Introduction
Pseudomonas aeruginosa (PA) is a
gram-negative bacteria that causes chronic lung infections in
individuals with various chronic lung diseases, including cystic
fibrosis (CF), bronchiectasis and chronic obstructive lung disease
(1,2). PA cells communicate in colonies via
quorum sensing and assemble into multicellular biofilms (3). Through interactions with pulmonary
epithelial cells, PA is resistant to various antimicrobials and is
able to cause chronic infections and persistent neutrophilic
inflammatory responses, which are followed by tissue damage, organ
injury, respiratory failure and mortality (4,5).
To the best of our knowledge, at present there is no effective
treatment against PA bacterial infections. Therefore, an effective
anti-inflammatory therapy that combats PA-induced neutrophilic
inflammation is urgently required.
A previous study reported that cluster of
differentiation (CD) 4+ T cells are associated with
specific adaptive immune responses directed against PA antigens
(6). T helper (Th)17 cells have
been identified as a subset of CD4+ T cells, which
predominately produce the cytokines interleukin (IL)-17A, IL-17F,
IL-21, IL-22 and IL-26 (7). Th17
differentiation is triggered by transforming growth factor-β and
IL-6 (8,9) and is controlled by transcription
factors, including retinoid-related orphan receptor (ROR)-α, ROR-γ
and the signal transducer and activator of transcription 3 (STAT3)
(10,11). IL-17A is a critical mediator of
neutrophil recruitment and activation via the CXC chemokine
(12). IL-17A is also able to
induce the expression of matrix metalloproteinases, which are
associated with tissue destruction (13). Dubin and Kolls (14) demonstrated that IL-23 mediates
inflammatory responses against PA lung infections in mice.
Additionally, they revealed that IL-23 deficient mice had
diminished levels of IL-17. These results indicate that Th17 cells
and their secretions serve a crucial role in inflammatory signaling
during PA infection.
The suppressors of cytokine signaling (SOCS) family
are important for the negative feedback inhibition of cytokines
(15). SOCSs are considered to be
strong inhibitors of Janus kinases (JAKs), cytokine receptors and
their downstream targets (16). A
recent study indicated that SOCS3 is a promising negative regulator
of Th17 differentiation and function (17). Additionally, Oshita et al
(18) demonstrated that enhanced
expression of SOCS3 significantly inhibited the differentiation of
T cells into Th17 cells. Chen et al (19) confirmed that SOCS3 inhibited
IL-23-mediated phosphorylation of STAT3 and thus prevented
interactions between phosphorylated (p)-STAT3 and the promoters of
IL-17A and IL-17F.
Based on these previous studies, it has been
hypothesized that SOCS3 may serve a key role in IL-17 mediated
neutrophilic airway inflammation during pulmonary PA infections. In
the present study lentivirus was used to deliver SOCS3 in a
well-established mouse model and PA lung infections were
subsequently introduced. The effects of SOCS3 on pulmonary Th17
responses and airway inflammation were investigated.
Materials and methods
Recombination of SOCS3 lentiviral and
cell transfection
In the present study a third generation lentiviral
system, including pGLV-EF1a-green fluorescent protein (GFP) vector,
pLV/helper-SL3 (gag/pol element), pLV/helper-SL4 (pRev element) and
pLV/helper-SL5 (pVSVG element) plasmids (Shanghai GenePharma Co.,
Ltd., Shanghai, China) was used to construct lentiviruses for the
overexpression of SOCS3. The lentivirus vectors with GFP were
purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China).
According to the information provided by National Center for
Biotechnology Information, the full length of the murine SOCS3 gene
(NM_007707.3) was screened and accurately inserted into the
pGLV-EF1a-GFP plasmid using polymerase chain reaction (PCR)
technology and DNA sequencing. Subsequently, plasmids
[pLV/helper-SL3, pLV/helper-SL4, pLV/helper-SL5, and
pGLV-EF1a-GFP-SOCS3 or pGLV-EF1a-GFP (negative control)] were
co-transfected into 293T cells using Lipofectamine™ 2000
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). At
12 h later, the 293T culture medium was replaced with fresh
Dulbecco's Modified Eagle Medium (DMEM; Thermo Fisher Scientific,
Inc.) containing 10% fetal bovine serum (FBS, Thermo Fisher
Scientific, Inc.). The lentivirus of SOCS3 and the negative control
were harvested with DMEM containing 10% FBS 48 h following
transfection. Following centrifugation at 4°C 750,000 × g for 90
min, the products were diluted to varying concentrations
(10−1–10−4) and used to infect fresh 293T
cells. The efficiency of 293T infections was assessed according to
the number of GFP positive cells using a fluorescence microscope
(magnification, ×400). The formula used to calculate virus titer
was as follows: Lentivirus titer [transducing units (TU)/ml] = GFP
positive cell number × dilution times/volume of lentivirus.
Construction of a chronic PA lung
infected mouse model
A total of 36 female C57/BL/6 specific pathogen-free
mice, 20–35 g and aged 8–12 weeks, were purchased from Shanghai
SLAC laboratory Animal Co. Ltd. (Shanghai, China). The mice were
housed in a temperature-controlled room (23°C and ~45% humidity)
with 12 h light/dark cycle and fed a standard mouse diet and water
with free access. PA cells (American Type Culture Collection 27863)
were purchased from China General Microbiological Culture
Collection Centre (Beijing, China) and embedded in agarose beads at
a final concentration of 2.0×106 colony-forming units
(CFU)/50 µl PBS as previously described (20). Following anesthesia, a total of 50
µl of PA-laden agarose beads were injected into the right
lung of each mouse. Experiments were performed according to the
guidelines for the Care and Use of Laboratory Animals and the
mortality rate during this procedure was 10%. All experiments were
approved by the Institution of Animal Care and Use Committee of
Shanghai Jiao Tong University School of Medicine (Shanghai,
China).
Lentivirus therapy in vivo
To avoid the increased mortality associated with a
single tail vein injection of a high dose of lentivirus, the doses
were divided into several injections of smaller doses and the
effects were investigated 3 days following the establishment of the
mouse model. In the present study 6 injections of
3.3×107 TU were administered over 12 h (a total of
2×108 TU/mouse). A total of 36 mice were randomly
allocated into 3 groups (12 mice/group) and treated as follows: i)
Blank control group, treated with the same volume of PBS; ii)
negative control group, treated with pGLV-EF1a-GFP and iii)
lentivirus LV-SOCS3 group, treated with pGLV-EF1a-GFP-SOCS3. The
weight, central and peripheral skin color, respiratory rate and
movement of mice were recorded daily. At 0, 4, 24, and 72 h
following lentivirus injection, the mice were euthanized 3 mice per
time point) and peripheral blood, lung tissue and bronchoalveolar
lavage (BAL) fluids were collected for subsequent analysis. To
investigate the kinetic profiles of lentiviruses, RNA from blood
cells and lung tissues was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). Lentivirus RNA
expression levels were measured using reverse
transcription-quantitative (RT-q) PCR and the number of lentivirus
copies per genome (C/G) was evaluated using RT-qPCR using
previously described primers and probes (21). Copies per genome (C/G) were
computed using the following equation: (ng lentivirus DNA/ng
endogenous DNA) × (no. of lentivirus integrations in the standard
curve).
