-operated mice; UUO,
unilateral ureteral obstruction-operated mice; Veh, vehicle; VPA,
valproic acid; ICAM-1, intercellular adhesion molecule-1; MCP-1,
macrophage chemoattractant protein-1; SD, standard deviation. |
For evaluation of cell adhesion molecules and
chemoattractant factor expression following ureteral obstruction,
ICAM-1 and MCP-1 expression was assessed in UUO kidneys via
immunohistochemistry. The expression of ICAM-1 was significantly
increased in the interstitial area from UUO kidneys. VPA treatment
significantly decreased the expression of ICAM-1 compared with
vehicle-treated UUO kidneys (Fig.
5B). ICAM-1 protein expression in was also examined in kidney
lysates via western blot analysis. Similar to the findings from
immunohistochemistry analysis, there was a significant decrease in
the UUO-induced increase in ICAM-1 expression by ~52.1% following
VPA treatment (Fig. 5C). The
expression of MCP-1 was significantly increased in the dilated
tubules of UUO kidney compared with sham-operated kidney. VPA
treatment significantly reduced the UUO-induced increase in MCP-1
expression (Fig. 5B). Tissue
MCP-1 protein levels in the UUO kidney were also evaluated by
ELISA. The level of MCP-1 was significantly increased in UUO
kidneys (7.35±1.67 pg/100 μg protein) compared with the
level in vehicle-treated kidneys (1±0.12 pg/100 μg protein)
or VPA-treated sham mice (1.04±0.02 pg/100 μg protein;
Fig. 5D). VPA treatment reduced
the UUO-induced increase in MCP-1 level (3.0±0.88 pg/100 μg
protein). These data suggest that VPA reduces UUO-induced renal
inflammation by regulation of macrophage infiltration and
proinflammatory cytokine expression.
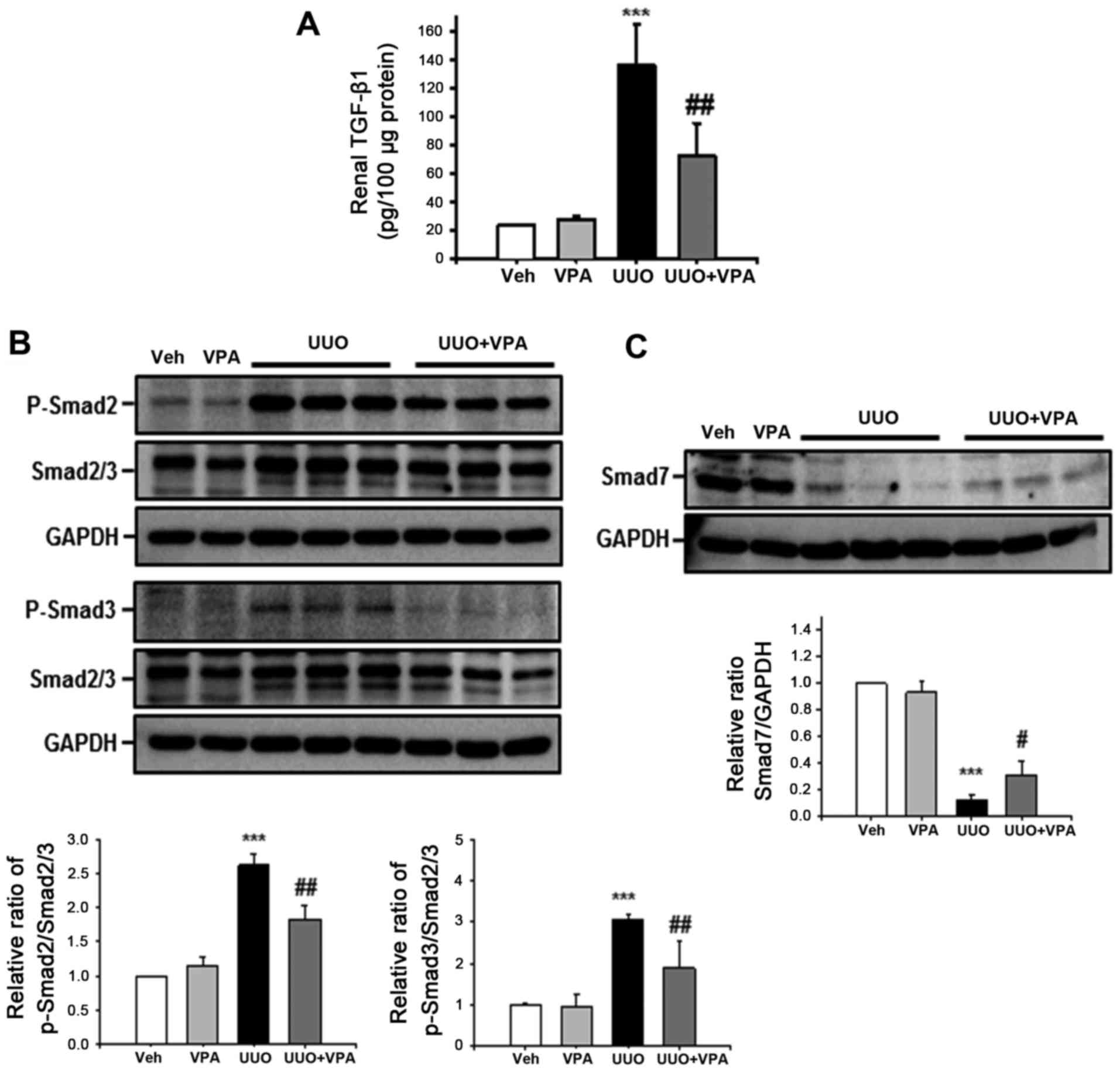
VPA regulates UUO-induced activation of
TGF-β1/Smad signaling
Among pro-fibrogenic cytokines, TGF-β1 is the major
cytokine that drives renal fibrosis (20). To address the effect of VPA on the
UUO-induced increase in TGF-β1 expression, tissue TGF-β1 level was
