Introduction
Colorectal carcinoma (CRC) is one of the most common
malignancies and remains the third leading cause of cancer-related
mortality worldwide (1). Although
novel molecular-based therapies are widely used for treating CRC,
the high recurrence and poor survival rate remain a risk for
several CRC patients (2). In
recent years, understanding the cancer-specific genes and potential
molecular mechanisms underlying the carcinogenesis and progression
of CRC has provided alternative approaches to diagnostic and
therapeutic evaluation.
B-cell CLL/lymphoma 6 member B (BCL6B), also
referred to as BAZF, ZNF62 and ZBTB28, belongs to the BCL6 gene
family and it acts as a sequence-specific transcriptional repressor
in the nucleus (3). As regards
its biological functions, BCL6B promotes differentiation into
stages or lineages in the human erythroleukemia cell line HEL, and
plays an important role in spermatogonial stem cell self-renewal
(4,5). BCL6B is essential for the secondary
responses of memory CD8+ T cells (6). Overexpression of BCL6B induces
apoptosis in NIH3T3 cells (7). In
recent years, loss of BCL6B expression due to promoter DNA
hypermethylation was identified in different types of tumors,
including chronic lymphocytic leukemia (8,9),
gastric cancer (10-12), hepatocellular carcinoma (13,14) and CRC (15). Methylation of BCL6B has been
implicated in tumor growth, angiogenesis, metastasis and invasion
(10,13-15). Thus, BCL6B is considered to be a
tumor suppressor, and its downregulation may contribute to the
development of cancer. However, the precise role and potential
molecular mechanism underlying the involvement of BCL6B in CRC
progression remain elusive.
The AKT serine/threonine kinase (also referred to as
protein kinase B), which comprises three highly homologous members,
namelyAKT1 (PKBα), AKT2 (PKBβ) and AKT3 (PKBγ), has emerged as a
critical signaling molecule within eukaryotic cells (16). AKT is activated in cells exposed
to diverse stimuli, such as hormones, growth factors and
extracellular matrix components. The activation mechanism occurs
downstream of phosphoinositide 3-kinase (PI3K), which generates
phosphatidylinositol-3,4,5-trisphosphate, a lipid second messenger
essential for the translocation of AKT to the plasma membrane,
where it is phosphorylated (17).
The PI3K/AKT pathway regulates the function of a number of cellular
proteins involved in metabolism, proliferation, apoptosis and
metastasis (18-20). Previous evidence revealed that the
PI3K/AKT pathway is frequently constitutively active in several
types of human cancer, including CRC (21,22). However, whether the PI3K/AKT
signaling pathway is involved in the BCL6B-induced effects on CRC
remains to be elucidated.
Based on the abovementioned studies, the aim of the
present study was to investigate the expression, biological role
and underlying molecular mechanisms of BCL6B in CRC in order to
provide alternative approaches to the treatment of CRC.
Materials and methods
Cell culture
The human CRC cell lines SW480 and LoVo and the
human normal intestinal epithelial cell line FHC were purchased
from the American Type Culture Collection (Manassas, VA, USA). The
cells were maintained in Dulbecco's modified Eagle's medium (DMEM;
Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Carlsbad, CA, USA) and 100
U/ml streptomycin/penicillin at 37°C in a humidified atmosphere
containing 5% CO2.
Reagents
Recombinant plasmid pcDNA3.1-BCL6B and pcDNA3.1 were
kindly provided by Dr Xiang (Epigenetics Laboratory, the First
Affiliated Hospital of Chongqing Medical University). TRIzol
reagent was purchased from Invitrogen; Thermo Fisher Scientific
(Carlsbad, CA, USA). Reverse transcription-polymerase chain
reaction (RT-PCR) reagents were purchased from Takara (Otsu,
Japan). Western blot detection reagents were purchased from the
Beyotime Institute of Biotechnology (Nanjing, China). The PI3K/AKT
inhibitor LY294002 was purchased from Sigma-Aldrich; Merck KGaA
(Saint Louis, MO, USA). The antibodies used were as follows: Rabbit
anti-BCL6B polyclonal antibody (cat no. AP20369c, Abgent, Wuxi,
China), mouse anti-β-actin monoclonal antibody (cat no. 47778,
Santa Cruz Biotechnology, Dallas, TX, USA), rabbit anti-cyclin D1
polyclonal antibody (cat no. 753, Santa Cruz Biotechnology), mouse
anti-matrix metalloproteinase (MMP)-9 monoclonal antibody (cat no.
13520, Santa Cruz Biotechnology), rabbit anti-E-cadherin polyclonal
antibody (cat no. YT1454, Immuno Way, TX, USA), rabbit anti-t-AKT
monoclonal antibody (cat no. 4691, Cell Signaling Technology, Inc.,
Danvers, MA, USA), rabbit anti-p-AKT (Ser473) monoclonal antibody
(cat no. 81283, Abcam, Boston, MA, USA), goat anti-mouse IgG (cat
no. 2305, Zhongshan Golden Bridge, Beijing, China), and goat
anti-rabbit IgG (cat no. 2301, Zhongshan Golden Bridge).
Plasmid transfection
The CRC cells were seeded in 6-well plates at a
density of 2×105 cells per well and transfected with
pcDNA3.1-BCL6B (4 μg/well) or the control vector pcDNA3.1 (4
μg/well) using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific). After 8 h, the medium was replaced with DMEM
containing 10% FBS followed by continued cell culturing for
subsequent experiments.
Cell viability assay
Cell viability was analyzed with the MTT assay. The
CRC cells were seeded at a density of 3×103 cells per
well in 96-well plates and incubated at 37°C. At 24, 48, 72 and 96
h after transfection, and with or without chemical inhibitor in
DMEM containing 4% FBS, 10 μl of the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide reagent
(MTT; Promega, Madison, WI, USA) was added to each well and
incubated for another 4 h at 37°C. The culture medium was removed
and the formazan was dissolved in DMSO (150 μl/well) for 10
min at room temperature. The color reaction was measured at 492 nm
with enzyme immunoassay analyzer (Bio-Rad, Hercules, CA, USA). Each
condition was carried out in quintuplicate and repeated in at least
three batches of independent experiments.
Colony-formation assay
Briefly, at 24 h post transfection, CRC cells were
collected and seeded in 6-well plates (500 cells/well) in 2 ml
complete growth medium for ~14 days, colonies (≥50 cells/colony)
were fixed with 4% paraformaldehyde for 15 min and stained with
0.1% crystal violet (Beyotime Institute of Biotechnology) for 20
min and then counted. The experiment was repeated thrice.
Cell apoptosis analysis
Cell apoptosis analysis was assessed by flow
cytometry(FCM). CRC cells were collected after transfection for 12
h and seeded in 6-well plates at a density of 2×105
cells/well and starved 12 h for synchronization, then re-stimulated
with 10% FBS for 24 h. Log-phase cells from each group were
harvested by centrifugation. After being washed twice with ice-cold
PBS, samples were added into apoptosis analysis solution and then
analyzed by a FACS Vantage SE flow cytometer (Becton-Dickinson, San
Jose, CA, USA). The experiment was performed three times.
