Introduction
Hyperglycemia, an important characteristic of
diabetes mellitus, is considered to be a dangerous factor of
diabetic complications. It is able to induce dysfunction of the
vascular endothelium through various pathophysiological mechanisms,
which are associated with atherosclerosis, leading to target
organic impairment, such as blindness (1), kidney failure (2), and cardiovascular diseases (3). Hence, it is crucially important to
protect endothelial cells against hyperglycemia-induced injury.
Numerous processes have been demonstrated to be
associated with injury induced by hyperglycemia, including
apoptosis (4), oxidative stress
(5), and mitochondrial
dysfunction (5). Traditionally,
cell death was divided into three categories: Apoptosis, necrosis
and autophagy. Apoptosis is an alternative term for programmed cell
death, which allows cells to commit suicide according to a
specifically designated program in the absence of inflammation.
Autophagy is another mechanism, which leads to formation of
autophagosomes in a dying cell to maintain homeostasis of the
organism. By contrast, necrosis is regarded as a passive and
unregulated form of cell death, which is characterized by early
plasma membrane destruction and swelling of the intracellular
organelles. However, necroptosis (also termed 'programmed
necrosis') (6), a newly
identified form of programmed cell death, has broken the
traditionally understood concept of cell death. In contrast with
apoptosis, necroptosis operates according to a caspase-independent
mechanism. Receptor-interacting protein (RIP)1 and RIP3 serve a
critical role as signal transducers in the pathway of necroptosis.
Recently, significant progress has been made in the study of
necroptosis. RIP3-mediated necroptosis has now been determined to
be a common cell-death pathway implicated in cytokine-, virus-, and
genotoxic stress-induced cell death, and has also been identified
to participate in a variety of pathological conditions, including
the immune response (7), multiple
tissue injury (8), the retinal
ischemia-reperfusion injury (9),
and myocardial infarction (10).
However, the role of necroptosis in hyperglycemia-induced injury of
endothelial cells has yet to be fully elucidated.
Hydrogen sulfide (H2S), previously
regarded as a poisonous gas emitted from rotten eggs, has been
demonstrated to be an endogenously produced, labile diffusible
mediator with multiple roles in the cardiovascular system
associated with health and disease (11–19), including vasodilation (11,12) and the stimulation of angiogenesis
(13). In recent years, more and
more attention has been paid to the endothelial protective effect
of H2S against injuries. Mani et al (20) reported that decreased endogenous
production of H2S accelerates atherosclerosis. Upon
application of H2S, the progression of atherosclerosis
was inhibited in fat-fed apoE−/− mice (21). Notably, it was shown that low
levels of H2S in the blood of patients with diabetes and
streptozotocin-treated diabetic rats may be associated with
vascular inflammation (22).
Absence of cystathionine γ-lyase, a synthase of H2S,
exacerbates hyperglycemic endothelial cell dysfunction (6). In addition, exogenous H2S
was shown to protect vascular endothelium against high glucose
(HG)-induced injuries, including an overabundant generation of
reactive oxygen species (ROS), a decrease in cell viability, and
DNA injury, by preserving mitochondrial function (6). Since cardiac RIP3 expression was
shown to be increased in diabetic rats (23), and RIP3 is involved in
atherosclerosis development (24), the present study aimed to explore
the influence of HG on the expression level of RIP3, and the role
of necroptosis in the HG-induced injury, and to examine whether
exogenous H2S protects against HG-induced injury by
inhibiting necroptosis in human umbilical vein endothelial cells
(HUVECs).
Materials and methods
Materials and reagents
Sodium hydrogen sulfide (NaHS), necrostatin-1
(Nec-1), Z-VAD-FMK, Hoechst 33258, 2′,7′-dichlorofluorescein
diacetate (DCFH-DA) and rhodamine 123 (Rh123) were purchased from
Merck KGaA (Darmstadt, Germany). Anti-cleaved caspase-3 antibody
(cat. no. 9664) was procured from Cell Signaling Technology, Inc.
(boston, MA, USA); anti-RIP3 (ab56164) was purchased from Abcam
(Cambridge, UK); and the anti-caspase-9 (10380-1-AP) and anti-GAPDH
(10494-1-AP) antibodies were purchased from Proteintech Group, Inc.
(Wuhan, China). The Cell Counting kit-8 (CCK-8) was obtained from
Dojindo Laboratories (Kumamoto, Japan). Fetal bovine serum (FBS)
and Gibco BRL™ Dulbecco's modified Eagle's medium (DMEM) were
obtained from Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Horseradish peroxidase-conjugated secondary antibody and the
bicinchoninic acid (BCA) protein assay kit were obtained from
KangChen Bio-tech, Inc. (Shanghai, China). Enhanced
chemiluminescence solution was purchased from Nanjing KeyGen
Biotech Co., Ltd. (Nanjing, China). Lysis buffer was purchased from
the Beyotime Institute of Biotechnology (Shanghai, China), and the
HUVECs were supplied by Guangzhou Jiniou Co., Ltd. (Guangzhou,
China).
Cell culture and treatments
HUVECs were cultured in DMEM medium at a
concentration of 1×106 cells/ml, supplemented with 10%
FbS at 37°C under an atmosphere of 5% CO2. To explore
the protective effects of H2S on HG-induced injury,
cells were pretreated with 400 µM NaHS (a well-known
H2S donor) for 30 min prior to HG treatment. To
determine the role of necroptosis in HG-induced injury, HUVECs were
pre-treated with Nec-1 (an inhibitor of necroptosis) prior to HG
treatment. The culture medium was replaced with fresh medium every
2–3 days, and expanded to new culture vessels when the cells had
reached ~80% confluence.
Cell viability assay
HUVECs were cultured in 96-well plates at a
concentration of 1×104 cells/ml. After incubation at
37°C for 24 h and receiving the specific treatments (as detailed in
the results section), the cells were washed with phosphate-buffered
saline (PBS), and 10 µl CCK-8 solution was added to each
well at a 10% dilution for an ~2 h incubation in the incubator.
Absorbance was measured at 450 nm with a microplate reader
(Multiskan MK3 Microplate reader; Thermo Fisher Scientific, Inc.).
The mean optical density (OD) of three wells in the indicated
groups was used to calculate the percentage of cell viability
according to the following formula: Percentage of cell viability
(%) = (ODtreatment group/ODcontrol group)
×100%. The experiment was repeated 5 times.
Hoechst 33258 nuclear staining for the
assessment of apoptosis
Apoptotic cell death was assessed using the Hoechst
33258 staining method. The HUVECs were plated in 60 mm dishes at a
density of 1×106 cells/well. After having performed the
indicated treatments, the cells were harvested and fixed with
paraformaldehyde in 0.1 mol/l PBS for 10 min. After rinsing 5 times
with PBS, the nuclear DNA was stained with 5 mg/ml Hoechst 33258
dye for 10 min, before being rinsed with PBS and then visualized
under a fluorescence microscope (bx50-FLA; Olympus Corporation,
Tokyo, Japan). Viable HUVECs exhibited a uniform blue fluorescence
throughout the nucleus, whereas apoptotic cells revealed fragmented
and condensed nuclei. The experiment was repeated 5 times.
Measurement of intracellular ROS
generation
Intracellular ROS generation was measured by
oxidation of DCFH-DA to fluorescent 2′,7′-dichlorofluorescein
(DCF). The HUVECs were cultured on a slide in DMEM, supplemented
with 10% FbS at 37°C under an atmosphere of 5% CO2.
Following the different treatments, the slides were washed 3 times
with PBS. DCFH-DA solution (10 µM) in serum-free medium was
added to the slides, and the cells were subsequently incubated at
37°C for 30 min in an incubator. The cells were washed 5 times with
PbS, and DCF fluorescence was measured over the entire field of
vision by using a fluorescence microscope connected to an imaging
system (bx50-FLA; Olympus Corporation). The mean fluorescence
intensity (MFI) from five random fields was measured using ImageJ
1.47i software, and the MFI was used as an index of the amount of
ROS. The experiment was performed 5 times.
Examination of the mitochondrial membrane
potential (MMP)
MMP was assessed using a fluorescent dye, Rh123. The
depolarization of MMP results in a loss of MMP, and a decrease in
green fluorescence. The cells were cultured on a slide with DMEM,
supplemented with 10% FbS at 37°C under an atmosphere of 5%
CO2. Following the indicated treatments, the slides were
washed 3 times with PBS. The cells were incubated with 1 µM
Rh123 at 37°C for 30 min in an incubator, and then washed briefly 5
times with PBS. Fluorescence was subsequently measured over the
whole field of vision using a fluorescence microscope connected to
an imaging system (bx50-FLA; Olympus Corporation). The MFI of Rh123
from five random fields was analyzed using ImageJ 1.47i software,
and the MFI was used as an index of the levels of MMP. The
experiment was performed 5 times.
