Introduction
It generally known that the central nervous system
(CNS) and the immune system are isolated with little interaction,
except during disease and/or trauma. The blood-brain barrier
prevents infiltration of immune cells and molecules into the CNS
(1,2). However, recent studies have reported
clear and convincing evidence of bidirectional communication
between these two systems. Neuronal transmitters and immune
cytokines are considered to mediate communication between CNS and
the immune system; however, since both immune receptors and neural
receptors may be co-expressed in neurons or innate immune cells,
such as microglia, whether the two types of receptors can interact
directly by protein-protein interactions has not been reported to
date.
Microglial cells have been described as the resident
macrophages of the brain that perform surveillance functions to
maintain the integrity of the CNS (3). In response to a wide range of
invading pathogens, microglia initiate innate immune responses
characterized by the production of cytokines and chemokines,
upregulation of cell surface molecules and expansion of local
immune responses (4,5). Neuron-microglia interactions play a
pivotal role in controlling microglial functions and restraining
their activation. Neurons are able to affect microglia through the
release of neurotransmitters, as well as through direct
cell-to-cell interactions mediated by membrane-bound antigens and
their cognate receptors. Similar to neurons, microglia express a
number of neurotransmitter receptors that enable microglia to
respond to the same signals acting on neurons.
Microglia endogenously express both neurotransmitter
and immune receptors, such as N-methyl-D-aspartate (NMDA) receptors
(6) and Toll-like receptor 4
(TLR4). NMDA receptors are glutamate-gated ion channels, which play
a key role in CNS function. NMDA receptor dysfunction is involved
in various neurological disorders, including stroke, pathological
pain, neurodegenerative diseases and neural inflammation. TLR4 is
considered to be a receptor essential for proper response to the
Gram-negative bacterial endotoxin lipopolysaccharide (LPS)
(7).
The interaction between neurotransmitter NMDA
receptor subunit 1 (GluN1) and immune receptor TLR4 in N9 and EOC
20 microglial cells was examined in the present study. LPS was
found to trigger direct binding of TLR4 to GluN1 in microglia,
whereas inhibition of the metabotropic glutamate receptor 5
(mGluR5) with its selective antagonist, MTEP, abolished LPS-induced
binding of TLR4 with GluN1. These results indicate that GluN1
directly interacts with TLR4 in response to LPS in N9 and EOC 20
microglial cells.
Materials and methods
Materials
The EOC 20 microglial cell line was purchased from
American Type Culture Collection (Manassas, VA, USA) and the N9
cell line was provided by Professor Yun Bai (Department of Medical
Genetics, Third Military Medical University, Chongqing, China).
DMEM/F12 medium, fetal bovine serum and trypsin were purchased from
HyClone (Logan, UT, USA). Goat anti-TLR4 antibody (cat. no.
sc-16240) was purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). β-actin antibody (cat. no. A2066), goat IgG
(purified immunogloblin) and LPS from Escherichia coli
0111:B4 were purchased from Sigma-Aldrich; Merck KGaA (St. Louis,
MO, USA). Pierce™ ECL Western Blotting substrate, goat anti-rabbit
IgG (cat. no. 31460) and rabbit anti-goat IgG (cat. no. A27014)
conjugated HRP, and immobilized protein A/G agarose were purchased
from Pierce (Rockford, IL, USA). Polyvinylidene fluoride (PVDF)
membranes were purchased from Millipore (Billerica, MA, USA).
Anti-GluN1 antibody (cat. no. 5704) was purchased from Cell
Signaling Technologies (Danvers, MA, USA). The rabbit anti-GluN1
antibody (cat. no. LS-B13901) used in immunochemistry was purchased
from LifeSpan BioSciences (Seattle, WA, USA). Donkey anti-goat IgG
Alexa Fluor 555 (cat. no. A-21432) and donkey anti-rabbit IgG Alexa
Fluor 488 (cat. no. A-21206) were purchased from Invitrogen; Thermo
Fisher Scientific (Carlsbad, CA, USA).
3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) was purchased
from Tocris (Ellisville, MO, USA).
Cell culture
The N9 and EOC 20 microglial cell lines were
maintained in DMEM/F12 with 10% fetal calf serum, 2 mM L-glutamine,
1X NEAA, 100 µg/ml penicillin and 100 µg/ml
streptomycin. The cultures were maintained at 37°C in 5%
CO2 and the medium was changed every other day.
Western blot and immunoprecipitation
analysis
The cells were washed thoroughly with cold
phosphate-buffered saline (PBS) and lysed in cold lysis buffer (1%
Triton X-100, 10 mM Tris pH 7.6, 50 mM NaCl, 30 mM sodium
pyrophosphate, 50 mM NaF, 5 mM EDTA and 0.1 mM
Na3VO4) with protease inhibitor cocktail
tablets on ice for 20 min. The lysates were centrifuged at 4°C at
16,000 × g for 30 min. Supernatant fractions containing equal
amounts of total protein were separated on sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred
onto PVDF membranes and analyzed by western blotting. Enhanced
chemiluminescence detection was performed according to the
manufacturer's protocol. For the co-immunoprecipitation assay,
total cell lysates were first incubated with protein A agarose with
gentle shaking at 4°C for 10 min, then centrifuged at 4°C at 16,000
× g for 15 min to remove non-specific binding. The cleared lysates
containing equal amounts of total protein were incubated with
anti-TLR4 antibody or goat IgG with gentle shaking at 4°C
overnight, then incubated with protein A/G agarose for a further 2
h at 4°C. After centrifugation, the agarose was washed three times
in cold lysis buffer. The proteins were eluted with Laemmli buffer
and separated by SDS-PAGE.
Cell surface biotinylation
The cells were rinsed thoroughly with ice-cold PBS
containing 0.1 mM CaCl2 and 1 mM MgCl2
(PBS2+) and incubated twice with 1 ml of 1.0 mg/ml
NHS-SS-biotin (Pierce) for 20 min (a total of 40 min) at 4°C.