Wound closure assay
Cell migration ability was analyzed with the wound
closure assay. Log-phase cells were collected, seeded in 6-well
plates and treated with pcDNA3.1-BCL6B or pcDNA3.1. After treating
for the indicated time, a wound was created at the center of the
culture using a pipette tip, and the cells were washed with
serum-free medium, cultured with 1% FBS. Images were captured at 0,
24, 48 and 72 h under a microscope after the incision was made. The
incision width of the different site was measured, and the average
wound closure rate was calculated as: (0 h incision width-72 h
incision width)/(0 h incision width) ×100%. Each experiment was
repeated three times.
Transwell assay
Migration: The CRC cells with different treatment
were suspended at a density of 5×104 in 300 μl
DMEM containing 10% FBS and seeded onto the upper chamber of
8.0-μm pore size Transwell chambers (Millipore Corporation,
Billerica, MA, USA). The lower chamber was filled with 700
μl of complete medium containing 20% FBS as a
chemoattractant. After incubation at 37°C for 24 h, the cells that
remained in the top chamber were removed with cotton swabs.
Invasion: The upper chamber was coated with Matrigel (BD
Biosciences). SW480 and LoVo cells were added to the upper chamber
at a density of 8×104/300 μl per chamber and
incubated for 36 h, followed by removal of the cells that remained
in the top chamber with cotton swabs. Cells that penetrated to the
lower membrane surface were washed with PBS, fixed in 4%
paraformaldehyde for 20 min and stained with 0.2% crystal violet
solution, then counted under an inverted microscope (magnification,
×100) and photographed. Independent Transwell assay was repeated
three times in triplicate.
RNA extraction, RT-PCR and RT-qPCR
analysis
Total RNA was extracted from CRC cells in different
groups using TRIzol (Invitrogen; Thermo Fisher Scientific),
according to the manufacturer's instructions. The RNA concentration
and purity were quantified using a NanoDrop 1000 spectrophotometer
(Thermo Fisher Scientific). First-strand DNA was synthesized using
the Reverse Transcriptase M-MLV (RNase H) kit with random hexamer
primers. The primers (Table I)
used for RT-PCR and RT-qPCR were designed using the Primer3 Input
0.4.0 (http://frodo.wi.mit.edu/). GAPDH was
used as an endogenous control. Touchdown PCR analysis determining
the gene expression level was performed under the following
conditions: 95°C × 5 min for one cycle, 9 cycles at 94°C × 30 sec,
62°C × 30 sec (with a decrease of 1 degree/cycle), 55°C × 30 sec
and 25 cycles at 94°C × 30 sec, 55°C × 30 sec, and 72°C × 30 sec,
72°C × 10 min for 1 cycle. The PCR products were separated by 2%
agarose gels. The results were recorded by the Gel imaging system
(Gel Doc 1000, Bio-Rad) and Quantity One Version 4.5.0 (Bio-Rad).
RT-qPCR was run in the Rotor-Gene 6000 Real-Time PCR machine
(Corbett Research, Sydney, Australia) using SYBR Premix Ex Taq
(Thermo Fisher Scientific) with the following protocol: 95°C for 10
min, followed by 40 cycles of denaturation at 95°C for 5 sec and an
annealing/elongation step at 60°C for 20 sec. Gene expression
results were normalized to GAPDH and analyzed according to the
2−ΔΔq method. Three separate experiments were performed
for each group.
 | Table IPrimer sequences in RT-PCR. |
Table I
Primer sequences in RT-PCR.
| Gene | Primer
sequences |
|---|
| GAPDH | F:
5′-CAGCGACACCCACTCCTC-3′ |
| R:
5′-TGAGGTCCACCACCCTGT-3′ |
| BCL6B | F:
5′-AAGCCGTATAAGTGTGAGACG-3′ |
| R:
5′-AGAATGTGGTAGTGCAC-3′ |
| Cyclin
D1 | F:
5′-CTGGCCATGAACTACCTGGA-3′ |
| R:
5′-GTCACACTTGATCACTCTGG-3′ |
|
E-cadherin | F:
5′-AAGGTGACAGAGCCTCTGGAT-3′ |
| R:
5′-CGTCTGTGGCTGTGACCT-3′ |
| MMP-9 | F:
5′-CCTGGAGCCTGAGAACCAATC-3′ |
| R:
5′-CCACCCGAGTGTAACCATGGC-3′ |
Western blot assay
Cells with different treatments were collected and
washed with ice-cold PBS, then lysed in ice-cold RIPA lysis buffer
(Beyotime Institute of Biotechnology, Shanghai, China) containing
protease inhibitor and phosphatase inhibitor (Roche, Mannheim,
Germany). Protein extracts were quantitated by the bicinchoninic
acid assay, denatured by boiling, separated by 10% sodium dodecyl
sulfate poly-acrylamide gel electrophoresis and blotted onto the
PVDF membranes. After blocking with 5% bovine serum albumin
(Solarbio, Beijing, China) at room temperature for 2 h in
Tris-buffered saline and Tween 20 (TBST), the membranes were probed
with the primary antibodies (1:1,000) and incubated at 4°C
overnight. Subsequently, the membranes were washed with TBST and
incubated with horseradish peroxidase-conjugated secondary
antibodies (1:5,000) for 1 h. After washing with TBST, the proteins
were detected using enhanced chemiluminescence (Millipore
Corporation), followed by exposure on the Gel Doc 1000
Electrophoresis Documentation (Bio-Rad). Three separate experiments
were performed for each group.
Statistical analysis
All quantitative data were calculated and are
presented as means ± standard deviation. The difference between two
different groups was determined by Student's t-test and the
differences between different groups were analyzed using one-way
analysis of variance followed by the Student-Newman-Keuls test. All
the statistical analyses were performed using GraphPad Prism 5
(GraphPad Software, La Jolla, CA, USA). Statistical significance
was set at P<0.05.