evaluated using ELISA. The tissue TGF-β1 level following ureteral
obstruction was significantly increased (135.4±29.2 pg/100
μg protein) compared with vehicle- (23.5±1.11 pg/100
μg protein) and VPA- (28.2±1.6 pg/100 μg protein)
treated sham mice (Fig. 6A).
However, VPA treatment significantly reduced the UUO-induced
increase in TGF-β1 level (72.7±22.5 pg/100 μg protein).
Phosphorylation of Smad2 and Smad3 mediates downstream TGF-β1
signaling, and protein binding to Smad-binding elements in DNA
promotes profibrotic gene transcription (21). The UUO kidney from vehicle-treated
mice had increased phosphorylation of Smad2 and Smad3 compared with
sham-operated kidney (Fig. 6B).
VPA-treated mice exhibited a significant reduction in the
UUO-induced increase in Smad2 and Smad3 phosphorylation compared
with vehicle-treated mice. By contrast, expression of inhibitory
Smad7 was significantly decreased following ureteral obstruction
(Fig. 6C). Smad7 expression was
increased in UUO kidneys from VPA-treated mice compared with the
UUO kidneys from vehicle-treated mice. These data suggest that VPA
treatment regulates the UUO-induced TGF-β1/Smad signaling pathway
in vivo.
VPA decreases TGF-β1-induced viability
and migration of NRK-49F cells
To determine the protective mechanism of VPA in
UUO-induced renal fibrosis, the effect of TGF-β1 on renal
interstitial fibroblast viability and migration was evaluated in
vitro using NRK-49F cells. Treatment with TGF-β1 increased
renal fibroblast viability by ~1.3-fold compared with
vehicle-treated cells. VPA treatment decreased TGF-β1-induced cell
proliferation in a dose-dependent manner (Fig. 7A). Cell migration was also
evaluated using a wound healing assay. TGF-β1-treated NRK-49F cells
exhibited increased migration compared with baseline or
vehicle-treated cells. VPA treatment significantly decreased the
TGF-β1-induced increase in cell migration (Fig. 7B). These data suggest that VPA
treatment regulates TGF-β1-induced renal fibroblast viability and
migration in vitro.
