Introduction
Tumor angiogenesis is crucial for cancer growth and
metastasis, as the new blood vessels support progression of the
primary tumor by providing oxygen and nutrients, which involves a
complex interaction among multiple cells and cytokines (1). The interaction between tumor cells
and vascular endothelial cells (VECs) participates in the process
of tumor angiogenesis. The formation of new blood vessels is
induced by proangiogenic factors secreted by tumor cells to
facilitate the migration and proliferation of VECs, and VEC
migration is a fundamental process of tumor angiogenesis (2). However, cancer cells degrade
endothelial basement membrane and extracellular matrix (ECM)
components in tumor blood vessels by releasing matrix
metalloproteinases; subsequently, cancer cells migrate and invade
into the circulation, ultimately forming new metastatic lesions in
surrounding tissues (3).
Therefore, preventing the migration of VECs and/or tumor cells may
be a valuable new approach to cancer therapy by inhibiting tumor
growth, invasion and metastasis.
Cell adhesion/migration is a complex and dynamic
multi-step process that involves a balance between assembly and
disassembly of matrix-cell adhesion sites. Focal adhesion kinase
(FAK) is a non-receptor tyrosine kinase that is overexpressed in
several types of tumors. FAK regulates cell adhesion and migration
signals in various cell lines, is involved in the engagement of
integrin and assembly of focal adhesions (FA) through catalyzing
several downstream signals, and mediates cell behavior. The
integrin family of cell adhesion receptors are major cellular
receptors of ECM, and they act as signaling centers orchestrating a
network of signaling pathways that mediate cell adhesion and
migration (4). The FA components,
including paxillin and talin, recruit FAK to FA. The
phosphorylation of paxillin specifically induces the recruitment of
the cytoskeletal adapter protein vinculin to FA (5). FAK directly binds to the β subunit
of integrins, and its FAT domain binds to talin and paxillin. The
initial autophosphorylation of Y397-FAK tyrosine may subsequently
result in the phosphorylation of additional multiple tyrosine sites
(Y-576, -577 and -925) (6). FAK
also interacts with the p85 subunit of phosphatidylinositol
3-kinase (PI3K) and then stimulates AKT, also referred to as
protein kinase B (PKB), which may regulate FAK indirectly through
interaction with a joint partner protein that can bind to both FAK
and AKT to form a triple complex (7,8).
FAK/PI3K- and Rho GTPase-mediated signaling is involved in signal
integration and crosstalk during cell adhesion and migration. The
Rho family of GTPases, including Rho, Rac and Cdc42, are crucial
for regulating the formation of stress fibers, lamelipodia and
filopodia, respectively, and control FA formation and various cell
functions, such as cell adhesion, cell migration and changes in
cell shape (9,10).
Endothelial FAK is required for adult pathological
tumor angiogenesis, and inhibition of FAK in both endothelial and
cancer cells is considered to be a possible target for
anti-angiogenic therapies. Endothelial FAK is required for tumor
angiogenesis (11), and the link
between FAK and cancer is supported by evidence of increased FAK
expression in several different types of cancer (12–17). However, the changes in those
crucial proteins associated with integrin-mediated FAK signaling,
and whether FAK acts in the same manner in VECs and hepatoblastoma
cells, remain poorly understood.
In the present study, the adhesion and migration
behavior of VECs (EA.hy926) and the human hepatoblastoma cells
HepG2 was examined following addition of a FAK inhibitor; the
expression of crucial proteins in the integrin-mediated FAK
signaling pathway was also investigated in order to determine
whether inhibition of FAK may be used for the treatment of cancer,
as well as that of other diseases, through inhibition of
angiogenesis.
Materials and methods
Cell lines
In this study, due to their higher affinity for the
tissue culture polystyrene without fibronectin or collagen
precoating, the human vascular EC line EA.hy926 (Jiangsu Institute
of Hematology, Suzhou, China) was used rather than original ECs.
EA.hy926 cells are a hybridoma cell line between human umbilical
vein eECs (HUVECs) and epithelioma A549 cells, and they retain most
of the characteristics of HUVECs, including the expression of
endothelial adhesion molecules and human factor Ⅷ-related antigen
(18), which has been previously
identified in our laboratory. Hepatoblastoma is the most common
pediatric malignant hepatic tumor worldwide. The human
hepatoblastoma HepG2 cell line (Institute of Biochemistry and Cell
Biology, Shanghai Institutes for Biological Sciences, Chinese
Academy of Sciences, Shanghai, China) is one of the most
extensively investigated in the literature, and was used as a
representative cancer cell line in the present study.
Antibodies
Detailed information on primary antibodies is
provided in Table I. The
secondary fluorescein isothiocyanate (FITC)-conjugated goat
anti-mouse immunoglobulin G (IgG) and PE-conjugated goat
anti-rabbit IgG were purchased from Beijing Biosynthesis
Biotechnology Co., Ltd. (Beijing, China). The BODIPY FL phallacidin
(Invitrogen Life Technologies; Thermo Fisher Scientific, Carlsbad,
CA, USA) was used for F-actin staining.
 | Table IDetailed information on the
antibodies used in the present study. |
Table I
Detailed information on the
antibodies used in the present study.
| Category | Antibody | Isotype | Blocking conditions
1 h room temperature | Primary Ab
incubation | Manufacturer | Cat. nos |
|---|
| Integrins | α2 (C-9) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA | sc-74466 |
| α5 (A-11) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-166665 |
| αV (H-2) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-376156 |
| β1 (A-4) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-374429 |
| β3 (B-7) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-46655 |
| FA components | Talin (8D4) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA | sc-59881 |
| Paxillin (D-9) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-365174 |
| Vinculin
(G-11) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-55465 |
| α-actinin
(H-2) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-17829 |
| Zyxin (H-200) | Rabbit pAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-15338 |
| pFAK | FAK (A-17) | Rabbit pAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA | sc-557 |
| pFAK (2D11) | Mouse mAb | 3% BSA in PBS | Overnight at 4°C,
1:200 dilution | | sc-81493 |
| PI3K/AKT | PI3K-C2β
(16L9) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA | sc-100407 |
| AKT1 (B-1) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-5298 |
| pAKT1 (Thr308) | Rabbit pAb | 3% BSA in PBS | Overnight at 4°C,
1:200 dilution | | sc-135650 |
| Rho GTPases | Rac1 (C-11) | Rabbit pAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA | sc-95 |
| pRac1 (Ser71) | Rabbit pAb | 3% BSA in PBS | Overnight at 4°C,
1:200 dilution | | sc-12924-R |
| RhoA (26C4) | Mouse mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:200 dilution | | sc-418 |
| C | dc42 | Rabbit mAb | 5% non-fat dry milk
in PBS | Overnight at 4°C,
1:100 dilution | Abcam®,
Cambridge, UK | ab64533 |
FAK inhibitor (Y15)
The FAK inhibitor 1,2,4,5-benzenetetramine
tetrahydrochloride (Y15) increases cell detachment and inhibits
cell adhesion in a dose- and time-dependent manner, as observed and
tested by Golubovskaya et al (19). In the present study, Y15
(Sigma-Aldrich; Merck KGaA, St. Louis, MO, USA) was used to
investigate whether the migration and adhesion of EA.hy926 and
HepG2 cells depend on FAK signaling, and to determine the
time-dependent expression of crucial proteins in the
integrin-induced FAK/Rho GTPases pathway.
First, Y15 at different concentrations (50, 100,
150, 200 and 250 µM) was added into 6-well polystyrene cell
culture plates for cell migration analysis, and an appropriate
concentration of the FAK inhibitor was determined for further
examination.
Cell culture
EA.hy926 cells were cultured in RPMI-1640 nutrient
mixture with 10% fetal bovine serum (FBS) (both from Gibco-BRL;
Thermo Fisher Scientific, Grand Island, NY, USA), HAT supplement
(Sigma-Aldrich; Merck KGaA), 100 U̸ml penicillin and streptomycin
(both from Beyotime Institute of Biotechnology, Shanghai, China),
and grown at 37°C in 95% air and 5% CO2. HepG2 cells
were cultured in Dulbecco's modified Eagle's medium-high glucose
(Gibco-BRL; Thermo Fisher Scientific) with 10% FBS, 100 U̸ml
penicillin and streptomycin, and grown at 37°C in 95% air and 5%
CO2.
Parallel cell migration-scratch wound
assay
The scratch wound assay was used to investigate the
migration ability of EA.hy926 and HepG2 cells in parallel
directions. Cells were seeded on a culture dish, the bottom of
which was previously marked with three parallel lines, and cells
with 100% confluence were starved without serum overnight. A
uniform scratch (~500 µm), perpendicular to the lines on the
bottom of the dish, was then made in the monolayer with a plastic
cell scraper to obtain three cross regions. The cell monolayer was
washed three times with phosphate-buffered saline (PBS); Y15 was
added to the serum-free medium and the three cross regions were
photographed with a reflective upright biological microscope.
Images of the wounds under culture were acquired and analyzed at 0,
2, 4, 8 and 24 h, the appropriate concentration of FAK inhibitor
was determined, and detailed images of the wounds were captured at
0, 5, 10, 30, 60 and 120 min under an inverted microscope.
Vertical cell migration-Transwell
assay
Cell migration was further determined by the
Transwell assay (8-µm pore size; BD Falcon™; BD Biosciences,
Franklin Lakes, NJ, USA). EA.hy926 and HepG2 cells were cultured
and adhered in the Transwell chamber for 4 h, and were
serum-starved overnight, followed by the addition of 50 µM
Y15 for 24 h at 37°C and 5% CO2. The inner chamber of
the Transwell unit was cleaned, and the migrated HepG2 cells on the
bottom of the chamber were fixed by 4% paraformaldehyde for 10 min
and stained with 0.1% crystal violet for 15 min. Each step was
followed by washing with PBS three times. The migrated cells were
photographed at 5 different areas under an inverted phase contrast
microscope (CK2; Olympus, Tokyo, Japan). The cell number of each
group in one vision field was counted based on obtained optical
images. At least three parallel experiments were performed, and
data are presented as mean ± standard deviation (SD).
Immunofluorescence staining
EA.hy926 and HepG2 cells were plated on small
circular slides with 50% confluence and serum starvation overnight,
and were incubated with FAK inhibitor (50 µM) for 5 min or 2
h. The cells were then washed twice with PBS for 8 min each time,
fixed with 4% paraformaldehyde in PBS for 15 min, washed three
times with PBS for 5 min each time, permeabilized for 5 min with 1%
(v/v) Triton X-100, and washed three times with PBS for 5 min each
time. Cells were pre-incubated with 1% bovine serum albumin (BSA)
in PBS for 15 min to decrease non-specific antibody binding, and
washed for 30 sec to 1 min with PBS. Primary antibody incubation
was performed at 4°C overnight, the slides were washed for 5 min
with PBS, further incubated for 90 min in FITC-conjugated goat
anti-mouse antibody and PE-conjugated goat anti-rabbit antibody,
and then incubated with 4′,6-diamidino-2-phenylindole (DAPI) for 30
min to stain the nuclei. The samples were sealed by 10% glycerol,
protected from light and examined under a laser scanning confocal
microscope.
