Introduction
Chronic glucocorticoid (GC) therapy is widely used
in clinical medicine as an effective way in treating skin disease,
inflammatory/autoimmune diseases, including rheumatoid arthritis
and chronic obstructive pulmonary disease, and preventing organ
transplantation rejection (1).
However, long-term use of GCs causes various side-effects, and the
most common one being GC-induced osteoporosis (GIO) (2). Approximately half of patients with
rheumatoid arthritis receiving chronic GCs therapy experience an
osteoporotic fracture; base-line data from randomized clinical
trials reported the prevalence of vertebral fractures is ~30%
(3). It was also reported that
long-term intake of GCs leads to bone infarction and osteonecrosis
of the femoral head, causing a significant public health issue
(4). Bone loss in GIO is a
biphasic process, bone resorption significantly increases within
the first 3 months, peaks at 6 months, followed by a slower annual
loss of ~3% for the duration of GC administration (5). This is caused by the accelerated
lifespan of osteoclasts in the early phase of bone resorption.
However, in the long term, osteoclastic activity is usually
inhibited by GCs (6). By
contrast, osteoblasts (OBs) are directly and consistently modulated
by GCs, whereby the production of new OBs precursors is decreased
and the mature, matrix-secreting OBs experience premature
apoptosis. Therefore, although both bone resorption and formation
are suppressed by chronic GCs therapy, bone formation is suppressed
more than bone resorption (7–9).
These histological characteristics are quite different from those
in other forms of osteoporosis in which osteoclasts have an
important role. Thus, more attention should be paid to OBs when
investigating GIO.
Autophagy is a lysosomal degradation pathway that is
essential for cell growth, survival, differentiation, development
and homeostasis. It is a highly regulated process that mainly
involves the formation of autophagosomes, which will fuse with the
lysosomes and form autolysosomes. Subsequently, degradation occurs
and the amino acids or other small molecules are delivered to the
cytoplasm for energy production or recycling (10). GC therapy leads to accumulation of
damaged proteins, oxygen radicals and degraded nucleic acids
(11). Autophagy is the only
intracellular degradative mechanism known to remove dysfunctional
organelles and/or oxidized proteins, and it is also activated as an
attempt to survive under stress (12,13). Despite the ability in which
autophagy acts as a survival strategy to preserve cell viability,
it may also lead to a programmed cell death process, because of the
excessive activation of this self-degrading system (14). Therefore, autophagy has been
reported as a 'double-edged sword' (15). It was previously reported that
autophagy was induced prior to apoptosis during lymphoid cell death
caused by dexamethasone (Dex) (16). Another study reported that GC
treatment could induce autophagy in mouse osteocytes. Inhibition of
autophagy led to augmentation of the effects of GCs on osteocyte
viability (11). The autophagic
process is also induced in OBs during mineralization, and autophagy
deficiency reduced mineralization capacity, which was proved by
knockdown of autophagy-essential genes and OB-specific
autophagy-deficient mice (17).
However, whether autophagy is involved in the effect
of GCs on OBs is unknown. We hypothesize that autophagy is one of
the mechanisms by which OBs respond to GC treatment. In this study,
it was investigated whether GC treatment of OBs induced autophagy.
In addition, as the association between autophagy and apoptosis was
unclear, as previously described, whether suppression of autophagy
altered the negative effects of exogenous GCs on OBs was also
examined. The present observations may offer a novel finding that
autophagy is a major mechanism by which OBs protect themselves
against the detrimental effect of GCs.
Materials and methods
Reagents and antibodies
The enhanced green fluorescent
protein-microtubule-associated protein 1 light chain 3β (eGFP-LC3B)
plasmid was provided by Dr Shaobo Xiao (Huazhong Agricultural
University, Wuhan, China) (18).
The antibodies used were: anti-microtubule-associated protein 1
light chain 3β (anti-LC3B; cat. no. L7543; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany); anti-Beclin 1 (cat. no. PD017; MBL
International Co., Woburn, MA, USA); anti-BAX apoptosis regulator
(Bax; cat. no. 50599-2-Ig; Proteintech, Chicago, IL, USA);
anti-apoptosis regulator Bcl-2 (Bcl-2; cat. no. PRS3335;
Sigma-Aldrich; Merck KGaA); anti-β-actin (cat. no. AA128; Beyotime
Institute of Biotechnology, Haimen, China). Dex (cat. no. D4902),
3-methyladenine (3-MA; cat. no. M9281) and rapamycin (cat. no.
R8781) were all obtained from Sigma-Aldrich (Merck KGaA). Dex,
rapamycin and 3-MA were all dissolved in dimethyl sulfoxide (cat.
no. D4540; Sigma-Aldrich; Merck KGaA). The stock solution
concentrations were 500 μmol/l for rapamycin, 100 mmol/l for
3-MA. The working solution concentrations were 500 nmol/l for
rapamycin and 400 μmol/l for 3-MA.
Cell culture
The MC3T3-E1 cell line was obtained from the Cell
Resource Center, Peking Union Medical College (Beijing, China; the
headquarters of National Infrastructure of Cell Line Resource). The
cell line was confirmed to have no mycoplasma contamination by
polymerase chain reaction (PCR). Its species origin was confirmed
by PCR. The identity of the cell line was authenticated with Short
Tandem Repeat profiling (CODIS; FBI). All the results can be viewed
at www.cellresource.cn. Cells were cultured
in α-minimum essential medium (cat. no. 12571-063) with 10% (v/v)
fetal calf serum (cat. no. 16140-071; both from Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), and antibiotics (100
IU/ml penicillin G and 100 Ag/ml streptomycin) in a humidified
incubator with 5% CO2 at 37°C. A total of
5×105 cells/well were cultured in 6-well plates and then
exposed to Dex of various doses at 10−8, 10−6
and 10−4 mol/l for 24, 48, 72 and 96 h. Autophagy
inhibitor 3-MA was added to cells 1.5 h before incubated with Dex
as negative control group. Autophagy agonist rapamycin was added 6
h ahead of each time-point as positive control group. Culture
medium was replaced with the same concentration GC at interval of
48 h.
Confocal microscopy
MC3T3-E1 cells were cultured on glass coverslips in
35-mm plates. For the detection of autophagosomes, eGFP-LC3B
plasmid (2.5 μg/well) was transfected into 1×105
cells/well using Lipofectamine® LTX and Plus reagent
(cat. no. 15338-100; Invitrogen; Thermo Fisher Scientific, Inc.)
according to manufacturer's instructions. Cells were then treated
with 10−8, 10−6 and 10−4 mol/l Dex
in complete culture medium for 24, 48, 72 and 96 h. Rapamycin (500
nmol/l) was added 6 h before each time-point as positive control,
and 250 μmol/l 3-MA was added 1.5 h before Dex as negative
control. Following fixation with phosphate-buffered saline (PBS)
containing 4% (w/v) paraformaldehyde at 37°C for 30 min and
staining with 0.1 μg/ml DAPI at 37°C for 1 min, glass
coverslips were observed under a Zeiss LSM 510 confocal
fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany). Cells
containing three or more GFP-LC3 puncta were defined as
autophagy-positive. The percentage of cells with characteristic
punctate GFP-LC3 staining relative to all GFP-positive cells was
calculated as previously described (19). Three random fields were counted,
and three independent experiments were performed. Analysis of
images was performed using ImageJ (National Institutes of Health,
Bethesda, MD, USA).