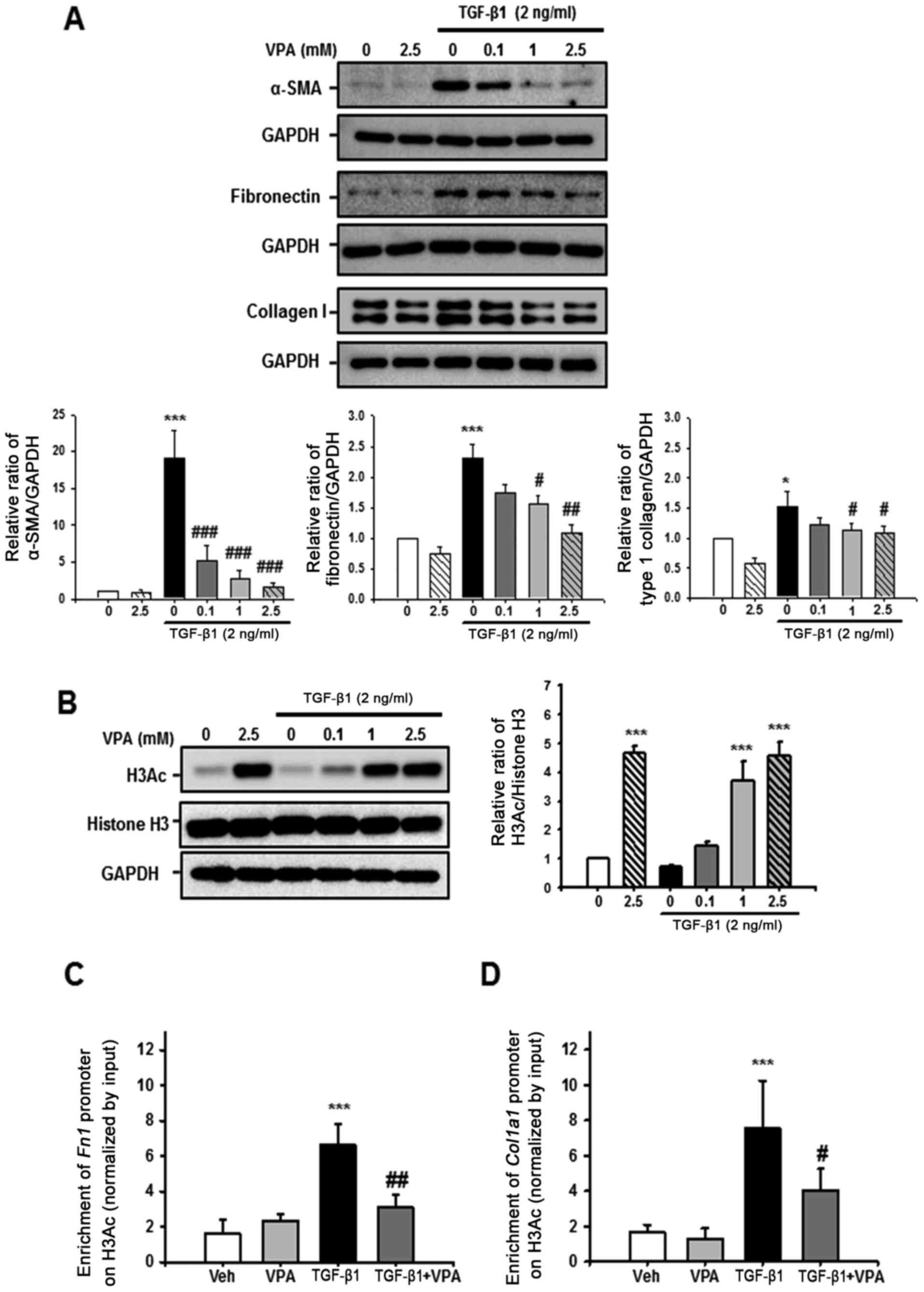
VPA suppresses TGF-β1-induced fibronectin
and Col1α1 promoter enrichment at a H3Ac site in NRK-49F cells
Whether VPA regulates TGF-β1-induced myofibroblast
activation and ECM production was evaluated in NRK-49F cells. After
24-h stimulation with TGF-β1, α-SMA, fibronectin and type I
collagen expression were significantly increased compared with the
control. However, VPA caused a dose-dependent decrease in α-SMA,
fibronectin and type I collagen expression in TGF-β1-induced cells
(Fig. 8A).