Results
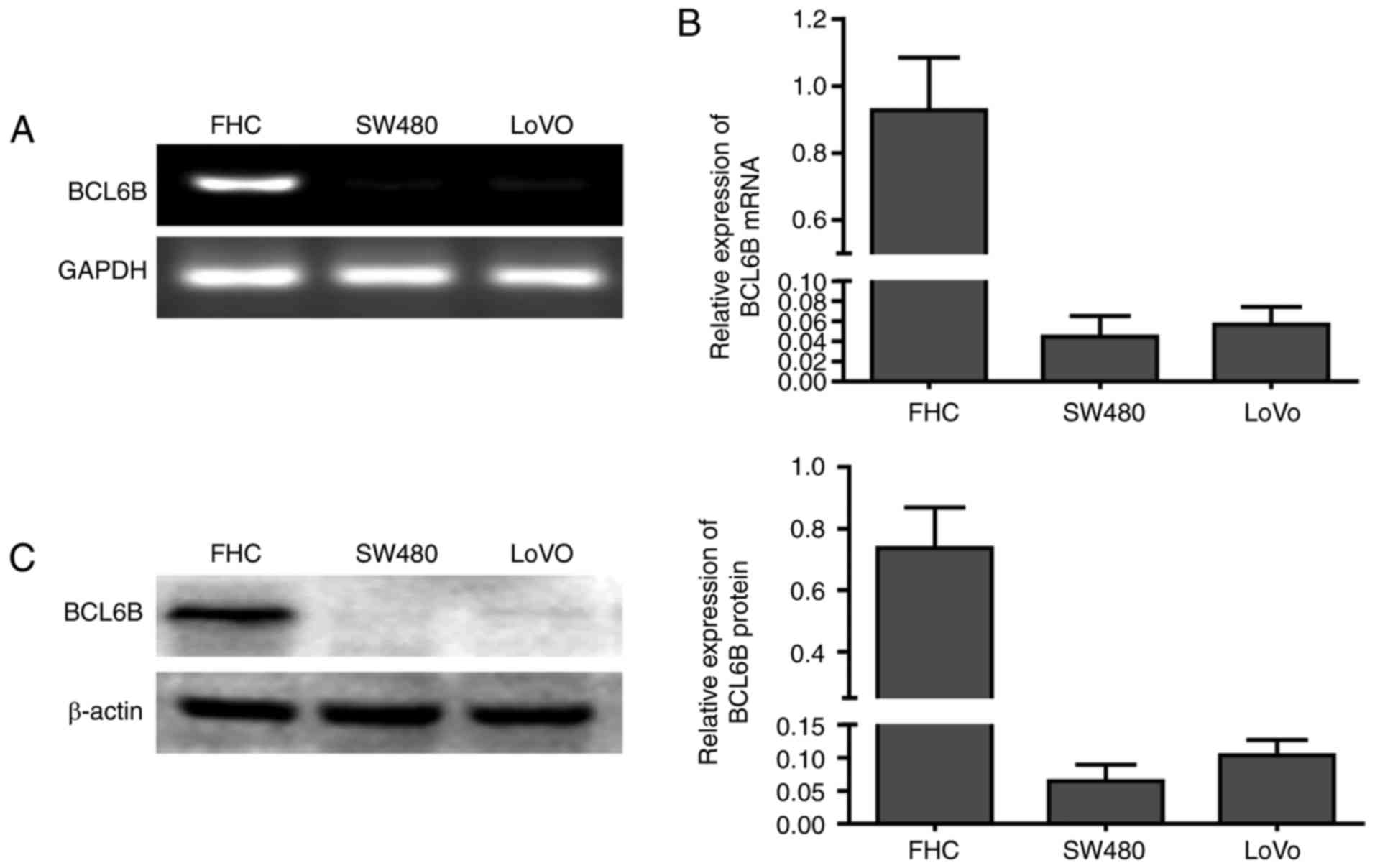
BCL6B is downregulated in human CRC
cells
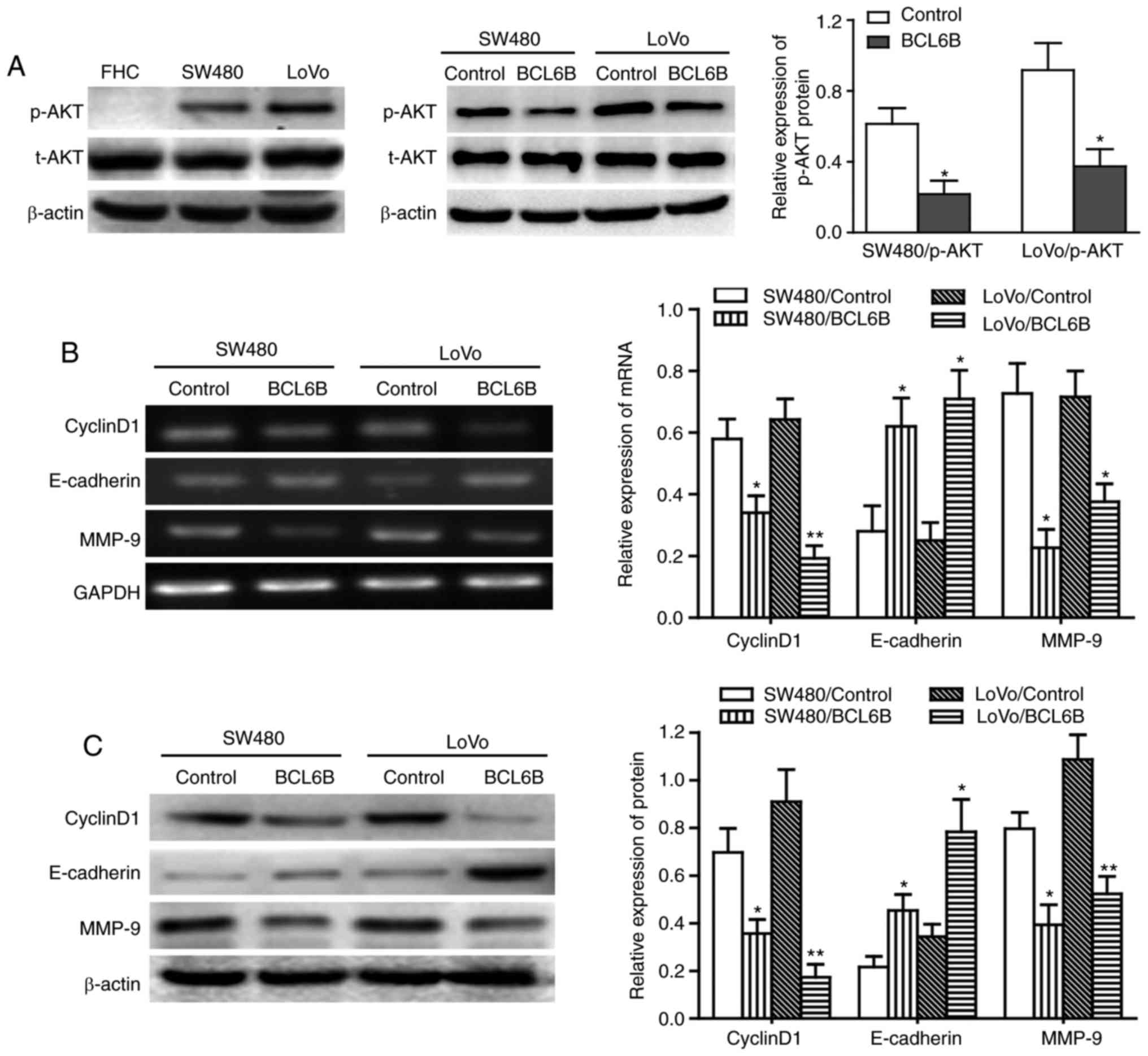
The mRNA expression of BCL6B was first compared
between the human normal intestinal epithelial FHC cells and the
two CRC cell lines SW480 and LoVo (Fig. 1A and B). BCL6B was readily
expressed in FHC cells, but was notably repressed in SW480 and LoVo
cells. Western blot analysis confirmed the loss of BCL6B protein
expression in SW480 and LoVo cells (Fig. 1C), suggesting that BCL6B may play
an important role in CRC.