Cell numbers in the BAL fluid
Following euthanasia, mouse tracheas were instilled
multiple times with 1.8 ml aliquots of aseptic PBS and the BAL
fluid was subsequently retrieved. Following centrifugation at 4°C,
650 × g for 10 min the supernatants of the retrieved BAL fluids
were sterile-filtered for cytokine analyses and the sediments were
resuspended in PBS. Leukocyte cell numbers were measured using a
hemocytometer and the numbers of neutrophils and lymphocytes were
quantified using a minimum of 100 consecutive hematoxylin and eosin
(H&E)-stained cells. Cell counting experiments were conducted
blindly and independently by two experienced pathologists.
Bacterial load evaluation
To quantify the number of bacteria, the right middle
lung lobes were harvested from sacrificed mice and homogenized in
saline solution. Serial dilutions were subsequently cultured on
sheep blood agar plates (BioTrading, Mijdrecht, The Netherlands)
overnight at 37°C. The bacterial colonies from the lung lobes of
each mouse were counted.
Histology
Following fixing with 10% neutral-buffered formalin
at 4°C for 24–48 h, tissues were embedded in paraffin and cut into
slices (5 µm). Sections of the right lower lobes were
stained with hematoxylin and eosin. Ten fields of each mouse were
analyzed using a light microscope at low power (magnification, ×40)
by two independently blinded pathologists. Each section was scored
using the following criteria: i) For intraluminal infiltrates, none
positive scored 0 points, <25% positive lumens scored 1 point,
25–50% positive lumens scored 2 points, 50–75% positive lumens
scored 3 points and diffuse staining scored 4 points; ii) For
peri-bronchial infiltrates, none positive scored 0 points, positive
area ≤4 cells thickness scored 1 point, positive area 5–10 cells
thickness scored 2 points, positive areas >10 cells thickness
but ≤50% of visualized lumens scored 3 points and diffuse cell
areas scored 4 points; iii) For alveolar involvement, no positive
cells scored 0 points, increased cellular characteristics scored 1
point, interlobular septal thickening scored 2 points, obliteration
≤a quarter of the visualized alveolar spaces scored 3 points and
obliteration >a quarter of the visualized alveolar spaces scored
4 points.
Myeloperoxidase (MPO) and cytokine
analysis in BAL fluids
Quantikine® ELISA kits (R&D Systems,
Inc., Minneapolis, MN, USA) were purchased for assays of MPO (cat.
no. DY3667), IL-6 (cat. no. M6000B), IL-8 (cat. no. MAB16081) and
IL-17A (cat. no. M1700) in the BAL fluids. Each assay was performed
twice according to manufacturer's protocol and the mean values were
calculated.
Isolation of lung CD4+ T
cells
Following homogenizing in 0.5 ml normal saline, the
lung tissues were digested using trypsin and filtered through a
40-µm nylon mesh. Subsequently, CD4+ T cell
isolation kits and Mini-MACS™ columns (both Miltenyi Biotec, Inc.,
Auburn, CA, USA) were used to isolate CD4+ T cells
according to manufacturer's protocol following centrifugation at
4°C, 1,100 × g for 10 min and three washes with PBS.
RT-qPCR assay for RNAs in CD4+
T cells
Total RNA from lung CD4+ T cells was
extracted using TRIzol reagent (Thermo Fisher Scientific, Inc.) and
RNA was reverse transcribed into cDNA using the Primer-ScriptTM one
step RT-PCR kit (Takara Bio, Inc., Otsu, Japan) according to
manufacturers' protocols. PCR was performed using the following
primers and probe sequences: Mouse SOCS3 sense,
5′-CTGCAGGAGAGCGGATTCTACT-3′ and antisense,
5′-GCTGTCGCGGATAAGAAAGG-3′; mouse SOCS3 probe,
5′-CTGCTGCTCAGCGCCGAGCC-3′; mouse STAT3 sense,
5′-GGGCCAGGCCAACCA-3′ and antisense, 5′-CCGGACATCCTGAAGATGCT-3′;
mouse STAT3 probe, 5′-CCAACAGCCGCCGTAGTGACAGAGT-3′; mouse RORγt
sense, 5′-TCTCTGCAAGACTCATCGACAAG-3′ and antisense,
5′-GCACAGGCTCCGGAGTTTT-3′; mouse RORγt probe,
5′-CTCCTAGCCAAGCTGCCACCCAAA-3′. PCR was performed on a
LightCycler® (Roche Diagnostics, Indianapolis, IN, USA)
using 2X SYBR-Green (Takara Bio, Inc.) according to manufacturer's
protocol with the following system: 95°C for 3 min and 40 cycles of
95°C for 10 sec and 55°C for 30 sec. The expression levels of the
target genes were normalized to the internal control β-actin using
the 2−ΔΔCq method (22).
Western blot analysis
Proteins from CD4+ T cells were extracted
using a Nuclear and Cytoplasmic Protein Extraction kit (Beyotime
Institute of Biotechnology, Haimen, China) and protein
concentrations were evaluated using a BCA protein assay. A total of
50 µg aliquots of protein samples were mixed with a loading
buffer and boiled for 10 min. Proteins (15 µg) were
separated by 10% SDS-PAGE. Proteins were transferred onto
polyvinylidene fluoride membranes and blocked with 5% non-fat milk
at room temperature for 1 h. The membranes were subsequently
incubated with rabbit anti-mouse SOCS3 polyclonal antibodies
(1:500; cat. no. sc-9023) rabbit anti-mouse β-actin polyclonal
antibodies (1:1,000; cat. no. sc-58673; both Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) or rabbit anti-mouse p-STAT3
monoclonal antibodies (1:2,000; cat. no. 9145; Cell Signaling
Technology, Inc., Danvers, MA, USA) at 4°C overnight. The membranes
were subsequently washed and incubated with horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:5,000; cat. no. 111035144;
Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) at
room temperature for 1 h. The protein bands were illuminated using
the enhanced chemiluminescence method (GE Healthcare Life Sciences,
Little Chalfont, UK), and quantified by densitometry analyses using
Quantity One software version 4.6.1 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) using β-actin as the internal reference.
Immunohistochemistry (IHC) detection of
activated STAT3 in lung tissues
Following deparaffinization and rehydrated in a
graded series of alcohol, and antigen retrieval was performed by
microwaving (650 W) for 10 min in sodium citrate buffer (pH 6.0).