Western blot analysis of proteins
associated with FAK signaling pathways
The western blot assays followed standard protocols.
Briefly, following treatment with the FAK inhibitor for 0, 5, 10,
30, 60 and 120 min, EA.hy926 and HepG2 cells were washed three
times with PBS and lysed by 200 µl cell lysis solution
(Beyotime Institute of Biotechnology). The RIPA lysis buffer
includes phosphatase inhibitor (Na3VO4),
protease inhibitor mixtures (BestBio Biotechnology Co., Ltd.,
Shanghai, China) and phenylmethanesulfonyl fluoride (PMSF; Amresco,
LLC, Solon, OH, USA), at a ratio of 100 (RIPA):1 (phosphatase
inhibitor):1 (protease inhibitor mixtures):1 (PMSF). The total
proteins were collected and centrifuged at 10,000 × g̸min at 4°C
for 10 min and the supernatant was collected. Following a protein
concentration assay by an ultraviolet spectrophotometer using the
bicinchoninic acid protein assay kit, the proteins (20
µg/lane) were separated by 10% SDS-PAGE and transferred to a
PVDF membrane. BSA (5%) powder in Tris-buffered saline with
Tween-20 (TBST) was used to block non-specific binding for 2 h at
37°C. Incubation with primary antibodies against integrin α2, α5,
αV, β1, β3, vinculin, paxillin, talin, pFAK, PI3K, pAKT, pRhoA,
pRac1 and Cdc42 overnight at 4°C was followed by incubation with
horseradish peroxidase-conjugated goat anti-mouse or goat
anti-rabbit antibody for 1 h at 37°C. After washing with TBST,
targeted proteins were detected using enhanced chemiluminescence
(Beyotime Institute of Biotechnology). Images of bands were
determined using Molecular Imager® ChemiDoc™ XRS+ with
Image Lab™ software (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Statistical analyses
Data obtained from the present study are expressed
as mean ± SD from at least three independent experiments. To reveal
differences among the groups, one-way analysis of variance followed
by Tukey's test was used. The differences were considered
significant at P<0.05.
Results
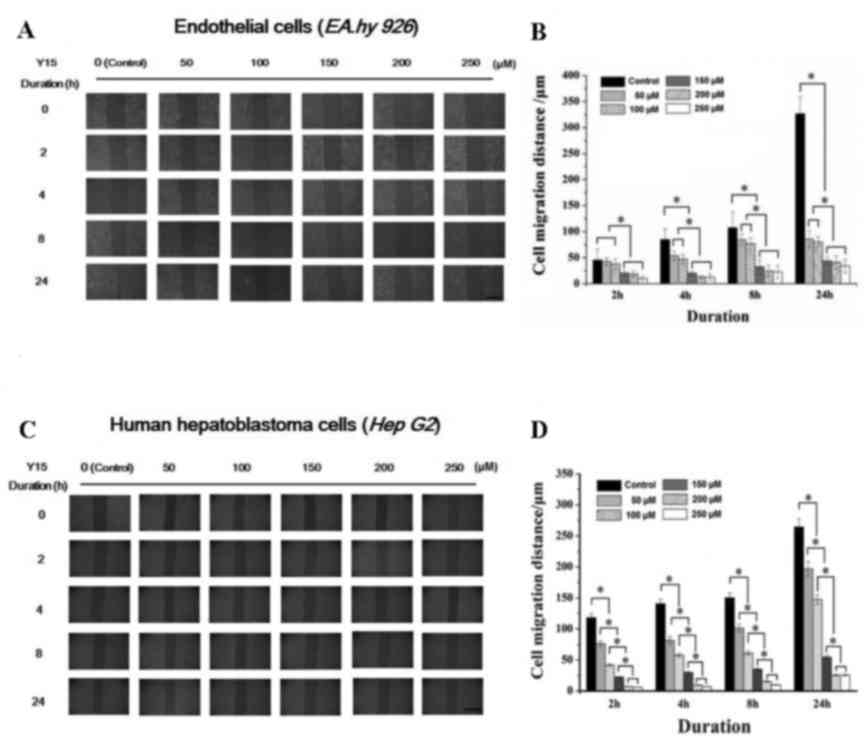
Effect of FAK inhibitor on cell migration
at various concentrations and durations
The effect of FAK inhibitor at various
concentrations (50–250 µM) on the migration behavior of
EA.hy926 and HepG2 cells was examined using scratch wound assays
(Fig. 1A and C), and the results
were calculated as shown in Fig. 1B
and D. It may be seen from Fig.
1 that the FAK inhibitor significantly inhibited the migration
of ECs, as well as that of tumor cells. Under high concentrations
of Y15 (>150 µM), the migration distance of ECs at 2 h
differed significantly from that of control cells (cells without
addition of Y15) and low concentration groups (50 and 100
µM). The migration distance at 24 h of all groups treated
with FAK inhibitor differed significantly from that of control
cells. As shown in Fig. 1A, the
ECs in the control group reached a ~90% confluence at 24 h, while
Y15 prevented EC migration. In HepG2 cells, the migration distance
in all Y15-treated groups differed significantly from that of
control cells throughout the duration of the assay (from the
initial 2 to 24 h). However, a confluence of only 50% was observed
in the control group at 24 h due to the weakened motility of HepG2
cells. The statistical results demonstrated that Y15 significantly
inhibited cell motility in a dose-dependent manner (Fig. 1B and D).
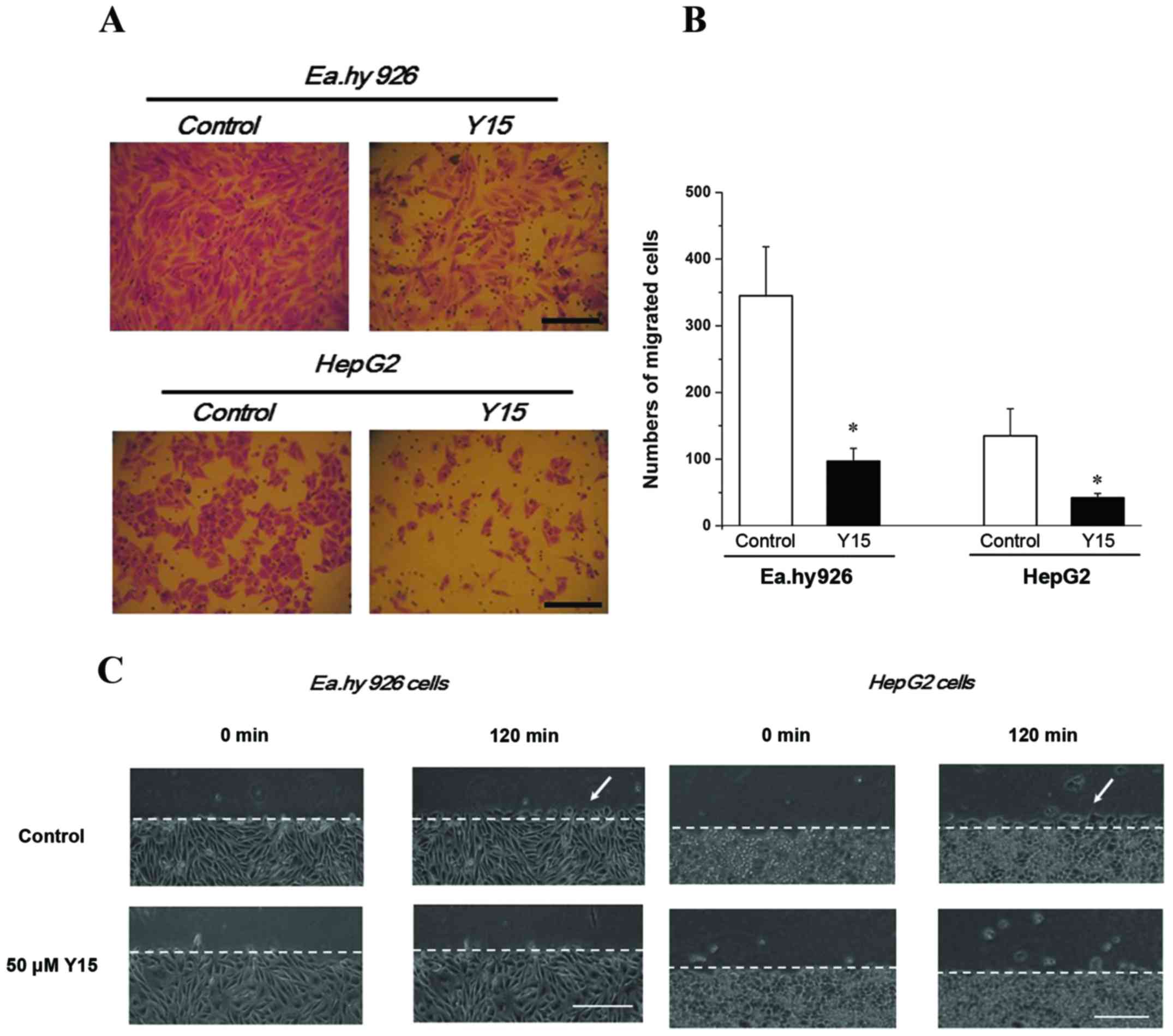
Based on the previous results, 50 µM Y15 was
selected to further investigate its inhibitory effect on cell
migration by the Transwell assay. It was observed that, with FAK
inhibition for 24 h, the migrating numbers of both endothelial
EA.hy926 cells and HepG2 cells decreased significantly compared
with the control group (P<0.05), suggesting that Y15 suppressed
migration in the two cell lines (Fig.
2A and B). Furthermore, 50 µM Y15 was also used to
examine its inhibitory effect on parallel cell migration by the
scratch wound assay within 2 h. The local changes of migrating
cells are described in Fig. 2C and
D. Migrating cells were clearly observed in the control group
(Fig. 2C and D, white arrows)
after 120 min, but could hardly be identified in the 50-µM
group of EA.hy926 and HepG2 cells. Therefore, migration in the two
types of cells was distinctly inhibited by 50 µM Y15.
