Introduction
Testicular ischemia/reperfusion (I/R) injury is a
common pathophysiological process that occurs with testicular
torsion (1). I/R results in
testis injury and spermatogenesis loss as well as damage to the
contralateral side (1). The
crucial underlying mechanism responsible for I/R in testicular
torsion is oxidative stress (2,3).
Caspase-independent apoptosis resulting from oxidative stress
mediates tissue injury; however, this mechanism is not solely
responsible for I/R-induced cell death (4). Ferroptosis is a recently recognized
type of non-apoptotic cell death that has been observed in various
pathological processes, including heart, kidney and liver I/R
injury (4–9). Developing a better understanding of
the signaling pathway that induces Sertoli cell death following
testicular I/R injury is important for the development of an
effective strategy to prevent I/R-induced spermatogenic arrest.
A previous study demonstrated that testicular I/R
injury-induced cell death of germ cells, but not Sertoli cells, was
inconsistent with spermatogenetic dysfunction (10). Spermatogenesis is a complex
process in which spermatogonial stem cells self-renew and
differentiate into spermatozoa under coordination of the testicular
microenvironment provided by Sertoli cells, which are the only
somatic cells located inside the seminiferous epithelium (11). Sertoli cells are major supportive
germ cells that regulate spermatogenic cells via a number of
paracrine factors (12,13). Germ cell and Sertoli cell
interactions are important for male fertility (14). Disrupted Sertoli cell signaling
results in germ cell loss and infertility (15). Germ cell loss following testicular
I/R injury may result in part from Sertoli cell damage-dependent
alterations in the microenvironment (16–18). The mechanism of testicular I/R
injury is lipid peroxidation (19). Previous studies have reported that
I/R induces ferroptosis in nephritic tubular cells and hepatocytes,
whereas other forms of regulated cell death are not observed
(7–9). It has been suggested that
ferroptosis is a latent mechanism associated with I/R injury and
oxidative toxicity in cells (7,20).
It was therefore hypothesized that oxidative damage may be
associated with I/R-induced ferroptosis or cell death in Sertoli
cells. The aim of the present study was to explore the potential
mechanisms and regulatory factors of cell death in testicular
Sertoli cells, which may reveal a novel therapeutic strategy for
the prevention of I/R-induced cell loss. To the best of our
knowledge, no previous studies have investigated the probability
and mechanism of I/R-induced ferroptosis in Sertoli cells.
Ferroptosis is distinct from apoptosis, autophagy
and necrosis at the biochemical, morphological and genetic levels
(4). Ferroptosis serves a major
role in acute tissue injury, I/R injury and neurotoxicity (4,21).
The major mediators of ferroptosis are lipid peroxidation and iron
metabolism signaling (4,22–24). In addition, inducers of
ferroptosis may be divided into two categories: i) Inhibitors of
cysteine import, which subsequently lead to glutathione (GSH)
depletion, and ii) inhibitors of GSH-dependent peroxidase 4 (GPX4).
A variety of inhibitors and inducers of ferroptosis have been
reported to regulate the accumulation of lethal lipid reactive
oxygen species (ROS) derived from iron metabolism turbulence
(4,25,26); however, the molecular mechanism
underlying the induction of ferroptosis in Sertoli cells remains
unknown.
In the present study, an in vitro I/R cell
model was established using TM4 cells to investigate the induction
of ferroptotic cell death and the associated signaling pathway
responsible for cell death regulation in Sertoli cells.
Materials and methods
Reagents
Ferrostatin-1 (Fer-1; cat. no. S7243),
liproxstatin-1 (cat. no. S7699),
carbobenzoxy-valyl-alanyl-aspartyl-[O-m ethyl]-fluoromethylketone
(Z-VAD-FMK; cat. no S7023), necrostatin-1 (cat. no. S8037),
3-methyladenine (3-MA; cat. no. S2767), SB203580 (cat. no. S1076),
SP600125 (cat. no. S1460), and SCH772984 (cat. no. S7101) were
obtained from Selleck Chemicals (Houston, TX, USA).
N-acetyl-cysteine (NAC; cat. no. A0737), deferoxamine (DFO; cat.
no. D9533), FeCl3 (cat. no. 157740),
buthionine-sulfoximine (BSO; cat. no. 19176), Iron Stain kit (cat.
no. HT20), GSH (cat. no. G6013), DNase I (cat. no. AMPD1),
hyaluronidase type III (cat. no. H3506), collagenase I (cat. no.