Endogenous peroxidase activity was blocked with 3% hydroperoxidase
at room temperature for 20 min and the sections were subsequently
incubated with rabbit anti-mouse p-STAT3 antibodies (1:400; cat.
no. 9145) at 4°C overnight followed by HRP-conjugated secondary
antibodies (1:1,000; cat. no. 111035144; Jackson ImmunoResearch
Laboratories, Inc.) at room temperature for 30 min and then
colorized using a 3,3′-diaminobenzidine substrate solution.
Finally, sections were counterstained with hematoxylin at room
temperature for 3–5 min to visualize the nuclei and analyzed by two
independently blinded pathologists using a light microscope
(magnification, ×40).
Flow cytometric analyses of
CD4+ and IL-17+ cells in the lungs
CD4+ cells derived from lung tissue were
permeabilized in 0.2% Triton X-100 and incubated with
allophycocyanin (APC)-conjugated IL-17 antibodies (1:200; cat. no.
45717782, eBioscience; Thermo Fisher Scientific, Inc.) in a final
volume of 100 µl buffer [PBS, pH 7.4 with 1% bovine serum
albumin (Thermo Fisher Scientific, Inc.)] overnight at 4°C. The
cells were subsequently washed 3 times in PBS and analyzed using a
FACSCalibur™ flow cytometer (Becton-Dickinson; BD Biosciences) in
the FL4 channel. Background fluorescence was assessed using a
non-specific rat immunoglobulin G (eBioscience; Thermo Fisher
Scientific, Inc.).
Statistical analysis
All data were analyzed using SAS version 8.1
statistical software (SAS Institute, Inc., Cary, NC, USA).
Normality analysis was conducted by a Shapiro-Wilk test. Data are
presented as the mean ± standard error of the mean and differences
were identified using analysis of variance. Non-parametric data
were analyzed using a Mann-Whitney U test with a Bonferroni
correction. P<0.05 was considered to indicate a statistically
significant difference.
Results
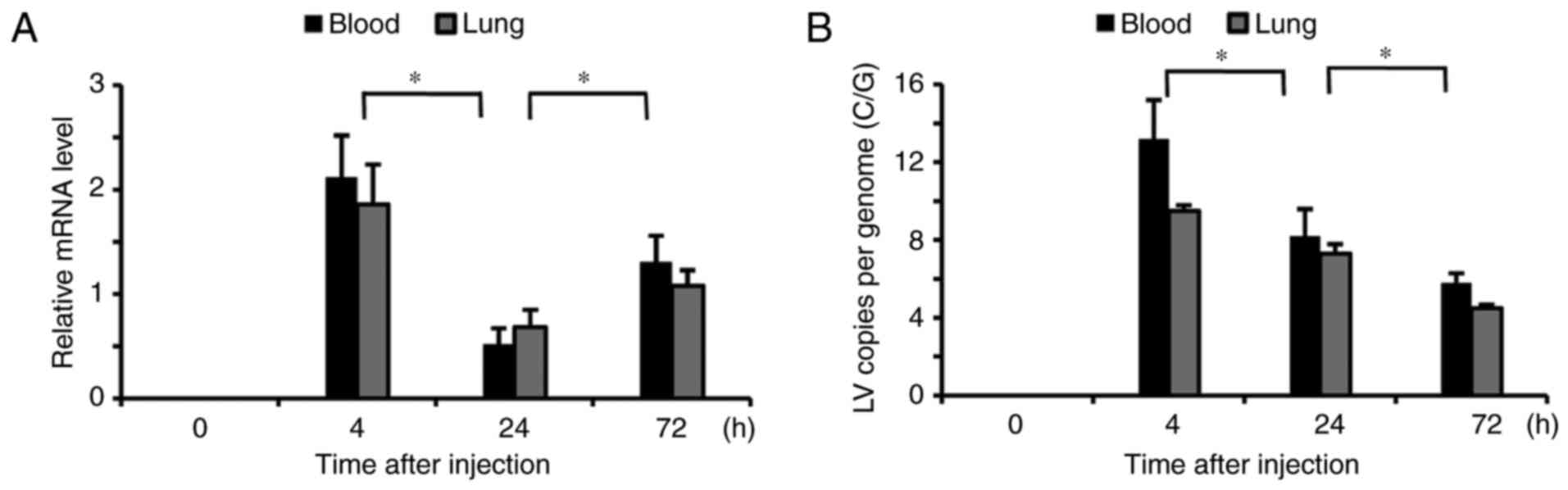
Kinetic profiles of lentivirus
Transfected kinetics were investigated using RT-qPCR
following six injections of lentivirus. Notably increased levels of
lentivirus were detected in the blood and lung tissues 4 h
following transfection and they significantly decreased at 24 h
(P<0.05; Fig. 1A). However, at
72 h following transfection the lentivirus RNA levels in the blood
and lung tissues were again significantly elevated (P<0.05)
compared with 24 h, but to a smaller degree that at 4 h. The number
of lentivirus C/G was significantly increased in the blood and lung
tissues at 4 h following the injection, but markedly decreased at
24 and 72 h compared with their previous time points (P<0.05;
Fig. 1B).