Western blot analysis
MC3T3-E1 cells were seeded into 6-well plates and
incubated at 37°C for 18 h. Cells were subsequently treated with
Dex at different concentrations for 24–96 h as previously
described. At each time-point, cells were washed with PBS twice and
incubated in a cold buffer containing 0.5% Triton X-100, 150 mmol/l
NaCl, 12.5 mmol/l β-glycerolphosphate, 1.5 mmol/l MgCl2,
2 mmol/l EDTA, 10 mmol/l NaF, 1 mmol/l
Na3VO4, 2 mmol/l dithiothreitol and protease
inhibitor cocktail (cat. no. P8340; Sigma-Aldrich). Centrifugation
was performed to obtain cell extracts at 10,000 × g at 4°C for 10
min, and protein concentration was determined using BCA Protein
Assay kit (cat. no. pp0101; Biomed, Beijing, China). Protein
samples were mixed with 30 μl 2X SDS-PAGE sample buffer,
boiled for 10 min, separated on 12% SDS-PAGE gel (30
μg/well), and electro-transferred onto a polyvinylidene
difluoride membrane. Blots were blocked for 1 h with Tris-buffered
saline (50 mmol/l Tris-HCl, pH 7.4, 150 mmol/l NaCl)-0.05% Tween-20
(TBST) supplemented with 5% non-fat milk at room temperature and
incubated overnight at 4°C with primary antibody [Bax (dilution,
1:1,000), Bcl-2 (dilution, 1:1,000), Beclin 1 (dilution, 1:1,000),
LC3 (dilution, 1:1,500) and β-actin (dilution, 1:2,000)]. Membranes
were than washed three times in 1X TBST buffer, and subsequently
incubated with horseradish peroxidase-conjugated secondary
antibodies at room temperature (cat. nos. ZF0136 and ZF0312;
OriGene Technologies, Inc., Beijing, China) for 45 min and washed
again prior to detection of signal with Western Lighting Plus-ECL
reagents (cat. no. P1010; Applygen Technologies, Inc., Beijing,
China). Densitometry analysis was performed using ImageJ 2×
(National Institutes of Health, Bethesda, MD, USA).
Measurement of apoptosis by flow
cytometry
MC3T3-E1 cells (1×106 per well) were
plated in a 6-well plate at 37°C. Following treatment with Dex,
3-MA and rapamycin, apoptosis incidence in OBs was detected by
using the Annexin V-fluorescein isothiocyanate (FITC) staining kit
(BD Pharmingen, Franklin Lakes, NJ, USA). Propidium iodide (PI)
staining also was performed at the same time using the same kit
(20). Apoptotic cells, including
those staining positive for Annexin V-FITC and negative for PI and
those were double positive, were counted and represented as a
percentage of the total cell count. Flow cytometry analysis was
performed by BD FACSDiva software (v 6.1.3; BD Biosciences, San
Jose, CA, USA).
Gene expression analysis
Total RNA was isolated using TRIzol reagent (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Total RNAs (1 μg) were then
reverse-transcribed using RayScript cDNA synthesis kit (GK8030;
Generay Biotech Co., Ltd., Shanghai, China), cDNAs were subjected
to reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) analysis in an ABI PRISM 7500 system (Applied Biosystems;
Thermo Fisher Scientific, Inc.). Reactions were performed in a 20
μl final volume using 8 μl diluted cDNAs and
AceQ™qPCR Probe Master Mix (Q112-02; Vazyme Biotech Co., Ltd.,
Nanjing, China). Amplification conditions were: 95°C for 5 min,
followed by 40 cycles of 95°C for 10 sec and 60°C for 34 sec.
Primer sequences were as follows: Runt-related transcription factor
2 (Runx2), 5′-CAG CAG CAG CAA CAA CAG-3′ (forward) and
5′-TGG TCC GCG ATG ATCTC-3′ (reverse), ACA GCA GCA GCA GCA GCA GCA
(probe); osteocalcin (Ocn), 5′-GAG CTT AAC CCT GCT TGTG-3′
(forward) and 5′-CTG CTG TGA CAT CCA TAC TTG-3′ (reverse), ATC AGA
CCA GTA TGG CTT GAA GAC CGC (probe); α-1 type 1 collagen
(Colla1), 5′-CTG GCA AGA ATG GAG ATG ATG-3′ (forward) and
5′-TCG GTG TCC CTT CAT TCC-3′ (reverse), TGT TCC AGG CAA TCC ACG
AGC ACC (probe); GAPDH, 5′-GCC TTC CGT GTT CCT ACC-3′
(forward) and 5′-CCT CAG TGT AGC CCA AGATG-3′ (reverse), TGC CTG
CTT GAC CAC CTT CTT GATGT (probe). Each gene was normalized to
GAPDH by relative quantification and the cycle quantification
(2-ΔΔCq) method was used to calculate relative gene
expression levels (21).
Statistical analysis
All data were expressed as the mean ± standard
deviation of at least three independent experiments. Differences
among the three groups were analyzed by one-way analysis of
variance followed by the least significant difference test or the
Student-Newman-Keuls test using the statistics package SPSS 19.0
(IBM Corp., Armonk, NY, USA). Other comparisons were performed
using the Student's t-test except for percentage comparison for
which the Kruskal-Wallis test was used. P<0.05 was considered to
indicate a statistically significant difference.
Results
Dex induces autophagy in a dose-dependent
manner in MC3T3-E1 cells
To determine whether GCs regulate autophagy, the
impact of Dex on formation of autophagosomes in MC3T3-E1 cells was
examined. MC3T3-E1 cells were transfected with an
eGFP-LC3B-expressing plasmid (22). At 24 h post-transfection, cells
were fixed, and examined for evidence of autophagosome formation by
confocal fluorescence microscopy. eGFP-LC3 fluorescence microscopy
is a well-accepted method for monitoring autophagosome formation
(23). As presented in Fig. 1A and B, MC3T3 cells had very low
basal autophagic activity, with <2% of cells expressed punctate
eGFP-LC3. However, treatment with 10−8, 10−6
and 10−4 mol/l Dex stimulated autophagy, leading to
eGFP-LC3 puncta formation in ~50% of cells. A clear redistribution
of eGFP-LC3B punctate was observed, indicating the formation of
autophagosomes (Fig. 1A).