Non-reactive biotin was quenched with 2X 20-min incubations at 4°C
in ice-cold PBS2+ containing 0.1 M glycine. After
incubation, the cells were washed with PBS2+ without 0.1
M glycine and solubilized for 1 h at 4°C with gentle shaking in
radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl, pH
7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium
deoxycholate and 0.1% SDS) containing protease inhibitors (1 mM
phenylmethylsulfonyl fluoride, 1 µg/ml each leupeptin,
aprotinin and pepstain). The cell lysate was centrifuged at maximum
speed for 20 min at 4°C to remove cell debris. Biotinylated and
non-biotinylated proteins were separated from equal amounts of
cellular protein (100 µg) by incubation with 100 µl
immobilized streptavidin (Pierce) for 12 h at 4°C. The mixture was
centrifuged at maximum speed for 2 min and the beads were washed
with 1 ml RIPA buffer four times. Proteins bound to streptavidin
beads were diluted in Laemmli sample buffer. Biotinylated proteins
were analyzed by SDS-PAGE.
Immunocytochemical examination
The cells were plated on poly-D-lysine-coated glass
coverslips and cultured for at least 24 h. After being treated with
1 µg/ml of LPS for different intervals, the cells were
washed with PBS and fixed with 4% formaldehyde for 15 min at room
temperature, then permeabilized in 0.1% Triton X-100 for 20 min,
and blocked with 5% goat serum for 1 h at room temperature.
Co-immunostaining with anti-TLR4 and anti-GluN1 antibody (1:1,000)
was performed at 4°C overnight; donkey anti-rabbit IgG Alexa Fluor
488 (1:1,000) and donkey anti-goat IgG Alex Fluor 555 (1:1,000)
were incubated for 1 h at room temperature and the nuclei were then
stained with DAPI. Images were acquired under a Leica TCS SP2
confocal microscope with a CCD camera (Leica Microsystems, Wetzlar,
Germany). Five to ten fields from each coverslip were randomly
selected and analyzed with CoLocalizer Pro software (8) and Protein Proximity Analysis
software (9).
Fluorescence resonance energy transfer
(FRET)
FRET was measured with the acceptor bleaching method
using a Leica confocal laser scanning microscope (Leica
Microsystems) according to the method described by König et
al (10) and modified by Liu
et al (11). Briefly,
microglia were seeded onto poly-D-lysine-coated glass coverslips
(24 mm in diameter). After a 24-h culture, cells were treated with
1 µg/ml LPS for 30 min, and fixed. Images were captured
using a 40X objective and a LUDL filter wheel that allows for rapid
exchange of filters (<100 msec). The system was equipped with
the following fluorescence filters: Donor filter (excitation, 488
nm; emission, 500–535 nm) and acceptor filter (excitation, 543 nm;
emission, 555–620 nm, 75% intensity). The acquisition of the images
was performed with Leica confocal software (Leica Microsystems).
Background fluorescence was subtracted from all images, and
fluorescence intensity was measured in different regions of
interest. The change in fluorescence intensity is expressed as FRET
efficiency (FRETeff), percentage of fluorescence
increase. To calculate FRETeff, the following equation
was used: FRETeff =
(Ipost−Ipre)/Ipost ×100, where
Ipost is the donor intensity after bleaching and
Ipre is the donor intensity before bleaching. Negative
control was performed with goat IgG and rabbit IgG antibody rather
than TLR4 and GluN1, respectively. One cell was selected as the
region of interest 1 (ROI1), which was bleached. Four other cells
and background were selected as controls, and were not bleached.
Five ROI1 regions were analyzed for each coverslip and the
experiment was repeated three times independently.
Statistical analysis
All statistical analyses were performed with the
SPSS 10.0 software (SPSS Inc., Chicago, IL, USA). The data are
expressed as mean ± standard error of the mean. Unpaired,
two-tailed Student's t-tests were performed to evaluate the
significance of the differences between two groups. For comparison
of multiple groups, analysis of variance was used. P<0.05 was
considered to indicate statistically significant differences.
Results
LPS triggers interactions of TLR4 and
GluN1 in N9 and EOC 20 microglial cells
LPS is the most frequently used model agent to study
microglial activation and inflammatory signaling (12). It is generally accepted that LPS