Western blot analysis assay
After the indicated treatments, the HUVECs were
harvested and lysed with cell lysis for 30 min at 4°C. The total
protein in the supernatant was quantified with a BCA protein assay
kit. Total protein (30 µg from each sample) was separated
using 12% SDS-PAGE. The protein in the gel was transferred to a
polyvinylidene difluoride membrane. The membrane was blocked with
5% fat-free milk for 60 min at room temperature, and then incubated
with primary antibodies specific to anti-cleaved caspase-3
(1:1,000), anti-caspase-9 (1:2,500), RIP3 (1:1,000), or GAPDH
(1:5,000), with slow agitation at 4°C overnight. Following three
washes with TbS/Tween 20 (TBS-T), the proteins were subsequently
incubated with the secondary antibodies for 60 min at room
temperature. Following three washes with TBS-T for 15 min,
membranes were visualized by enhanced chemiluminescence and
exposure to X-ray films. To quantify protein expression, the X-ray
films were scanned and analyzed using ImageJ 1.47i software. Each
experiment was repeated 3 times.
Statistical analysis
All data are presented as the means ± SEM.
Differences between groups were analyzed using one-way analysis of
variance using SPSS 20.0 software (IBM Corp., Armonk, NY, USA),
followed by the least significant difference (LSD) post hoc
comparison test. P<0.05 was considered to indicate a
statistically significant difference.
Results
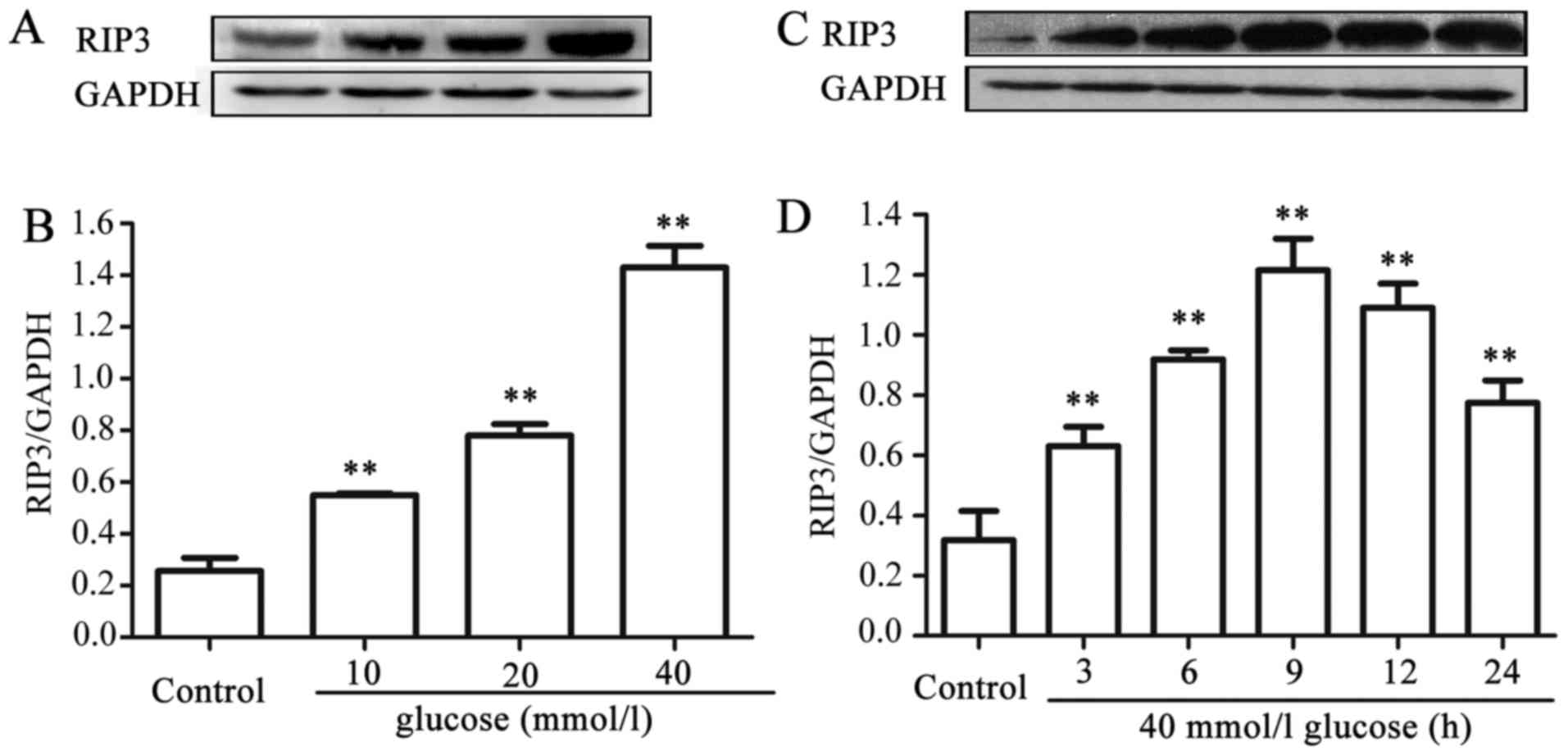
HG upregulates the expression level of
RIP3 in HUVECs
In order to explore the effects of HG on necroptosis
in HUVECs, dose- and time-response experiments to determine the
effects of different concentrations of glucose on the expression
level of RIP3, a kinase promoting necroptosis, were performed. As
shown in Fig. 1A and B, after the
cells were exposed to different concentrations (10, 20 and 40 mM)
of HG for 24 h, the expression levels of RIP3 were increased in a
dose-dependent manner, reaching a peak at 40 mM glucose. Therefore,
glucose at 40 mM was used in the subsequent time-response
experiment (Fig. 1C and D). The
cells were exposed to 40 mM HG for 0, 3, 6, 9, 12 and 24 h,
respectively. Following exposure of the cells to HG for 3 h, the
expression level of RIP3 began to increase significantly
(P<0.01), and the maximum increase in expression level of RIP3
was observed at 9 h. These results suggested that HG induces
necroptosis in HUVECs.
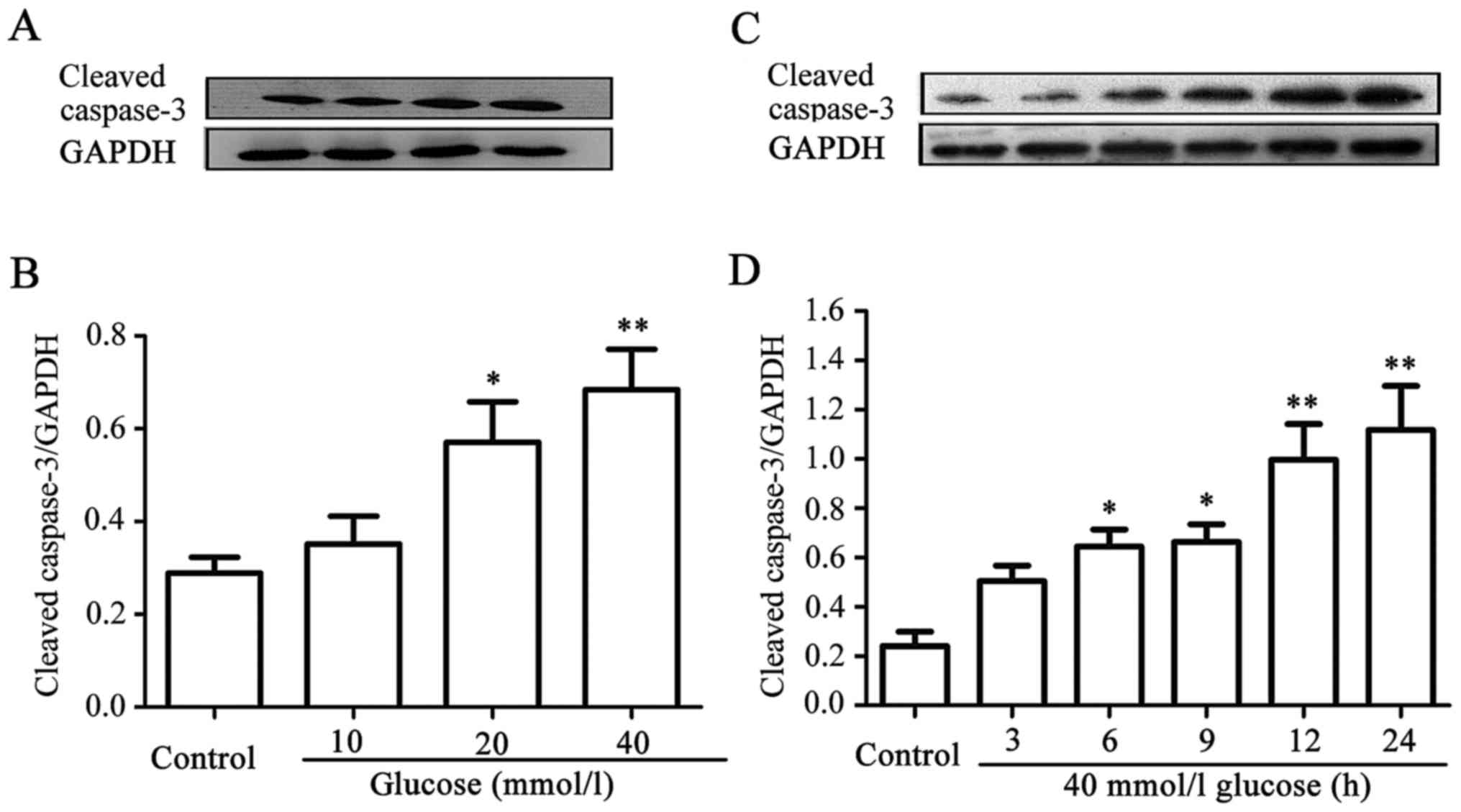
HG enhances the expression level of
cleaved caspase-3 in HUVECs
Similarly, dose-and time-response experiments to
explore the effects of different concentrations of glucose on the
expression levels of cleaved caspase-3 in HUVECs were performed.