C0130), insulin-transferrin-selenium (ITS; cat. no. 00031626),
epidermal growth factor (EGF; cat. no. E4127), testosterone (cat.
no. T6147), follicle stimulating hormone (FSH; cat. no. I9149),
ponasterone (cat. no. P3490), neopterin (cat. no. N3386),
diphenyleneiodonium chloride (DPI; cat. no. D2926),
diethyldithiocarbamate (DETC; cat. no. 228680),
3-amino-1,2,4-triazole (ATZ; cat. no. A8056), Lipid Peroxidation
malondialdehyde (MDA) Assay kit (cat. no. MAK085), Glutathione
Peroxidase Cellular Activity Assay kit (cat. no. CGP1),
Quantification kit for oxidized and reduced GSH (cat. no. 38185),
Iron Assay kit (cat. no. MAK 025), and Caspase 3 Assay kit (cat.
no. CASP3C) were obtained from Sigma Aldrich (Merck KGaA,
Darmstadt, Germany) The following reagents were purchased from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA): Ferroportin-1
short interfering (si)RNA (cat. no. sc-60634), XCT siRNA (cat. no.
sc-76934), GPx-4 siRNA (cat. no. sc-63302), control siRNA (cat. no.
sc-37007), siRNA Reagent System (cat. no. sc-45064), ferroportin-1
CRISPR Activation Plasmid (cat. no. sc-424774-ACT), GPx-4 CRISPR
Activation Plasmid (cat. no. sc-436949-ACT), GPx-1 CRISPR
Activation Plasmid (cat. no. sc-420662-ACT), anti-rabbit/mouse IgG
horseradish peroxidase conjugate (HRP; cat. no. sc-2379 and
sc-2380) and antibodies against transferrin (Tf; cat. no.
sc-393595), transferrin receptor 1/cluster of differentiation 71
(TFR1; cat. no. sc-32272), divalent metal transporter 1/natural
resistance-associated macrophage proteins (DMT1; cat. no.
sc-166884), ferritin (cat. no. sc-25617), GPX4 (cat. no.
sc-166120), ferroportin-1 (Fpn; cat. no. sc-49668), and β-actin
(cat. no. sc-58671). Antibodies against p38 mitogen-activated
protein kinase (MAPK; cat. no. 9212), extracellular
signal-regulated kinase1/2 (ERK1/2; cat. no. 9102), c-Jun
NH2-terminal kinase (JNK; cat. no. 9252), phosphorylated (p)-p38
MAPK (cat. no. 4511), p-ERK1/2 (cat. no. 9101), p-JNK (cat. no.
9255), and XCT (SLC7A11; cat. no. 12691) were purchased from Cell
Signaling Technology (Danvers, MA, USA). TRIzol reagent was
obtained from Invitrogen (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). The reverse transcription (RT) system and One Step SYBR
PrimeScript RT-quantitative polymerase chain reaction (PCR) kit
(cat. no. RR066) were purchased from Takara Bio, Inc. (Otsu,
Japan). CellTiter 96 AQueous Non-Radioactive Cell Proliferation
Assay kit (G cat. no. 5421) was obtained from Promega Corp.
(Madison, WI, USA).
TM4 cell culture and establishment of
cell I/R injury model
TM4 mouse Sertoli cells (cat. no. CM-1912; American
Type Culture Collection, Manassas, VA, USA) were cultured in
Dulbecco's modified Eagle medium/nutrient mixture F-12 (DMEM/F-12;
Gibco; Thermo Fisher Scientific, Inc.) medium supplemented with 5%
fetal bovine serum, 0.1% ITS, 1 µg/ml EFG, 0.1 µM
testosterone, 25 U/l FSH, 100 units/ml penicillin, and 100
µg/ml streptomycin at 37°C in an atmosphere containing 5%
CO2 for 72 h. The I/R cell model was established as
previously described (27,28).