activates microglia through TLR4 (7), one of LPS's interaction
proteins.
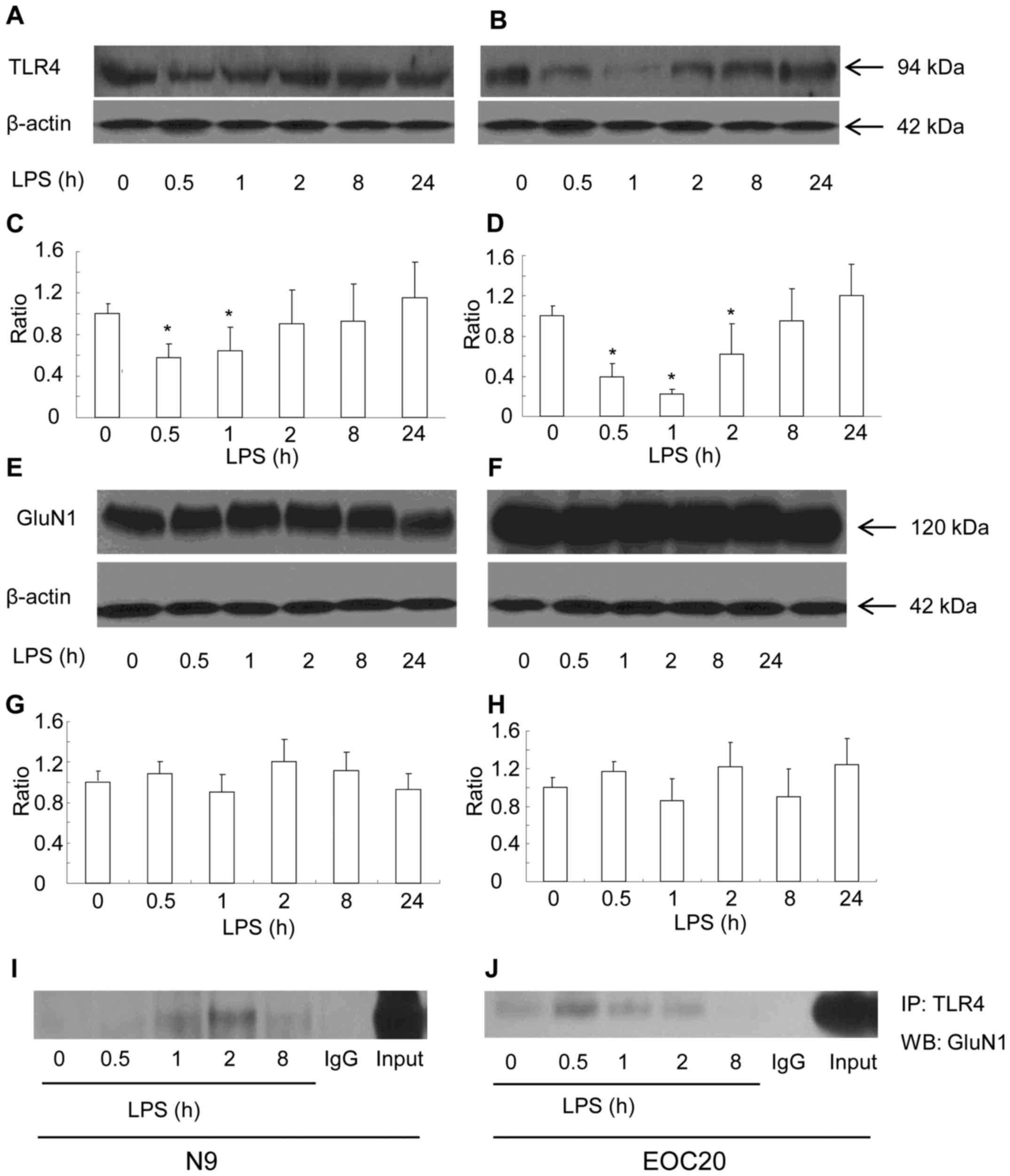
First, the surface expression of TLR4 was measured
with the biotinylation method in response to LPS in N9 microglial
cells. Briefly, cell surface proteins were biotinylated, and
cleared total cell lysates were incubated with streptavidin beads
to pull down biotinylated proteins originally localized on the cell
surface. The surface expression of TLR4 was found to be gradually
reduced in the N9 microglial cells when treated with 1 µg/ml
of LPS within the first 2 h. The reduction at 30 min and 1 h after
exposure to LPS was significantly lower compared to that prior to
LPS treatment (Fig. 1A and C)
(P<0.05 vs. prior to LPS treatment). At 2 h after exposure to
LPS, the surface expression of TLR4 had recovered to the level
before treatment (Fig. 1A and C).
Thereafter and until 24 h after exposure to LPS, the surface
expression of TLR4 remained at a similar level. The total TLR4
expression was not altered at all the tested timepoints in response
to LPS (data not shown). The expression of GluN1 was also observed
in N9 microglial cell. The surface expression of GluN1 (data not
shown) and total GluN1 expression were not affected by LPS
treatment (Fig. 1E and G). The
cellular level of β-actin did not differ among the LPS-treated
groups. We tested whether TLR4 was bound to GluN1. The total cell
lysates of the N9 microglia following LPS treatment for various
intervals were immunoprecipitated with the TLR4 antibody.
Anti-GluN1 western blot analysis of the TLR4 immunoprecipitates
revealed that GluN1 was co-immunoprecipitated with TLR4 (Fig. 1I). The co-immunoprecipitates of
TLR4 with GluN1 appeared at 1 h, reached a maximum level at 2 h,
and declined 8 h after LPS treatment. The N9 microglial cell line
is a type of primary mouse microglial cells immortalized with the
v-myc and v-mil oncogenes of the avian retrovirus MH2, which share
many phenotypical characteristics with primary mouse microglia and
express functional TLR4 receptor (13,14). To elucidate whether LPS-induced
co-immunoprecipitation of GluN1 and TLR4 is dependent on TLR4
receptor, TLR4-mutant EOC 20 microglial cells were utilized
(15). EOC 20 microglial cells
were derived from the C3H/HeJ mouse brain, which carries a missense
mutation in the TLR4 gene. This destructive mutation in the TLR4
receptor leads to a defective response to LPS in EOC 20 microglia
(15,16). As shown in Fig. 1B and D, LPS treatment
significantly reduced the surface expression of TLR4 in EOC 20
microglia within the first 8 h (P<0.05 vs. before LPS
treatment). The surface expression of TLR4 reached a minimum at 1 h
and gradually returned back to the level before LPS treatment at 8
h after LPS treatment. Compared with N9 microglia, the recovery of
surface TLR4 was slow in EOC 20 microglia. However, similar to N9
microglia, the total TLR4 expression at all tested timepoints was
at a similar level (data not shown). The surface and total
expression of GluN1 in EOC 20 microglia were not affected by LPS
treatment, and the total expression of GluN1 is shown in Fig. 1F and H. Similar to N9 microglial
cells, co-immunoprecipitates of TLR4 and GluN1 were detected in EOC
20 microglial cells. The co-immunoprecipitates of TLR4 and GluN1
reached a maximum level at 0.5 h after LPS treatment, were
gradually reduced, and diminished at 8 h after LPS treatment. These
results demonstrated that LPS induced co-immunoprecipitation of
TLR4 and GluN1 in both N9 and EOC 20 microglial cells, which is not
affected by TLR4-destructive missense mutations.
Immunocytochemistry shows co-localization
of TLR4 and GluN1 in response to LPS in N9 and EOC 20 microglial
cells
The LPS-induced co-immunoprecipitation of TLR4 and
GluN1 in microglia suggests the possibility of co-localization of
the two proteins. The co-localization of TLR4 and GluN1 was
examined with immunocytochemistry. N9 microglia were treated with 1
µg/ml LPS, and then cells were fixed at various times. As
shown in Fig. 2A–L, the
co-localization of TLR4 and GluN1 was observed at 30 min (Fig. 2D) after LPS treatment. These
images were analyzed with CoLocalizer Pro software (8) and protein proximity index (PPI)
(9) and the results are presented
in Fig. 2I–L. Pearson's
correlation coefficient (Rr) in Fig.