When the cells were treated with 10, 20, and 40 mM glucose for 24
h, the expression level of cleaved caspase-3 gradually increased,
peaking at 40 mM glucose (Fig. 2A and
B). Furthermore, after the cells had been exposed to 40 mM HG
for 3, 6, 9, 12, and 24 h, respectively, the expression level of
cleaved caspase-3 began to evidently increase at 6 h, reaching a
maximum level at 24 h (Fig. 2C and
D). These data indicated that HG induces apoptosis in
HUVECs.
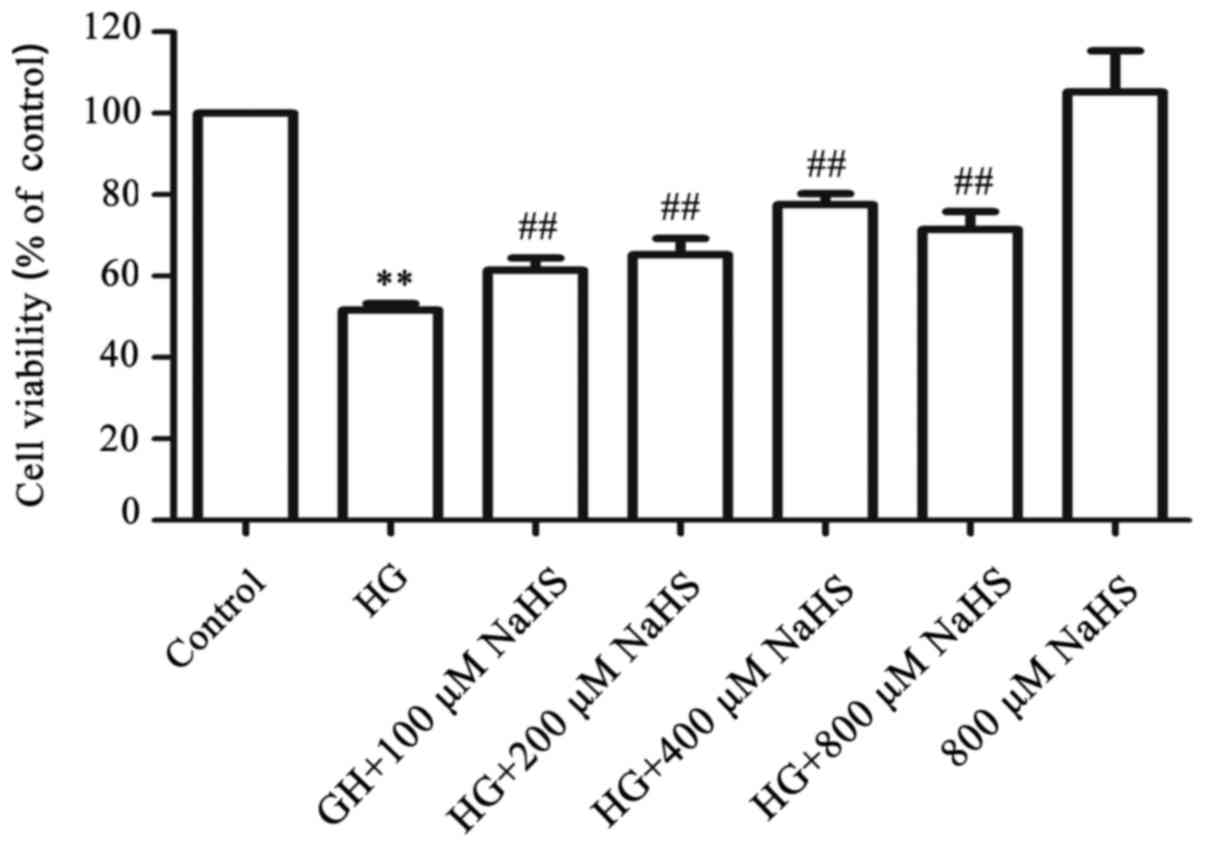
Exogenous H2S attenuates the
HG-induced cytotoxicity in HUVECs
To examine the cytoprotective effects of
H2S against HG-induced cytotoxicity in HUVECs, a
dose-response study with different concentrations of NaHS (a donor
of H2S) was performed. As shown in Fig. 3, exposure of HUVECs to 40 mM
glucose (HG) for 24 h induced considerable cytotoxicity, leading to
a decrease in cell viability to 51.61±1.63% (P<0.01) compared
with the control group. However, treatment of the cells with NaHS
at 100, 200, 400, and 800 µM, respectively, for 30 min prior
to exposure to HG markedly ameliorated the HG-induced cytotoxicity,
resulting in an increase in cell viability. The maximum protective
effect was observed with 400 µM NaHS. When administered
alone, 800 µM NaHS failed to markedly affect the cell
viability of HUVECs. Based on these results, the cells were treated
with 400 µM NaHS for 30 min prior to exposure to HG in the
following experiments.
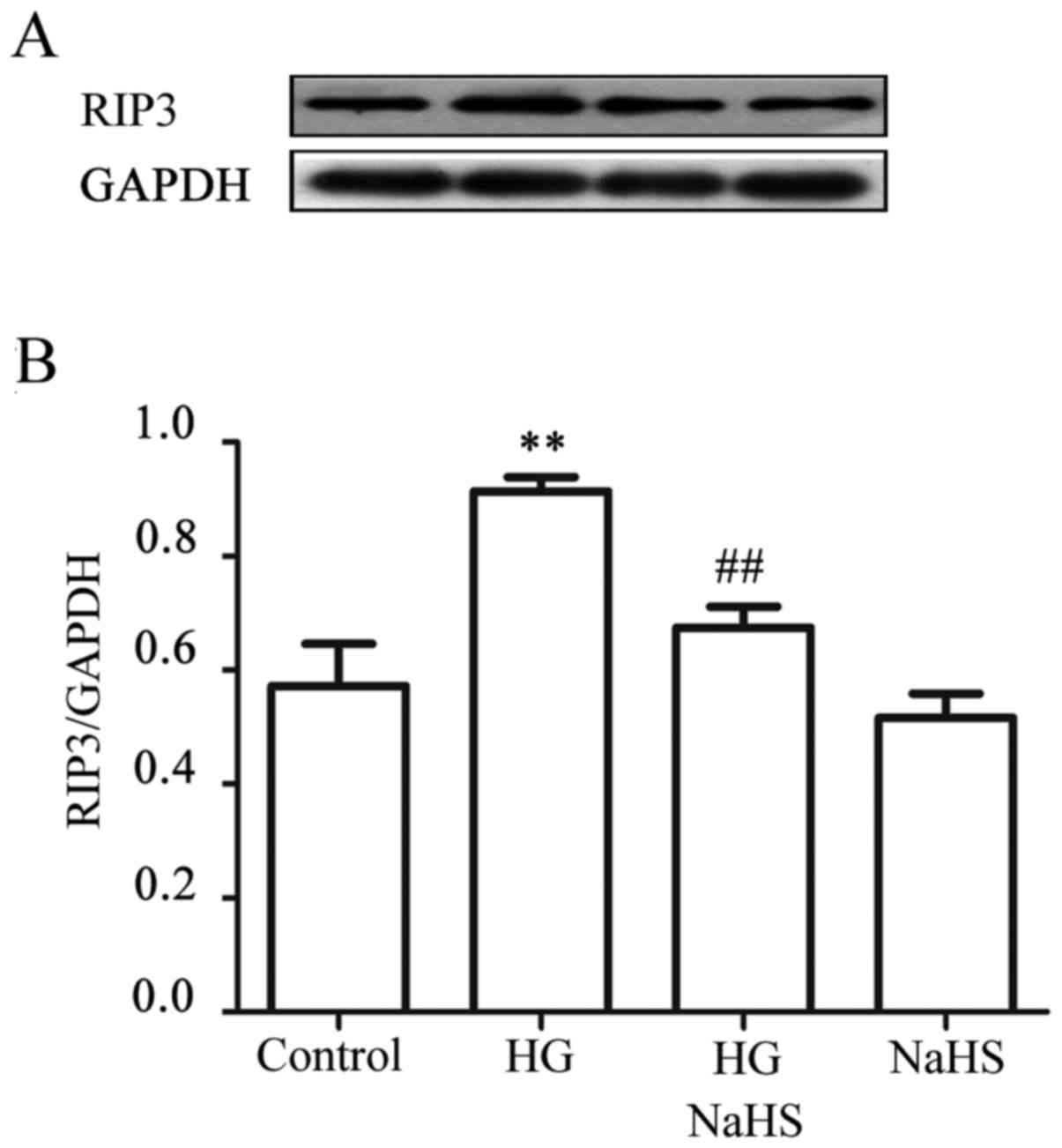
Exogenous H2S alleviates the
HG-induced increase in expression of RIP3 in HUVECs
To observe the effect of exogenous H2S on
HG-induced necroptosis, HUVECs were treated with 400 µM NaHS
for 30 min prior to exposure to HG for 9 h. As shown in Fig. 4, treatment of the cells with 40 mM
HG for 9 h markedly increased the expression level of RIP3.