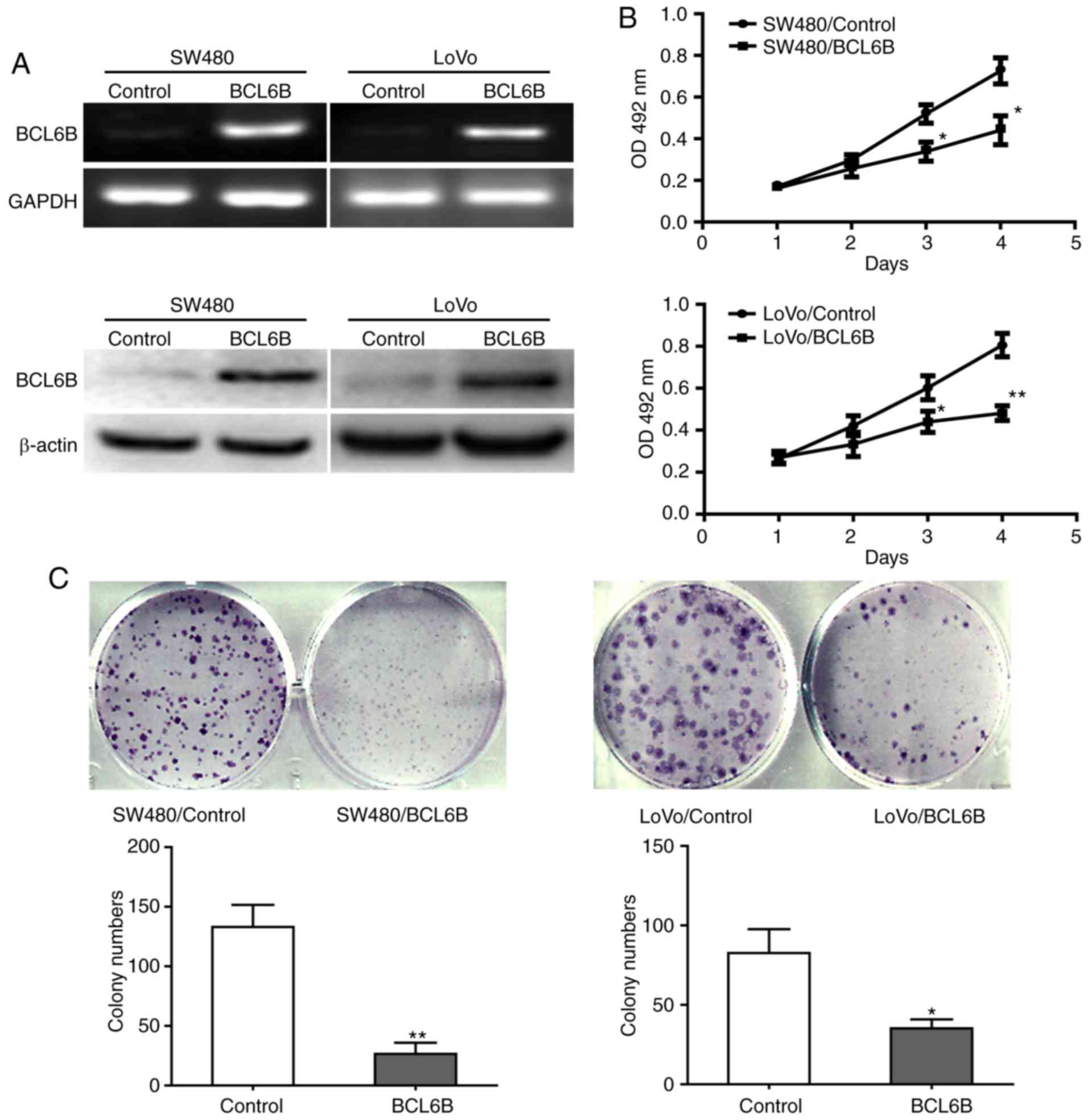
BCL6B overexpression suppresses
proliferation of CRC cells
To investigate the biological role of BCL6B in CRC,
SW480 and LoVo cells were transfected with pcDNA3.1-BCL6B for BCL6B
overexpression (Fig. 2A). Cell
viability and proliferation were analyzed with the MTT assay and
the colony formation assay. Compared with the control group, BCL6B
overexpression strongly decreased the viability of SW480 and LoVo
cells at 72 and 96 h (Fig. 2B).
In addition, the colony numbers of SW480 or LoVo cells in the BCL6B
group were significantly lower compared with those in the control
group (Fig. 2C).
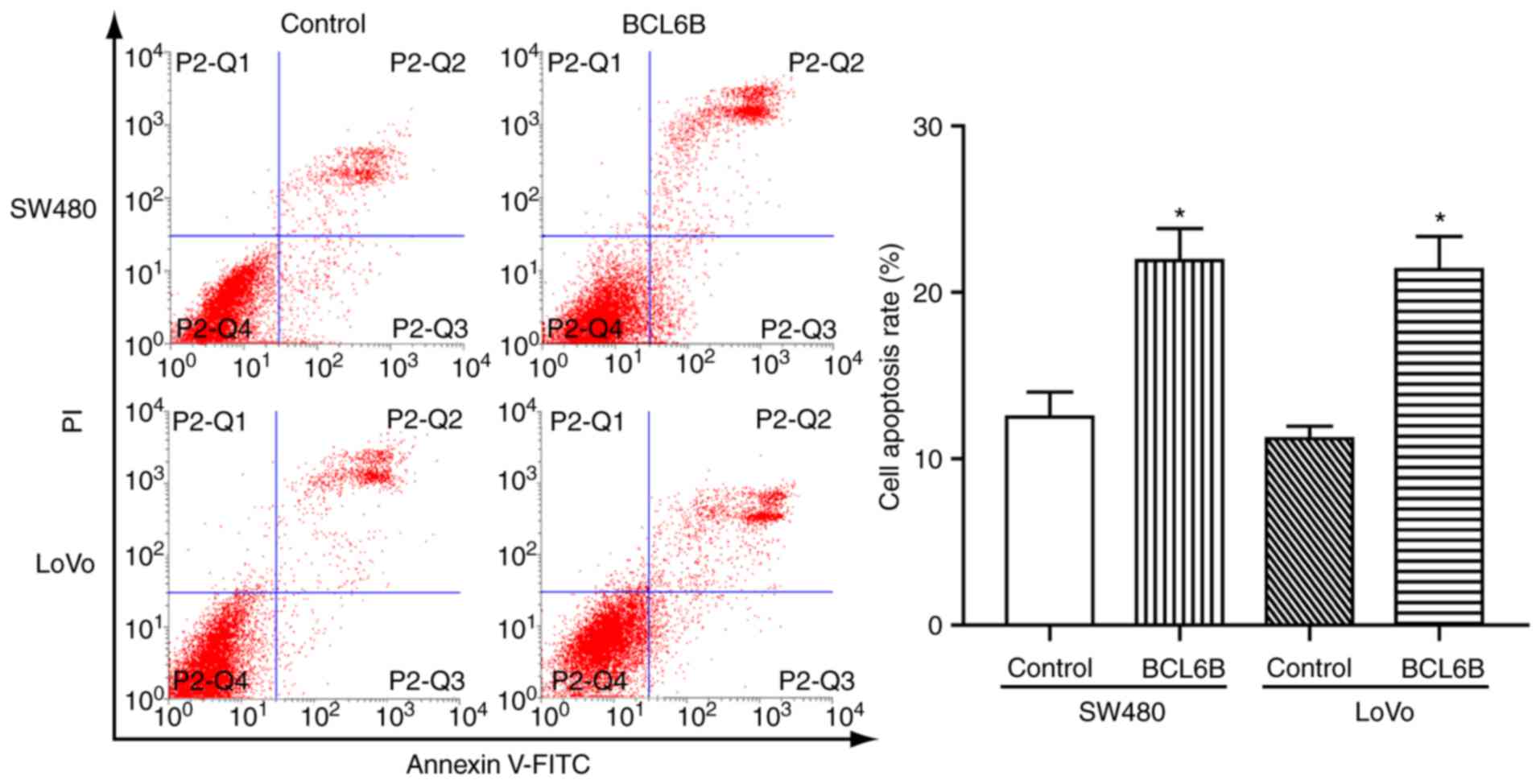
BCL6B overexpression induces apoptosis of
CRC cells
To determine whether the observed growth inhibition
induced by BCL6B was associated with cell apoptosis, FCM was
applied. As a result, the rate of apoptotic cells was 21.92±3.32
vs. 12.51±2.61% (P<0.05), and 21.37±3.46 vs. 11.21±1.28%
(P<0.05) in SW480 and LoVo cells with and without overexpression
of BCL6B, respectively (Fig. 3).