2I describes the correlation of the intensity distributions
between two channels, TLR4 (red) and GluN1 (green). The 1.0 value
indicates complete co-localization. Overlap coefficients K1 and K2
(AbsK1-K2) in Fig. 2J split the
value of co-localization into a pair of separate parameters. The
lower the absolute value of K1-K2, the stronger the
co-localization. PPI analysis provides a separate value for each
channel to characterize the contribution of each protein to the
overall co-localization. TLR4 and GluN1 contribute equally to the
co-localization based on the PPI analysis (Fig. 2K and L). As shown in Fig. 2D–G, I and J, the co-localization
of TLR4 and GluN1 appeared at 30 min, reached a maximum at 1 h, and
decreased gradually until 4 h after LPS treatment. Without LPS
treatment, co-localization of TLR4 and GluN1 was not observed
(Fig. 2C). When TLR4 and GluN1
antibodies were replaced with goat IgG and rabbit IgG,
respectively, no specific staining was observed (Fig. 2H). Similar results were obtained
when EOC 20 microglia were treated with LPS (Fig. 2M–X). These results suggest that
TLR4 and GluN1 are located in the same organelles in microglia
following exposure to LPS, and the co-localization of TLR4 and
GluN1 was not affected by destructive TLR4 missense mutations.
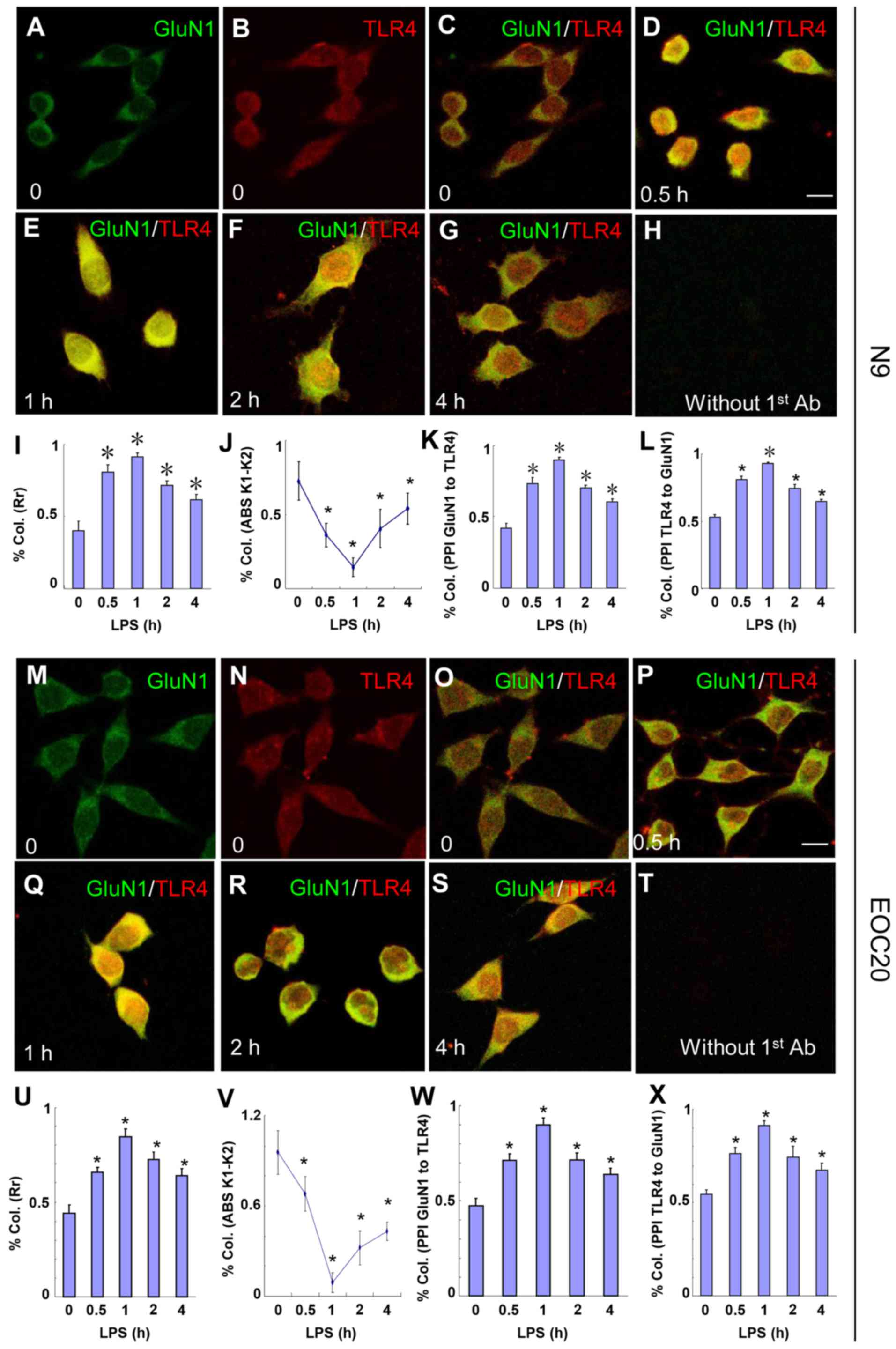 | Figure 2Toll-like receptor 4 (TLR4) and
glutamate receptor N-methyl-D-aspartate subunit 1 (GluN1)
co-localize in N9 and EOC 20 microglial cells. (A–L) N9 and (M–X)
EOC 20 microglial cells were treated with 1 µg/ml LPS for (D
and P) 30 min, (E and Q) 1 h, (F and R) 2 h or (G and S) 4 h, and
stained for GluN1 (green) and TLR4 (red). Without LPS treatment, (C
and O) no co-localization of TLR4 and GluN1 was observed. Single
GluN1 and TLR4 images are shown in (A and M) and (B and N),
respectively, in the group without LPS treatment. No signal was
observed if TLR4 and GluN1 antibodies were replaced with goat IgG
and rabbit IgG, respectively. Five to ten fields from each
condition were randomly selected and analyzed with CoLocalizer Pro
software and Protein Proximity Analysis software. The results are
shown in (I–L) for N9 and (U–X) for EOC 20 microglial cells.