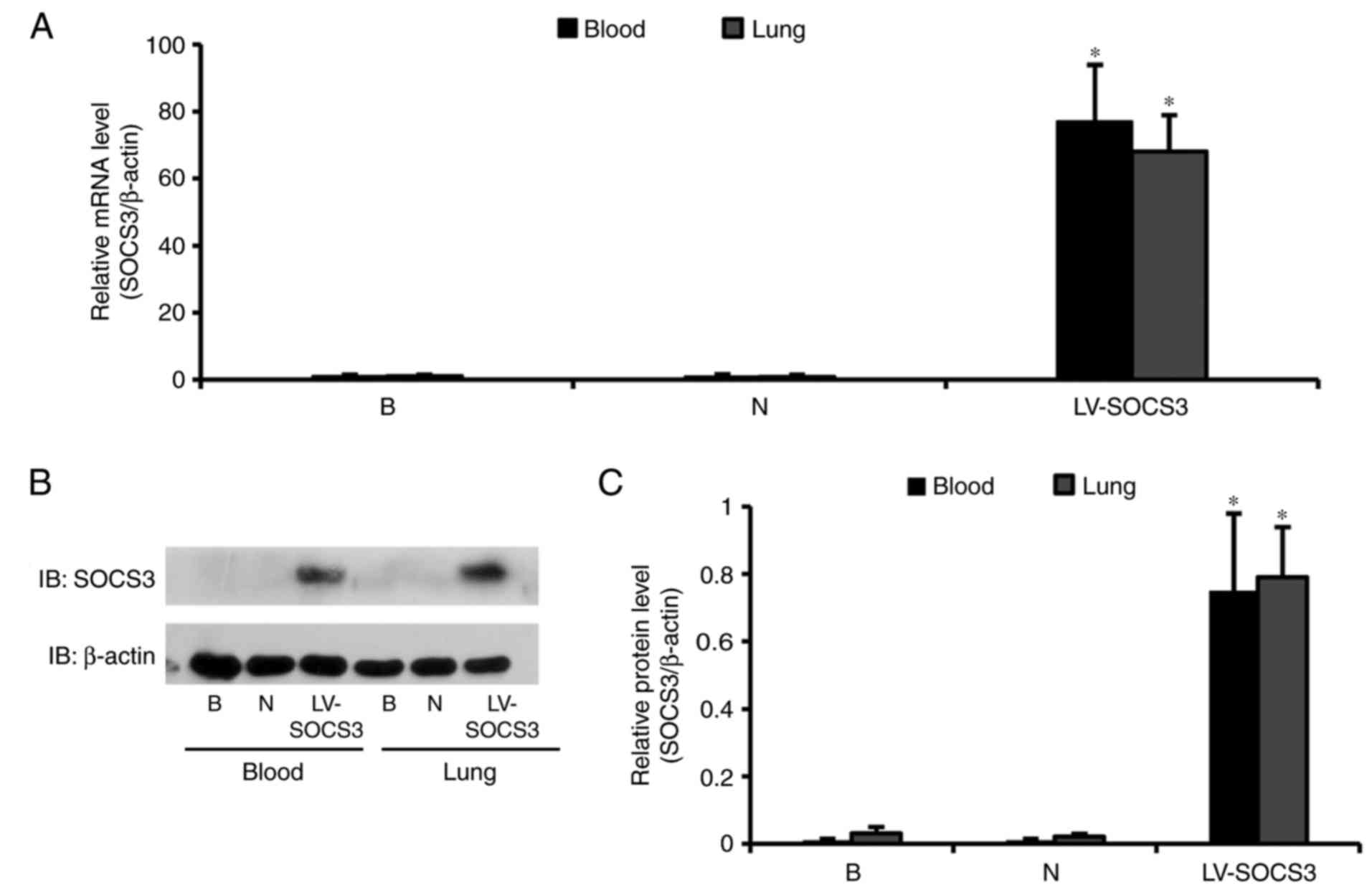
SOCS3 gene overexpression in alveolar
CD4+ T cells following lentivirus injection
SOCS3 expression in lung CD4+ T cells was
confirmed by RT-qPCR and western blot analysis. The relative mRNA
expression of SOCS3 in the LV-SOCS3 group was significantly
increased compared with the blank and negative control groups (both
P<0.05); however, no notable differences in SOCS3 mRNA
expression were observed between the blank and negative control
groups (Fig. 2A). Western blot
analyses revealed a significantly higher SOCS3 expression in the
LV-SOCS3 group compared with the blank or negative control groups
(both P<0.05; Fig. 2B and
C).
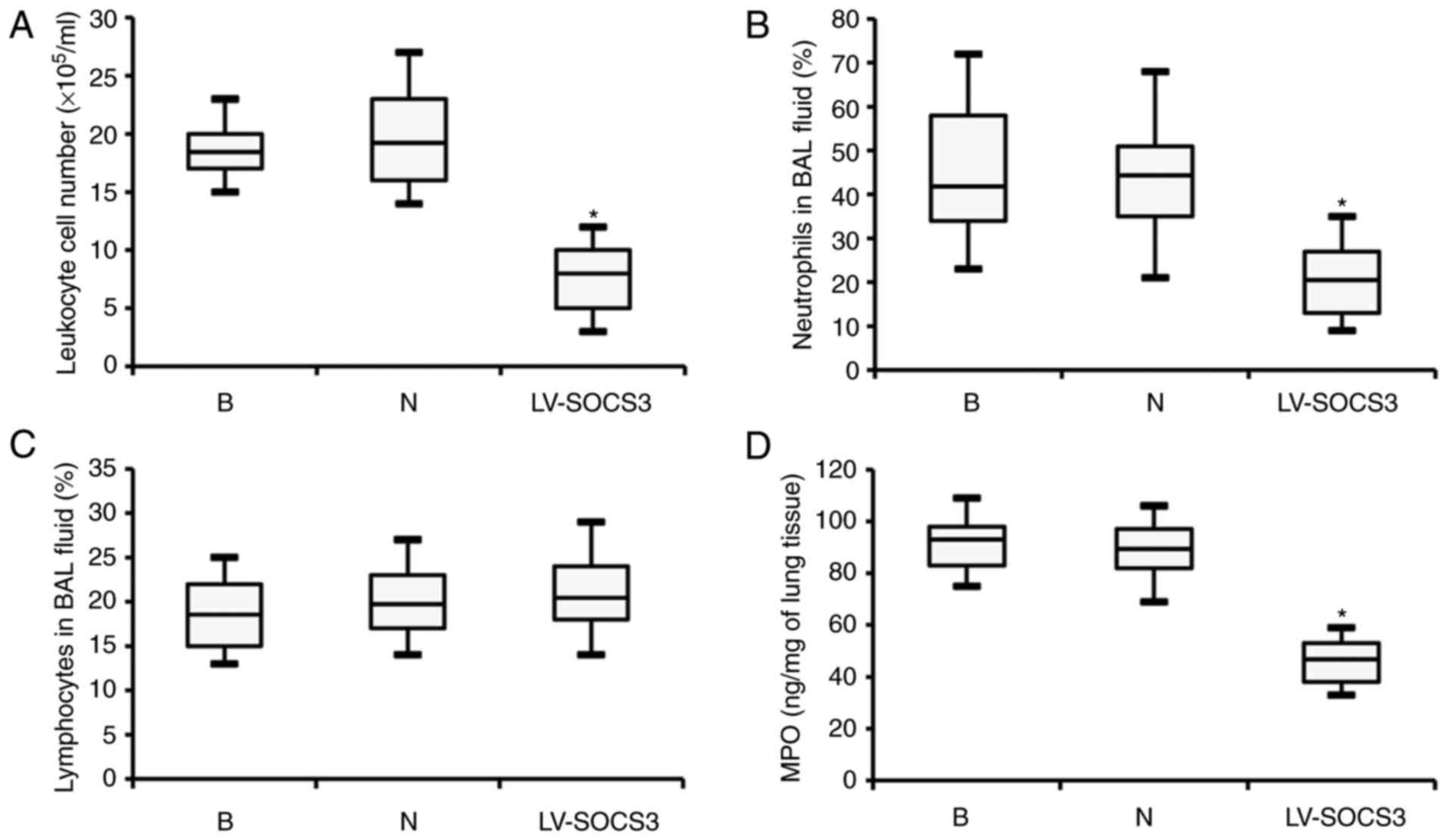
Overexpression of SOCS3 inhibits
neutrophil airway infiltration but does not affect the bacterial
load
Following treatment with lentivirus, the severity of
dyspnea and weight loss in the LV-SOCS3 group was significantly
reduced compared with the blank and negative control groups (data
not shown). However, the leukocyte cell number was significantly
reduced in the BAL fluids of the LV-SOCS3 group compared with the
blank and negative control groups (P<0.05; Fig. 3A). Similarly, the percentage of
neutrophils in the BAL fluid was also significantly reduced in the
LV-SOCS3 group compared with the blank and negative control groups
(P<0.05; Fig. 3B), however,
the percentages of lymphocytes was only marginally increased
(Fig. 3C). Additionally, ELISA
analysis revealed significantly lower levels of MPO in the BAL
fluid of the LV-SOCS3 group compared with the blank and negative
control groups (P<0.05; Fig.