However, treatment of the cells with 400 µM NaHS for 30 min
prior to exposure to HG significantly reduced the increased
expression of RIP3 induced by HG. NaHS at 400 µM alone did
not affect the basal expression level of RIP3. These findings
revealed that exogenous H2S inhibits the HG-induced
necroptosis in HUVECs.
Exogenous H2S and a
necroptosis inhibitor attenuate HG-induced oxidative stress in
HUVECs
As shown in Fig. 5Ab
and B, exposure of the cells to HG for 24 h markedly enhanced
ROS generation. However, the increase in ROS generation was
attenuated by pre-treatment of the cells with 400 µM NaHS
for 30 min prior to exposure to HG (Fig. 5Ac and B). Similarly, treatment of
the cells with 100 µM Nec-1 (an inhibitor of necroptosis)
for 24 h prior to exposure to HG also clearly blocked the increase
in ROS generation (Fig. 5Ad and
B). When administered alone, 400 µM NaHS (Fig. 5Ae and B) or 100 µM Nec-1
(Fig. Af and B) did not affect the basal ROS generation. These
results indicated that exogenous H2S protects HUVECs
against the HG-induced oxidative stress by inhibiting
necroptosis.
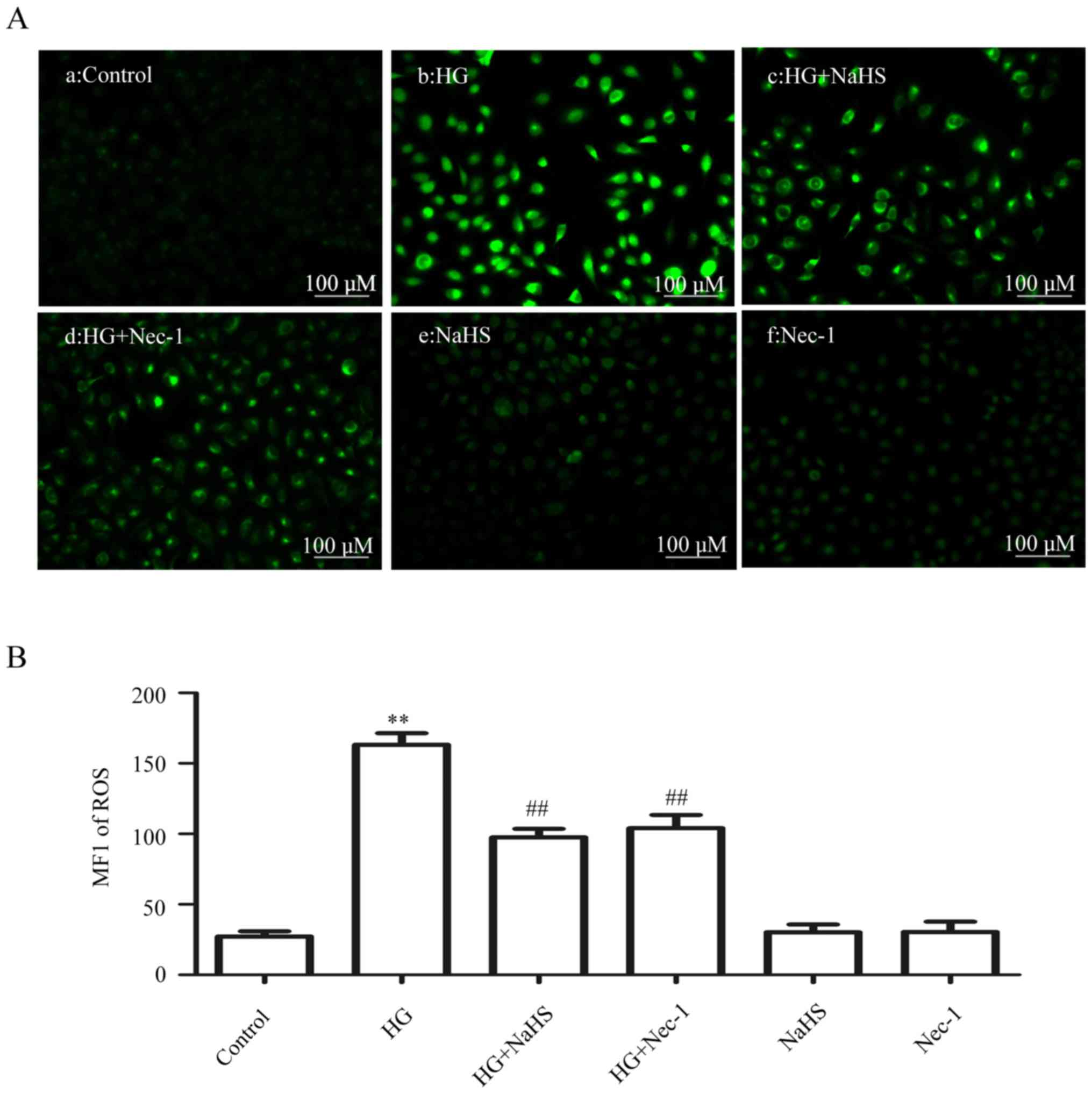 | Figure 5Exogenous H2S attenuates
HG-induced ROS generation by inhibiting necroptosis in HUVECs.
(Aa–f) After the indicated treatments, the intracellular ROS level
was assessed by DCFH-DA staining followed by photofluorography.
(Aa) The control group. HUVECs were treated with (Ab) 40 mM glucose
for 24 h, (Ac) 400 µM NaHS for 30 min, or (Ad) with 100
µM Nec-1 for 24 h, prior to exposure to HG. (Ae) HUVECs were
treated with 400 µM NaHS for 30 min, followed by 24 h of
culture. (Af) HUVECs were treated with 100 µM Nec-1 for 24
h, followed by 24 h of culture. (B) Quantitative analysis for the
MFI of DCFH-DA in (Aa–f) with the ImageJ 1.41o software. Data are
presented as mean ± SEM (n=5). **P< 0.01 vs. the
control group; ##P<0.01 vs. the HG-treated group.
H2S, hydrogen sulfide; HG, high glucose; ROS, reactive
oxygen species; NaHS, sodium hydrogen sulfide; MFI, mean
fluorescence intensity; Nec-1, necrostatin-1; HUVECs, human
umbilical vein endothelial cells; DCFH-DA,
2′,7′-dichlorofluorescein diacetate. |
Exogenous H2S and a
necroptosis inhibitor lower the HG-induced dissipation of MMP in
HUVECs
As shown in Fig.
6, treatment of cells with HG for 24 h induced a marked
dissipation of the MMP (P<0.01) compared with the control group
(Fig. 6Aa and B). However,
dissipation of the MMP was ameliorated by treatment of the cells
with 400 µM NaHS for 30 min (Fig. 6Ac and B) or 100 µM Nec-1
for 24 h (Fig. 6Ad and B) prior
to exposure to HG for 24 h. When administered alone, 400 µM
NaHS (Fig. 6Ae and B) or 100
µM Nec-1 (Fig. 6Af and B)
failed to elicit a loss of MMP. These data demonstrated that
exogenous H2S protects HUVECs against the HG-induced
dissipation of MMP by inhibiting necroptosis.