*P<0.05 vs. 0 (without LPS treatment). Scale bar, 20
µM. The experiments were repeated at least three times with
similar results, and two coverslips were examined for each
condition in one test (n=6). Col., colocalization; Rr, Pearson's
correlation coefficient; LPS, lipopolysaccharide. |
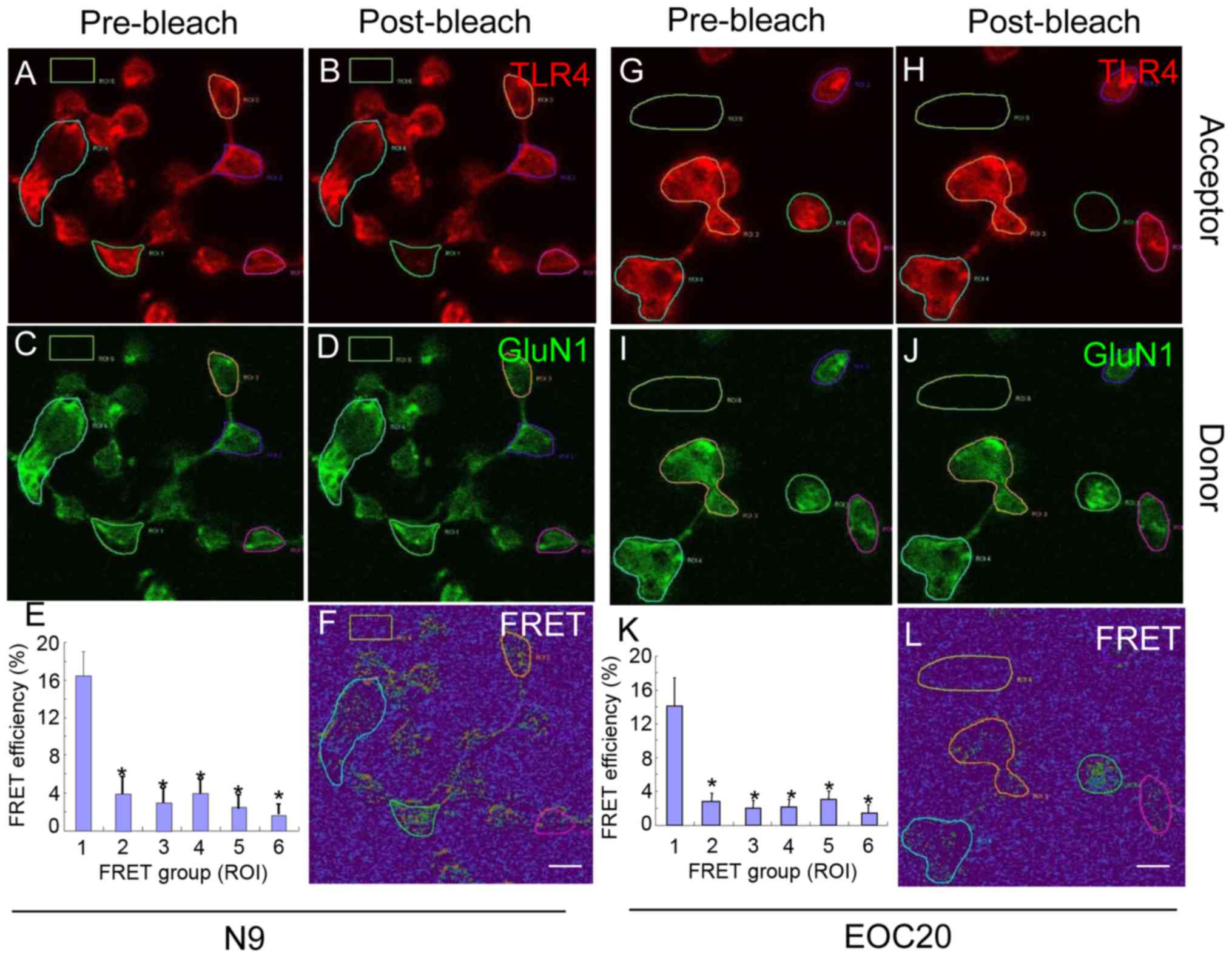
To further validate our findings, antibody-based
FRET technology was utilized. N9 microglia were treated with 1
µg/ml LPS for 30 min and co-stained with antibodies against
TLR4 (red) and GluN1 (green). Images pre- and post-bleach are shown
for TLR4 in Fig. 3A and B, and
for GluN1 in Fig. 3C and D. As
shown in Fig. 3E and F, the
average FRET efficiency in ROI1 was 16.38±2.63%, which was
significantly higher compared with that in ROI2-ROI6 (P<0.05 vs.
ROI1). Cells at ROI2-5 were not bleached and ROI6 was background
control. When the experiment was performed with EOC 20 microglia,
similar results were obtained (Fig.
3G–L). Thus, the FRET analysis provided further evidence that
LPS triggers direct binding of TLR4 to GluN1 in both N9 and EOC 20
microglial cells, and that the binding of TLR4 with GluN1 was not
affected by TLR4 missense mutations.