3D). No notable differences were observed between the blank and
negative control groups in any of the categories listed above.
In pathological assessments of mice from the
presented treatment groups, H&E staining analyses revealed that
the pathological condition in the LV-SOCS3 group was less severe
than in the blank and negative control groups (Fig. 4A). This indicates reduced airway
inflammation and fewer neutrophils present in the LV-SOCS3 group.
Histology scores of the images revealed a significantly lower
intraluminal infiltration, peribronchial infiltration, and alveolar
hemorrhage in the LV-SOCS3 group compared with blank and negative
control groups (P<0.05; Fig.
4B). The bacterial loads (×104 CFU/g lung tissue)
did not significantly differ between the LV-SOCS3 group
(3.10±0.56), the blank (2.80±0.78) and negative control (2.50±0.96)
groups (data not shown).
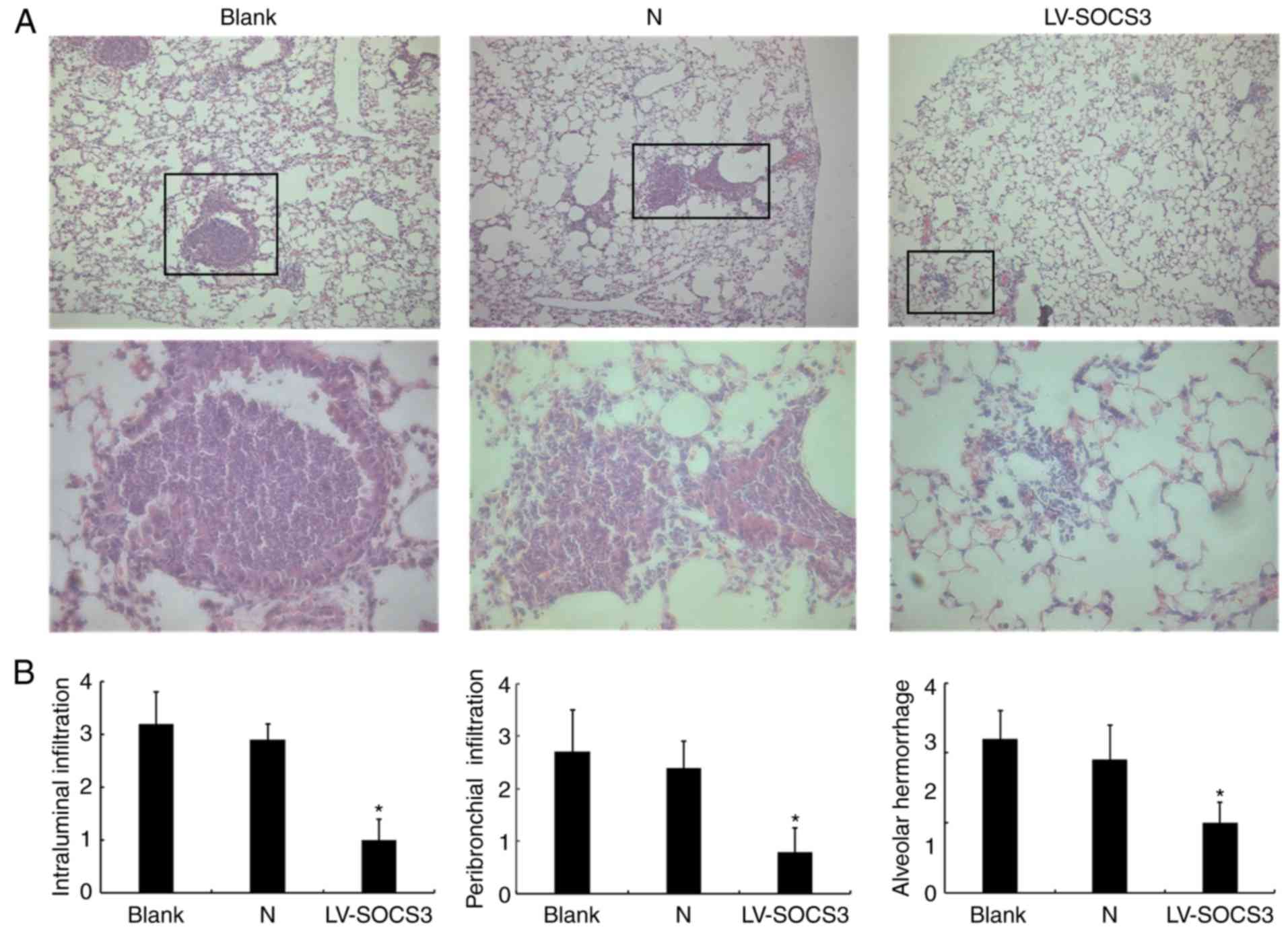 | Figure 4H&E staining of lung tissue
following the overexpression of SOCS3. (A) Representative images of
the H&E staining of lung tissue from the blank control group,
negative control group and LV-SOCS3 group. The boxes in the upper
image are enlarged in the image below. Upper image magnification,
×40; scale bar, 500 µm. Lower image magnification, ×400;
scale bar, 50 µm. (B) Histology scores of the intraluminal
infiltration, peribronchial infiltration and alveolar hemorrhage.