These data suggested that BCL6B promoted apoptosis in CRC
cells.
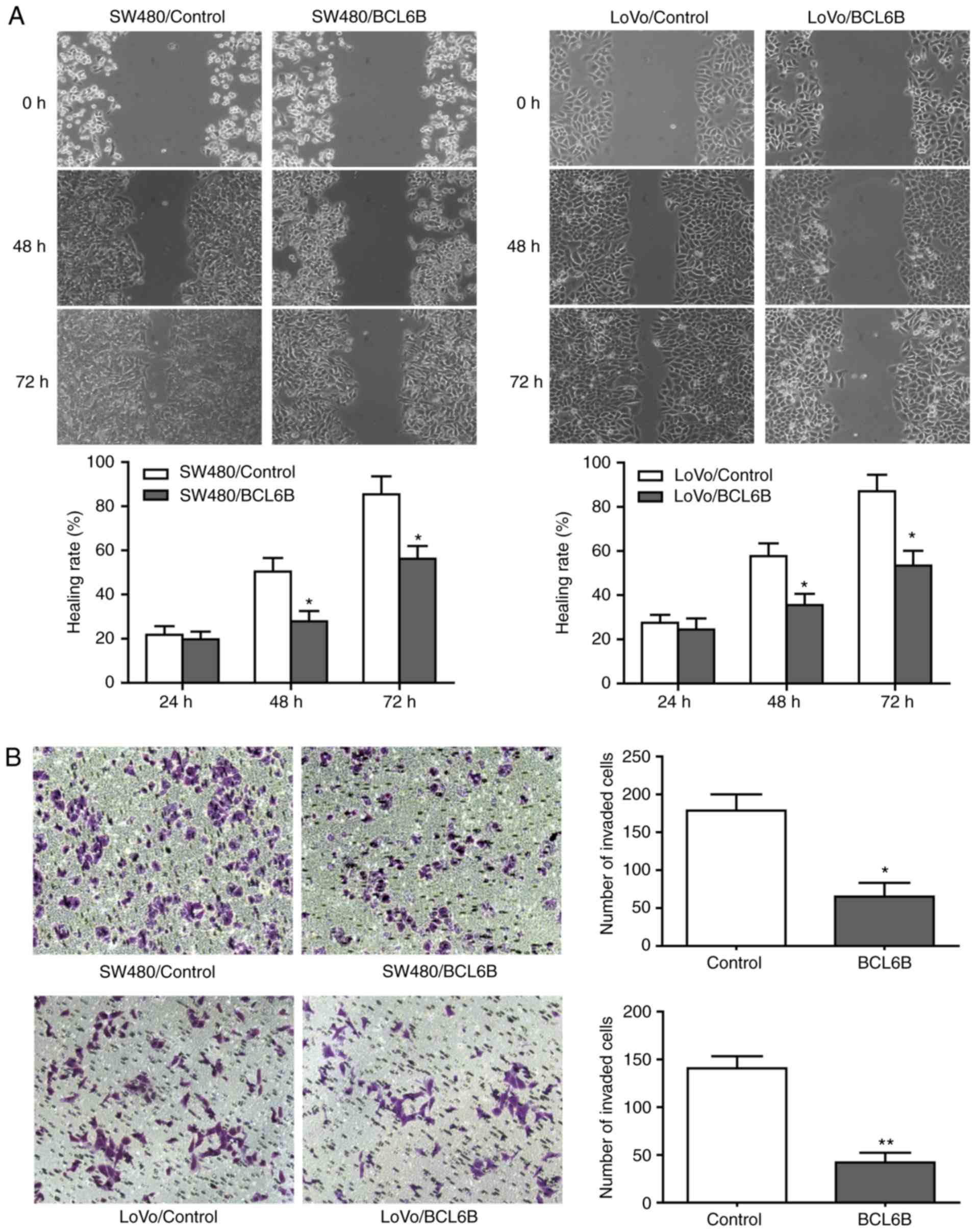
BCL6B overexpression inhibits migration
and invasion of CRC cells
As shown in Fig.
4A, the wound closure rate of SW480 and LoVo cells in the BCL6B
overexpression group was decreased by 38.89% (P<0.05) and 31.17%
(P<0.05), respectively, at 48 h. In addition, 36 h after
transfection, the numbers of invading SW480 and LoVo cells in the
BCL6B overexpression group were decreased by 63.69% (P<0.05) and
70.71% (P<0.01), respectively (Fig. 4B).
BCL6B inhibits the PI3K/AKT signaling
pathway in CRC cells
We next sought to determine the signaling mechanism
involved in BCL6B-induced decrease in the proliferation and
migration of CRC cells by focusing on the PI3K/AKT signaling
pathway, which plays a critical role in cell proliferation,
migration and cancer progression. Our data demonstrated that the
basal phosphorylation level of AKT in the CRC cell lines SW480 and
LoVo was significantly higher compared with that in the normal
intestinal epithelial cell line FHC, and that the phosphorylation
level of AKT was markedly decreased in these two CRC cell lines
with BCL6B overexpression (P<0.05), whereas there was no
significant change in the total AKT level (Fig. 5A).