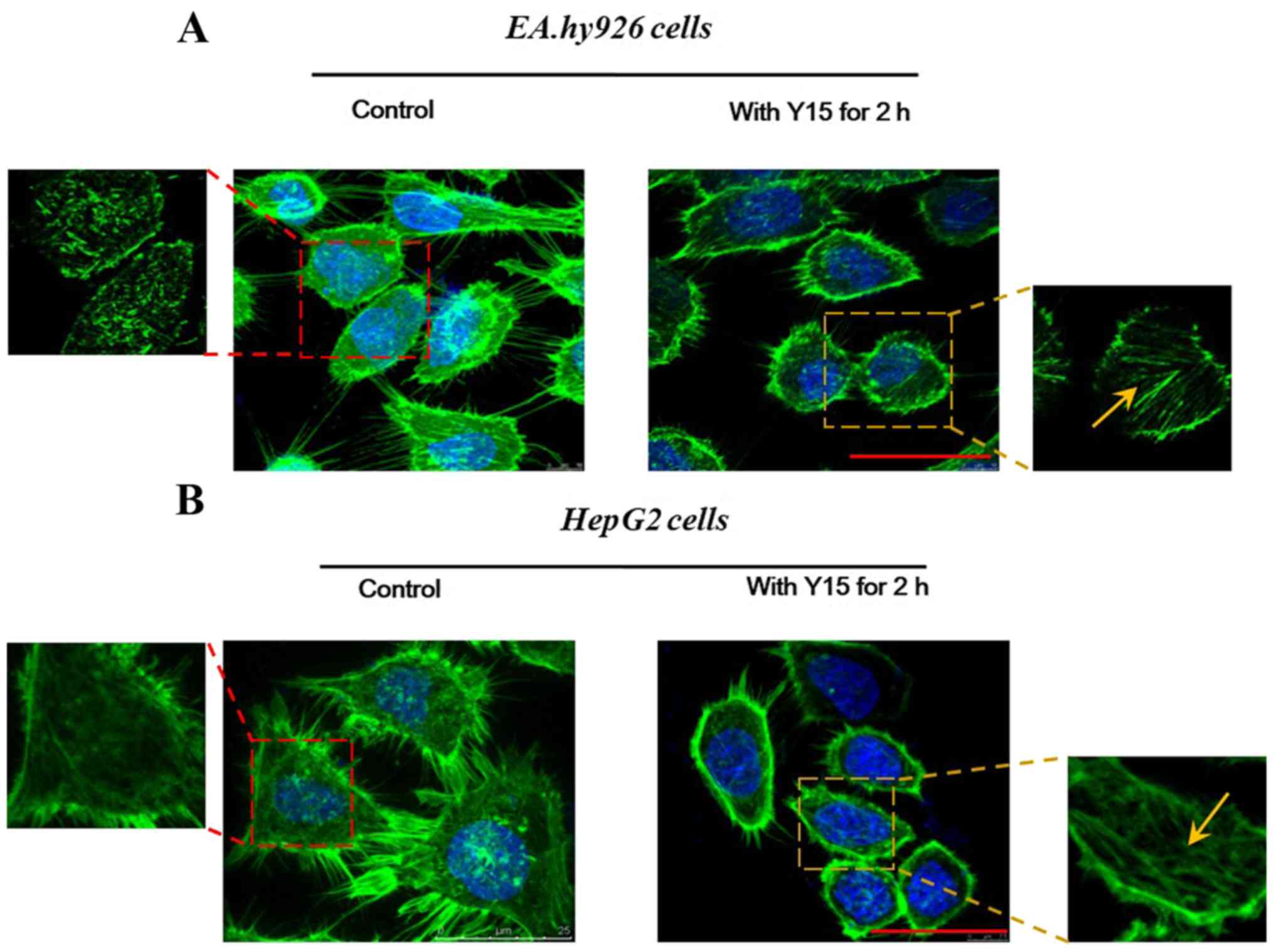
Effect of FAK inhibition on the
cytoskeleton
Furthermore, changes in the localization of F-actin
were examined in order to determine the effect of FAK inhibition on
cell adhesion. Cell adhesion is closely coupled with the
protrusions of the leading edge of migrating cells (filopodia and
lamellipodia). It was observed from confocal immunofluorescence
images of F-actin staining that the formation of filopodia was
markedly impaired by FAK in EA.hy926 (Fig. 3A) as well as in HepG2 cells
(Fig. 3B). In the control groups
of the two cell lines (Fig. 3),
all spreading cells displayed polymerized F-actin in the
protrusions, and more obvious spike-like filopodia and
intercellular filaments were observed among cells. However, in the
treated groups (2-h FAK inhibition; right panels in Fig. 3), endothelial as well as
hepatoblastoma cells exhibited decreased cell spreading areas, and
fewer filopodia were present at the edge of any single cell. In
HepG2 cells (Fig. 3B), in
particular, it was observed that the cells became round and lost
their filopodia and lamellipodia, whereas cell adhesion was
evidently inhibited. However, the enlarged images revealed a
distinct filamentous stress fiber structure (Fig. 3, yellow arrows) in single cells
following addition of Y15 for 2 h, while there was no ordered
stress fiber structure found in the control group (without Y15
treatment). These results suggested that inhibition of FAK reduced
the adhesion sites between the cells and the matrix, which may
result in promotion of cell detachment and inhibition of cell
adhesion. Furthermore, the inhibition of FAK induced retraction of
protrusive filopodia and lamellipodia, which was responsible for
actin polymerization and stress fiber formation in the retractile
cell bodies.
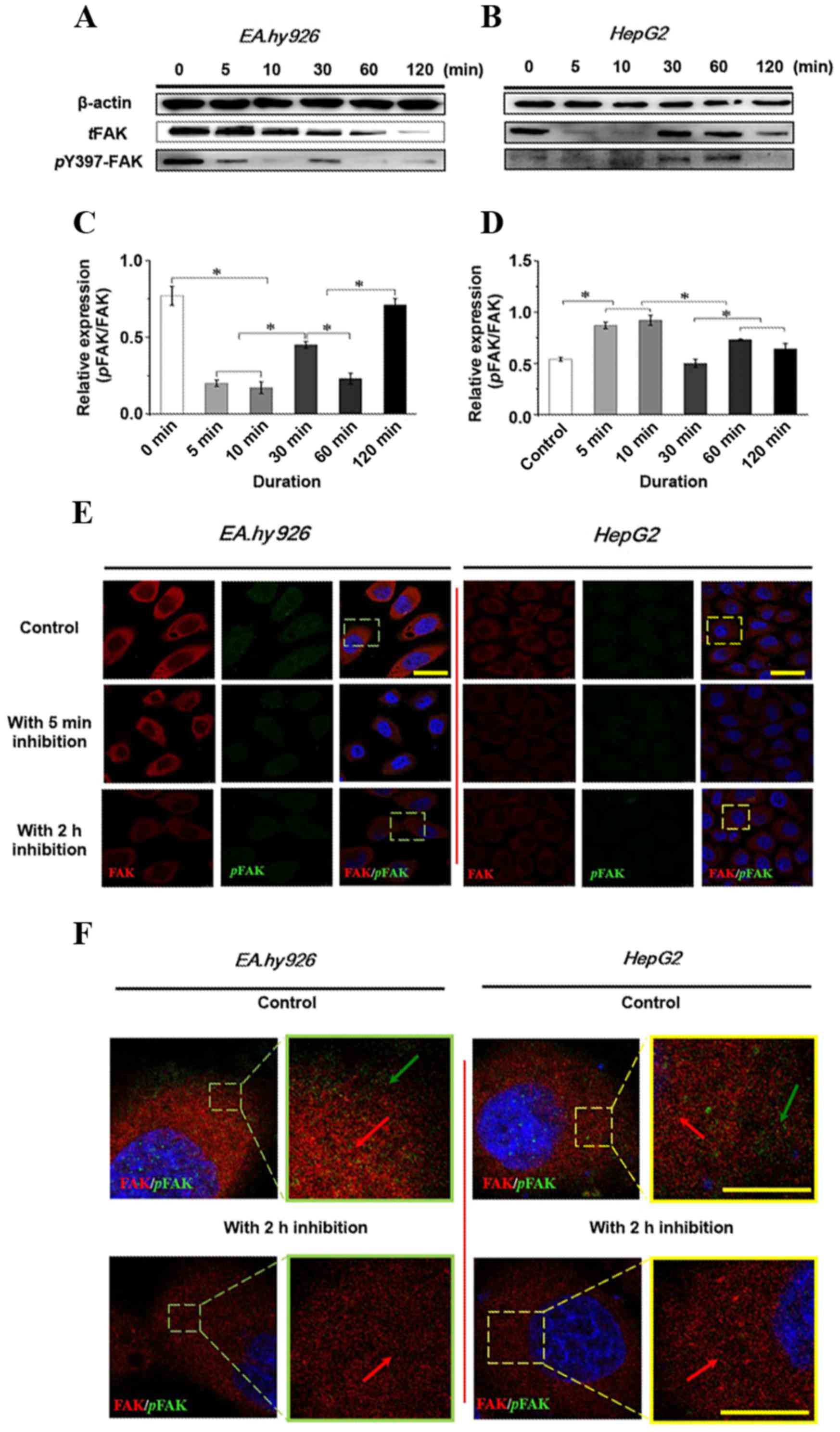
Expression and distribution of FAK with
the addition of the FAK inhibitor Y15
The initial autophosphorylation of FAK at Y397
results in phosphorylation of additional multiple tyrosine sites
(Y-576, -577 and -925) in FAK, which regulate a number of cell
functions, including cell adhesion and cell migration. Therefore,
the effect of 50 µM Y15 on total phosphorylation and
phosphorylation at the Y397 site of FAK within 2 h was first
examined by western blot analysis (Fig. 4A and B), and the relative
quantification of the phosphorylated FAK (pFAK) level against total
FAK (tFAK) expression is presented in Fig. 4C and D in EA.hy926 and HepG2
cells, respectively. Increased duration of FAK inhibition resulted
in a significant decrease of the total and phosphorylated FAK level
in ECs (Fig. 4A). tFAK exhibited
a gradual reduction tendency. However, pFAK decreased initially,
followed by a transient increase over 10–30 min, suggesting that
inhibition of FAK may activate compensatory effects by other
signaling molecules. The statistical result of the relative
quantification of the pFAK level in tFAK (Fig. 4C) revealed that the pFAK/tFAK
ratio at 5 and 10 min was significantly decreased compared with the
control (P<0.05); subsequently, the increased pFAK/tFAK ratio at
30 min was significantly different compared with that at 5 and 10
min (P<0.05), indicating a temporarily higher expression of pFAK
at 10–30 min with a continuous decline of tFAK. The pFAK/tFAK ratio
at 120 min was relatively higher, suggesting a weak effect of FAK
inhibition on pFAK, but a strong effect on tFAK. By contrast, tFAK
in HCC cells was greatly inhibited at the initial stage, both tFAK
and pFAK increased at 10–30 min, and then decreased at 120 min.
Initially, tFAK sharply decreased, but pFAK was not inhibited;
therefore, the relative pFAK/tFAK ratio at 5 and 10 min was
significantly increased compared with the control (Fig. 4D; P<0.05), and the ratio
decreased significantly at 30 min compared with that at 5 and 10
min and at 60 and 120 min (P<0.05), indicating that the
expression of pFAK was not inhibited initially with the changes in
tFAK in HCC cells. The results also indicated that inhibition of
FAK in various cell lines exerted different time-dependent effects
on tFAK and pFAK expression.