n=3. *P<0.05 vs. Blank group and
*P<0.05 vs. N group. H&E, hematoxylin and eosin;
SOCS3, suppressor of cytokine signaling protein 3; LV-SOCS3,
lentiviral vector-mediated SOCS3 group; N, negative control
group. |
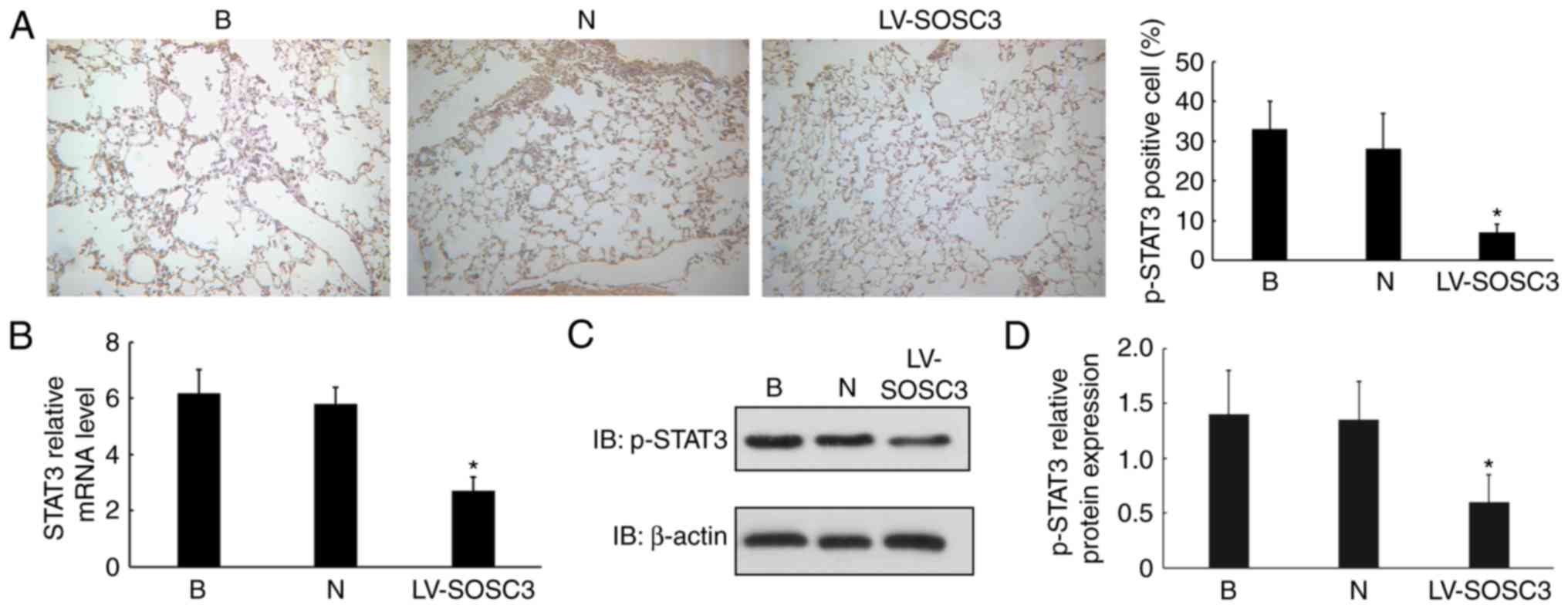
Overexpression of SOCS3 decreases p-STAT3
in pulmonary CD4+ T cells
To investigate the mechanism of SOCS3, p-STAT3
expression was detected by IHC. Significantly fewer p-STAT3
positive cells were observed in the LV-SOCS3 group compared with
the blank and negative control groups (P<0.05; Fig. 5A). To further explore whether
SOCS3 directly regulates p-STAT3 in alveolar CD4+
positive cells, the mRNA level of SOCS3 was measured. These
experiments demonstrated a significantly lower expression of STAT3
in CD4+ cells from the LV-SOCS3 group compared with
those from the blank and negative control groups (P<0.05;
Fig. 5B). p-STAT3 protein levels
were significantly downregulated in the LV-SOCS3 group compared
with the blank and negative control groups (P<0.05; Fig. 5C and D).
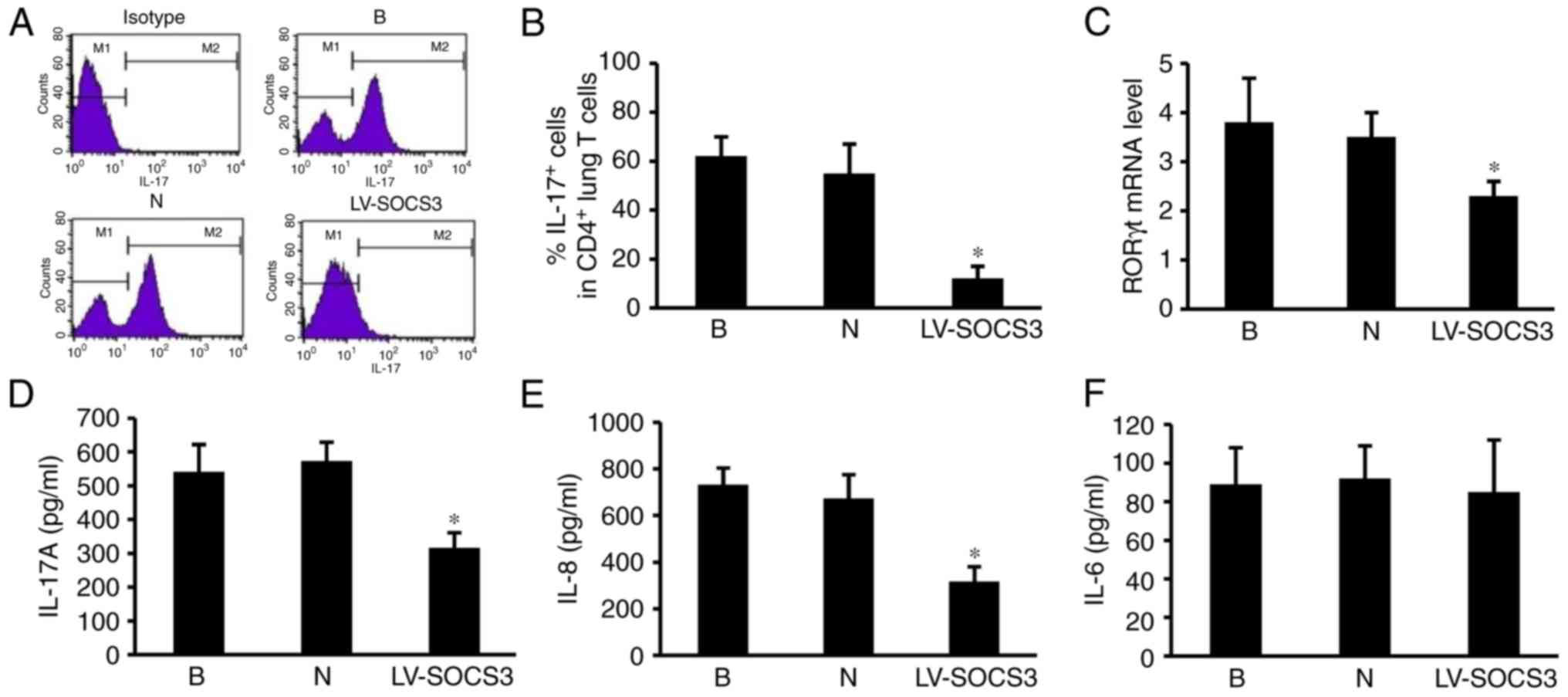
Overexpression of SOCS3 suppresses the
pulmonary Th17 response via RORγt, IL-17A and IL-8
To evaluate the effect of the SOCS3 gene on
pulmonary Th17 responses in the lung, the percentages of
CD4+ IL-17+ cells were measured using flow
cytometry (Fig. 6A). The results
revealed a markedly lower percentage of CD4+
IL-17+ positive cells in the LV-SOCS3 group compared
with the blank and negative control groups (P<0.05; Fig. 6B). The levels of RORγt, IL-17A,
IL-8 and IL-6 mRNA were also measured, as they are important
contributors to the pulmonary Th17 response. The results revealed
significantly lower RORγt mRNA expression in the LV-SOCS3 group
compared with the blank and negative control groups (P<0.05;
Fig. 6C). ELISA assays
demonstrated significantly lower levels of IL-17A and IL-8 in the
LV-SOCS3 group compared with the blank and negative groups
(P<0.05; Fig. 6D and E). No
significant differences were identified in the expression of IL-6
were detected between the three groups (Fig. 6F).