BCL6B upregulates E-cadherin expression
and downregulates cyclin D1 and MMP-9 expression in CRC cells
Previous studies demonstrated that the expression of
cyclin D1, E-cadherin and MMP-9 was regulated by the PI3K/AKT
signaling pathway, and cyclin D1, E-cadherin and MMP-9 have been
found to be involved in the proliferation and migration of various
tumor cells (23-26). We next examined the effect of
BCL6B on the expression of cyclin D1, E-cadherin and MMP-9. The
results revealed that BCL6B overexpression upregulated E-cadherin
and downregulated cyclin D1 and MMP-9 in SW480 and LoVo cells, at
the mRNA as well as the protein levels (Fig. 5B and C).
BCL6B suppresses cell proliferation and
migration indirectly via inhibition of PI3K
To further investigate how BCL6B acts on the
PI3K/AKT signaling pathway, BCL6B-overexpressing LoVo cells were
treated with 10 μM of the PI3K/AKT inhibitor LY294002, and
cell viability and migration were measured at the indicated time
points by the MTT assay and Transwell chamber assay, respectively.
As a result, our data revealed that the decreased proliferation and
migration abilities and the repressed AKT activity caused by either
BCL6B overexpression or LY294002 in LoVo cells were both notably
enhanced by combined treatment with LY294002 and BCL6B (Fig. 6A–C). Furthermore, the upregulated
expression of E-cadherin and downregulated expression of cyclin D1
and MMP-9 induced by BCL6B in LoVo cells were enhanced by LY294002
(Fig. 6D). These findings
suggested that BCL6B suppresses proliferation and migration of CRC
cells indirectly, via inhibition of PI3K.
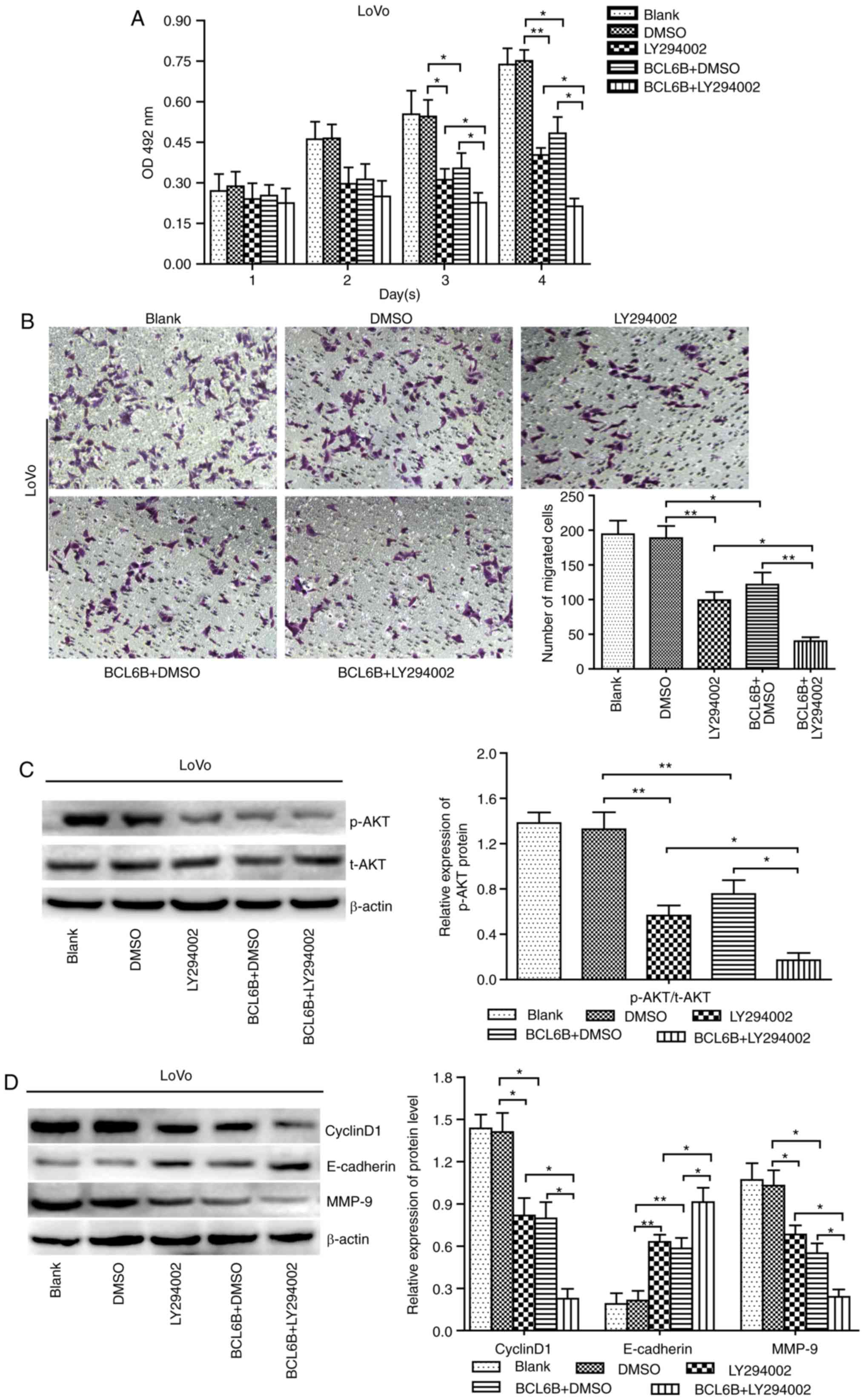 | Figure 6LY294002 enhances BCL6B-induced
inhibition of the proliferation and migration of LoVo cells. (A)
Effect of LY294002 on BCL6B-induced inhibition of viability of LoVo
cells. Cell viability was measured with the MTT assay. The
inhibitory role of BCL6B in cell proliferation was enhanced by
LY294002. The results show the mean absorbance ± SD of three
independent experiments (*P<0.05 vs. LY294002 group,
**P<0.01 vs. BCL6B + DMSO group). (B) Effect of
LY294002 on BCL6B-induced inhibition of migration of LoVo cells,
which was measured by the Transwell assay (magnification, ×100).
The inhibitory role of BCL6B in cell migration was enhanced by
LY294002 (*P<0.05 vs. LY294002 group,
**P<0.01 vs. BCL6B + DMSO group). (C) Effect of
LY294002 on BCL6B-induced suppression of AKT in LoVo cells was
detected by western blot analysis. The decreased expression of
p-AKT induced by BCL6B was enhanced by LY294002. Densitometric
quantification data are on the right panel. Data are shown as mean
values ± SD of three individual measurements
(**P<0.01 vs. DMSO group, *P<0.05 vs.