Furthermore, the distribution of tFAK and pFAK at 5
min and 2 h was examined by double immunofluorescence staining
(Fig. 4E). The expression levels
of tFAK and pFAK were consistent with the results of the western
blot analysis. With a 2-h inhibition of FAK, the levels of tFAK and
pFAK decreased in both endothelial and hepatoblastoma cells. It was
observed that pFAK expression was significantly inhibited in ECs;
in addition, the intensity of tFAK was also reduced. The expression
level of tFAK in hepatoblastoma cells decreased, and pFAK exhibited
a lower intensity after 5 min of treatment with the inhibitor. When
combining the distribution of FAK and its phosphorylated
counterpart, pFAK (green) is clearly more highly expressed at the
leading edge of the cell in the control groups compared with the
groups undergoing FAK inhibition for 2 h (Fig. 4E and F). Compared with the control
groups of both cell lines, weak green fluorescence (pFAK) was
observed at the leading edge of the cells, suggesting that the FAK
inhibitor used in the present study clearly inhibited the
expression of pFAK and also decreased the level of tFAK. In
conclusion, the results demonstrated that the response time of FAK
differs among various cells treated with FAK inhibitor, resulting
in time-dependent differences in protein expression in the entire
signaling cascade. Furthermore, the inhibitory effect of FAK on the
integrin-induced signaling pathway involved in cell adhesion and
migration was further investigated.
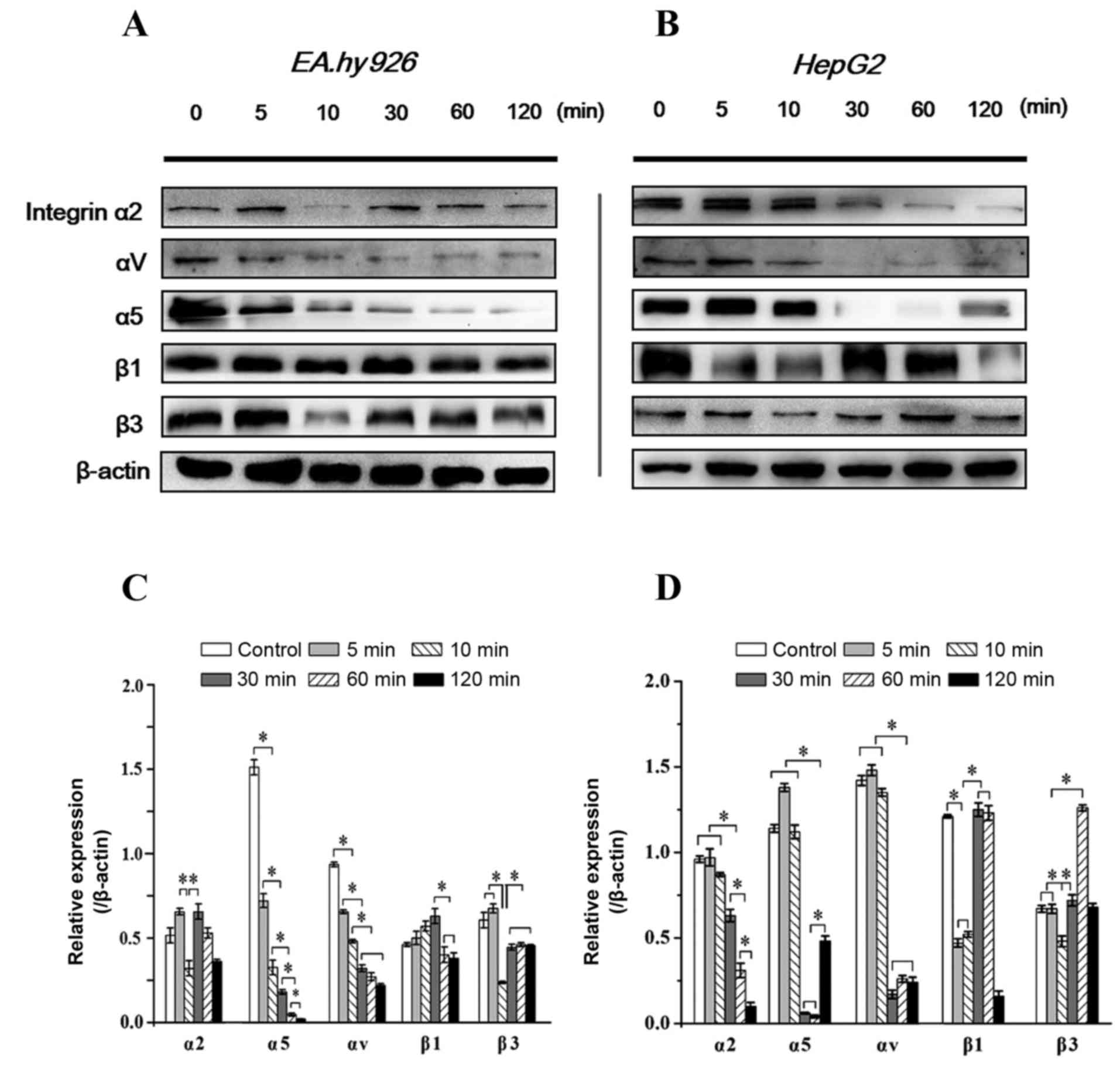
Time-dependent expression of integrins
with inhibition of FAK
Integrins are transmembrane ECM receptors containing
α and β subunits that bind to cytoskeletal proteins and transduce
signals from extracellular stimuli to intracellular events
(20). It has been reported that
pFAK regulates cell adhesion and migration via binding activated
integrin (16). In the present
study, the expression levels of integrin α2, α5, αV, β1 and β3
subunits in response to FAK inhibition were investigated. It was
observed that, as in EA.hy926 cells, integrin α2 and β3
significantly decreased at 5–10 min with inhibition of FAK, and
were restored to a higher level at 10–30 min, consistently with the
recovery of the pFAK level at 10–30 min; the β1 subunit exhibited a
stronger expression at 10–30 min; a gradually weakened expression
level of the α5 subunit was associated with increasing duration of
Y15 treatment, whereas the expression of αV exhibited a gradual
decrease within 30 min and then remained stable (Fig. 5A and C). In HepG2 cells, β3
exhibited a similar tendency to that in EA.hy926 cells within 60
min, but exhibited a sharp increase at 1 h, while the expression of
α2 was gradually downregulated after 30 min. The expression of α5
decreased sharply after 10 min, but appeared to recover after 2 h.
The expression of αV exhibited an acute decrease at 30 min, and
then remained stable, similar to ECs (Fig. 5B and D). Of note, with inhibition
of FAK within 2 h, the expression of the β1 subunit appeared to be
correlated with the level of tFAK in HepG2 cells (decreased
initially at 5 min, was upregulated at 10–30 min, and reached a
maximum level at 1 h). Since FAK directly and indirectly binds to
the β tails of integrins, these results suggest that inhibition of
FAK initiated a feedback signal to upstream events and decreased
the expression of integrins, blocked integrin-induced loss of
clustering and resulted in cell detachment. However, the responses
of integrin subunits in various cell lines with FAK inhibition were
different.
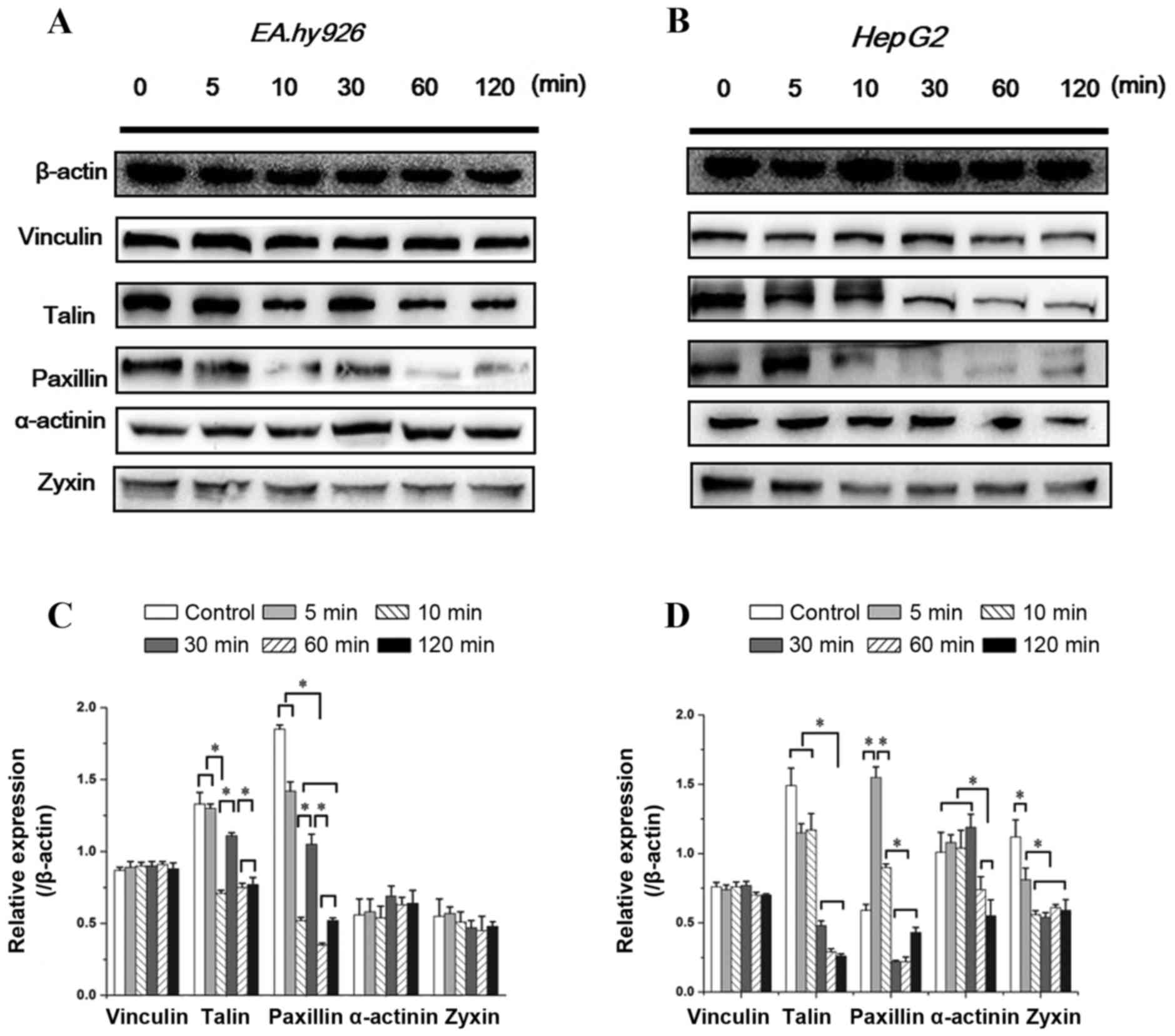
Expression of FA components with FAK
inhibition
Integrins bind to the intracellular actin
cytoskeleton through a complex network of regulatory proteins
referred to as FA. The FA complex consists of several proteins,
such as talin, vinculin and paxillin, and is located at the
cytosolic face of the cell membrane. As a ligand of β integrin
cytoplasmic domains, talin plays a key role in the first step of
focal complex formation following initial integrin engagement, and
talin degradation plays a crucial role in FA turnover (4). The integrin-binding proteins
paxillin and talin recruit FAK and vinculin to focal contacts.