Discussion
As a member of the SOCS protein family, SOCS3
negatively regulates the cytokine signaling pathway (23) and may be induced by selected
inflammatory cytokines (19). In
the present study, SOCS3 expression was enhanced by lentivirus
infection in mice with chronic PA lung infections and the ensuing
effects were investigated. Overexpression of SOCS3 significantly
suppressed pulmonary Th17 responses and neutrophilic infiltration
by inhibiting the expression of RORγt, IL-17A and IL-8 in pulmonary
CD4+ T cells.
A previous study reported that SOCS3 serves a
critical role in restricting inflammation and regulating protective
immune responses against infection in multiple mouse models
(24). A significant
downregulation in airway inflammation with no concurrent change in
bacterial load was observed in the present study following the
lentivirus overexpression of SOCS3 in mice with chronic PA lung
infection. Dubin and Kolls (14)
previously eliminated Th17 responses by attenuating IL-23
expression, this also had no influence on the bacterial load and
dissemination in mice with chronic PA lung infections.
Additionally, IL-23 and IL-17 signaling has been confirmed as
dispensable for the establishment of host mycobacteria, which are
also prevalent opportunistic pathogens in patients with CF
(25). It has been demonstrated
that SOCS3 regulates STAT3-mediated chemokine and chemokine
receptor interactions in the bone marrow and serves a crucial role
in the neutrophil mobilization response (26). Taken together these results
indicate that SOCS3 suppresses airway inflammation by aborting Th17
signal responses and that STAT3 is associated with this process.
However, in the present study SOCS3 did not significantly influence
PA infection loads in mouse lungs.
STAT3 mRNA expression significantly decreased with
lentivirus-mediated overexpression of SOCS3. p-STAT3 protein levels
were also significantly downregulated in the lung tissue and
CD4+ T cells under these conditions. Phosphorylated
cascades of the JAK/STAT signaling pathway are associated with
multiple biological processes in health and disease, including DNA
binding, histone deacetylase recruiting and inflammatory
development (27,28). Therefore the negative effect of
SOCS3 on inflammation may deregulate the expression and activation
of STAT3. Shouda et al (29) and Suzuki et al (30) have reported that SOCS3 inhibits
the STAT3 inflammatory pathway; furthermore, Chen et al
(19) reported that SOCS3 is a
major regulator of IL-23 mediated phosphorylation of STAT3, which
binds directly to IL-17A and IL-17F promoters in CD4+ T
cells. Additionally, STAT3 was observed to be a major regulator of
Th17 responses, including Th17 differentiation and cytokine
production (31). Accumulating
evidence indicates that SOCS3 suppresses Th17 responses via STAT3,
which is an important mediator of signaling transduction and
inhibits airway inflammation in CD4+ T cells in the
lungs.
IL-17A and IL-8 are produced by Th17 cells, whilst
RORγt and STAT3 act as important regulators during Th17 cell
differentiation (32,33). IL-17A is produced by activated
Th17 cells in response to infected antigens (34,35). Gram-negative bacteria appear to
induce the expression of IL-17A via IL-23 and toll-like receptor
4-dependent signaling pathways (36,37). In the present study, the
percentage of IL-17A+ cells and the levels of IL-17A and
IL-8 mRNA were significantly decreased in CD4+ T cells
following the upregulation of SOCS3. However, RORγt mRNA expression
significantly declined with increasing SOCS3 levels. These results
suggest that SOCS3 may suppress Th17 responses by inhibiting RORγt,
which causes a decline in Th17 differentiation and production, thus
limiting the proinflammatory contribution of STAT3 and p-STAT3 and
attenuating PA-mediated inflammation.
In summary, overexpressed SOCS3 using lentivirus
vectors significantly suppresses Th17 responses in alveolar
CD4+ T cells by inhibiting the expression and activation
of STAT3. These results indicate that SOCS3 and STAT3 may be
potential targets for relieving airway inflammation during PA lung
infection.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81300005).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Hengzhuang W, Song Z, Ciofu O, Onsøyen E,
Rye PD and Høiby N: OligoG CF-5/20 disruption of mucoid Pseudomonas
aeruginosa biofilm in a murine lung infection model. Antimicrob
Agents Chemother. 60:2620–2626. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mayer-Hamblett N, Kloster M, Rosenfeld M,
Gibson RL, Retsch-Bogart GZ, Emerson J, Thompson V and Ramsey BW:
Impact of sustained eradication of new pseudomonas aeruginosa
infection on long-term outcomes in cystic fibrosis. Clin Infect
Dis. 61:707–715. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sharma G, Rao S, Bansal A, Dang S, Gupta S
and Gabrani R: Pseudomonas aeruginosa biofilm: Potential
therapeutic targets. Biologicals. 42:1–7. 2014. View Article : Google Scholar
|
|
4
|
Darch SE, McNally A, Harrison F, Corander
J, Barr HL, Paszkiewicz K, Holden S, Fogarty A, Crusz SA and Diggle
SP: Recombination is a key driver of genomic and phenotypic
diversity in a Pseudomonas aeruginosa population during cystic
fibrosis infection. Sci Rep. 5:76492015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Armstrong DS, Hook SM, Jamsen KM, Nixon
GM, Carzino R, Carlin JB, Robertson CF and Grimwood K: Lower airway
inflammation in infants with cystic fibrosis detected by newborn
screening. Pediatr Pulmonol. 40:500–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Regamey N, Tsartsali L, Hilliard TN, Fuchs
O, Tan HL, Zhu J, Qiu YS, Alton EW, Jeffery PK, Bush A and Davies
JC: Distinct patterns of inflammation in the airway lumen and
bronchial mucosa of children with cystic fibrosis. Thorax.