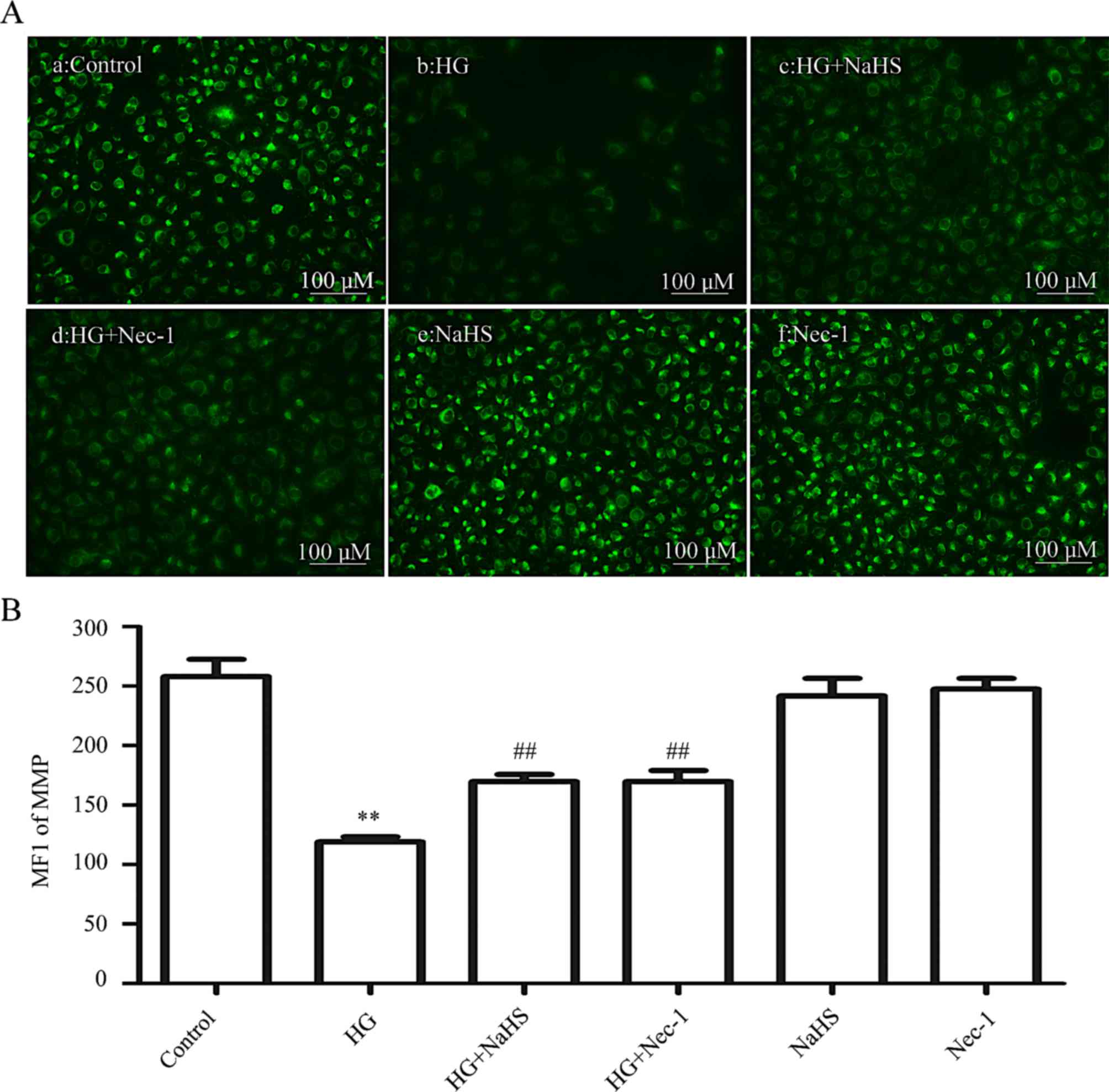 | Figure 6Exogenous H2S alleviates
the HG-induced reduction in the MMP by inhibiting necroptosis in
HUVECs. (Aa–f) After the indicated treatments, the MMP was assessed
by Rh123 staining, followed by photofluorography. (Aa) The control
group. HUVECs were treated with (Ab) 40 mM glucose for 24 h, (Ac)
400 µM NaHS for 30 min, or (Ad) with 100 µM Nec-1 for
24 h prior to exposure to HG. (Ae) HUVECs were treated with 400
µM NaHS for 30 min, followed by 24 h of culture. (Af) HUVECs
were treated with 100 µM Nec-1 for 24 h, followed by 24 h of
culture. (B) Quantitative analysis for the MFIs of Rh123 in (Aa–f)
using ImageJ 1.41i software. Data are presented as the means ±
standard error of the mean (n=5). **P<0.01 vs. the
control group; ##P<0.01 vs. the HG-treated group.
H2S, hydrogen sulfide; HG, high glucose; MMP,
mitochondrial membrane potential; Rh123, rhodamine 123; ROS,
reactive oxygen species; Nec-1, necrostatin-1; NaHS, sodium
hydrogen sulfide; MFI, mean fluorescence intensity. |
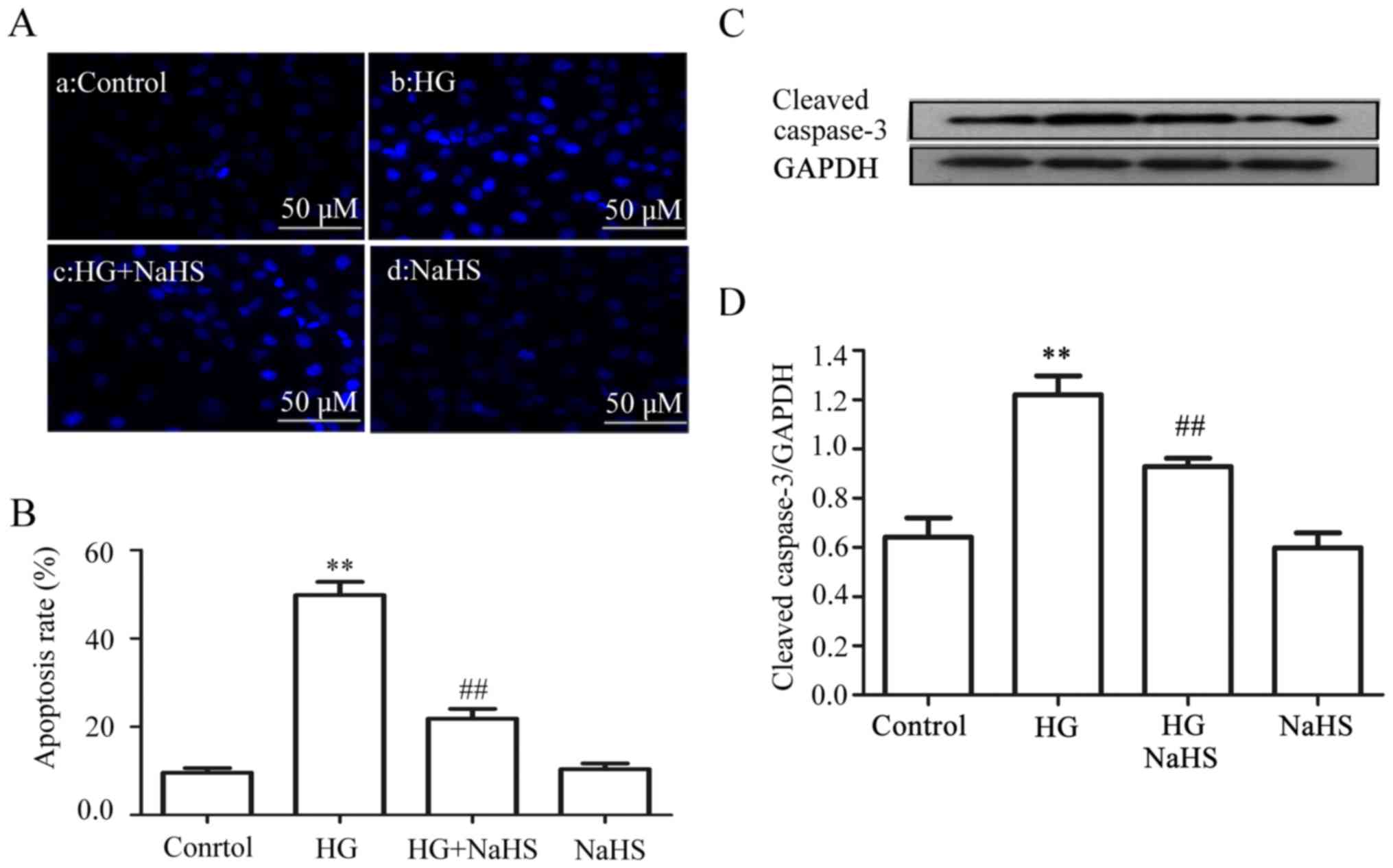
Exogenous H2S reduces
HG-induced apoptosis in the HUVECs
The data obtained from the experiment of Hoechst
33258 staining followed by photofluorography revealed that exposure
of the cells to HG for 24 h markedly increased the number of
apoptotic cells (Fig. 7Ab and B)
(P<0.01) compared with the control group (Fig. 7Aa and B). However, the increased
number of apoptotic cells was reduced by treatment of the cells
with 400 µM NaHS for 30 min prior to exposure to HG
(Fig. 7Ac and B). Similarly,
pre-treatment with 400 µM NaHS also inhibited the HG-induced
increase in the expression level of cleaved caspase-3 (Fig. 7C and D). NaHS at 400 µM
alone did not affect the number of apoptotic cells or the basal
expression level of cleaved caspase-3 (Fig. 7Ad, B–D).