Histone acetylation has an important role in
chromatin remodeling and regulation of gene transcription. The
effect of VPA on H3Ac and Fn1 and Col1α1 promoter
enrichment at acetyl-histone H3 was also evaluated. Treatment with
VPA for 1 h significantly increased H3Ac at lysine 9 and 14 in a
dose-dependent manner in NRK-49F cells however, TGF-β1 did not have
an effect on histone H3 acetylation (Fig. 8B). Subsequently, ChIP assay was
performed to address whether VPA-induced H3Ac was associated with
ECM promoter enrichment in NRK-49F cells. Following H3Ac
immunoprecipitation, Fn1 and Col1α1 promoter
enrichment on H3Ac was increased by treatment with TGF-β1 compared
with vehicle or VPA-treated NRK-49F cells. VPA significantly
decreased the TGF-β1-induced increase in Fn1 and
Col1α1 promoter enrichment (Fig. 8B and C). These data suggest that
VPA promotes H3 acetylation at lysine 9 and 14, and regulates
Fn1 and Col1α1 promoter enrichment at the
acetyl-histone H3. Thus, VPA decreases the TGF-β1-induced increase
of Fn1 and Col1α1 gene expression via histone
modification at ECM promoters.
Discussion
Following tissue injury, inflammation and fibrosis
are precisely regulated by the immune system for tissue repair.
However, in most cases, the inflammatory response and myofibroblast
activation stimulate an inappropriate pro-fibrotic response
(22). Therefore, regulation of
the inflammatory response and myofibroblast activation are
potential targets for anti-fibrotic therapy. The results of the
current study suggest that HDAC1 inhibition by VPA suppresses
UUO-induced inflammation, myofibroblast activation and
proliferation, and ECM deposition. VPA also inhibits TGF-β1-induced
renal fibroblast proliferation and migration, and ECM gene promoter
enrichment at acetylated histone H3 in NRK-49F cells. These results
suggest that VPA has therapeutic potential for preventing renal
fibrosis by regulation of myofibroblast activation and ECM
production.
Fibrosis is the final common pathological feature in
chronic inflammatory disease and is characterized by accumulation
of ECM proteins, including fibronectin and collagens (23). There are various factors that
contribute to the development of fibrotic disease. Among them,
activation of myofibroblasts is a hallmark of fibrogenesis
(23). Although the origin of
myofibroblasts is controversial, activation of local fibroblasts,
phenotypic changes from epithelial, endothelial, pericyte and bone
marrow-derived fibroblasts have been suggested as origins of
myofibroblasts in fibrotic tissues (24). The results of the current study
demonstrated that VPA treatment inhibited the UUO-induced
myofibroblast infiltration and proliferation based on double
immunofluorescence staining for α-SMA and Ki-67. Furthermore, VPA
decreased the TGF-β1-induced increase in fibroblast activation,
proliferation, and migration in NRK-49F cells. Therefore, the in
vivo and in vitro data suggest that regulation of
myofibroblast activation is an important therapeutic target for
progressive renal fibrosis.
In the UUO model, there is robust infiltration of
inflammatory cells around tubulointerstitial areas. Cell adhesion
molecules, chemokines and their receptors and cytokines regulate
the inflammatory cells transmigration via the vascular endothelium
and differentiation into macrophages and dendritic cells (25). The in vivo data of the
current study indicated that there was increased infiltration of
F4/80(+) macrophages accompanied by increased expression of ICAM-1
in the interstitial area, and increased expression of MCP-1 at the
dilated tubules following ureteral obstruction. VPA treatment
ameliorated UUO-induced renal inflammation via regulation of
inflammatory cell infiltration and proinflammatory cytokine
expression. Therefore, regulation of the renal inflammatory process
is one suggested protective mechanism for VPA in UUO-induced renal
injury.
TGF-β1 is an important mediator of renal fibrosis,
and the downstream Smad signaling pathway is crucial in regulating
TGF-β1-induced fibrosis (7,26).
Following activation of TGF-β1 signaling, the Smad effector
proteins have distinct roles in renal fibrosis (21). The in vivo data in the
present study demonstrate that VPA decreases the UUO-induced
increase in Smad2 and Smad3 phosphorylation, which has a
profibrotic role in kidney injury. By contrast, Smad7, an
inhibitory Smad, is decreased following ureteral obstruction. VPA
restores the expression of Smad7, which reduces the activation of
TGF-β1/Smad signaling. These results suggest that VPA could
potentially alter TGF-β1/Smad signaling pathway in renal
fibrosis.