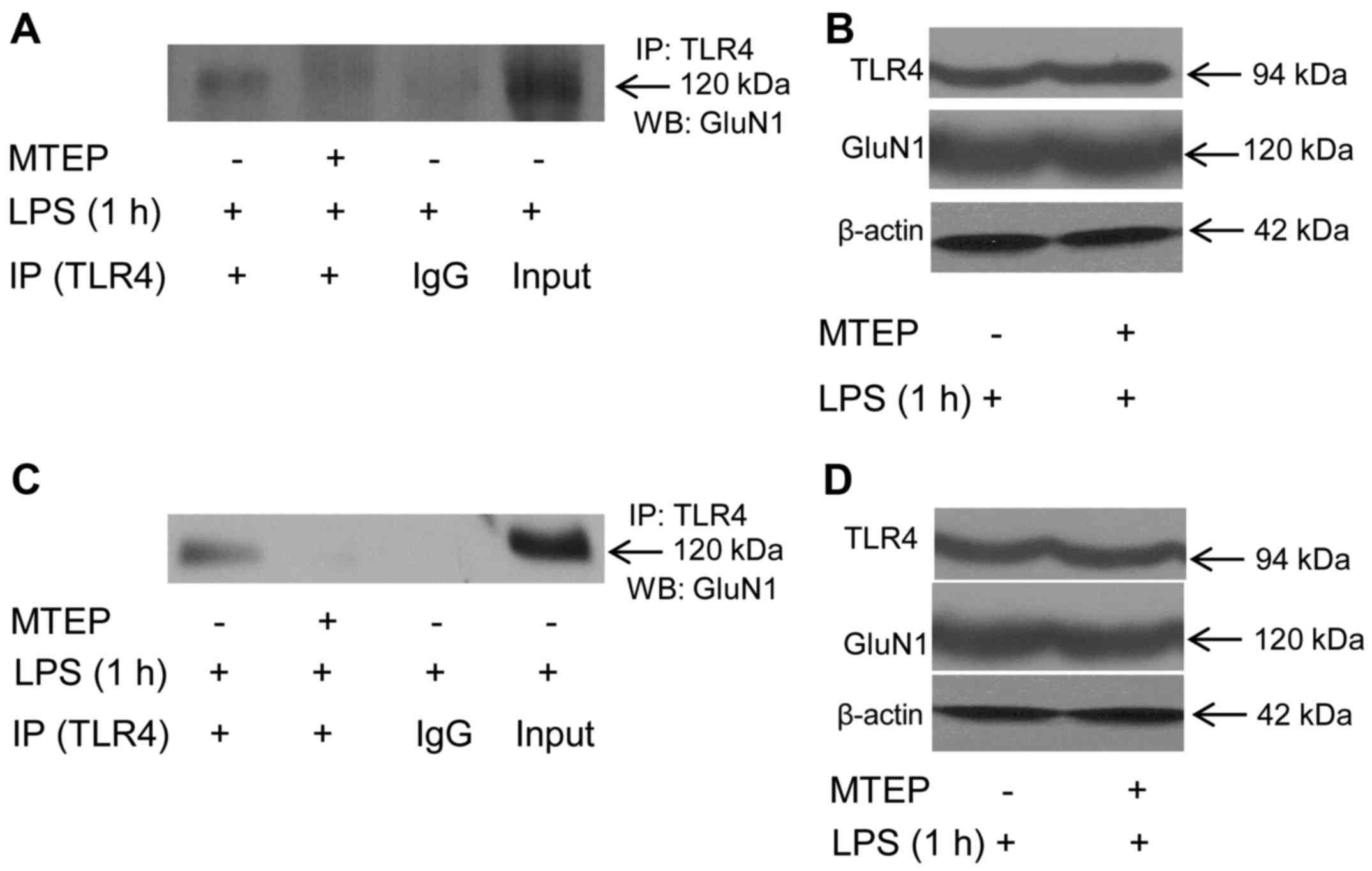
LPS-triggered binding of TLR4 and GluN1 is dependent
on mGluR5 in N9 and EOC 20 microglial cells. We have previously
demonstrated that LPS triggers calcium
[Ca2+]i oscillation through directly binding
to mGluR5 in microglial cells (11). A large number of studies have
indicated a regulatory role of mGluR5 in the GluN1 responses to its
ligand activation (17–19). We tested whether mGluR5 is
involved in the direct binding of TLR4 to GluN1 in response to
LPS.
As shown in Fig.
4A, when N9 microglia were first treated with the mGluR5
selective antagonist MTEP (0.1 mM) for 30 min, and then treated
with 1 µg/ml LPS for another 1 h, the co-immunoprecipitates
of TLR4 and GluN1 was significantly reduced. The cellular level of
β-actin did not differ between groups with and without MTEP
treatment (Fig. 4B), and the
total cellular levels of TLR4 and GluN1 were also similar (Fig. 4B), suggesting that the decreased
co-immunoprecipitation of TLR4 and GluN1 by MTEP pretreatment was
not due to the change of turnover of TLR4 or GluN1. Similarly, MTEP
abolished LPS-triggered co-immunoprecipitation of TLR4 and GluN1 in
TLR4-mutant EOC 20 microglia (Fig. 4C
and D). Co-localization of TLR4 and GluN1 was not observed in
N9 or EOC 20 microglial cells when MTEP was applied prior to LPS
treatment (data not shown). These results indicate that inhibition
of mGluR5 abolished LPS-induced direct binding of TLR4 to GluN1 in
N9 and EOC 20 microglial cells.
Discussion
Microglia, the primary immune cells in the brain,
have been implicated as the predominant cells regulating
inflammation-mediated neuronal damage. In response to immunological
challenges, such as LPS, microglia are activated and subsequently
the inflammatory process is initiated as evidenced by the release
of pro-inflammatory chemokines and cytokines. Activated microglia
accumulate around brain lesions that evident in neurodegenerative
disorders, such as Alzheimer's disease and Parkinson's disease, and
likely play a role in the neuronal damage that occurs in these
diseases (20,21). The effective function of
microglial cells is critical for controlling neuroinflammation and
alleviating neuropathogenesis. In the present study, it was
observed that LPS may trigger interactions between TLR4 and GluN1
in N9 and EOC 20 microglial cells. Immunocytochemistry demonstrated
co-localization of TLR4 and GluN1 in response to LPS, and the
direct binding of TLR4 and GluN1 was further validated by
antibody-based FRET technology. Inhibition of mGluR5 with its
selective antagonist, MTEP, abolished LPS-induced direct binding of
TLR4 with GluN1.
TLR4, a member of the Toll-like receptor family, has
been shown to serve as the main upstream sensor for response to LPS
(7). TLR4 is a type I
transmembrane protein characterized by an extracellular domain
containing leucine-rich repeats (LRRs) and a cytoplasmic tail that
contains a conserved region referred to as the Toll/IL-1 receptor
(TIR) domain. After TLR4 encounters LPS, the first signaling
pathway mediated by a pair of proteins, namely TIRAP and MyD88, is
activated from the plasma member (22,23). Subsequently, TLR4 undergoes
internalization into the endosomal network where the second
signaling pathway is triggered through the adaptors TRAM and TRIF
(24,25). Thus, LPS-induced endocytosis of
TLR4 is essential for its signaling functions. We observed that the
surface expression of TLR4 was significantly reduced at 30 min
after exposure to 1 µg/ml LPS in both N9 and EOC 20
microglial cells. At 2 h after LPS treatment, the surface
expression of TLR4 returned back to the level prior to treatment.