Semi-quantitative analysis as indicated that the increase in
puncta-positive cells was dose-dependent, and rapamycin could
increase the number of positive cells, while 3-MA effectively
reduced it (Fig. 1B).
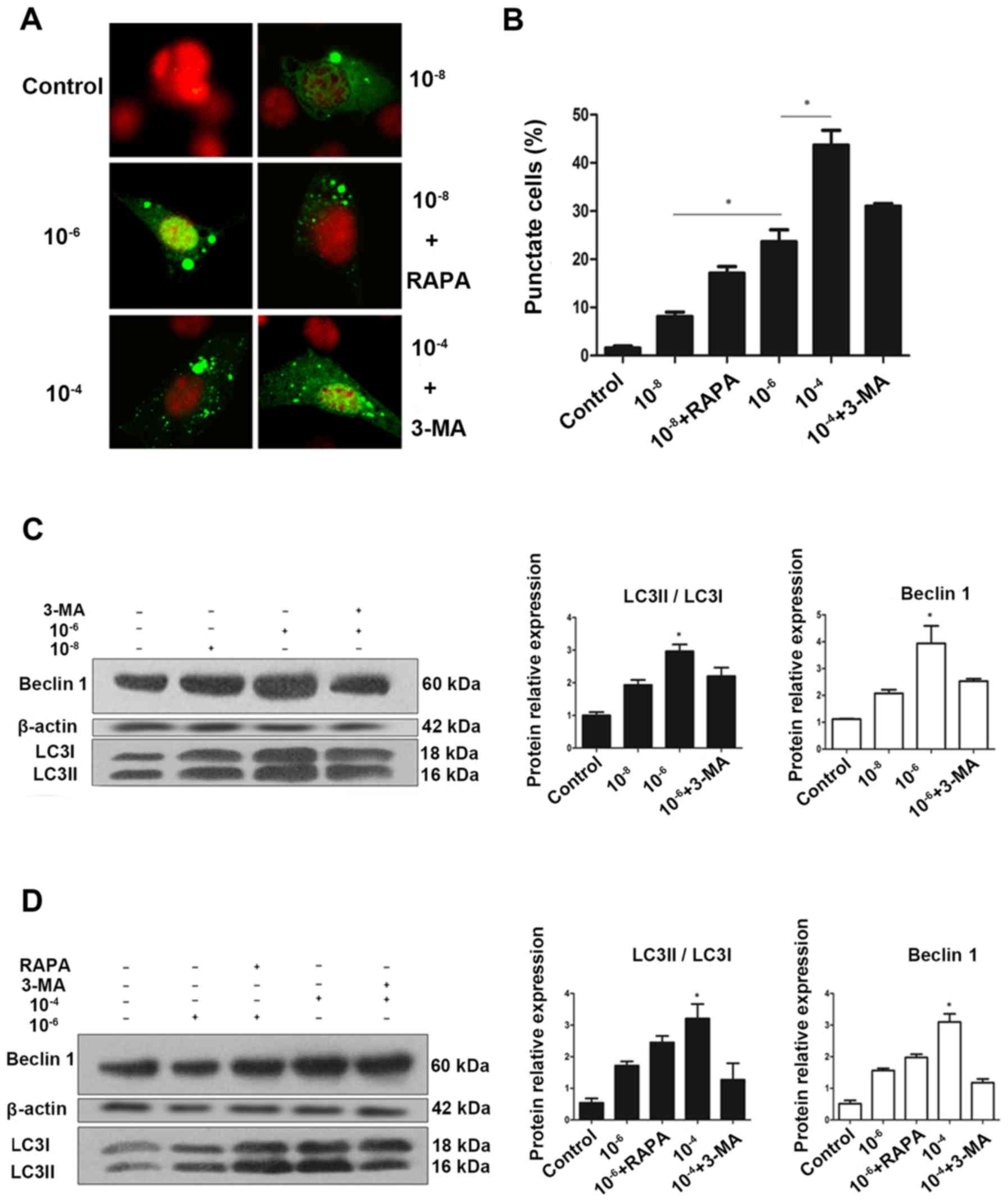 | Figure 1Autophagy is activated during
glucocorticoid treatment. (A) Cells transiently transfected plasmid
expressing eGFP-LC3B fusion protein, were exposed to Dex or control
(no Dex) for 24 h. Representative fluorescence microscopy images
are presented (magnification, ×100). (B) Control group had <2%
of cells expressing punctate eGFP-LC3. Whereas, 24 h treatment with
10−8, 10−6 and 10−4 mol/l Dex led
to eGFP-LC3 puncta formation in ~50% of cells.
*P<0.05 vs. control, 10−8, and
10−6 + 3-MA groups. Cells were exposed to culture medium
containing 10−8, 10−6 and 10−4
mol/l Dex for 24 h with (C) 3-MA (400 μmol/l) added 1.5 h
before the experiment plus (D) RAPA (500 nmol/l) added during the
last 6 h of the experiment. Autophagy markers were evaluated by
western blotting. Positive blots (right) and corresponding
semiquantitative analysis (left) are presented. Values are
presented as the mean ± standard deviation (n=3).
*P<0.05 vs. control, 10−6, 10−6
+ RAPA, and 10−4 + 3-MA groups. Dex, dexamethasone;
eGFP, enhanced green fluorescent protein; RAPA, rapamycin; 3-MA,
3-methyladenine; LC3, microtubule-associated protein 1 light chain
3β. |
To validate this finding, western blotting was
performed to examine the endogenous expression of LC3, which is a
protein involved in the formation of autophagic phagophore
membranes. Activation of autophagy is associated with the
conversion of LC3 from the cytosolic, soluble form (LC3-I), to the
membrane-bound form (LC3-II). This ubiquitin-like modification,
termed lipidation, can be illustrated by a change in migration on
SDS-PAGE gel and is a sensitive marker for monitoring autophagy
(23,24). For further validation, Beclin 1,
another marker of autophagy was examined using western blotting.
Beclin 1 is involved in the initial step of the autophagic process
and regulates both the formation and maturation of autophagosomes
(25,26). Collectively, consistent with the
eGFP-LC3 fluorescence data, endogenous expression of LC3II and
Beclin 1 suggested that Dex increased the autophagic activity in
MC3T3-E1 cells in a dose-dependent manner following treatment with
Dex for 24 h (Fig. 1C–F).