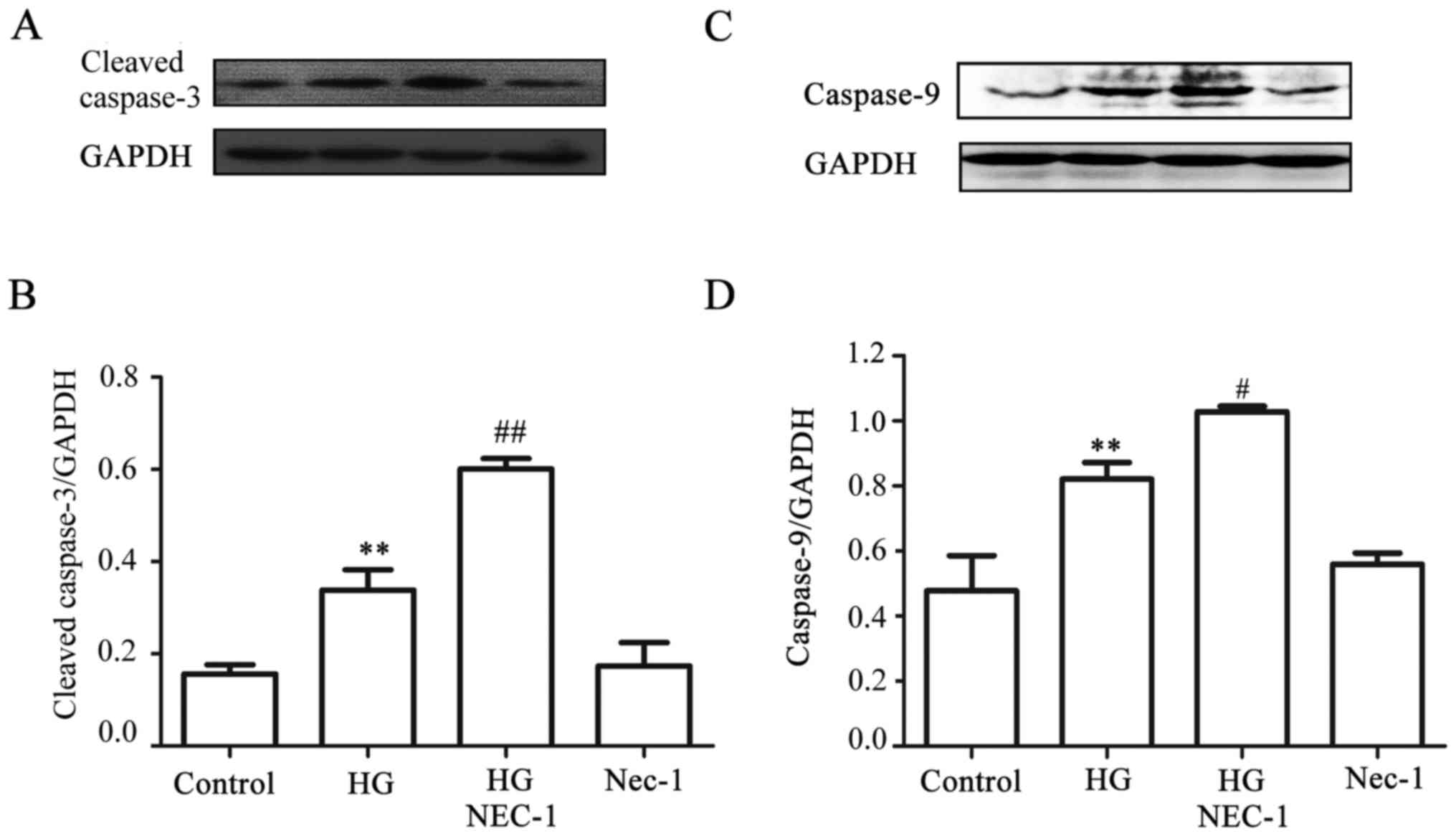
A negative interaction exists between
necroptosis and apoptosis in the HG-treated HUVECs
Since it has been reported that Nec-1, an inhibitor
of necroptosis, converts shikonin-induced necroptosis into
apoptosis in HL60 cells (25), it
appeared reasonable to explore whether, in the presence of Nec-1,
HG-induced necroptosis was reverted to apoptosis in HUVECs. As
indicated in Fig. 8A and C,
exposure of the cells to HG for 24 h markedly increased the
expression levels of cleaved caspases-3 and -9. Of note, treatment
of the cells with 100 µmol Nec-1 for 24 h prior to exposure
to HG further promoted the increase in the expression of cleaved
caspases-3 and -9. When administered alone, 100 µmol Nec-1
did not affect the basal expression level of cleaved caspases-3 and
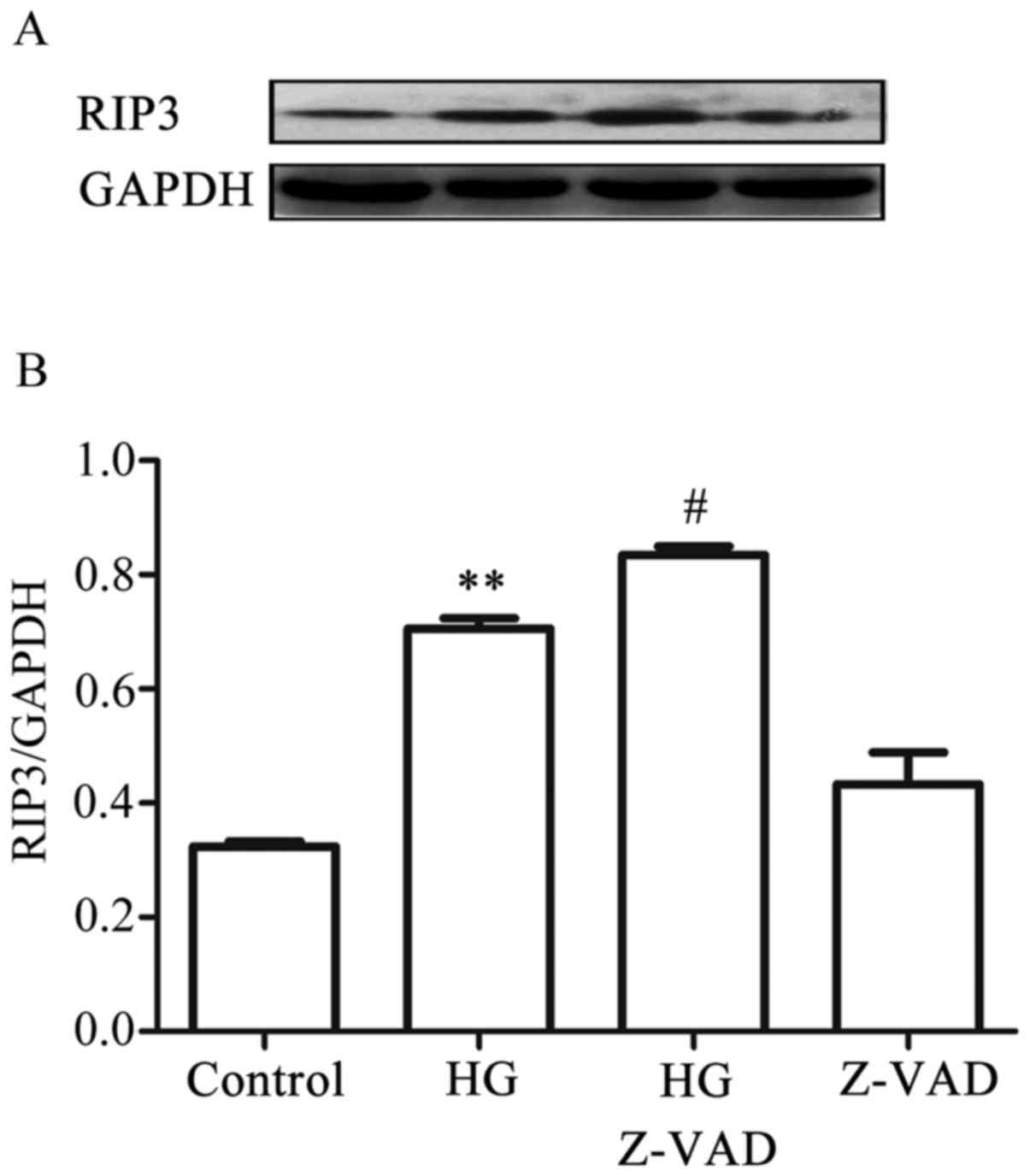
-9. On the other hand, treatment of the cells with 20 µmol
Z-VAD-FMK, a pan-caspase inhibitor, for 30 min prior to exposure to
HG for 9 h further promoted the increased expression level of RIP3
induced by HG. When administered alone, Z-VAD-FMK did not affect
the basal expression level of RIP3 (Fig. 9A and B). Collectively, these
results suggested that there was a negative interaction between
necroptosis and apoptosis in the HG-treated HUVECs: On the one
hand, necroptosis inhibited apoptosis, whereas on the other,
apoptosis suppressed necroptosis.