However, the total TLR4 expression was not altered by LPS
treatment. These results indicate that LPS treatment accelerates
TLR4 internalization and reduces its surface expression, but does
not disturb TLR4 turnover, which is in agreement with previous
studies in murine macrophages (24,26), suggesting that TLR4 undergoes
internalization in response to LPS in microglial cells.
NMDA receptors (NMDARs) are the major subtype of
glutamate-gated ion channels and are crucial for neuronal
communication. NMDARs are assembled as heterotetramers composed by
multiple subunits, which fall into three subfamilies according to
sequence homology, one GluN1 subunit, four distinct GluN2 subunits,
and two GluN3 subunits (27,28). NMDARs are typically composed of
GluN1 subunits and GluN2 subunits or a mixture of GluN2 and GluN3
subunits. The NMDA receptor is densely distributed in the brain,
and the main subunit GluN1 is ubiquitously expressed from embryonic
E14 to adulthood (29,30). We observed that GluN1 is expressed
in N9 and EOC 20 microglial cells, and its expression is not
altered by LPS treatment. NMDARs have been shown to be involved in
the LPS-induced innate immune response (31–33). NMDAR antagonists attenuate
LPS-induced fever and increase in hypothalamic hydroxyl radicals in
rabbits (34) and LPS-enhanced
cerebral infarcts in rats (33).
Accumulating evidence also indicates that LPS modulates NMDAR
subunits, including GluN1 and GluN2 expression (35–37). Our results (Fig. 1I and J) indicate that TLR4 is
co-immunoprecipitated with GluN1, the main NMDAR subunit, in
response to LPS in N9 and EOC 20 microglial cells. The
co-immunoprecipitates of TLR4 and GluN1 emerged at 30 min, reached
a maximum at 2 h and diminished at 8 h after LPS application. This
time frame is similar to LPS-induced TLR4 internalization,
suggesting the possibility that the internalized TLR4 interacts
with GluN1. The immunocytochemistry results in the present study
(Fig. 2) further verify that TLR4
co-localizes with GluN1 in response to LPS treatment. Similar to
co-immunoprecipitates, the co-localization of TLR4 and GluN1
reached a maximum at 1 h after LPS application. Antibody-based FRET
analysis (Fig. 3) revealed that
TLR4 directly binds to GluN1 in vivo in response to LPS. The
binding of TLR4 with GluN1 was not altered by destructive TLR4
missense mutations, as similar binding was observed in both N9 and
EOC 20 microglial cells. These results suggest that LPS-induced
binding of TLR4 to GluN1 is not mediated by the TLR4 pathway.
The mGluR5 has been shown to be an alternative
critical receptor in response to LPS in microglial cells (11). The mGluR5 selective antagonist
MTEP abolished LPS-triggered binding of TLR4 with GluN1, suggesting
that [Ca2+]i oscillation mediated by mGluR5
in response to LPS may be involved in this process. It has been
demonstrated that mGluR5 agonists increase GluN1 phosphorylation in
rats (17). It remains unknown
whether the phosphorylation status of GluN1 affects its binding
with TLR4, which domains of TLR4 and GluN1 are responsible for
their binding, and where the binding localizes in the microglial
cells. The investigation is still undergoing.
In conclusion, the results of the present study
demonstrated that TLR4 directly binds to GluN1 in response to LPS,
and mGluR5 modulates LPS-induced binding of TLR4 and GluN1 in N9
and EOC 20 microglial cells. Thus, GluN1 may be a potential target
for modulating LPS-induced neuroinflammation.
References
|
1
|
Goncharova LB and Tarakanov AO: Molecular
networks of brain and immunity. Brain Res Brain Res Rev.
55:155–166. 2007. View Article : Google Scholar
|
|
2
|
Gallowitsch-Puerta M and Pavlov VA:
Neuro-immune interactions via the cholinergic anti-inflammatory
pathway. Life Sci. 80:2325–2329. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garden GA and Möller T: Microglia biology
in health and disease. J Neuroimmune Pharmacol. 1:127–137. 2006.
View Article : Google Scholar
|
|
4
|
Aloisi F: The role of microglia and
astrocytes in CNS immune surveillance and immunopathology. Adv Exp
Med Biol. 468:123–133. 1999. View Article : Google Scholar
|
|
5
|
Kielian T, Mayes P and Kielian M:
Characterization of microglial responses to Staphylococcus aureus:
Effects on cytokine, costimulatory molecule, and Toll-like receptor
expression. J Neuroimmunol. 130:86–99. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hirayama M and Kuriyama M: MK-801 is
cytotoxic to microglia in vitro and its cytotoxicity is attenuated
by glutamate, other excitotoxic agents and atropine. Possible
presence of glutamate receptor and muscarinic receptor on
microglia. Brain Res. 897:204–206. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hoshino K, Takeuchi O, Kawai T, Sanjo H,
Ogawa T, Takeda Y, Takeda K and Akira S: Cutting edge: Toll-like
receptor 4 (TLR4)-deficient mice are hyporesponsive to
lipopolysaccharide: evidence for TLR4 as the Lps gene product. J
Immunol. 162:3749–3752. 1999.PubMed/NCBI
|
|
8
|
Zinchuk V, Zinchuk O and Okada T:
Quantitative colocalization analysis of multicolor confocal
immunofluorescence microscopy images: Pushing pixels to explore
biological phenomena. Acta Histochem Cytochem. 40:101–111. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zinchuk V, Wu Y, Grossenbacher-Zinchuk O
and Stefani E: Quantifying spatial correlations of fluorescent
markers using enhanced background reduction with protein proximity
index and correlation coefficient estimations. Nat Protoc.
6:1554–1567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
König P, Krasteva G, Tag C, König IR,
Arens C and Kummer W: FRET-CLSM and double-labeling indirect
immunofluorescence to detect close association of proteins in
tissue sections. Lab Invest. 86:853–864. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu F, Zhou R, Yan H, Yin H, Wu X, Tan Y
and Li L: Metabotropic glutamate receptor 5 modulates calcium
oscillation and innate immune response induced by
lipopolysaccharide in microglial cell. Neuroscience. 281:24–34.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hoffmann A, Kann O, Ohlemeyer C, Hanisch
UK and Kettenmann H: Elevation of basal intracellular calcium as a
central element in the activation of brain macrophages (microglia):
Suppression of receptor-evoked calcium signaling and control of
release function. J Neurosci. 23:4410–4419. 2003.PubMed/NCBI
|
|
13
|
Righi M, Mori L, De Libero G, Sironi M,
Biondi A, Mantovani A, Donini SD and Ricciardi-Castagnoli P:
Monokine production by microglial cell clones. Eur J Immunol.