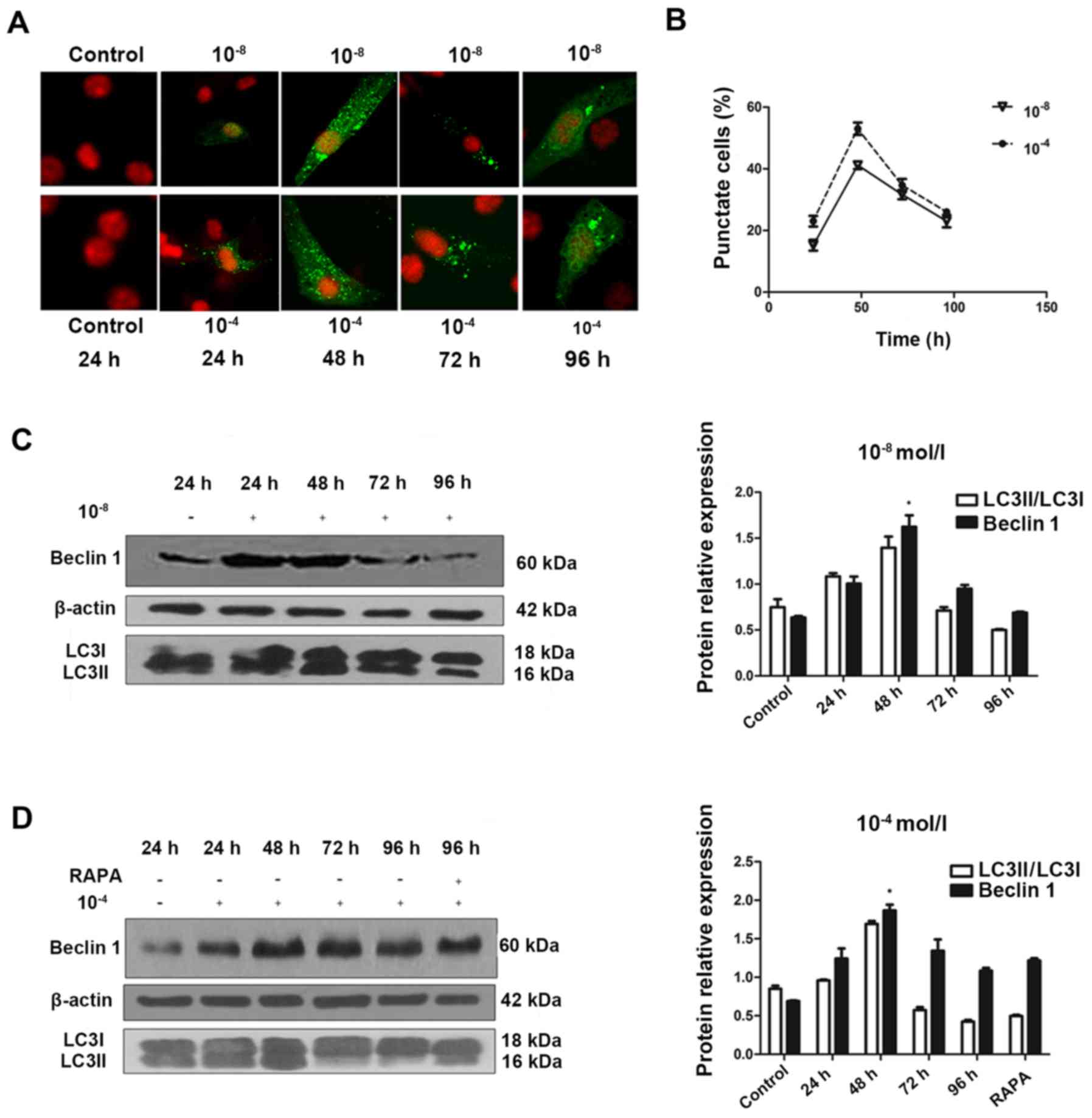
Dex-induced autophagic activity peaks at
48 h and decreases over time
To understand the temporal characteristics
Dex-induced autophagy, eGFP-LC3B puncta formation in MC3T3-E1 cells
transfected with eGFP-LC3B was monitored in the 10−8 and
10−4 mol/l Dex-treated groups over time. eGFP-LC3B
puncta formation was observed in cytoplasm as early as 24 h
post-transfection (Fig. 2A). In
addition, eGFP-LC3B puncta increased at 48 h post-transfection
(Fig. 2A and B), in accordance
with an increase in LC3II protein expression levels (Fig. 2C). In the 10−8 mol/l
Dex group, ~15% of cells exhibited cytoplasmic eGFP-LC3B puncta at
24 h post-transfection, and the number increased to 40% at 48 h.
The positive rate was 20% at 24 h, and increased to >50% at 48 h
in the 10−4 mol/l Dex group. However, this trend was not
persistent. At 48 h post-transfection, in the 10−8 and
10−4 mol/l groups, the percentage of cells with
eGFP-LC3B puncta decreased and the eGFP-LC3B fluorescence was
decreased (Fig. 2B). This was
also verified by the LC3II expression levels at 48 h
post-transfection. In accordance with these data, the endogenous
Beclin 1 protein expression was upregulated in the first 48 h, then
reduced substantially afterwards (Fig. 2C and D). Rapamycin was also added
to the cells at 96 h. The results showed that the level of Beclin 1
was not significantly elevated, which suggested that autoghagy was
a transient process in MC3T3-E1 cells (Fig. 2D). This result provided additional
support for the feature of Dex-induced autophagic activity in
MC3T3-E1 cells.