LY294002 group and BCL6B + DMSO group). (D) Effects of LY294002 on
BCL6B-induced changes in the protein levels of cyclin D1,
E-cadherin and MMP-9 in LoVo cells were detected by western blot
analysis. The expression levels of cyclin D1, E-cadherin and MMP-9
was enhanced by LY294002. Densitometric quantification data are
presented on the right panel. Data are shown as mean values ± SD of
three individual measurements (*P<0.05 vs. LY294002
group, *P<0.05 vs. BCL6B+DMSO group). BCL6B, B-cell
CLL/lymphoma 6 member B; OD, optical density; SD, standard
deviation; DMSO, dimethyl sulfoxide; MMP, matrix
metalloproteinase. |
Discussion
In the present study, the mRNA and protein
expression of BCL6B were found to be markedly repressed in the CRC
cell lines SW480 and LoVo compared with that in the normal
intestinal epithelial cell line FHC. In addition, BCL6B
overexpression inhibited cancer cell proliferation, colony
formation, migration and invasion, whereas it promoted cell
apoptosis. Mechanistically, these findings provide evidence showing
that the basal phosphorylation level of AKT in the SW480 and LoVo
cells was observably elevated compared with that in FHC cells, and
that BCL6B overexpression reduced AKT phosphorylation, cyclin D1
and MMP-9, and increased E-cadherin. More importantly, these
effects of BCL6B on CRC cells were enhanced by the PI3K/AKT
inhibitor LY294002. These results suggest that BCL6B suppresses the
proliferation and migration of CRC cells through inhibition of the
PI3K, in an indirect manner.
Over the past decades, molecular genetic studies
have revealed that epigenetic alterations, particularly
inactivation of tumor suppressor genes or tumor-related genes
through promoter hypermethylation, play an important role in the
tumorigenesis and development of CRC (27-30). Emerging evidence suggests that
BCL6Bwas silenced or downregulated due to promoter DNA
hypermethylation in a variety of human cancers (8). Clinical evidences indicated that
loss of BCL6B expression was associated with cancer progression,
metastasis and survival (10-15). Consistent with previous reports,
we found that the endogenous expression of BCL6B in the CRC cell
lines SW480 and LoVo was significantly lower compared with that in
the human normal intestinal epithelial cell line FHC. Therefore,
SW480 and LoVo cells were transfected with pcDNA3.1-BCL6B to
investigate the biological role and molecular basis of BCL6B in
CRC.
Our data revealed that SW480 and LoVo cells with
BCL6B overexpression exhibited significantly decreased viability
and colony-forming ability. A possible explanation for the growth
inhibition is cell cycle arrest or increase in apoptosis. The
results derived from FCM demonstrated that BCL6B induced apoptosis
of SW480 and LoVo cells. In addition, we observed that cyclin D1,
an important regulator of the cell cycle, was downregulated at both
the mRNA and protein levels, further confirming that BCL6B inhibits
CRC cell proliferation. Previous studies have reported that BCL6B
can suppress cancer cell growth, invasion and metastasis (10,13,15). For example, BCL6B inhibits
hepatocellular carcinoma metastasis and angiogenesis through the
upregulation of E-cadherin and downregulation of vascular
endothelial growth factor (13).
Restoration of BCL6B expression suppresses CRC cell growth and
migration by activating P53 signaling (15). In agreement with these studies,
the results of the wound closure assay and Transwell assay revealed
that SW480 and LoVo cells with BCL6B overexpression exhibited
decreased migration and invasion abilities. In addition, we
observed that the mRNA and protein level of the EMT-related marker
E-cadherin was increased and that the expression of the tumor
metastasis-related gene MMP-9 was decreased. Upregulation of
E-cadherin and downregulation of MMP-9 by BCL6B may enhance
intercellular adhesions, thus inhibiting tumor cell migration
(31).
The PI3K/AKT signaling pathway plays a pivotal role
in cell proliferation, apoptosis, survival, migration and invasion
(21). A number of studies
indicated that aberrant activation of the PI3K/AKT signaling
pathway is associated with human CRC (22,32). AKT kinase activity is induced
following activation of PI3K in growth factor receptor-mediated
signaling cascades (33).
Previous findings demonstrated frequent alterations inPI3K/AKT
signaling in human CRC (33,34). The PI3K/AKT pathway regulates
cyclin D1, E-cadherin and MMP-9 expression, thus affecting cell
proliferation and migration in different tumors (24,35,36). In the present study, the data
demonstrated that the basal AKT activity in SW480 and LoVo cells
was significantly higher compared with that in the normal
intestinal epithelial cell line FHC, and that BCL6B overexpression
effectively reduced the phosphorylation level of AKT in SW480 and
LoVo cells. Moreover, the decreased proliferation and migration
ability and repressed AKT activity caused by either BCL6B
overexpression or the PI3K/AKT inhibitor LY294002 in LoVo cells
were both notably enhanced by combined treatment with LY294002 and
BCL6B. Furthermore, the upregulation of E-cadherin and
downregulation of cyclin D1 and MMP-9 by BCL6B in LoVo cells were
enhanced by LY294002. These results demonstrated that BCL6B
suppresses proliferation and migration of CRC cells indirectly, via
inhibition of PI3K, and the mechanism underlying the effects of
BCL6B on the PI3K/AKT pathway requires further investigation. In
conclusion, the present study demonstrated that BCL6B, which is
downregulated in CRC cells, acts as a tumor suppressor in CRC by
suppressing cell proliferation, migration and invasion through the
inhibition of PI3K/AKT signaling pathway. Therefore, BCL6B
overexpression may prove to be a valuable tool for inhibiting
cancer growth. However, future studies are required to elucidate
the association between BCL6B and the PI3K/AKT pathway in CRC.
Acknowledgments
The authors would like to thank Professor Xiang
(Epigenetics Laboratory, The First Affiliated Hospital of Chongqing
Medical University) for her kind provision of the recombinant
plasmids pcDNA3.1-BCL6B and pcDNA3.1.
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2010. CA Cancer J Clin. 61:10–29. 2011.