TM4 cells were cultured in airtight bottles with ischemic buffer
(5.37 mM KCl, 136.89 mM NaCl, 0.44 mM KH2PO4,
0.338 mM Na2HPO4, 4.166 mM NaHCO3,
and 5 mM D-glucose; pH 6.8) at 37°C in a hypoxic incubator with 5%
CO2, 1% O2 and 94% N2 for 2 h. The
medium was subsequently removed, normal routine culture was resumed
and reoxygenation was initiated, as described above. The I/R cell
model was the OGD/R model of TM4 cells. The cells of OGD/R model
were set as OGD/R group and normal TM4 cells were set as control
group.
Cell transfection
Ferroportin-1 and GPx-4 knockdown was achieved by
transfection with siRNA targeting ferroportin-1 and GPx-4
(si-ferroportin-1 and si-GPx-4; Santa Cruz Biotechnology, Inc.)
using an siRNA Transfection Reagent (Santa Cruz Biotechnology,
Inc.) with a control siRNA as the control (Santa Cruz
Biotechnology, Inc.). Overexpression of ferroportin-1 and GPx-4
were achieved by transfection with ferroportin-1, GPx-4 and GPx-1
overexpression vectors (ferroportin-1, GPx-4, and GPx-1 CRISPR
Activation Plasmid; Santa Cruz Biotechnology, Inc.) using Plasmid
Transfection Medium (Santa Cruz Biotechnology, Inc.), with the
CRISPR/Cas9 Plasmid used as the control. At 7 h post-transfection,
the medium was changed to whole medium and cells were cultivated
for an additional 24 h. The changes in ferroportin-1, GPx-4, and
GPx-1 were measured using RT-qPCR, as described below.
Cell viability assay
Cell viability was analyzed using the CellTiter 96
AQueous Non-Radioactive Cell Proliferation Assay kit according to
the manufacturer's protocol. TM4 cells were seeded onto 96-well
plates (1×103 cells/well), and 20 µl
MTS/phenazine methosulfate was pipetted into each well. The plate
was incubated for 2 h at 37°C, and then the absorbance was measured
at 490 nm using a Spectramax M2 Multi-Mode Microplate Reader
(Molecular Devices, LLC, Sunnyvale, CA, USA) to calculate the
percentage of surviving cells.
Caspase-3 activity assay
Caspase-3 activity in TM4 cells was assessed using
the Caspase-3 Activity Assay kit according to the manufacturer's
protocol.
Lipid ROS measurement
TM4 cells were lysed with MDA lysis buffer from the
Lipid Peroxidation malondialdehyde (MDA) Assay kit and centrifuged
at 8,000 × g for 10 min at 4°C to remove insoluble material. The
level of MDA in TM4 cell lysates was assessed using a Lipid
Peroxidation MDA Assay kit according to the manufacturer's
protocol. The absorbance was measured at 532 nm using a
spectrophotometer.
GSH content and GPXs activity
measurement
The expression of reduced and oxidized forms of GSH
was analyzed using a quantification kit for oxidized and reduced
GSH according to the manufacturer's protocol. Cultured TM4 cells
were washed three times with PBS, 5 min each time mixed with assay
buffer and substrate solution from the Quantification kit for
oxidized and reduced GSH, and incubated at room temperature in a
fluorometer. Absorbance was assessed at 412 nm and the results were
expressed as a percent of the control.
GPX activity in the cell lysates was measured using
the Glutathione Peroxidase Cellular Activity Assay kit according to
the manufacturer's protocol. The absorbance was measured at 405 or
415 nm using a microplate reader. The values were expressed as a
percentage of non-treated cells.
Intracellular iron measurement
The intracellular iron concentration was measured
using an Iron Assay kit according to the manufacturer's protocol.
An iron reducer was added to cell lysates to reduce ferric iron
(Fe3+) to ferrous iron (Fe2+) and incubated
for 30 min at room temperature in the dark. The iron reaction
produced a colored complex and absorbance was measured at 593
nm.