Vinculin, as a binding partner of talin, promotes and stabilizes
initial integrin clustering, which may promote cell spreading by
mechanically coupling integrins with the cytoskeleton, with the
migration velocity of cells lacking vinculin being twice that of
wild-type cells (21). Zyxin is a
zinc-binding phosphoprotein that concentrates at FAs and along the
actin cytoskeleton, and α-actinin is an actin-binding protein that
binds actin to the membrane. The assembly of these cytoskeletal
proteins form actin-rich structures of FA, and contribute to
support stable cell-matrix adhesion. Therefore, the time-dependent
changes in the levels of talin, vinculin, paxillin, zyxin and
α-actinin with inhibition of FAK within 2 h were investigated.
As seen in Fig.
6A, the vinculin level in ECs exhibited no significant
differences with increased duration of Y15 treatment. However, the
expression of talin and paxillin in ECs showed an identical
tendency (decreased at 5–10 min, but was upregulated at 30 min and
subsequently decreased). The expression of zyxin exhibited a slight
decrease, while that of α-actinin exhibited a slight increase at
10–30 min, but there were no significant differences among groups
(Fig. 6A).
Similar to ECs, increased duration of 50 µM
Y15 treatment did not affect the vinculin level in HepG2 cells
(Fig. 6B). The expression of
talin and paxillin in HepG2 cells exhibited a similar tendency,
remaining at a lower level after 30 min. There was no significant
change in talin expression within 10 min, but the level of paxillin
initially increased at 5 min compared with the control (P<0.05;
Fig. 6B). Another difference
between the two cell types is that inhibition of FAK significantly
affected zyxin and α-actinin expression in HepG2 cells, with a
significant decrease in zyxin expression at 10 min and the
α-actinin level at 60 min (Fig.
6B). These results demonstrated that partial FA complexes
disassembled with inhibition of FAK, resulting in cell detachment
and inhibition of cell adhesion. Furthermore, inhibition of FAK
exerted different time-dependent effects on FA components in the
two cell lines.
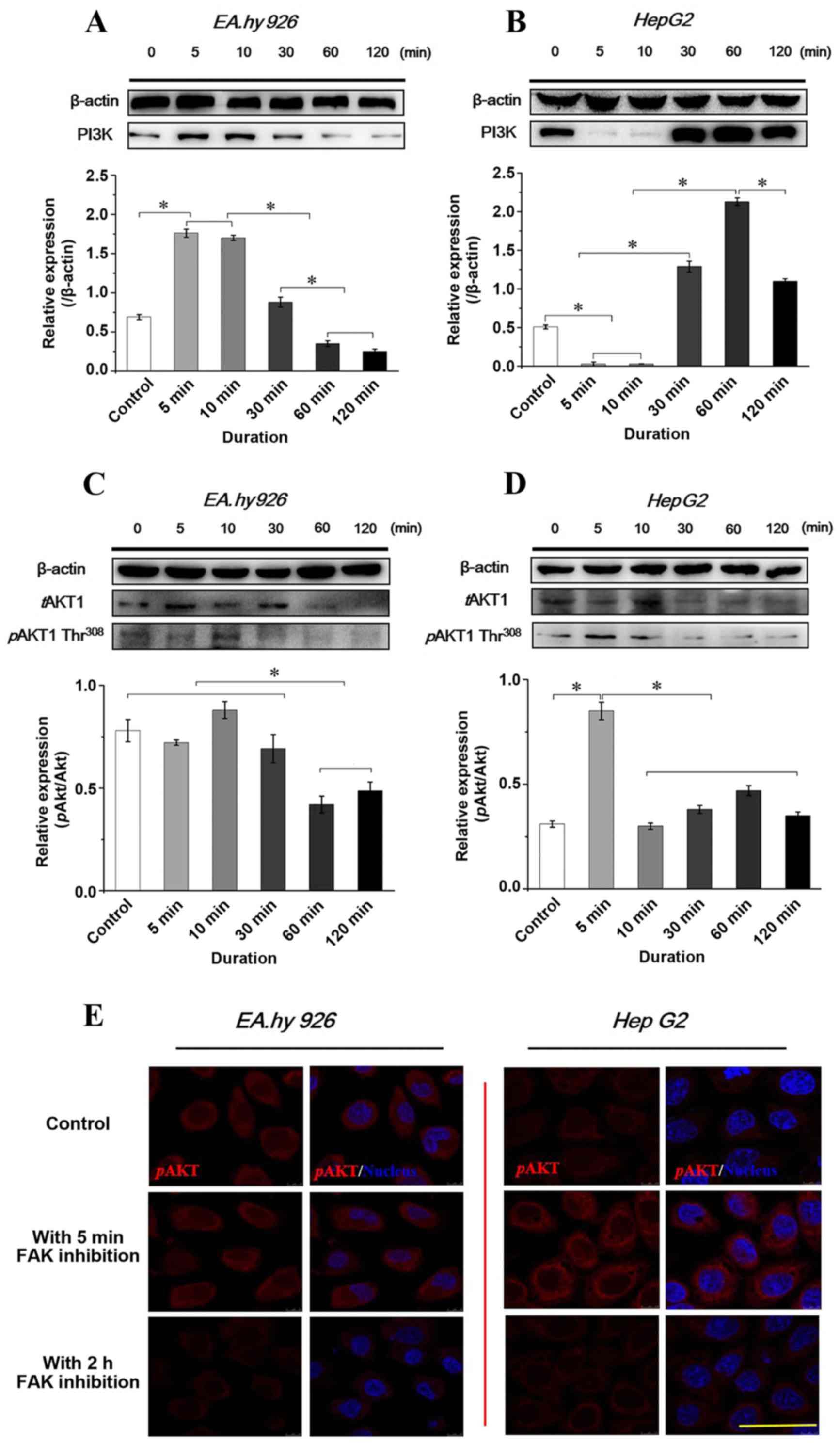
Expression and distribution of PI3K/AKT
signaling with FAK inhibition
The autophosphorylation of FAK at Y397 is a binding
site for Src and PI3K and, as downstream molecules of FAK,
activated PI3K/AKT signaling events are crucial in regulating cell
viability and migration. The SH2 domain phosphorylation of the p85
subunit of PI3K binding to FAK at Y397 may activate the p110
catalytic subunit of the PI3K/AKT signaling pathway. Therefore,
PI3K̸AKT signaling was investigated to determine whether inhibition
of FAK induces changes in the PI3K/AKT pathway in the two cell
lines. The PI3K/AKT expression levels with inhibition of FAK are
shown in Fig. 7. Of note, the
results demonstrated that the PI3K expression in response to FAK
inhibitor treatment was opposite in the two cell lines (Fig. 7A and B). In endothelial EA.hy926
cells, blocking FAK increased the PI3K level during the initial 5
min, which then gradually decreased to a relatively lower level.
However, weaker expression of PI3K was found during the first 5–10
min in HepG2 cells when FAK was inhibited, subsequently increasing
at 30 min and reaching a maximum level at 1 h. The result was
opposite in terms of PI3K expression in the two cell lines, but it
was consistent with the expression of tFAK (Fig. 4A and B).
AKT1 may be activated and phosphorylated at Thr308
and Ser473 in a PI3K-dependent as well as -independent manner
(22). Therefore, tAKT1 and pAKT1
were examined, and the relative ratio (pAKT/tAKT) was calculated in
the two cell lines according to the results of western blot
analyses. The results revealed a statistically significant
difference of pAKT/tAKT in EA.hy926 cells during the initial 30 min
compared with that after 30 min, while the pAKT/tAKT ratio at 5 min
in HepG2 cells exhibited a significant difference compared with
other groups (P<0.05; Fig. 7C and
D). Furthermore, the distribution of pAKT was examined by
immunofluorescence staining (Fig.
7E). With the 2-h inhibition of FAK, the expression levels of
pAKT decreased in both endothelial and hepatoblastoma cells,
consistently with the western blot results.
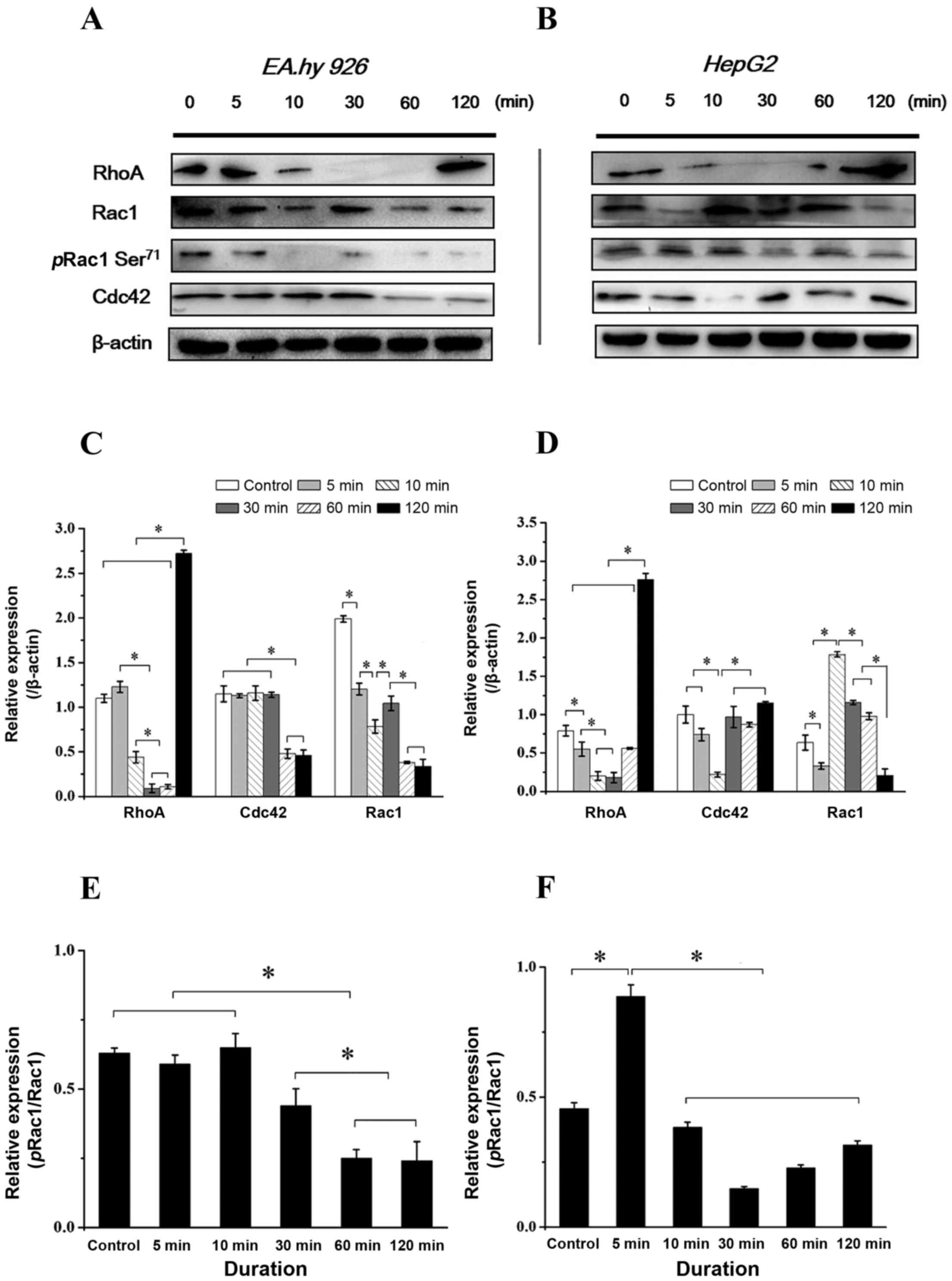
Time-dependent expression and
distribution of Rho GTPases with addition of Y15
Rho family GTPases (RhoA, Rac1 and Cdc42) result in
direct local actin assembly to form stress fibres, lamellipodia or
filopodia, respectively, and regulate cell polarity and motility
through their effects on the cytoskeleton, membrane trafficking and
cell adhesion (23). The Rho
family small GTPases are important regulators of actin dynamics
that potentiate cell migration. Cdc42 and Rac1 act at the leading
edge of the cell to induce cell polarization and lamellipodia
formation, respectively, whereas RhoA acts in the cell body to
facilitate contraction (10). The
phosphorylation of Rac1 at Ser71 (pRac1) predominantly modulates
affinity to guanine nucleotide dissociation inhibitor and
subcellular localization of GTPases, negatively regulating
activation (24). As the
downstream molecules of the integrin-FAK signaling pathway
associated with cell adhesion and migration, the expression of
RhoA, pRac1 and Cdc42 was examined following inhibition of FAK.