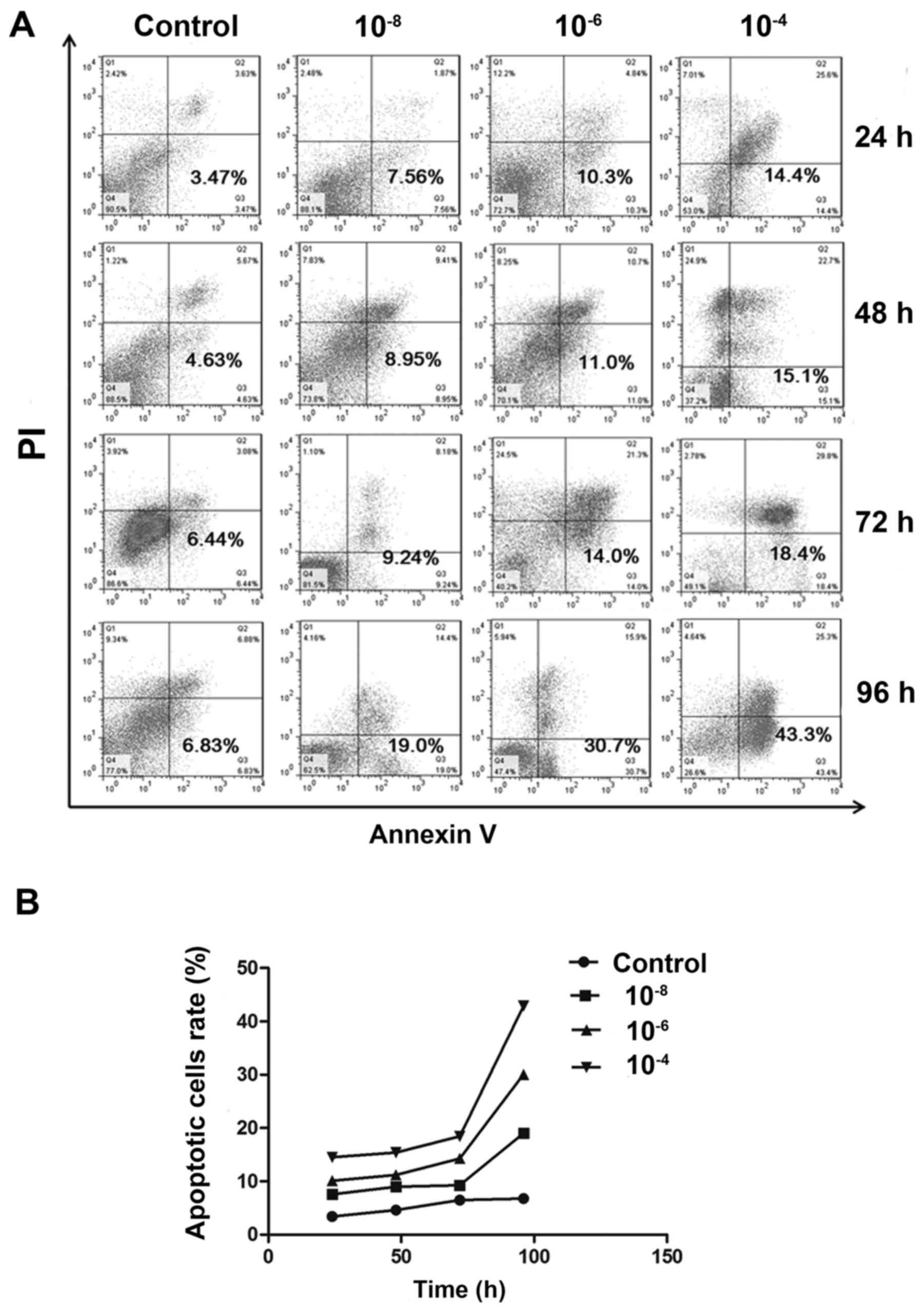
Dex induced MC3T3-E1 cell apoptosis
Following treatment with various concentrations of
Dex, cell apoptosis was detected by Annexin V/PI double staining.
Flow cytometry assays indicated a substantial increase in the
FITC-positive population among cells treated with 10−6
and 10−4 mol/l Dex, compared to non-treated group
(P<0.05). And the difference between 10−8 mol/l and
non-treated group has no statistical significance (P>0.05). But
in general, the apoptotic population was increased in a
dose-dependent manner (Fig. 3A and
B). Similarly, when time was taken into account, it was
demonstrated that the apoptotic population was also increased in
all Dex treated groups over time, which indicates Dex exerted
time-dependent effects on MC3T3-E1 cells.
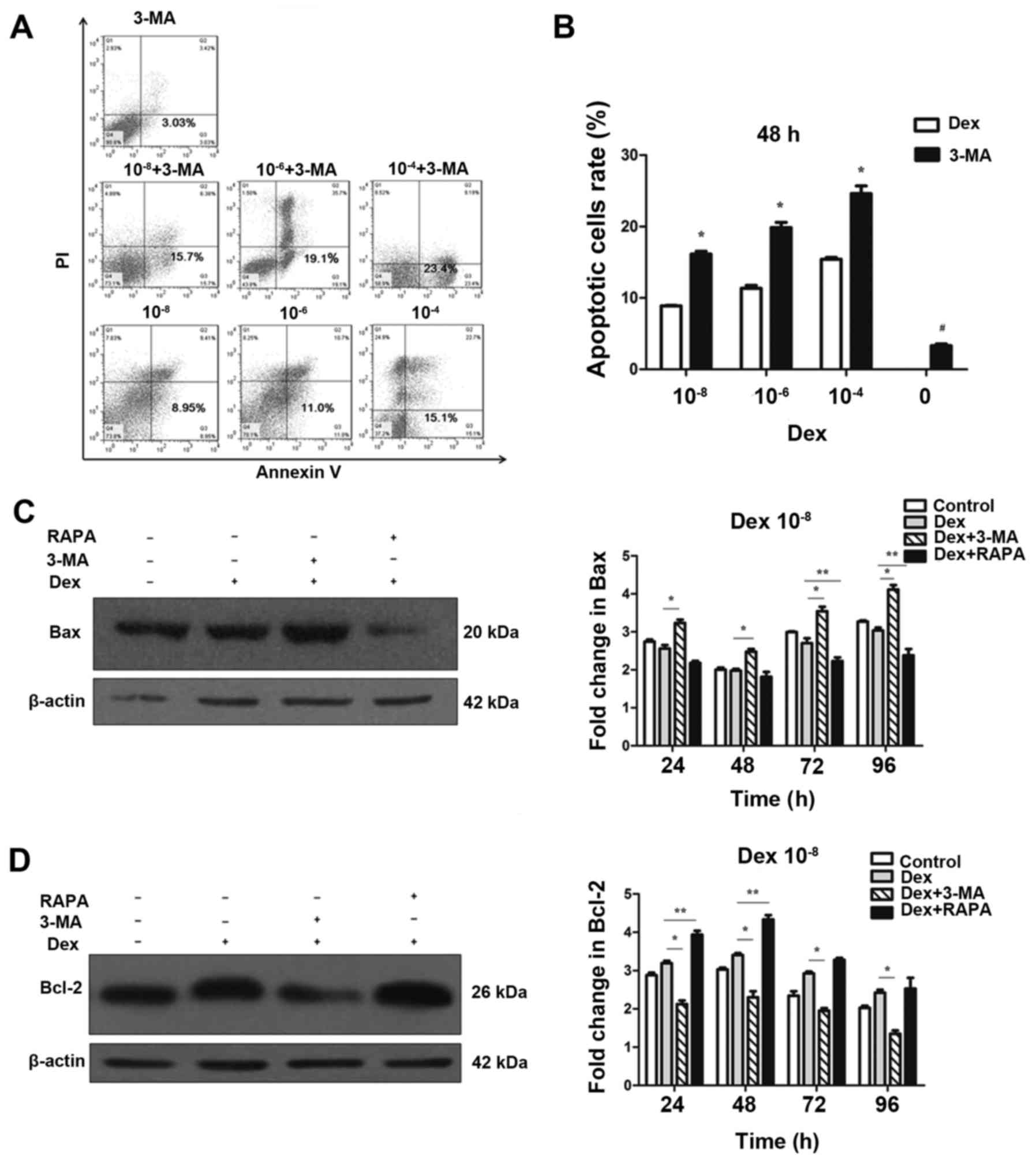
Dex-induced autophagy protects MC3T3-E1
cells from undergoing apoptosis
To investigate the effect of autophagy on apoptosis,
3-MA, a potent pharmacological inhibitor of autophagy, was used to
suppress Dex-induced autophagy. By contrast, rapamycin was used to
increase Dex-induced autophagy. The flow cytometry analysis
demonstrated that 3-MA alone did not cause a marked increase in
apoptosis and cell death (Fig.