Discussion
Necroptosis (programmed necrosis) represents a newly
identified mechanism of cell death, combining features of both
apoptosis and necrosis. Increasing evidence has indicated the
involvement of necroptosis in a variety of pathological conditions,
including diabetic cardiac injury (23) and development of atherosclerosis
(24). However, the roles of
necroptosis in HG-induced vascular endothelial injury, and in the
protection afforded by exogenously added H2S are, at
present, unclear. The present study clearly demonstrated the
following effects of HG-treatment of the HUVECs: i) HG
significantly enhanced the expression level of RIP3, a kinase
promoting necroptotic cell death; ii) Nec-1, an inhibitor of
necroptosis, attenuated the HG-induced injury, including ROS
generation and a loss of MMP; iii) a negative interaction between
necroptosis and apoptosis was observed; on the one hand, Nec-1
increased the expression level of cleaved caspase-3, whereas on the
other, Z-VAD-FMK, a pan-caspase inhibitor, upregulated the
expression level of RIP3; iv) exogenous H2S ameliorated
the HG-induced increase in the expression level of RIP3 and
endothelial injury, leading to an increase in cell viability and a
decrease in the number of apoptotic cells and the expression level
of cleaved caspase-3, ROS generation, as well as dissipation of
MMP. Taken together, the findings of the present study have
provided, to the best of our knowledge, the first evidence that
necroptosis mediates HG-induced injury, and that the inhibition of
necroptosis contributes to the protective effect of exogenous
H2S against HG-induced injury in HUVECs.
Hyperglycemia is a risk factor associated with
vascular dysfunction, resulting in various vascular diseases, which
may be a contributor towards morbidity and mortality in diabetic
patients (26,27). Thus, further exploration of the
mechanisms underlying hyperglycemia-induced vascular endothelial
injury is very important for the prevention and treatment of
diabetic complications, such as retinopathy (28) and nephropathy (29). Multiple factors have been revealed
to contribute towards hyperglycemia-elicited vascular endothelial
damage, for example, ROS generation (5), mitochondrial damage (5), and cell death, including apoptosis
and necrosis (30). Recently, a
novel mechanism termed necroptosis or 'programmed necrosis' has
been suggested as another critical mediator of cell death in the
heart (31). In vitro
studies have demonstrated that tumor necrosis factor-α-dependent
formation of a complex between RIP1 and RIP3 is an important step
for the induction of necroptosis (32,33). In this process, RIP3 appears to
exert a key role, controlling RIP1 phosphorylation (33,34). However, although a recent study
has indicated that RIP3 is overexpressed in diabetic myocardial
tissue (23), the effect of
hyperglycemia on RIP3 in the vascular endothelial cells remains
unclear. To investigate this, dose- and time-response experiments
to determine the expression level of RIP3 were performed in the
HG-treated HUVECs. The data from these experiments revealed that
exposure of the cells to HG significantly upregulated the
expression level of RIP3, suggesting the induction of necroptosis
in the HG-treated HUVECs. Combining our results with the findings
reported by Liu et al (23), it was revealed that hyperglycemia
may be a strong stimulus for inducing necroptosis in the
cardiovascular tissues.
That necroptosis has been demonstrated to be
implicated in ischemia-induced cardiac lesions (10,35) and the development of
atherosclerosis (24) inspired us
to explore the role of necroptosis in HG-triggered injury in the
endothelial cells. Consistently with a previously published study
(6), the results of the present
study demonstrated that exposure of HUVECs to HG induced multiple
injuries, as indicated by a decrease in cell viability and an
increase in the number of apoptotic cells, the expression level of
cleaved caspase-3, ROS generation, as well as dissipation of MMP.
However, treatment of the cells with Nec-1 (an inhibitor of
necroptosis) prior to exposure to HG markedly attenuated HG-induced
injury, leading to a decrease in ROS generation, as well as a loss
of MMP. These results suggested that necroptosis is involved in
HG-induced injury in HUVECs, which may be a novel mechanism
responsible for hyperglycemia-induced vascular endothelial
injury.
It has been indicated previously that necroptosis
may function as a cellular 'backup' mechanism to ensure the
elimination of damaged cells under certain conditions when
apoptosis is inhibited (6,36).
RIP3 has been reported to participate in mediation of apoptosis and
necrosis (37). In addition,
results from a recent study revealed that the presence of Nec-1
shifts shikonin-induced necroptosis to apoptosis (25). Based on other published studies
(6,36–38), the present study aimed to explore
further the interaction between necroptosis and apoptosis in the
HG-treated HUVECs. The results demonstrated that pre-treatment with
Z-VAD-FMK (an inhibitor of caspase) prior to the exposure of HUVECs
to HG may markedly enhance the expression level of RIP3. It is
noteworthy that pre-treatment with Nec-1 prior to exposure of the
cells to HG clearly upregulated the expression of cleaved
caspases-3 and -9. These findings suggested that there is a
negative interaction between necroptosis and apoptosis in the
HG-treated HUVECs. The results of the present study are supported
by other recent studies (6,36,38). However, Zhang et al
(37) demonstrated that Nec-1
attenuates the expression level of cleaved caspase-3 in
11′-deoxyverticillin A (C42)-treated human colon cell lines. The
reasons underlying this different conclusion are complex, although
one possible explanation may be concerned with the different
experimental models and different cell lines used.
Another novel finding of the present study is
associated with the contribution of necroptosis to the protective
effect of exogenous H2S against HG-induced injury in
HUVECs. H2S, as an endogenously produced gas
transmitter, is cardiovascular-protective (6,11–22,38,39). Accumulating evidence has indicated
that exogenous H2S protects cardiac cells against the
HG-induced injury (16,18). H2S replacement also
improves endothelial function in hyperglycemic endothelial cells
and in diabetic rats (5). In
agreement with a previously published study (5), the findings of the present study
demonstrated that exposure of HUVECs to HG induced multiple
injuries, including cytotoxicity, an increase in the number of
apoptotic cells and the expression level of cleaved caspase-3, ROS
generation, as well as a loss of MMP. However, exogenous
H2S protected the cells against the above injuries.
Importantly, the results of the present study demonstrated that
exogenous H2S markedly alleviated the increased
expression of RIP3 by HG, suggesting an inhibitory effect of
exogenous H2S on HG-induced necroptosis. Also taking
into consideration with these results our finding that necroptosis
mediates HG-induced HUVEC injury, the present study supports a
novel hypothesis that inhibition of necroptosis may be one of the
key mechanisms underlying the protective effect of H2S
against the HG-induced endothelial injury.
In conclusion, the present study has highlighted the
role of necroptosis in HG-induced endothelial cell injury, and its
potential clinical significance. Hence, necroptosis may become a
novel therapeutic target when establishing treatment strategies.
Crucially, it has been demonstrated, to the best of our knowledge
for the first time, that exogenous H2S protects HUVECs
against HG-induced injury by inhibiting the necroptosis pathway.
Furthermore, in spite of the identification of the interaction
between necroptosis and apoptosis, it may not be assumed that the
inhibition of necroptosis or apoptosis does not mean prevention of
the cells from mortality. Rather, it has been demonstrated that
H2S may inhibit both apoptosis and necroptosis,
suggesting that it is a favorable protective agent. Thus, exogenous
H2S may be a major factor to take into consideration
when developing novel therapeutic strategies for the prevention and
treatment for diabetic cardiovascular complications.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81450062) and
Natural Science Foundation of Guangdong Province, China (no.
2015A030313872).