With FAK inhibition for 2 h, the expression level of RhoA decreased
in both endothelial and hepatoblastoma cells at 5–10 min, but had
increased at 2 h (Fig. 8A and B),
which is consistent with previous results, indicating that
inhibition of FAK induced the retraction of lamellipodia at the
cell protrusions and the formation of filamentous stress fibers in
the cell body (Fig. 3). The total
expression and phosphorylation levels at Ser71 of Rac1 were
different between the two cell types; the expression of Rac1 in
EA.hy926 cells decreased during the initial 5 min, and pRac1 was
also lower after 5 min (Fig. 8A).
While the expression of Rac1 in HepG2 cells decreased with FAK
inhibition during the initial 5 min, and immediately increased and
remained at a higher level thereafter, pRac1 exhibited a lower
level at 10 min (Fig. 8B). The
results revealed a statistically significant difference of
pRac1/Rac1 in EA.hy926 cells during the initial 10 min compared
with that thereafter, while the pRac1/Rac1 ratio at 5 min in HepG2
cells exhibited significant differences compared with other groups
(P<0.05; Fig. 8C and D). As
regards the Cdc42 level in ECs with inhibition of FAK, it was
similar compared with that in the control group from the initial 5
min up to 30 min, but decreased from 30 min to 1 h (Fig. 8A). In HepG2 cells, the expression
of Cdc42 fluctuated, exhibiting a relative lower level at 10 min,
which recovered thereafter and was maintained at a higher level
after 30 min (Fig. 8B). The
time-dependent difference in Rho GTPase expression indicates
different mechanisms of FAK-mediated migration behavior in various
types of cells.
Distribution of FA proteins (talin) and
Rho GTPases (Rac1) with FAK inhibition
To further investigate the distribution of FA
complexes and Rho GTPases, the cytoskeletal protein talin, which
binds to integrin β tails to form intercellular FA plaques
associated with cell adhesion, and the Rho family GTPase Rac1,
which mediates lamellipodia formation and regulates cell migration
behavior, were selected to study their respective distribution by
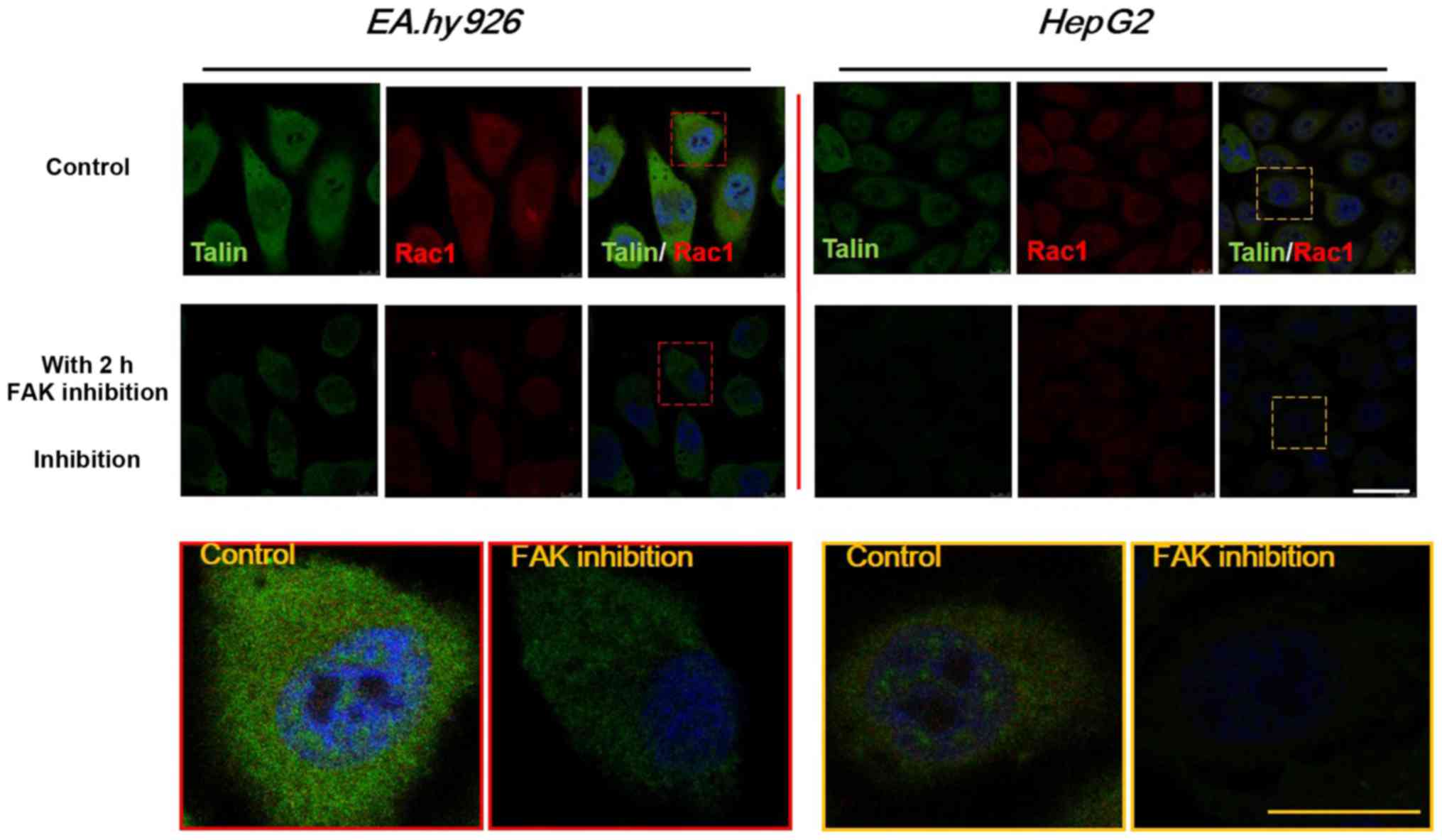
double-labeled immunofluorescence analysis. As shown in Fig. 9, talin (green) and Rac1 (red) were
mainly distributed in the entire cytoplasm. The expression levels
of talin and Rac1 were downregulated with FAK inhibition for 2 h,
consistently with the western blot results for talin (Fig. 6) and Rac1 (Fig. 8). However, the expression of talin
exhibited a more prominent decrease, with weakness of fluorescence
intensity in HepG2 cells after 2 h of Y15 treatment, while the
expression of talin in EA.hy926 cells exhibited a relatively mild
downregulation. It was suggested that inhibition of total and
phosphorylated FAK provided feedback signals to FA components,
which may result in cell detachment, and also exert stronger
effects on downstream Rho GTPases, which may weaken cell
motility.
Discussion
Tumor-induced angiogenesis results from
neovascularization of migrated ECs in solid tumors. Once adequate
vasculature has been developed and established, the tumor invades
the vessel wall and spreads to distant organs to form dormant
micrometastases (25). Therefore,
controlling tumor-associated angiogenesis is a promising strategy
in limiting cancer progression.
The migration of endothelial and cancer cells plays
a key role in the process of tumor-induced angiogenesis. Cell
migration is an integrated process requiring continuous,
coordinated assembly and disassembly of adhesion structures. To
investigate the time-dependent differences in FAK-dependent signals
in various cell lines, VECs and hepatoblatoma cells were selected
for the experiments. In the present study, EA.hy926 cells were used
rather than original VECs, as they exhibit a higher affinity to
substrate, they express EC markers to the same extent as HUVECs,
particularly vWF, PECAM-1, ICAM-1, E-selectin, and uptake of
DiI-Ac-LDL, and have no tumorigenic potential, whereas they have
been widely used to investigate different aspects of EC biology. By
using the EA.hy926 cells as a model cell line, the expression level
of integrins could be accurately studied, and it may help to better
describe the direct effect of FAK inhibition on integrin signals
(primary ECs, such as HUVECs, exhibit low affinity to tissue
culture polystyrene without fibronectin or collagen pre-coating).
However, the marked phenotype differences between EA.hy926 cells
and primary VECs should be noted (26). The human hepatoblastoma cell line,
HepG2, was selected as an in vitro model system for the
study of adhesion and migration of tumor cells. Integrin- and
growth factor-stimulated migratory cues were considered as
significant events for activating FAK (27).
It has been proved that endothelial FAK is crucial
for vascular network stability, cell survival and lamellipodia
formation (28). Increased level
of FAK was found in a variety of human tumors, and inhibition of
FAK reduced tumor formation in the early stages (29), suggesting that FAK may be a new
therapeutic target in cancer (30). The FAK inhibitor used in the
present study was selected by Golubovskava et al (19), who reported that treatment with
Y15 effectively decreased Y397 phosphorylation in BT474 breast
carcinoma cells at 8 h. Their studies also reported that this
small-molecule FAK inhibitor increased cancer cell apoptosis and
decreased tumor growth (31,32). Thus, targeting the Y397 site may
be an effective therapeutic approach to developing novel FAK
inhibitors (33–36).