4B), thus that pretreatment with 3-MA was able to block
autophagy in MC3T3-E1 cells without significant cytotoxicity.
However, 3-MA significantly induced apoptosis at 48 h when combined
with Dex exposure (P<0.05; Fig. 4A
and B). This result indicated that suppression of autophagy by
3-MA was able to increase Dex-induced damage in MC3T3-E1 cells. To
further confirm that apoptosis could be modulated by the regulation
of autophagy, western blotting was performed to detect the
expression of Bax and Bcl-2. As displayed in Fig. 4C and D, the expression of Bax was
not decreased in the 10−8 mol/l Dex groups compared with
the control group (P>0.05). However, when co-treated with 3-MA,
Bax expression was significantly upregulated compared with Dex
treatment alone. By contrast, Bax was significantly decreased
compared with Dex only treatment when co-treated with rapamycin
(P<0.05; Fig. 4C). Unlike Bax,
which a pro-apoptosis protein, Bcl-2 is an anti-apoptosis protein.
As shown in Fig. 4D, compared
with Dex-only treatment, Bcl-2 expression was increased when cells
were co-treated with rapamycin, but was reduced by co-treatment
with 3-MA (P<0.05). These results indicated that the blockage of
autophagy enhanced the apoptotic effects of Dex in MC3T3-E1 cells.
Autophagy induced by Dex served a protective function, and it can
be enhanced by promotion of this process.
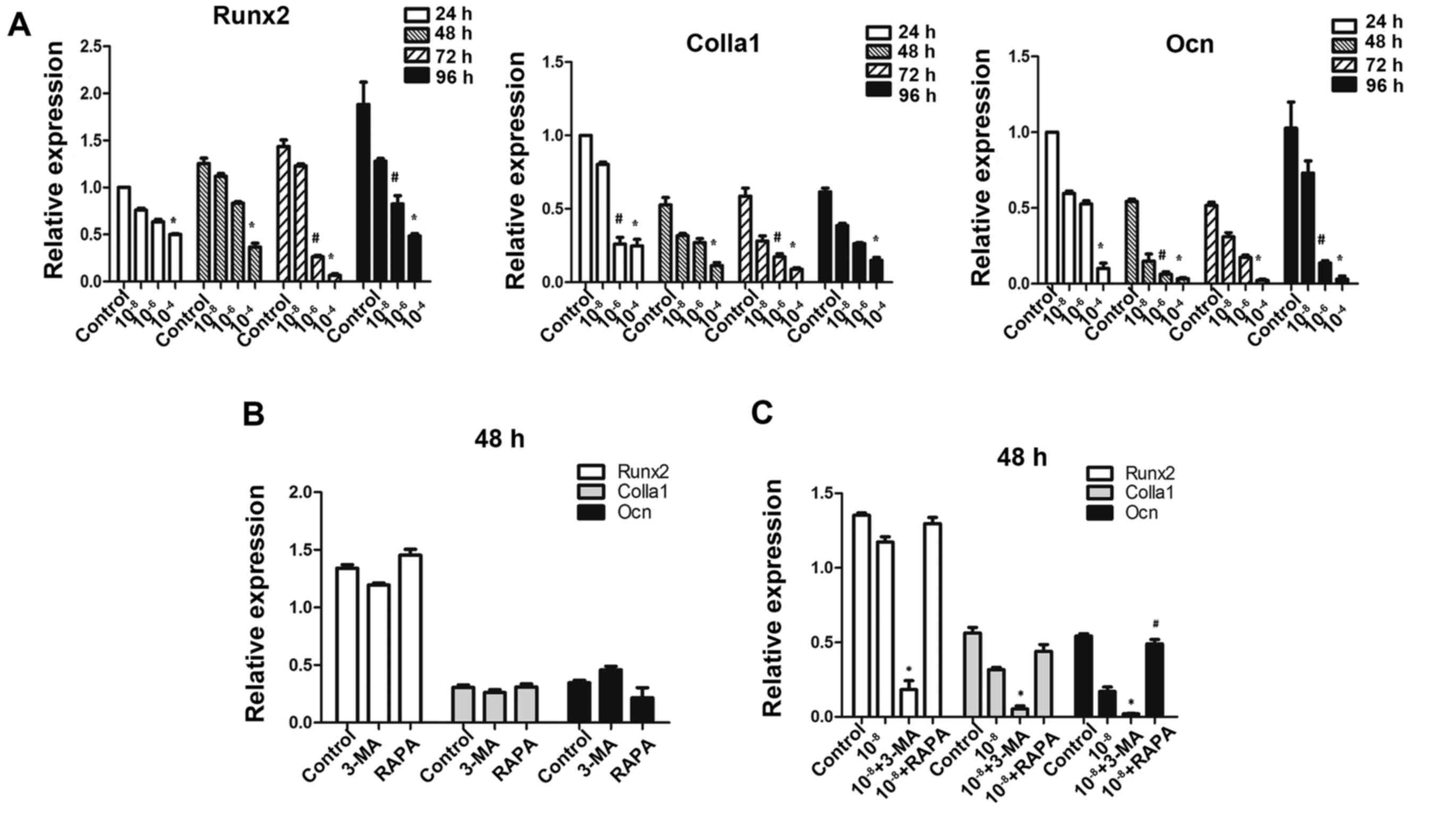
Autophagy deficiency interferes with
osteoblastic function in MC3T3-E1 cells
To further investigate whether autophagic deficiency
may aggravate the inhibitory action of Dex by reducing the
osteoblastic function in MC3T3-E1 cells, the expression of
osteoblastic-associated genes was determined. As expected on the
basis of previous findings in these osteoblastic cells (27), exposure to GCs was downregulated
the expression of Runx2, Colla1 and Ocn (Fig. 5A), which are markers of bone
matrix synthesis ability. Treatment of MC3T3-E1 cells with 3-MA
alone did not reduce the expression of these genes (P>0.05;
Fig. 5B), and treatment with
rapamycin alone did not promote their expression, thus making 3-MA
and rapamycin practical tools as autophagy-inhibitor and
autophagy-inducer in this study (Fig.
5B). The results demonstrated that pretreatment with 3-MA
significantly reduced Runx2, Colla1 and Ocn gene expressions in
cells exposed to Dex. Additionally, rapamycin promoted the
expression of Ocn compared to Dex treatment (Fig. 5C).