References
|
1
|
Mohsen L, Abou-Alam M, El-Dib M, Labib M,
Elsada M and Aly H: A prospective study on hyperglycemia and
retinopathy of prematurity. J Perinatol. 34:453–457. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ghosh SS, Righi S, Krieg R, Kang L, Carl
D, Wang J, Massey HD, Sica DA, Gehr TW and Ghosh S: High fat high
cholesterol diet (western diet) aggravates atherosclerosis,
hyperglycemia and renal failure in nephrectomized LDL receptor
knockout mice: role of intestine derived lipopolysaccharide. PLoS
One. 10:e01411092015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Laakso M and Kuusisto J: Insulin
resistance and hyperglycaemia in cardiovascular disease
development. Nat Rev Endocrinol. 10:293–302. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cal L, Li W, Wang G, Guo L, Jiang Y and
Kang YJ: Hypergly cemia-induced apoptosis in mouse myocardium:
Mitochondrial cytochrome C-mediated caspase-3 activation pathway.
Diabetes. 51:1938–1948. 2002. View Article : Google Scholar
|
|
5
|
Suzuki K, Olah G, Modis K, Coletta C, Kulp
G, Gerö D, Szoleczky P, Chang T, Zhou Z, Wu L, et al: Hydrogen
sulfide replacement therapy protects the vascular endothelium in
hyperglycemia by preserving mitochondrial function. Proc Natl Acad
Sci USA. 108:13829–13834. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar
|
|
7
|
Han J, Zhong CQ and Zhang DW: Programmed
necrosis: Backup to and competitor with apoptosis in the immune
system. Nat Immunol. 12:1143–1149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Smith CC and Yellon DM: Necroptosis,
necrostatins and tissue injury. J Cell Mol Med. 15:1797–1806. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dvoriantchikova G, Degterev A and Ivanov
D: Retinal ganglion cell (RGC) programmed necrosis contributes to
ischemia-reperfusion-induced retinal damage. Exp Eye Res. 123:1–7.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oerlemans MI, Liu J, Arslan F, den Ouden
K, van Middelaar BJ, Doevendans PA and Sluijter JP: Inhibition of
RIP1-dependent necrosis prevents adverse cardiac remodeling after
myocardial ischemia-reperfusion in vivo. Basic Res Cardiol.
107:2702012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao W, Zhang J, Lu Y and Wang R: The
vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP)
channel opener. EMBO J. 20:6008–6016. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang G, Wu L, Jiang B, Yang W, Qi J, Cao
K, Meng Q, Mustafa AK, Mu W, Zhang S, et al: H2S as a
physiologic vasorelaxant: Hypertension in mice with deletion of
cystathionine gammalyase. Science. 322:587–590. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Papapetropoulos A, Pyriochou A, Altaany Z,
Yang G, Marazioti A, Zhou Z, Jeschke MG, Branski LK, Herndon DN,
Wang R, et al: Hydrogen sulfide is an endogenous stimulator of
angiogenesis. Proc Natl Acad Sci USA. 106:21972–21977. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Calvert JW, Elston M, Nicholson CK,
Gundewar S, Jha S, Elrod JW, Ramachandran A and Lefer DJ: Genetic
and pharmacologic hydrogen sulfide therapy attenuates
ischemia-induced heart failure in mice. Circulation. 122:11–19.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang X, Wang Q, Guo W and Zhu YZ: Hydrogen
sulfide attenuates cardiac dysfunction in a rat model of heart
failure: A mechanism through cardiac mitochondrial protection.
Biosci Rep. 31:87–98. 2011. View Article : Google Scholar
|
|
16
|
Xu W, Wu W, Chen J, Guo R, Lin J, Liao X
and Feng J: Exogenous hydrogen sulfide protects H9c2 cardiac cells
against high glucose-induced injury by inhibiting the activities of
the p38 MAPK and ERK1/2 pathways. Int J Mol Med. 32:917–925. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang XY, Yang CT, Zheng DD, Mo LQ, Lan AP,
Yang ZL, Hu F, Chen PX, Liao XX and Feng JQ: Hydrogen sulfide
protects H9c2 cells against doxorubicin-induced cardiotoxicity
through inhibition of endoplasmic reticulum stress. Mol Cell
Biochem. 363:419–426. 2012. View Article : Google Scholar
|
|
18
|
Xu W, Chen J, Lin J, Liu D, Mo L, Pan W,
Feng J, Wu W and Zheng D: Exogenous H2S protects H9c2
cardiac cells against high glucose-induced injury and inflammation
by inhibiting the activation of the NF-κB and IL-1β pathways. Int J
Mol Med. 35:177–186. 2015. View Article : Google Scholar
|
|
19
|
Liang W, Chen J, Mo L, Ke X, Zhang W,
Zheng D, Pan W, Wu S, Feng J, Song M, et al: ATP-sensitive
K+ channels contribute to the protective effects of
exogenous hydrogen sulfide against high glucose-induced injury in
H9c2 cardiac cells. Int J Mol Med. 37:763–772. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mani S, Li H, Untereiner A, Wu L, Yang G,
Austin RC, Dickhout JG, Lhoták Š, Meng QH and Wang R: Decreased
endogenous production of hydrogen sulfide accelerates
atherosclerosis. Circulation. 127:2523–2534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang H, Guo C, Wu D, Zhang A, Gu T, Wang
L and Wang C: Hydrogen sulfide inhibits the development of
atherosclerosis with suppressing CX3CR1 and CX3CL1 expression. PLoS
One. 7:e411472012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jain SK, Bull R, Rains JL, Bass PF, Levine
SN, Reddy S, McVie R and Bocchini JA Jr: Low levels of hydrogen
sulfide in the blood of diabetes patients and
streptozotocin-treated rats causes vascular inflammation? Antioxid
Redox Signal. 12:1333–1337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu YS, Huang ZW, Wang L, Liu XX, Wang YM,
Zhang Y and Zhang M: Sitagliptin alleviated myocardial remodeling
of the left ventricle and improved cardiac diastolic dysfunction in
diabetic rats. J Pharmacol Sci. 127:260–274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin J, Li H, Yang M, Ren J, Huang Z, Han
F, Huang J, Ma J, Zhang D, Zhang Z, et al: A role of RIP3-mediated
macrophage necrosis in atherosclerosis development. Cell Reports.
3:200–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han W, Xie J, Li L, Liu Z and Hu X:
Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis.
Apoptosis. 14:674–686. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ruderman NB, Williamson JR and Brownlee M:
Glucose and diabetic vascular disease. FASEB J. 6:2905–2914. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
van Dieren S, Beulens JW, van der Schouw
YT, Grobbee De and Neal B: The global burden of diabetes and its
complications: An emerging pandemic. Eur J Cardiovasc Prev Rehabil.
17(Suppl 1): S3–S8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kern TS, Tang J, Mizutani M, Kowluru RA,
Nagaraj RH, Romeo G, Podesta F and Lorenzi M: Response of capillary
cell death to aminoguanidine predicts the development of
retinopathy: Comparison of diabetes and galactosemia. Invest
Ophthalmol Vis Sci. 41:3972–3978. 2000.PubMed/NCBI
|
|
29
|
Zhang X, Liang D, Lian X, Jiang Y, He H,
Liang W, Zhao Y and Chi ZH: Berberine activates Nrf2 nuclear
translocation and inhibits apoptosis induced by high glucose in
renal tubular epithelial cells through a phosphatidylinositol
3-kinase/Akt-dependent mechanism. Apoptosis. 21:721–736. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Frustaci A, Kajstura J, Chimenti C,
Jakoniuk I, Leri A, Maseri A, Nadal-Ginard B and Anversa P:
Myocardial cell death in human diabetes. Circ Res. 87:1123–1132.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kung G, Konstantinidis K and Kitsis RN:
Programmed necrosis, not apoptosis, in the heart. Circ Res.
108:1017–1036. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Declercq W, Vanden Berghe T and
Vandenabeele P: RIP kinases at the crossroads of cell death and
survival. Cell. 138:229–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Smith CC, Davidson SM, Lim SY, Simpkin JC,
Hothersall JS and Yellon DM: Necrostatin: A potentially novel
cardioprotective agent. Cardiovasc Drugs Ther. 21:227–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Holler N, Zaru R, Micheau O, Thome M,
Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B and Tschopp
J: Fas triggers an alternative, caspase-8-independent cell death
pathway using the kinase RIP as effector molecule. Nat Immunol.
1:489–495. 2000. View
Article : Google Scholar
|
|
37
|
Zhang N, Chen Y, Jiang R, Li E, Chen X, Xi
Z, Guo Y, Liu X, Zhou Y, Che Y, et al: PARP and RIP 1 are required
for autophagy induced by 11′-deoxyverticillin A, which precedes
caspase-dependent apoptosis. Autophagy. 7:598–612. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu S, Liu Z and Liu P: Targeting hydrogen
sulfide as a promising therapeutic strategy for atherosclerosis.
Int J Cardiol. 172:313–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mani S, Untereiner A, Wu L and Wang R:
Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxid
Redox Signal. 20:805–817. 2014. View Article : Google Scholar
|