67:164–170. 2012. View Article : Google Scholar
|
|
7
|
Kolls JK: CD4+ T-cell subsets
and host defense in the lung. Immunol Rev. 252:156–163. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Veldhoen M, Hocking RJ, Atkins CJ,
Locksley RM and Stockinger B: TGFbeta in the context of an
inflammatory cytokine milieu supports de novo differentiation of
IL-17-producing T cells. Immunity. 24:179–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mangan PR, Harrington LE, O'Quinn DB,
Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR and
Weaver CT: Transforming growth factor-β induces development of the
TH17 lineage. Nature. 441:231–234. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang XO, Pappu BP, Nurieva R, Akimzhanov
A, Kang HS, Chung Y, Li M, Shah B, Panopoulos AD, Schluns KS, et
al: T helper 17 lineage differentiation is programmed by orphan
nuclear receptors ROR alpha and ROR gamma. Immunity. 28:29–39.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ivanov II, Mckenzie BS, Liang Z, Tadokoro
CE, Lepelley A, Lafaille JJ, Cua DJ and Littman DR: The orphan
nuclear receptor RORgammat directs the differentiation program of
proinflammatory IL-17+ T helper cells. Cell.
126:1121–1133. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kolls JK and Lindén A: Interleukin-17
family members and inflammation. Immunity. 21:467–476. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lubberts E: The role of IL-17 and family
members in the pathogenesis of arthritis. Curr Opin Investig Drugs.
4:572–577. 2003.PubMed/NCBI
|
|
14
|
Dubin PJ and Kolls JK: IL-23 mediates
inflammatory responses to mucoid Pseudomonas aeruginosa lung
infection in mice. Am J Physiol Lung Cell Mol Physiol.
292:L519–L528. 2007. View Article : Google Scholar
|
|
15
|
Linossi EM, Babon JJ, Hilton DJ and
Nicholson SE: Suppression of cytokine signaling: The SOCS
prespective. Cytokine Growth Factor Rev. 24:241–248. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoshimura A and Yasukawa H: JAK's SOCS: A
mechanism of inhibition. Immunity. 36:157–159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu X, Ren S, Qu X, Ge C, Cheng K and Zhao
RC: Mesenchymal stem cells inhibit Th17 cells differentiation via
IFN-γ-mediated SOCS3 activation. Immunol Res. 61:219–229. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tanaka K, Ichiyama K, Hashimoto M, Yoshida
H, Takimoto T, Takaesu G, Torisu T, Hanada T, Yasukawa H, Fukuyama
S, et al: Loss of suppressor of cytokine signaling 1 in helper T
cells leads to defective Th17 differentiation by enhancing
antagonistic effects of IFN-gamma on STAT3 and Smads. J Immunol.
180:3746–3756. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen Z, Laurence A, Kanno Y,
Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L and
O'Shea JJ: Selective regulatory function of Socs3 in the formation
of IL-17-secreting T cells. Proc Natl Acad Sci USA. 103:8137–8142.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Heeckeren AM and Schluchter MD: Murine
models of chronic Pseudomonas aeruginosa lung infection. Lab Anim.
36:291–312. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brown BD, Venneri MA, Zingale A, Sergi
Sergi L and Naldini L: Endogenous microRNA regulation suppresses
transgene expression in hematopoietic lineages and enables stable
gene transfer. Nat Med. 12:585–591. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔC T method. Methods. 25:402–408.
2011. View Article : Google Scholar
|
|
23
|
Dalpke A and Heeg K: Suppressors of
cytokine signaling proteins in innate and adaptive immune
responses. Arch Immunol Ther Exp. 51:91–103. 2003.
|
|
24
|
Carow B and Rottenberg ME: SOCS3, a major
regulator of infection and inflammation. Front Immunol. 5:582014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Khader SA, Pearl JE, Sakamoto K, Gilmartin
L, Bell GK, Jelley-Gibbs DM, Ghilardi N, deSauvage F and Cooper AM:
IL-23 compensates for the absence of IL-12p70 and is essential for
the IL-17 response during tuberculosis but is dispensable for
protection and antigen-specific IFN-gamma responses if IL-12p70 is
available. J Immunol. 175:788–795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nguyen-Jackson H, Panopoulos AD, Zhang H,
Li HS and Watowich SS: STAT3 controls the neutrophil migratory
response to CXCR2 ligands by direct activation of G-CSF-induced
CXCR2 expression and via modulation of CXCR2 signal transduction.
Blood. 115:3354–3363. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu D and Qu CK: Protein tyrosine
phosphatases in the JAK/STAT pathway. Front Biosci. 13:4925–4932.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shuai K and Liu B: Regulation of JAK-STAT
signalling in the immune system. Nat Rev Immunol. 3:900–911. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shouda T, Yoshida T, Hanada T, Wakioka T,
Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, et
al: Induction of the cytokine signal regulator SOCS3/CIS3 as a
therapeutic strategy for treating inflammatory arthritis. J Clin
Invest. 108:1781–1788. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suzuki A, Hanada T, Mitsuyama K, Yoshida
T, Kamizono S, Hoshino T, Kubo M, Yamashita A, Okabe M, Takeda K,
et al: Cis3/Socs3/Ssi3 plays a negative regulatory role in STAT3
activation and intestinal inflammation. J Exp Med. 193:471–481.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harris TJ, Grosso JF, Yen HR, Xin H,
Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV,
Maris CH, et al: Cutting edge: An in vivo requirement for STAT3
signaling in TH17 development and TH17-dependent autoimmunity. J
Immunol. 179:4313–4317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ernst M, Najdovska M, Grail D,
Lundgren-May T, Buchert M, Tye H, Matthews VB, Armes J, Bhathal PS,
Hughes NR, et al: STAT3 and STAT1 mediate IL-11-dependent and
inflammation- associated gastric tumorigenesis in gp130 receptor
mutant mice. J Clin Invest. 118:1727–1738. 2008.PubMed/NCBI
|
|
33
|
Brender C, Tannahill GM, Jenkins BJ,
Fletcher J, Columbus R, Saris CJ, Ernst M, Nicola NA, Hilton DJ,
Alexander WS and Starr R: Suppressor of cytokine signaling 3
regulates CD8 T-cell proliferation by inhibition of interleukins 6
and 27. Blood. 110:2528–2536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moseley TA, Haudenschild DR, Rose L and
Reddi AH: Interleukin-17 family and IL-17 receptors. Cytokine
Growth Factor Rev. 14:155–174. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ye P, Garvey PB, Zhang P, Nelson S, Bagby
G, Summer WR, Schwarzenberger P, Shellito JE and Kolls JK:
Interleukin-17 and lung host defense against Klebsiella pneumoniae
infection. Am J Respir Cell Mol Biol. 25:335–340. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Happel KI, Zheng M, Young E, Quinton LJ,
Lockhart E, Ramsay AJ, Shellito JE, Schurr JR, Bagby GJ, Nelson S
and Kolls JK: Cutting edge: Roles of Toll-like receptor 4 and IL-23
in IL-17 expression in response to Klebsiella pneumoniae infection.
J Immunol. 170:4432–4436. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lindén A and Adachi M: Neutrophilic airway
inflammation and IL-17. Allergy. 57:769–775. 2002. View Article : Google Scholar : PubMed/NCBI
|