Discussion
GCs are effective anti-inflammatory and autoimmune
modulating agents, but excessive use or chronic GC therapy alters
bone metabolism and produces GIO, leading to bone fragility and
bone fractures (28). Unlike
senile and postmenopausal osteoporosis, in which osteolysis is
activated by the increased population and function of osteoclasts,
GIO is predominantly caused by the impaired function of
osteogenesis (29). OBs have a
central role in bone formation, and GCs directly inhibited cellular
proliferation and differentiation of OB lineage cells (30), and reduce OB maturation and
activity (31). GCs also induce
OB and osteocyte apoptosis in vivo (32). In the current study, flow
cytometry was used to demonstrated that apoptosis of MC3T3-E1 cells
was induced by Dex in a dose-dependent manner. However, the
mechanism of apoptosis was not clear. The expression of Bax is
closely associated with mitochondrial apoptosis, which is an
important factor in the regulation of cell apoptosis (33). The present study demonstrated that
selective gene deletion of Bak and Bax in OBs increased their
lifespan and thereby cancellous bone mass in the femur (34). Therefore, we hypothesized that Bax
may be involved in the osteoblastic apoptosis caused by Dex. The
current study demonstrated that Dex induced apoptosis and also
induced autophagy in MC3T3-E1 cells. As shown in Fig. 1, LC3II, Beclin 1 and GFP-LC3
puncta were upregulated by Dex in a dose-dependent manner. The
process of apoptosis is often accompanied by the occurrence of
autophagy (35), however, the
association between apoptosis and autophagy is complex (36).
Several studies have reported that inhibition of
autophagy results in dysfunction and death of cells, including
hematopoietic stem cells, pancreatic β-cells, kidney epithelium,
myocardial cells and neurons (37–40). Previous studies suggested
autophagy provided a mechanism for osteocytes to survive the stress
after GCs exposure in vitro (12,41), thus we hypothesized that
suppression of autophagy in MC3T3-E1 cells may also aggravate the
impact of GCs on osteoblastic function. The findings of the current
study demonstrated that the suppression of autophagy by 3-MA was
able to increase Dex-induced apoptosis in MC3T3-E1 cells.
Furthermore, the expression of pro-apoptotic protein Bax was also
upregulated following co-treatment with 3-MA and Dex, whereas it
was downregulated following co-treatment with the autophagy-inducer
rapamycin. To further validate the role of autophagy in
mitochondrial apoptosis, the expression of Bcl-2 was examined.
Bcl-2 is located in mitochondria and can stop cytochrome c
release into the cytoplasm, and the subsequent activation of
caspase-3, by reducing mitochondrial membrane permeability
(42). The results of the current
study indicated that 3-MA significantly reduced the expression of
Bcl-2 compared with the group treated with Dex only. By contrast,
Bcl-2 expression was increased by co-treatment with rapamycin.
These data indicated that autophagy is a pro-survival mechanism in
Dex-treated MC3T3-E1 cells to reduce the incidence of apoptosis and
facilitate the survival of the cells. The potential mechanism may
be via regulation of Bax/Bcl-2 by autophagic process through the
mitochondrial pathway. A notable finding produced by the flow
cytometry analysis was that there was a marked increase in the
apoptotic cell population after 72 h of treatment with Dex
(Fig. 3), in comparison to the
relative slower increase prior to 72 h. This may be due to the
decline of the activity of autophagy following the peak at 48 h, as
indicated by the confocal fluorescence microscopy data, resulting
in a decreased ability to relieve the negative effect of Dex on
MC3T3-E1 cells. This result suggested that autophagy may not be
consistently protective if stress lasts for a prolonged time,
particularly under chronic GC therapy. Therefore, this indicates
that the suppression of autophagy may have an important role in the
dysfunction of osteogenesis during long-term intake of GCs.
Furthermore, in accordance with the finding that
autophagy facilitated the survival of cells, osteogenic gene
expression as also examined by RT-qPCR in order to determine
whether autophagy altered the effect of Dex on osteoblastic
function. Dex reduced the gene expression of Runx2, Colla1 and Ocn,
particularly at relatively higher concentrations. The expression of
these genes was also determined in cells following co-treatment
with 3-MA and rapamycin, with the results suggesting that
osteogenic gene expression was suppressed by 3-MA and induced by
rapamycin. These results indicated that autophagy not only altered
the process of apoptosis, but also the function of osteoblastic
cells.
In conclusion, the findings of the current study
demonstrate that excess GC stimulates the process of autophagy in
MC3T3-E1 cells, and inhibition of this phenomenon mediates the
negative effects of Dex on cell proliferation and osteoblastic
function. These results indicate that activated autophagy is a
self-protective mechanism used by OBs to oppose the stress induced
by excess GC.
Acknowledgments
This study was funded by two grants from China,
National High-tech R&D Program (863 Program) (grant no.
SS2012AA041604) and National Natural Science Foundation of China
(grant no. 31271000).
References
|
1
|
Kanis JA, Johansson H, Oden A, Johnell O,
de Laet C, Melton LJ III, Tenenhouse A, Reeve J, Silman AJ, Pols
HA, et al: A meta-analysis of prior corticosteroid use and fracture
risk. J Bone Miner Res. 19:893–899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Van Staa TP, Laan RF, Barton IP, Cohen S,
Reid DM and Cooper C: Bone density threshold and other predictors
of vertebral fracture in patients receiving oral glucocorticoid
therapy. Arthritis Rheum. 48:3224–3229. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weinstein RS: Clinical practice.
Glucocorticoid-induced bone disease. N Engl J Med. 365:62–70. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mankin HJ: Nontraumatic necrosis of bone
(osteonecrosis). N Engl J Med. 326:1473–1479. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
LoCascio V, Bonucci E, Imbimbo B, Ballanti
P, Adami S, Milani S, Tartarotti D and DellaRocca C: Bone loss in
response to long-term glucocorticoid therapy. Bone Miner. 8:39–51.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jia D, O'Brien CA, Stewart SA, Manolagas
SC and Weinstein RS: Glucocorticoids act directly on osteoclasts to
increase their life span and reduce bone density. Endocrinology.
147:5592–5599. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hong JM, Teitelbaum SL, Kim TH, Ross FP,
Kim SY and Kim HJ: Calpain-6, a target molecule of glucocorticoids,
regulates osteoclastic bone resorption via cytoskeletal
organization and microtubule acetylation. J Bone Miner Res.
26:657–665. 2011. View
Article : Google Scholar
|
|
8
|
Li H, Qian W, Weng X, Wu Z, Li H, Zhuang
Q, Feng B and Bian Y: Glucocorticoid receptor and sequential 53
activation by dexamethasone mediates apoptosis and cell cycle
arrest of osteoblastic MC3T3E1 cells. PLoS One. 7:e370302012.