RT-qPCR
Total cellular RNA was isolated from the cells in
the control and OGD/R groups using TRIzol (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The RNA was reverse transcribed (42°C for 10 min, 95°C for 5 min,
4°C for 3 min, 5 cycles) into cDNA using an RT system. qPCR was
performed using a One Step SYBR Green PrimeScript RT-qPCR kit with
denaturation at 95°C for 15 min, 50 cycles at 95°C for 15 sec, 72°C
for 15 sec, 95°C for 30 sec and 60°C for 15 sec. PCR was performed
in triplicate and normalized using the ABI 7500 Real-Time PCR
System (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
primers used were as follows: Fpn, forward
5′-AGCCAAGATGGCACTAAGCAC-3′ and reverse
5′-TCTATGTTATCGAACAGACAT-3′; GPX4, forward
5′-AGCCAAGACCGAAGTAAACTACAC-3′ and reverse
5′-GGATCTTCATCCAGTTCCACAG-3′; GAPDH, forward
5′-CAAGGTCATCCATCCATGACAACTTTG-3′ and reverse
5′-GTCCACCACCCTGTTGCTGTAG-3′. GAPDH was used as the reference gene.
Relative gene expression was analyzed by 2−ΔΔCq method
(28).
Western blot analysis
Cells in the control and OGD/R groups were lysed in
cell lysis solution containing 1 mM phenyl-methane
sulfonyl-fluoride. The total protein concentration was determined
with bicinchoninic acid assay kit (Thermo Fisher Scientific, Inc.).
Protein lysates (40 µg/lane) were separated by 10–12%
SDS-PAGE and transferred onto 0.22 µm polyvinyli-dene
difluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with 3% bovine serum albumin (Sigma-Aldrich;
Merck KGaA) in Tris-buffered saline-0.05% Tween (TBST) for 1 h at
37°C. Membranes were subsequently incubated with antibodies against
Fpn (1:100), GPX4 (1:200), p38 (1:500), p-p38 MAPK (1:500), ERK1/2
(1:500), p-ERK1/2 (1:500), JNK (1:500), p-JNK (1:500), Tf (1:200),
TFR1 (1:200), DMT1 (1:200), ferritin (1:100), SLC7A11 (1:100) and
β-actin (1:1,000) at 4°C overnight. The membranes were washed three
times with TBST and incubated with horseradish
peroxidase-conjugated goat anti-rabbit/mouse IgG for 1 h at room
temperature the following morning. The blots were visualized using
an enhanced chemiluminescence detection kit (Thermo Fisher
Scientific, Inc.). The band density of target proteins was
quantified and normalized to that of β-actin using Quantity One
software (version 4.6.2, Bio-Rad Corporation, Hercules, CA,
USA).
Statistical analysis
All data were processed with the using SPSS 19.0