|
|
2
|
Meyerhardt JA, Li L, Sanoff HK, Carpenter
W IV and Schrag D: Effectiveness of bevacizumab with first-line
combination chemotherapy for Medicare patients with stage IV
colorectal cancer. J Clin Oncol. 30:608–615. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Okabe S, Fukuda T, Ishibashi K, Kojima S,
Okada S, Hatano M, Ebara M, Saisho H and Tokuhisa T: BAZF, a novel
Bcl6 homolog, functions as a transcriptional repressor. Mol Cell
Biol. 18:4235–4244. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sakashita C, Fukuda T, Okabe S, Kobayashi
H, Hirosawa S, Tokuhisa T, Miyasaka N, Miura O and Miki T: Cloning
and characterization of the human BAZF gene, a homologue of the
BCL6 oncogene. Biochem Biophys Res Commun. 291:567–573. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Oatley JM, Avarbock MR, Telaranta AI,
Fearon DT and Brinster RL: Identifying genes important for
spermatogonial stem cell self-renewal and survival. Proc Natl Acad
Sci USA. 103:9524–9529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Manders PM, Hunter PJ, Telaranta AI, Carr
JM, Marshall JL, Carrasco M, Murakami Y, Palmowski MJ, Cerundolo V,
Kaech SM, et al: BCL6b mediates the enhanced magnitude of the
secondary response of memory CD8+ T lymphocytes. Proc Natl Acad Sci
USA. 102:7418–7425. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang H, Okada S, Hatano M, Okabe S and
Tokuhisa T: A new functional domain of Bcl6 family that recruits
histone deacetylases. Biochim Biophys Acta. 1540:188–200. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ying J, Srivastava G, Hsieh WS, Gao Z,
Murray P, Liao SK, Ambinder R and Tao Q: The stress-responsive gene
GADD45G is a functional tumor suppressor, with its response to
environmental stresses frequently disrupted epigenetically in
multiple tumors. Clin Cancer Res. 11:6442–6449. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Greipp PT, Smoley SA, Viswanatha DS,
Frederick LS, Rabe KG, Sharma RG, Slager SL, Van Dyke DL, Shanafelt
TD, Tschumper RC and Zent CS: Patients with chronic lymphocytic
leukaemia and clonal deletion of both 17p13.1 and 11q22.3 have a
very poor prognosis. Br J Haematol. 163:326–333. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu L, Li X, Chu ES, Zhao G, Go MY, Tao Q,
Jin H, Zeng Z, Sung JJ and Yu J: Epigenetic inactivation of BCL6B,
a novel functional tumour suppressor for gastric cancer, is
associated with poor survival. Gut. 61:977–985. 2012. View Article : Google Scholar
|
|
11
|
Yang Q, Gao J, Xu L, Zeng Z, Sung JJ and
Yu J: Promoter hypermethylation of BCL6B gene is a potential plasma
DNA biomarker for gastric cancer. Biomarkers. 18:721–725. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Deng J, Han L, Dong Q, Hou Y, Xie X, Yu J,
Fan D and Hao X: The survival decrease in gastric cancer is
associated with the methylation of B-cell CLL/lymphoma 6 member B
promoter. Open Biol. 4:1400672014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang J, Dong L, Xu L, Chu ES, Chen Y, Shen
J, Li X, Wong CC, Sung JJ and Yu J: B cell CLL/lymphoma 6 member B
inhibits hepatocellular carcinoma metastases in vitro and in mice.
Cancer Lett. 355:192–200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X, Yu J, Brock MV, Tao Q, Herman JG,
Liang P and Guo M: Epigenetic silencing of BCL6B inactivates p53
signaling and causes human hepatocellular carcinoma cell resist to
5-FU. Oncotarget. 6:11547–11560. 2015.PubMed/NCBI
|
|
15
|
Hu S, Cao B, Zhang M, Linghu E, Zhan Q,
Brock MV, Herman JG, Mao G and Guo M: Epigenetic silencing BCL6B
induced colorectal cancer proliferation and metastasis by
inhibiting P53 signaling. Am J Cancer Res. 5:651–662.
2015.PubMed/NCBI
|
|
16
|
Franke TF: PI3K/Akt: Getting it right
matters. Oncogene. 27:6473–6488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nicholson KM and Anderson NG: The protein
kinase B/Akt signalling pathway in human malignancy. Cell Signal.
14:381–395. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Michl P and Downward J: Mechanisms of
disease: PI3K/AKT signaling in gastrointestinal cancers. Z
Gastroenterol. 43:1133–1139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kawauchi K, Ogasawara T, Yasuyama M,
Otsuka K and Yamada O: The PI3K/Akt pathway as a target in the
treatment of hematologic malignancies. Anticancer Agents Med Chem.
9:550–559. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Manfredi GI, Dicitore A, Gaudenzi G,
Caraglia M, Persani L and Vitale G: PI3K/Akt/mTOR signaling in
medullary thyroid cancer: A promising molecular target for cancer
therapy. Endocrine. 48:363–370. 2015. View Article : Google Scholar
|
|
21
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rychahou PG, Jackson LN, Silva SR,
Rajaraman S and Evers BM: Targeted molecular therapy of the PI3K
pathway: Therapeutic significance of PI3K subunit targeting in
colorectal carcinoma. Ann Surg. 243:833–844. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao N, Zhang Z, Jiang BH and Shi X: Role
of PI3K/AKT/mTOR signaling in the cell cycle progression of human
prostate cancer. Biochem Biophys Res Commun. 310:1124–1132. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song G, Ouyang G and Bao S: The activation
of AKT/PKB signaling pathway and cell survival. J Cell Mol Med.
9:592005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zang M, Zhang B, Zhang Y, Li J, Su L, Zhu
Z, Gu Q, Liu B and Yan M: CEACAM6 promotes gastric cancer invasion
and metastasis by inducing epithelial-mesenchymal transition via
PI3K/AKT signaling pathway. Plos One. 9:e1129082014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ptak A, Hoffmann M, Gruca I and Barć J:
Bisphenol A induce ovarian cancer cell migration via the MAPK and
PI3K/Akt signalling pathways. Toxicol Lett. 229:357–365. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang W, Glöckner SC, Guo M, Machida EO,
Wang DH, Easwaran H, Van Neste L, Herman JG, Schuebel KE, Watkins
DN, et al: Epigenetic inactivation of the canonical Wnt antagonist
SRY-box containing gene 17 in colorectal cancer. Cancer Res.
68:2764–2772. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ying J, Poon F, Yu J, Geng H, Wong AH, Qiu
GH, Goh HK, Rha SY, Tian L, Chan AT, et al: DLEC1 is a functional
3p22.3 tumour suppressor silenced by promoter CpG methylation in
colon and gastric cancers. Br J Cancer. 100:663–669. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hanada N, Takahata T, Zhou Q, Ye X, Sun R,
Itoh J, Ishiguro A, Kijima H, Mimura J, Itoh K, et al: Methylation
of the KEAP1 gene promoter region in human colorectal cancer. BMC
Cancer. 12:662012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Migheli F and Migliore L: Epigenetics of
colorectal cancer. Clin Genet. 81:312–318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Conacci-Sorrell M, Zhurinsky J and
Ben-Ze'ev A: The cadherin-catenin adhesion system in signaling and
cancer. J Clin Invest. 109:987–991. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Parsons DW, Wang TL, Samuels Y, Bardelli
A, Cummins JM, DeLong L, Silliman N, Ptak J, Szabo S, Willson JK,
et al: Colorectal cancer: Mutations in a signalling pathway.
Nature. 436:7922005. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Burgering BM and Coffer PJ: Protein kinase
B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction.
Nature. 376:599–602. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ekstrand AI, Jönsson M, Lindblom A, Borg Å
and Nilbert M: Frequent alterations of the PI3K/AKT/mTOR pathways
in hereditary nonpolyposis colorectal cancer. Fam Cancer.
9:125–129. 2010. View Article : Google Scholar
|
|
35
|
Lau MT and Leung PC: The PI3K/Akt/mTOR
signaling pathway mediates insulin-like growth factor 1-induced
E-cadherin down-regulation and cell proliferation in ovarian cancer
cells. Cancer Lett. 326:191–198. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dilly AK, Ekambaram P, Guo Y, Cai Y,
Tucker SC, Fridman R, Kandouz M and Honn KV: Platelet-type
12-lipoxygenase induces MMP9 expression and cellular invasion via
activation of PI3K/Akt/NF-κB. Int J Cancer. 133:1784–1791. 2013.
View Article : Google Scholar : PubMed/NCBI
|