19:1443–1448. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stansley B, Post J and Hensley K: A
comparative review of cell culture systems for the study of
microglial biology in Alzheimer's disease. J Neuroinflammation.
9:1152012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Poltorak A, He X, Smirnova I, Liu MY, Van
Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al:
Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations
in Tlr4 gene. Science. 282:2085–2088. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takeuchi O, Takeda K, Hoshino K, Adachi O,
Ogawa T and Akira S: Cellular responses to bacterial cell wall
components are mediated through MyD88-dependent signaling cascades.
Int Immunol. 12:113–117. 2000. View Article : Google Scholar
|
|
17
|
Choe ES, Shin EH and Wang JQ: Regulation
of phosphorylation of NMDA receptor NR1 subunits in the rat
neostriatum by group I metabotropic glutamate receptors in vivo.
Neurosci Lett. 394:246–251. 2006. View Article : Google Scholar
|
|
18
|
Pisani A, Calabresi P, Centonze D and
Bernardi G: Enhancement of NMDA responses by group I metabotropic
glutamate receptor activation in striatal neurones. Br J Pharmacol.
120:1007–1014. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Contractor A, Gereau RW IV, Green T and
Heinemann SF: Direct effects of metabotropic glutamate receptor
compounds on native and recombinant N-methyl-D-aspartate receptors.
Proc Natl Acad Sci USA. 95:8969–8974. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu B and Hong JS: Role of microglia in
inflammation-mediated neurodegenerative diseases: Mechanisms and
strategies for therapeutic intervention. J Pharmacol Exp Ther.
304:1–7. 2003. View Article : Google Scholar
|
|
21
|
Town T, Nikolic V and Tan J: The
microglial 'activation' continuum: From innate to adaptive
responses. J Neuroinflammation. 2:242005. View Article : Google Scholar
|
|
22
|
Latz E, Visintin A, Lien E, Fitzgerald KA,
Monks BG, Kurt-Jones EA, Golenbock DT and Espevik T:
Lipopolysaccharide rapidly traffics to and from the Golgi apparatus
with the toll-like receptor 4-MD-2-CD14 complex in a process that
is distinct from the initiation of signal transduction. J Biol
Chem. 277:47834–47843. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kagan JC and Medzhitov R:
Phosphoinositide-mediated adaptor recruitment controls Toll-like
receptor signaling. Cell. 125:943–955. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kagan JC, Su T, Horng T, Chow A, Akira S
and Medzhitov R: TRAM couples endocytosis of Toll-like receptor 4
to the induction of interferon-beta. Nat Immunol. 9:361–368. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tanimura N, Saitoh S, Matsumoto F,
Akashi-Takamura S and Miyake K: Roles for LPS-dependent interaction
and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys
Res Commun. 368:94–99. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Józefowski S, Czerkies M, Sobota A and
Kwiatkowska K: Determination of cell surface expression of
Toll-like receptor 4 by cellular enzyme-linked immunosorbent assay
and radiolabeling. Anal Biochem. 413:185–191. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Traynelis SF, Wollmuth LP, McBain CJ,
Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ and
Dingledine R: Glutamate receptor ion channels: Structure,
regulation, and function. Pharmacol Rev. 62:405–496. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cull-Candy SG and Leszkiewicz DN: Role of
distinct NMDA receptor subtypes at central synapses. Sci STKE.
2004:re162004.PubMed/NCBI
|
|
29
|
Watanabe M, Inoue Y, Sakimura K and
Mishina M: Developmental changes in distribution of NMDA receptor
channel subunit mRNAs. Neuroreport. 3:1138–1140. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Monyer H, Burnashev N, Laurie DJ, Sakmann
B and Seeburg PH: Developmental and regional expression in the rat
brain and functional properties of four NMDA receptors. Neuron.
12:529–540. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Glezer I, Zekki H, Scavone C and Rivest S:
Modulation of the innate immune response by NMDA receptors has
neuropathological consequences. J Neurosci. 23:11094–11103.
2003.PubMed/NCBI
|
|
32
|
Maroso M, Balosso S, Ravizza T, Liu J,
Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi
AA, et al: Toll-like receptor 4 and high-mobility group box-1 are
involved in ictogenesis and can be targeted to reduce seizures. Nat
Med. 16:413–419. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cho GS, Lee JC, Ju C, Kim C and Kim WK:
N-Methyl-D-aspartate receptor antagonists memantine and MK-801
attenuate the cerebral infarct accelerated by intracorpus callosum
injection of lipopolysaccharides. Neurosci Lett. 538:9–14. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kao CH, Kao TY, Huang WT and Lin MT:
Lipopolysaccharide- and glutamate-induced hypothalamic hydroxyl
radical elevation and fever can be suppressed by
N-methyl-D-aspartate-receptor antagonists. J Pharmacol Sci.
104:130–136. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Weaver-Mikaere L, Gunn AJ, Mitchell MD,
Bennet L and Fraser M: LPS and TNF alpha modulate AMPA/NMDA
receptor subunit expression and induce PGE2 and glutamate release
in preterm fetal ovine mixed glial cultures. J Neuroinflammation.
10:1532013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Harré EM, Galic MA, Mouihate A, Noorbakhsh
F and Pittman QJ: Neonatal inflammation produces selective
behavioural deficits and alters N-methyl-D-aspartate receptor
subunit mRNA in the adult rat brain. Eur J Neurosci. 27:644–653.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yeh SH, Hung JJ, Gean PW and Chang WC:
Hypoxia-inducible factor-1alpha protects cultured cortical neurons
from lipopolysaccharide-induced cell death via regulation of NR1
expression. J Neurosci. 28:14259–14270. 2008. View Article : Google Scholar : PubMed/NCBI
|