(IBM Corp., Armonk, NY, USA). Data are presented as the mean ±
standard deviation from three independent experiments. One-way
analysis of variance with Tukey's multiple comparison post hoc test
was employed for comparison among groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
Ferroptosis is induced following I/R
injury in TM4 cells
Testicular I/R injury has been reported to induce
cell death in germ cells, but not in Sertoli cells (19,28,29). It was therefore examined whether
I/R was able to induce cell death in Sertoli cells. TM4 cells in
control and OGD/R groups were analyzed and a notable increase in
cell death was observed in the OGD/R group compared with the
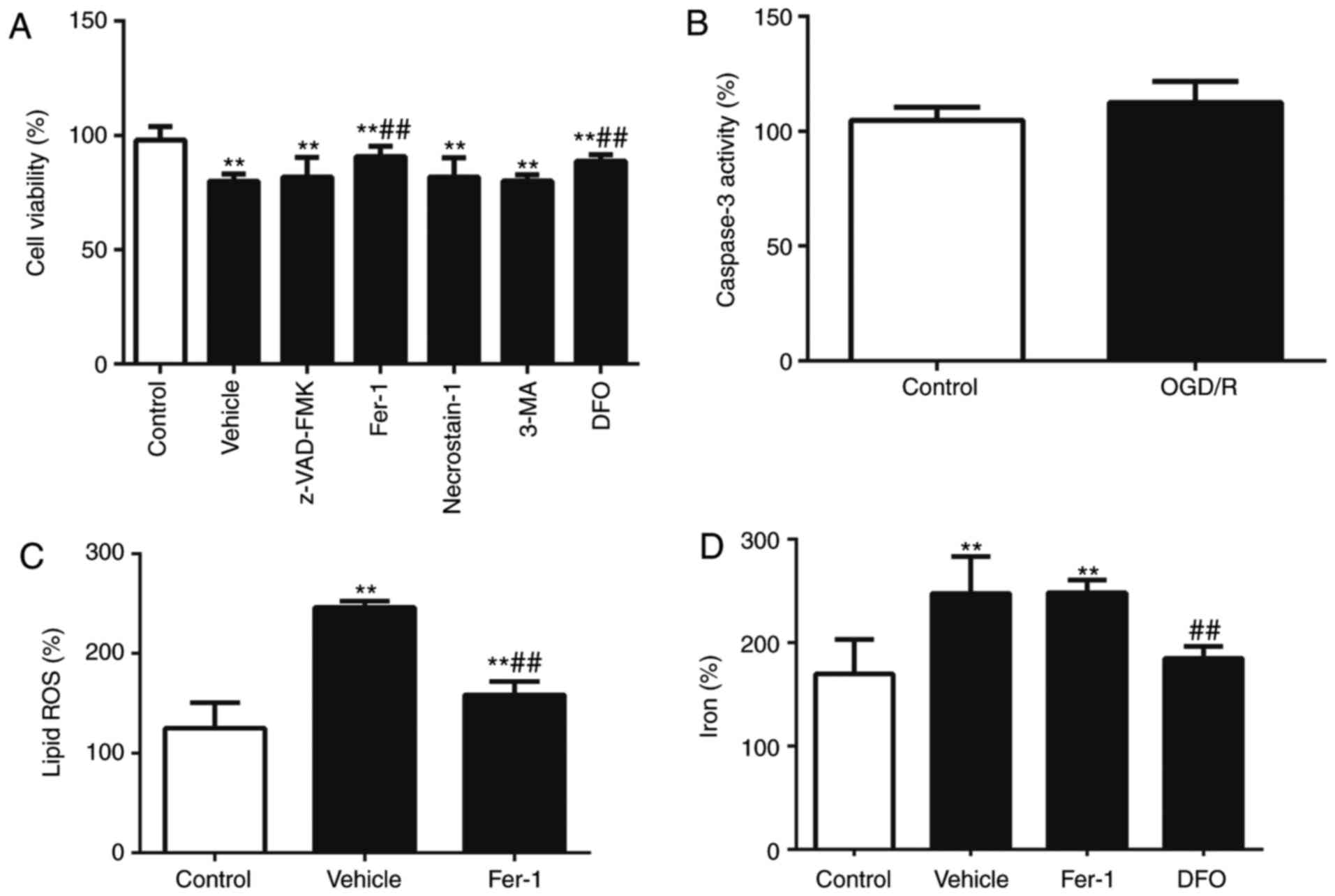
control (P<0.01; Fig. 1A). I/R
stress activates various types of cell death, including apoptosis,
necrosis, autophagy, and ferroptosis-mediated cell death (30). Recent studies have indicated that
I/R induces ferroptosis, but not other forms of cell death, in
nephrotic tubular cells and hepatocytes (8,31,32). Cells were incubated with the cell
death inhibitor Z-VAD-FMK, ferroptosis inhibitor Fer-1, necrosis
inhibitor necrostatin-1 and autophagy inhibitor 3-MA and cell death
was evaluated. The results indicated that OGD/R-induced cell death
was ameliorated by Fer-1 compared with the control group
(P<0.01; Fig. 1A); however, no
significant effect was observed with inhibitors of apoptosis,
necrosis or autophagy. OGD/R did not significantly promote the
activity of apoptotic marker caspase 3 (Fig. 1B). The results indicate that
ferroptosis serves a role in the accumulation of lipid peroxidation
and iron; lipid ROS levels were increased in the OGD/R group
compared with the control, and this effect was ameliorated by Fer-1
(P<0.01; Fig. 1C). To evaluate
the effect of iron on OGD/R-induced cell death, the iron chelator
DFO was used to treat Sertoli cells; the results demonstrated that
cell death and iron levels were reduced (P<0.01; Fig. 1A and D). Taken together, these
results suggest that ferroptosis contributes to OGD/R-induced
growth inhibition in Sertoli cells, whereas cell death, necroptosis
and autophagy do not.
Lipid peroxidation regulates
OGD/R-induced ferroptotic cell death
Prior studies have reported that ferroptosis is
characterized by an increase in lethal lipid peroxidation products,
which exhibit cytotoxicity (4,33).