However, the role of FAK in cell migration and
metastasis remains controversial in various cell types (37). It has been demonstrated that the
overexpression of wild-type FAK in various different cell types may
enhance cell migration (17). In
HeLa cells, reduced expression level of FAK by siRNA and FAK-null
were associated with increased cell motility, while FAK-null
fibroblasts derived from FAK−/− embryos exhibited
reduced cell motility. FAK-deficient ECs exhibit defective but not
reduced motility (38). Although
in different cancer cell types, a substantial body of evidence
demonstrated the contradictory roles of FAK as a positive or
negative regulator of tumor cell migration and metastasis (37). In addition, elevated expression of
FAK in cancer cells had been correlated with increased migration
and invasiveness. Hauck et al (39) found that inhibition of FAK
expression reduced the migration and invasion of EGF-stimulated
human carcinoma cells (A549). Our results also supported the
conclusion that inhibition of FAK was able to decrease the
migration behavior of both ECs and hepatoblastoma cells.
Additionally, inhibition of FAK exerted different time-dependent
effects on tFAK and pFAK expression in ECs and hepatoblastoma
cells. The tFAK level in ECs gradually decreased with increased
duration of Y15 treatment. Of note, the tFAK level rapidly declined
in HepG2 cells, suggesting that these cells are sensitive to this
type of small-molecule compound FAK inhibitor, and FAK signaling is
possibly the most dominant pathway in regulating HepG2 cell
functions. However, other evidence supported that dephosphorylation
of FAK increased tumor cell motility, invasion and metastasis in
various human carcinoma cells (40). Furthermore, FAK signaling has been
shown to participate in the process of transforming growth
factor-β-induced epithelial-to-mesenchymal transition in
hepatocytes (41). Kallergi et
al (42) demonstrated that
activation of FAK̸PI3K̸Rac1 signaling regulated actin
reorganization and inhibited cell motility in A375 melanoma cells.
In the present study, a simple inhibitor technique and scratch
wound assays were used for cell migration analysis, and some
crucial proteins in integrin-induced signaling pathways were
extensively investigated to reveal possible alterations in the
mechanisms of cell adhesion and migration under FAK inhibition.
The effect of integrins on the phosphorylation of
FAK and the downstream signaling events associated with cell
adhesion and migration in various cells has been widely
investigated. Vinculin and the actin-binding protein talin, as the
ligands of β integrin cytoplasmic tails, assemble around enriched
integrins at cell adhesion sites to form nascent FA plaques
(43). The maturation of
intracellular nascent FAs involves a sequential cascade of
compositional changes. At the early phase of integrin activation,
the adapter protein paxillin is recruited and integrins are
clustered into FAs. The detailed sequence of events leading to the
assembly of adhesion complexes remains unclear (44). The cluster of FA complex recruits
and binds FAK via the C-terminal FA targeting (FAT) domain. The
initial tyrosine phosphorylation at the Y397 site of FAK may
subsequently result in phosphorylation of additional multiple
tyrosine sites (Y-576, -577 and -925). FAK preferentially interacts
with the tyrosine phosphorylated paxillin (p-pax), and its
recruitment is implicated in the high turnover of focal complexes
and FA translocation. The p-pax is present in focal complexes, but
not in fibrillar adhesions (45).
Recently, Lawson et al (46) demonstrated that FAK promotes talin
recruitment to nascent adhesions occurring independently from talin
binding to β1 integrins. In the present study, we observed that
gradually decreasing FAK in ECs by an inhibitor significantly
downregulated the expression levels of integrin α2, α5 and β3 at
5–10 min; α2 and β3 returned to higher levels with longer duration
of treatment, but the α5 subunit continued to decrease with
increasing treatment duration. The inhibition of FAK in HepG2 cells
has different inhibition level in different treatment durations,
and resulted in relatively lower levels of α2 and α5, but a higher
level of β1 over 30–60 min (Fig.
5). These results demonstrated that inhibition of FAK
transmitted feedback signals to upstream integrins in both types of
cells. The difference in tFAK and pFAK expression suggested the
presence of diverse mechanisms in the two cell lines. Some evidence
indicated that lack of FAK (by knockout, FAK siRNA or inhibition)
in cells may activate compensatory effects by other signaling
molecules. The compensatory switch from FAK to Pyk2 (FAK-related
proline-rich tyrosine kinase 2) was reported in angiogenesis in
adult mice lacking endothelial FAK, suggesting the adaptive
capacity of cells to switch to Pyk2-dependent signaling following
deletion or inhibition of FAK (47,48). Therefore, some potential signals
possibly compensate for the loss of FAK, and induce transient
upregulation of pFAK in ECs. In HepG2 cells, the Y15 inhibitor
sharply decreased total FAK and β1 expression during the initial 5
min, indicating rapid degradation of FAK, as it directly binds to
the β1 cytoplasmic domain. We hypothesized that HepG2 cells were
sensitive to this type of small-molecule FAK inhibitor, and FAK
signaling was possibly the most dominant pathway in regulating
HepG2 cell functions. Furthermore, similar changes in the α5 and β3
subunits were found in the two different cell types. Integrin α5 is
primarily regarded as the fibronectin receptor, while β3
non-covalently connecting to the αV subunit regulates cell
migration speed. The different expression levels of the α2 and β1
subunits in the two cell types suggested changes in different ECM
proteins with FAK inhibition, which may control cell adhesion,
spreading, migration and detachment with specified treatment
durations.
As mentioned above, the FA components recruit actin
filaments to the cytoplasmic domain of integrins, and contribute to
stabilization of cell adhesion. As is known, the recruitment of
paxillin, which contains binding sites at its LIM domains that
directly bind to the FAT domain of FAK (49), and talin, plays a central role in
the first step of focal complex formation following initial
integrin engagement, while vinculin acts in linking the actin
cytoskeleton to FAs as a paxillin-binding partner. Our results
demonstrated that both talin and paxillin in the two cell lines
exhibited a gradually downregulated tendency, while vinculin
maintained a stable expression level over the duration of FAK
inhibition (Fig. 6). Long-lasting
inhibition of FAK possibly resulted in the degradation of paxillin
and talin, and turnover of FA, which regulate morphological changes
and detachment in various cells (Fig.
3). However, the decrease in FAK expression with inhibition did
not significantly affect vinculin, in part because it indirectly
binds to FA as a paxillin-binding partner, participating in the
formation of FAs.
FAK is considered to be upstream of PI3K regulating
cell apoptosis and other functions (50). Activated FAK, serving as a
scaffold or adaptor, provides a binding site and recruits a number
of signaling molecules for Src family kinases and PI3K at SH2
domains, activating a cascade of downstream signaling pathways. The
phosphorylation of FAK at the Y397 site is known to serve as a
binding site for the p85 subunit of PI3K, and activated PI3K can
stimulate several intermediates, including AKT, which is required
for FAK to promote cell migration (51). Inhibition of FAK pharmacologically
or by dominant-negative FAK, attenuated the phosphorylation of p85
subunit of PI3K and AKT (50),
and it is consistent with our results from EA.hy926 cells (Figs. 4 and 7A and C). However, the expression of
AKT1 was downregulated in HepG2 cells, which was in contrast to
PI3K (Fig. 7B and D), indicating
that AKT was likely activated independently of PI3K. Thamilselvan
et al (7) reported that
FAK siRNA did not affect the activation of PI3K induced by
pressure, but blocked AKT phosphorylation. The difference between
EA.hy926 and HepG2 cells in response to the same stimuli, suggested
the diverse functional role of FAK/PI3K/pAKT1 signaling in
controlling the motility of various types of cells.
Rho family GTPases, including RhoA, Rac1 and Cdc42,
are key regulators of actin dynamics and cell adhesion/migration in
a variety of cellular processes. The contribution of FAK to actin
remodeling, which is required for cell migration, is mediated
through binding to Rho protein effectors and subsequent effects on
Rho GTPases. FAK may up- and downregulate the Rho GTPases by
modulating various upstream regulators (52). Ren et al (53) indicated that FAK is required for
Rho inhibition to promote FA turnover and cell migration. In the
present study, the expressions of RhoA, Rac1 and Cdc42 under FAK
inhibition were generally downregulated, but reactivated in
different treatment durations. RhoA was reactivated and exhibited a
higher level at 2 h in both endothelial and hepatoblastoma cells,
while Rac1 and Cdc42 in hepatoblastoma cells exhibited high levels
after 10 and 30 min of FAK inhibition (Fig. 8A and B). Combined with previous
cell migration analysis findings, cell motility was significantly
inhibited by long-lasting FAK inhibition (24 h) compared with
control cells (Fig. 1). This
time-dependent effects of Rho GTPases in response to FAK inhibition
prompted us to investigate the mechanisms of blocking cell
migration in our future study. However, it should be noted that
only total expression and phosphorylated levels of Rac and Rho were
examined in the present study, while the activation of Rho GTPases
should also be addressed.
In the present study, the time-dependent effects of
FAK inhibition on integrin-induced signaling pathways associated
with endothelial and hepatoblastoma cell migration in tumor
angiogenesis were investigated. Our results demonstrated that the
inhibition of FAK promoted cell detachment by decreasing the
expression of FA components (talin and paxillin), and inhibiting
cell motility by reducing the levels of Rho GTPases (Rac1, Cdc42
and RhoA). Most signaling proteins in the two cell lines exhibited
a different time-dependent expression within 2 h of FAK inhibitor
treatment, suggesting that the mechanisms underlying FAK-mediated
migration behaviors differ between various types of cells.
Elucidating the potential mechanism may help design novel drugs
targeting FAK to inhibit endothelial and cancer cell migration in
tumor angiogenesis.
Acknowledgments
Not applicable.
Notes
[1]
Funding
This study was supported in part by grants from the
National Natural Science Foundation of China (nos. 11372203,
11172189 and 31300775), the program for New Century Excellent
Talents in the University of China (no. NCET-06-0791), the Visiting
Scholar Foundation of Key Laboratory of Biorheological Science and
Technology (Chongqing University), the Specialized Research Fund
for the Doctoral Program of Higher Education (no. 20120181120058)
and the National Science Foundation for Fostering Talents in Basic
Research of the National Natural Science Foundation of China (no.
J1103604).
[2] Authors'
contributions
YS and XL conceived and designed this project, and
provided financial support. HY and MG performed the experiments. YM
and LW contributed to the materials/analysis tools and analyzed the
data. HY drafted the manuscript. All authors read and approved the
final manuscript.
[3] Availability
of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Weis SM and Cheresh DA: Tumor
angiogenesis: molecular pathways and therapeutic targets. Nat Med.
17:1359–1370. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lamalice L, Le Boeuf F and Huot J:
Endothelial cell migration during angiogenesis. Circ Res.
100:782–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Friedl P and Wolf K: Tumour-cell invasion
and migration: diversity and escape mechanisms. Nat Rev Cancer.