The final fibrotic process involves the accumulation
and dysregulated remodeling of ECM proteins (27). According to Seet et al
(28), VPA suppresses type I
collagen synthesis via the TGF-β1 regulatory pathway in
conjunctival fibroblasts and mouse glaucoma model. The results of
the current study also demonstrate that VPA decreases the
TGF-β1-induced increase in ECM, fibronectin and type I collagen
expression in NRK-49F cells and UUO kidneys. In contrast to the
study of Seet et al (28),
ChIP results from the current study indicated that VPA increases
H3Ac and regulates enrichment of Fn1 and Col1α1
promoter at acetylated histone H3. Thus, VPA may inhibit
TGF-β1-induced increases in ECM gene expression via increased H3
acetylation at lysine 9 and 14.
Tissue inhibitor of metalloproteinases have
important roles in inhibiting ECM proteolysis and controlling ECM
turnover, which is dependent on metalloproteinase inhibition
(29). However, in the current
study, the focus was on H3Ac and regulation of transcription from
ECM promoters via histone acetylation. This is a limitation of the
present study and further studies are required to determine the
effect of VPA on regulation of metalloproteinase activities in
kidney fibrosis. Another limitation is the known toxicity of VPA
when used in the treatment of patients with epilepsy, including
side-effects such as cerebral edema (30) and hyperammonemic encephalopathy
(31), hepatotoxicity (32) and electrolyte imbalances (33). These toxicities are usually
associated with overdose of VPA. Therefore, caution would be
required if using VPA for the treatment of renal fibrosis.
In conclusion, the findings of the current study
indicate that VPA may have a protective effect against UUO-induced
tubulointerstitial fibrosis via regulation of inflammation, renal
fibroblast activation and TGF-β1/Smad signaling. In addition, the
data suggest that VPA decreased UUO-induced ECM production via
regulation of chromatin remodeling and Fn1 and Col1α1
promoter enrichment at the H3Ac sites. Therefore, targeting HDAC1
inhibition in renal fibrosis may be a novel therapeutic approach
for preventing and treating CKD.
Acknowledgments
We thank Kieu Thi Thu Trang for the excellent
technical assistance. This study was supported by the National
Research Foundation of Korea (NRF) funded by the Korean government
(NRF-2015R1D1A3A03015653, to K.P.K and NRF-2014R1A1A4A01003832, to
S.K.P) and by research funds of Chonbuk National University in 2014
(to K.P.K).
References
|
1
|
Eckardt KU, Coresh J, Devuyst O, Johnson
RJ, Köttgen A, Levey AS and Levin A: Evolving importance of kidney
disease: From subspecialty to global health burden. Lancet.
382:158–169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sharif MU, Elsayed ME and Stack AG: The
global nephrology workforce: Emerging threats and potential
solutions. Clin Kidney J. 9:11–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Couser WG, Remuzzi G, Mendis S and Tonelli
M: The contribution of chronic kidney disease to the global burden
of major noncommunicable diseases. Kidney Int. 80:1258–1270. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
No authors listed. The global issue of
kidney disease. Lancet. 382:1012013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Braun L, Sood V, Hogue S, Lieberman B and
Copley-Merriman C: High burden and unmet patient needs in chronic
kidney disease. Int J Nephrol Renovasc Dis. 5:151–163. 2012.
|
|
6
|
Liu Y: Cellular and molecular mechanisms
of renal fibrosis. Nat Rev Nephrol. 7:684–696. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He W and Dai C: Key Fibrogenic Signaling.
Curr Pathobiol Rep. 3:183–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xia J, He LQ and Su X: Interventional
mechanisms of herbs or herbal extracts on renal interstitial
fibrosis. J Integr Med. 14:165–173. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morikawa M, Derynck R and Miyazono K:
TGF-β and the TGF-β family: Context-dependent roles in cell and
tissue physiology. Cold Spring Harb Perspect Biol. 8:a0218732016.
View Article : Google Scholar
|
|
10
|
Kim D, Lee AS, Jung YJ, Yang KH, Lee S,
Park SK, Kim W and Kang KP: Tamoxifen ameliorates renal
tubulointerstitial fibrosis by modulation of estrogen receptor
α-mediated transforming growth factor-β1/Smad signaling pathway.