Lipid ROS levels were assessed and the results revealed that lipid
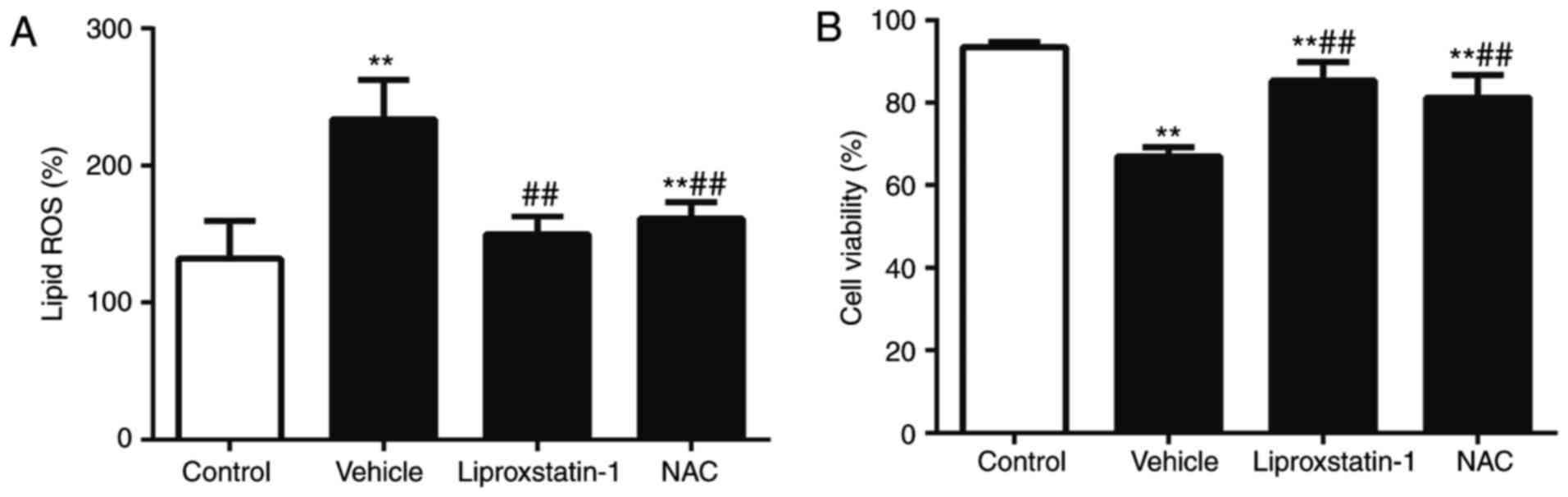
ROS were significantly increased in the OGD/R group compared with
the control group (P<0.01; Fig.
2A). Lipid ROS generation was inhibited by liproxstatin-1 and
NAC. Liproxstatin-1 and NAC both significantly reduced ROS
generation (P<0.01; Fig. 2A).
It was also observed that NAC and liproxstatin-1 effectively
inhibited cell death (P<0.01; Fig.
2B). These results suggest that lipid ROS is required for
OGD/R-induced ferroptotic cell death.