3:362–374. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Baker EL and Zaman MH: The biomechanical
integrin. J Biomech. 43:38–44. 2010. View Article : Google Scholar :
|
|
5
|
Pasapera AM, Schneider IC, Rericha E,
Schlaepfer DD and Waterman CM: Myosin II activity regulates
vinculin recruitment to focal adhesions through FAK-mediated
paxillin phosphorylation. J Cell Biol. 188:877–890. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hsia DA, Mitra SK, Hauck CR, Streblow DN,
Nelson JA, Ilic D, Huang S, Li E, Nemerow GR, Leng J, et al:
Differential regulation of cell motility and invasion by FAK. J
Cell Biol. 160:753–767. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thamilselvan V, Craig DH and Basson MD:
FAK association with multiple signal proteins mediates
pressure-induced colon cancer cell adhesion via a Src-dependent
PI3K/Akt pathway. FASEB J. 21:1730–1741. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gurney SM, Forster P, Just U and
Schwanbeck R: Suppression of the PI3K subunit p85α delays embryoid
body development and inhibits cell adhesion. J Cell Biochem.
112:3573–3581. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Clark EA, King WG, Brugge JS, Symons M and
Hynes RO: Integrin-mediated signals regulated by members of the Rho
family of GTPases. J Cell Biol. 142:573–586. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Raftopoulou M and Hall A: Cell migration:
Rho GTPases lead the way. Dev Biol. 265:23–32. 2004. View Article : Google Scholar
|
|
11
|
Tavora B, Batista S, Reynolds LE, Jadeja
S, Robinson S, Kostourou V, Hart I, Fruttiger M, Parsons M and
Hodivala-Dilke KM: Endothelial FAK is required for tumour
angiogenesis. EMBO Mol Med. 2:516–528. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao X and Guan JL: Focal adhesion kinase
and its signaling pathways in cell migration and angiogenesis. Adv
Drug Deliv Rev. 63:610–615. 2011. View Article : Google Scholar :
|
|
13
|
Yom CK, Noh DY, Kim WH and Kim HS:
Clinical significance of high focal adhesion kinase gene copy
number and overexpression in invasive breast cancer. Breast Cancer
Res Treat. 128:647–655. 2011. View Article : Google Scholar
|
|
14
|
Miyazaki T, Kato H, Nakajima M, Sohda M,
Fukai Y, Masuda N, Manda R, Fukuchi M, Tsukada K and Kuwano H: FAK
overexpression is correlated with tumour invasiveness and lymph
node metastasis in oesophageal squamous cell carcinoma. Br J
Cancer. 89:140–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao J and Guan JL: Signal transduction by
focal adhesion kinase in cancer. Cancer Metastasis Rev. 28:35–49.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wozniak MA, Modzelewska K, Kwong L and
Keely PJ: Focal adhesion regulation of cell behavior. Biochim
Biophys Acta. 1692:103–119. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schlaepfer DD and Mitra SK: Multiple
connections link FAK to cell motility and invasion. Curr Opin Genet
Dev. 14:92–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu ZJ, Ren YQ, Wang GP, Song Q, Li M,
Jiang SS, Ning T, Guan YS, Yang JL and Luo F: Biological behaviors
and proteomics analysis of hybrid cell line EAhy926 and its parent
cell line A549. J Exp Clin Cancer Res. 28:162009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Golubovskaya VM, Nyberg C, Zheng M, Kweh
F, Magis A, Ostrov D and Cance WG: A small molecule inhibitor,
1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397
site of focal adhesion kinase decreases tumor growth. J Med Chem.
51:7405–7416. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moissoglu K and Schwartz MA: Integrin
signalling in directed cell migration. Biol Cell. 98:547–555. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ezzell RM, Goldmann WH, Wang N,
Parashurama N and Ingber DE: Vinculin promotes cell spreading by
mechanically coupling integrins to the cytoskeleton. Exp Cell Res.
231:14–26. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Engelman JA: Targeting PI3K signalling in
cancer: opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ridley AJ: Rho GTPases and actin dynamics
in membrane protrusions and vesicle trafficking. Trends Cell Biol.
16:522–529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schwarz J, Proff J, Hävemeier A, Ladwein
M, Rottner K, Barlag B, Pich A, Tatge H, Just I and Gerhard R:
Serine-71 phosphorylation of Rac1 modulates downstream signaling.
PLoS One. 7:e443582012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hoeben A, Landuyt B, Highley MS, Wildiers
H, Van Oosterom AT and De Bruijn EA: Vascular endothelial growth
factor and angiogenesis. Pharmacol Rev. 56:549–580. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Unger RE, Krump-Konvalinkova V, Peters K
and Kirkpatrick CJ: In vitro expression of the endothelial
phenotype: comparative study of primary isolated cells and cell
lines, including the novel cell line HPMEC-ST1.6R. Microvasc Res.
64:384–397. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sieg DJ, Hauck CR, Ilic D, Klingbeil CK,
Schaefer E, Damsky CH and Schlaepfer DD: FAK integrates
growth-factor and integrin signals to promote cell migration. Nat
Cell Biol. 2:249–256. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Braren R, Hu H, Kim YH, Beggs HE,
Reichardt LF and Wang R: Endothelial FAK is essential for vascular
network stability, cell survival, and lamellipodial formation. J
Cell Biol. 172:151–162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
van Nimwegen MJ, Verkoeijen S, van Buren
L, Burg D and van de Water B: Requirement for focal adhesion kinase
in the early phase of mammary adenocarcinoma lung metastasis
formation. Cancer Res. 65:4698–4706. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McLean GW, Carragher NO, Avizienyte E,
Evans J, Brunton VG and Frame MC: The role of focal-adhesion kinase
in cancer - a new therapeutic opportunity. Nat Rev Cancer.
5:505–515. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Beierle EA, Ma X, Stewart J, Nyberg C,
Trujillo A, Cance WG and Golubovskaya VM: Inhibition of focal
adhesion kinase decreases tumor growth in human neuroblastoma. Cell
Cycle. 9:1005–1015. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hochwald SN, Nyberg C, Zheng M, Zheng D,
Wood C, Massoll NA, Magis A, Ostrov D, Cance WG and Golubovskaya
VM: A novel small molecule inhibitor of FAK decreases growth of
human pancreatic cancer. Cell Cycle. 8:2435–2443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Infusino GA and Jacobson JR: Endothelial
FAK as a therapeutic target in disease. Microvasc Res. 83:89–96.
2012. View Article : Google Scholar
|
|
34
|
Golubovskaya VM and Cance W: Focal
adhesion kinase and p53 signal transduction pathways in cancer.
Front Biosci (Landmark Ed). 15:901–912. 2010. View Article : Google Scholar
|
|
35
|
Lim ST, Mikolon D, Stupack DG and
Schlaepfer DD: FERM control of FAK function: implications for
cancer therapy. Cell Cycle. 7:2306–2314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Eleniste PP and Bruzzaniti A: Focal
adhesion kinases in adhesion structures and disease. J Signal
Transduct. 2012:2964502012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zheng Y and Lu Z: Paradoxical roles of FAK
in tumor cell migration and metastasis. Cell Cycle. 8:3474–3479.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yano H, Mazaki Y, Kurokawa K, Hanks SK,
Matsuda M and Sabe H: Roles played by a subset of integrin
signaling molecules in cadherin-based cell-cell adhesion. J Cell
Biol. 166:283–295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hauck CR, Sieg DJ, Hsia DA, Loftus JC,
Gaarde WA, Monia BP and Schlaepfer DD: Inhibition of focal adhesion
kinase expression or activity disrupts epidermal growth
factor-stimulated signaling promoting the migration of invasive
human carcinoma cells. Cancer Res. 61:7079–7090. 2001.PubMed/NCBI
|
|
40
|
Lu Z, Jiang G, Blume-Jensen P and Hunter
T: Epidermal growth factor-induced tumor cell invasion and
metastasis initiated by dephosphorylation and downregulation of
focal adhesion kinase. Mol Cell Biol. 21:4016–4031. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cicchini C, Laudadio I, Citarella F,
Corazzari M, Steindler C, Conigliaro A, Fantoni A, Amicone L and
Tripodi M: TGFbeta-induced EMT requires focal adhesion kinase (FAK)
signaling. Exp Cell Res. 314:143–152. 2008. View Article : Google Scholar
|
|
42
|
Kallergi G, Agelaki S, Markomanolaki H,
Georgoulias V and Stournaras C: Activation of FAK/PI3K/Rac1
signaling controls actin reorganization and inhibits cell motility
in human cancer cells. Cell Physiol Biochem. 20:977–986. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Tadokoro S, Shattil SJ, Eto K, Tai V,
Liddington RC, de Pereda JM, Ginsberg MH and Calderwood DA: Talin
binding to integrin beta tails: a final common step in integrin
activation. Science. 302:103–106. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Baumann K: Cell adhesion: FAK or talin:
who goes first. Nat Rev Mol Cell Biol. 13:138–139. 2012.
|
|
45
|
Zaidel-Bar R, Milo R, Kam Z and Geiger B:
A paxillin tyrosine phosphorylation switch regulates the assembly
and form of cell-matrix adhesions. J Cell Sci. 120:137–148. 2007.
View Article : Google Scholar
|
|
46
|
Lawson C, Lim ST, Uryu S, Chen XL,
Calderwood DA and Schlaepfer DD: FAK promotes recruitment of talin
to nascent adhesions to control cell motility. J Cell Biol.
196:223–232. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Weis SM, Lim ST, Lutu-Fuga KM, Barnes LA,
Chen XL, Göthert JR, Shen TL, Guan JL, Schlaepfer DD and Cheresh
DA: Compensatory role for Pyk2 during angiogenesis in adult mice
lacking endothelial cell FAK. J Cell Biol. 181:43–50. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lim Y, Lim ST, Tomar A, Gardel M,
Bernard-Trifilo JA, Chen XL, Uryu SA, Canete-Soler R, Zhai J, Lin
H, et al: PyK2 and FAK connections to p190Rho guanine nucleotide
exchange factor regulate RhoA activity, focal adhesion formation,
and cell motility. J Cell Biol. 180:187–203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Schaller MD: Paxillin: a focal
adhesion-associated adaptor protein. Oncogene. 20:6459–6472. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xia H, Nho RS, Kahm J, Kleidon J and Henke
CA: Focal adhesion kinase is upstream of phosphatidylinositol
3-kinase/Akt in regulating fibroblast survival in response to
contraction of type I collagen matrices via a beta 1 integrin
viability signaling pathway. J Biol Chem. 279:33024–33034. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Reiske HR, Kao SC, Cary LA, Guan JL, Lai
JF and Chen HC: Requirement of phosphatidylinositol 3-kinase in
focal adhesion kinase-promoted cell migration. J Biol Chem.
274:12361–12366. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Myers JP, Robles E, Ducharme-Smith A and
Gomez TM: Focal adhesion kinase modulates Cdc42 activity downstream
of positive and negative axon guidance cues. J Cell Sci.
125:2918–2929. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ren XD, Kiosses WB, Sieg DJ, Otey CA,
Schlaepfer DD and Schwartz MA: Focal adhesion kinase suppresses Rho
activity to promote focal adhesion turnover. J Cell Sci.
113:3673–3678. 2000.PubMed/NCBI
|