View Article : Google Scholar
|
|
9
|
Naves MA, Pereira RM, Comodo AN, de
Alvarenga EL, Caparbo VF and Teixeira VP: Effect of dexamethasone
on human osteoblasts in culture: involvement of β1 integrin and
integrin-linked kinase. Cell Biol Int. 35:1147–1151. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizushima N: Physiological functions of
autophagy. Curr Top Microbiol Immunol. 335:71–84. 2009.
|
|
11
|
Xia X, Kar R, Gluhak-Heinrich J, Yao W,
Lane NE, Bonewald LF, Biswas SK, Lo WK and Jiang JX:
Glucocorticoid-induced autophagy in osteocytes. J Bone Miner Res.
25:2479–2488. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martinet W, Agostinis P, Vanhoecke B,
Dewaele M and De Meyer GR: Autophagy in disease: a double-edged
sword with therapeutic potential. Clin Sci (Lond). 116:697–712.
2009. View Article : Google Scholar
|
|
14
|
Tsujimoto Y and Shimizu S: Another way to
die: autophagic programmed cell death. Cell Death Diffe. 12(Suppl
2): 1528–1534. 2005. View Article : Google Scholar
|
|
15
|
Czaja MJ: Autophagy in health and disease.
2. Regulation of lipid metabolism and storage by autophagy:
pathophysiological implications. Am J Physiol Cell Physiol.
298:C973–C978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laane E, Tamm KP, Buentke E, Ito K,
Kharaziha P, Oscarsson J, Corcoran M, Björklund AC, Hultenby K,
Lundin J, et al: Cell death induced by dexamethasone in lymphoid
leukemia is mediated through initiation of autophagy. Cell Death
Differ. 16:1018–1029. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nollet M, Santucci-Darmanin S, Breuil V,
Al-Sahlanee R, Cros C, Topi M, Momier D, Samson M, Pagnotta S,
Cailleteau L, et al: Autophagy in osteoblasts is involved in
mineralization and bone homeostasis. Autophagy. 10:1965–1977. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kimura S, Noda T and Yoshimori T:
Dissection of the autophagosome maturation process by a novel
reporter protein, tandem fluorescent-tagged LC3. Autophagy.
3:452–460. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J,
Ding H and Yuan Z: Subversion of cellular autophagy machinery by
hepatitis B virus for viral envelopment. J Virol. 85:6319–6333.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang YH, Chen K, Li B, Chen JW, Zheng XF,
Wang YR, Jiang SD and Jiang LS: Estradiol inhibits osteoblast
apoptosis via promotion of autophagy through the ER-ERK-mTOR
pathway. Apoptosis. 18:1363–1375. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Yang YH, Li B, Zheng XF, Chen JW, Chen K,
Jiang SD and Jiang LS: Oxidative damage to osteoblasts can be
alleviated by early autophagy through the endoplasmic reticulum
stress pathway - implications for the treatment of osteoporosis.
Free Radic Biol Med. 77:10–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matsunaga K, Saitoh T, Tabata K, Omori H,
Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe
T, et al: Two Beclin 1-binding proteins, Atg14L and Rubicon,
reciprocally regulate autophagy at different stages. Nat Cell Biol.
11:385–396. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Itakura E, Kishi C, Inoue K and Mizushima
N: Beclin 1 forms two distinct phosphatidylinositol 3-kinase
complexes with mammalian Atg14 and UVRAG. Mol Biol Cell.
19:5360–5372. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alm JJ, Heino TJ, Hentunen TA, Väänänen HK
and Aro HT: Transient 100 nM dexamethasone treatment reduces inter-
and intraindividual variations in osteoblastic differentiation of
bone marrow-derived human mesenchymal stem cells. Tissue Eng Part C
Methods. 18:658–666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dalle Carbonare L, Arlot ME, Chavassieux
PM, Roux JP, Portero NR and Meunier PJ: Comparison of trabecular
bone microarchitecture and remodeling in glucocorticoid-induced and
postmenopausal osteoporosis. J Bone Miner Res. 16:97–103. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mazziotti G, Angeli A, Bilezikian JP,
Canalis E and Giustina A: Glucocorticoid-induced osteoporosis: an
update. Trends Endocrinol Metab. 17:144–149. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Canalis E, Bilezikian JP, Angeli A and
Giustina A: Perspectives on glucocorticoid-induced osteoporosis.
Bone. 34:593–598. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Weinstein RS: Glucocorticoid-induced
osteoporosis. Rev Endocr Metab Disord. 2:65–73. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weinstein RS, Jilka RL, Parfitt AM and
Manolagas SC: Inhibition of osteoblastogenesis and promotion of
apoptosis of osteoblasts and osteocytes by glucocorticoids.
Potential mechanisms of their deleterious effects on bone. J Clin
Invest. 102:274–282. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chipuk JE, McStay GP, Bharti A, Kuwana T,
Clarke CJ, Siskind LJ, Obeid LM and Green DR: Sphingolipid
metabolism cooperates with BAK and BAX to promote the mitochondrial
pathway of apoptosis. Cell. 148:988–1000. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jilka RL, O'Brien CA, Roberson PK,
Bonewald LF, Weinstein RS and Manolagas SC: Dysapoptosis of
osteoblasts and osteocytes increases cancellous bone formation but
exaggerates cortical porosity with age. J Bone Miner Res.
29:103–117. 2014. View Article : Google Scholar
|
|
35
|
Zhang T, Li Y, Park KA, Byun HS, Won M,
Jeon J, Lee Y, Seok JH, Choi SW, Lee SH, et al: Cucurbitacin
induces autophagy through mitochondrial ROS production which
counteracts to limit caspase-dependent apoptosis. Autophagy.
8:559–576. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rangaraju S, Verrier JD, Madorsky I, Nicks
J, Dunn WA Jr and Notterpek L: Rapamycin activates autophagy and
improves myelination in explant cultures from neuropathic mice. J
Neurosci. 30:11388–11397. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jiang M, Wei Q, Dong G, Komatsu M, Su Y
and Dong Z: Autophagy in proximal tubules protects against acute
kidney injury. Kidney Int. 82:1271–1283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Warr MR, Binnewies M, Flach J, Reynaud D,
Garg T, Malhotra R, Debnath J and Passegué E: FOXO3A directs a
protective autophagy program in haematopoietic stem cells. Nature.
494:323–327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jung HS, Chung KW, Won Kim J, Kim J,
Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al:
Loss of autophagy diminishes pancreatic beta cell mass and function
with resultant hyperglycemia. Cell Metab. 8:318–324. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jia J, Yao W, Guan M, Dai W, Shahnazari M,
Kar R, Bonewald L, Jiang JX and Lane NE: Glucocorticoid dose
determines osteocyte cell fate. FASEB J. 25:3366–3376. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xia HF, Jin XH, Cao ZF, Shi T and Ma X:
miR-98 is involved in rat embryo implantation by targeting Bcl-xl.
FEBS Lett. 588:574–583. 2014. View Article : Google Scholar : PubMed/NCBI
|