Nephrol Dial Transplant. 29:2043–2053. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chateauvieux S, Morceau F, Dicato M and
Diederich M: Molecular and therapeutic potential and toxicity of
valproic acid. J Biomed Biotechnol. 479364:2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Blaheta RA, Michaelis M, Driever PH and
Cinatl J Jr: Evolving anticancer drug valproic acid: Insights into
the mechanism and clinical studies. Med Res Rev. 25:383–397. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Göttlicher M, Minucci S, Zhu P, Krämer OH,
Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG,
et al: Valproic acid defines a novel class of HDAC inhibitors
inducing differentiation of transformed cells. EMBO J.
20:6969–6978. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu N and Zhuang S: Treatment of chronic
kidney diseases with histone deacetylase inhibitors. Front Physiol.
6:1212015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Khan S, Jena G and Tikoo K: Sodium
valproate ameliorates diabetes-induced fibrosis and renal damage by
the inhibition of histone deacetylases in diabetic rat. Exp Mol
Pathol. 98:230–239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Van Beneden K, Geers C, Pauwels M,
Mannaerts I, Verbeelen D, van Grunsven LA and Van den Branden C:
Valproic acid attenuates proteinuria and kidney injury. J Am Soc
Nephrol. 22:1863–1875. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zheng Q, Liu W, Liu Z, Zhao H, Han X and
Zhao M: Valproic acid protects septic mice from renal injury by
reducing the inflammatory response. J Surg Res. 192:163–169. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chevalier RL, Forbes MS and Thornhill BA:
Ureteral obstruction as a model of renal interstitial fibrosis and
obstructive nephropathy. Kidney Int. 75:1145–1152. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
Inflammatory processes in renal fibrosis. Nat Rev Nephrol.
10:493–503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kovacs EJ and DiPietro LA: Fibrogenic
cytokines and connective tissue production. FASEB J. 8:854–861.
1994.PubMed/NCBI
|
|
21
|
Meng XM, Nikolic-Paterson DJ and Lan HY:
TGF-β: The master regulator of fibrosis. Nat Rev Nephrol.
12:325–338. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lupher ML Jr and Gallatin WM: Regulation
of fibrosis by the immune system. Adv Immunol. 89:245–288. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wynn TA and Ramalingam TR: Mechanisms of
fibrosis: Therapeutic translation for fibrotic disease. Nat Med.
18:1028–1040. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Grande MT and López-Novoa JM: Fibroblast
activation and myofibroblast generation in obstructive nephropathy.
Nat Rev Nephrol. 5:319–328. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Alikhan MA and Ricardo SD: Mononuclear
phagocyte system in kidney disease and repair. Nephrology
(Carlton). 18:81–91. 2013. View Article : Google Scholar
|
|
26
|
Boor P, Ostendorf T and Floege J: Renal
fibrosis: Novel insights into mechanisms and therapeutic targets.
Nat Rev Nephrol. 6:643–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Genovese F, Manresa AA, Leeming DJ,
Karsdal MA and Boor P: The extracellular matrix in the kidney: A
source of novel non-invasive biomarkers of kidney fibrosis.
Fibrogenesis Tissue Repair. 7:42014. View Article : Google Scholar
|
|
28
|
Seet LF, Toh LZ, Finger SN, Chu SW,
Stefanovic B and Wong TT: Valproic acid suppresses collagen by
selective regulation of Smads in conjunctival fibrosis. J Mol Med
(Berl). 94:321–334. 2016. View Article : Google Scholar
|
|
29
|
Arpino V, Brock M and Gill SE: The role of
TIMPs in regulation of extracellular matrix proteolysis. Matrix
Biol. 44–46. 247–254. 2015.
|
|
30
|
Dupuis RE, Lichtman SN and Pollack GM:
Acute valproic acid overdose. Clinical course and pharmacokinetic
disposition of valproic acid and metabolites. Drug Saf. 5:65–71.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dealberto MJ: Valproate-induced
hyperammonaemic encephalopathy: Review of 14 cases in the
psychiatric setting. Int Clin Psychopharmacol. 22:330–337. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bryant AE III and Dreifuss FE: Valproic
acid hepatic fatalities. III. U.S. experience since 1986.
Neurology. 46:465–469. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zaki EL and Springate JE: Renal injury
from valproic acid: Case report and literature review. Pediatr
Neurol. 27:318–319. 2002. View Article : Google Scholar : PubMed/NCBI
|