Lipid peroxidation results from the
accumulation of iron and depletion of GSH
The production of lipid ROS is required for
ferroptosis and is associated with multiple processes, including
NADPH-mediated lipid peroxidation, iron-dependent ROS generation
through the Fenton reaction and GSH depletion (4,23,25). To ascertain whether NADPH oxidases
contribute to lipid ROS production, TM4 cells were treated with
NADPH oxidase inhibitors, neopterin and DPI; lipid ROS levels were
unaffected. However, treatment with the iron chelator DFO
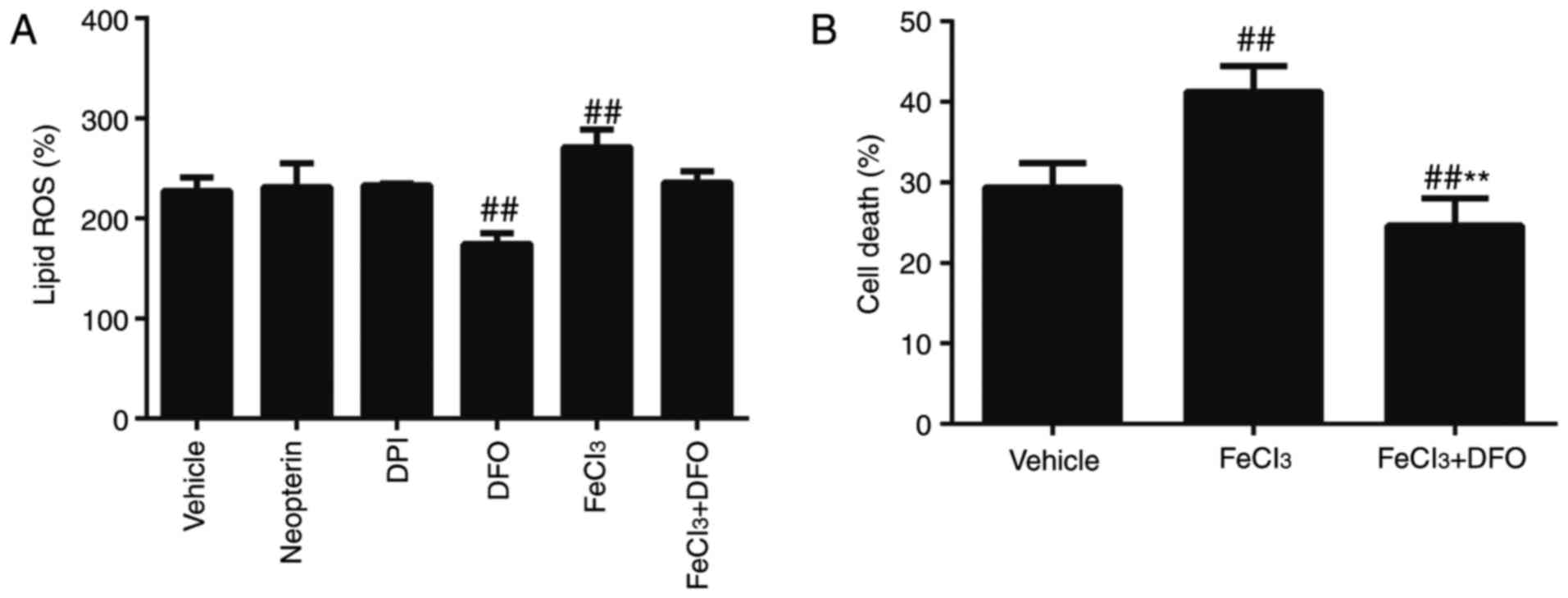
effectively blocked OGD/R-induced lipid ROS production (P<0.01;
Fig. 3A) compared with untreated
cells, suggesting that lipid ROS generation is associated with iron
content. Recent studies have demonstrated that iron overload
injures Sertoli cells (34).
Cells were subsequently treated with exogenous Fe
(FeCl3) to confirm that iron affected OGD/R-induced
ferroptosis and the results revealed that lipid ROS levels and cell
death were further increased (P<0.01; Fig. 3A and B). A reduction in lipid ROS
levels and cell death was also observed in cells cultured with DFO
compared with the vehicle (P<0.01; Fig. 3A and B). These data suggest that
ROS production results from iron accumulation and that iron may
serve a crucial role in OGD/R-induced ferroptosis.
To explore whether lipid ROS generation partially
reflects GSH depletion, GSH levels in cells were assessed. GSH
content was significantly reduced in the OGD/R group compared with
the control (P<0.01; Fig. 4).
Cells were treated with GSH or the GSH precursor NAC and cell
viability and lipid ROS levels were assessed. The results revealed
that GSH or NAC treatment significantly prevented OGD/R-induced
lipid ROS generation (P<0.01), whereas GSH had no significant
effect on OGD/R-induced cell death (Fig. 4B and C). It was also observed that
the protein level of XCT (SLC7A11), a cysteine transport receptor,
was reduced following OGD/R injury (P<0.01; Fig. 4D). Furthermore, siRNA-mediated
knockdown of XCT, the upstream activator of GSH, reduced the GSH
content and markedly increased cell death and lipid ROS levels
(P<0.01; Fig. 4A–C). These
data indicate that GSH depletion is consistent with OGD/R-induced
production of lipid ROS, resulting in ferroptosis. These findings
suggest that ROS generation results from iron accumulation and GSH
depletion.
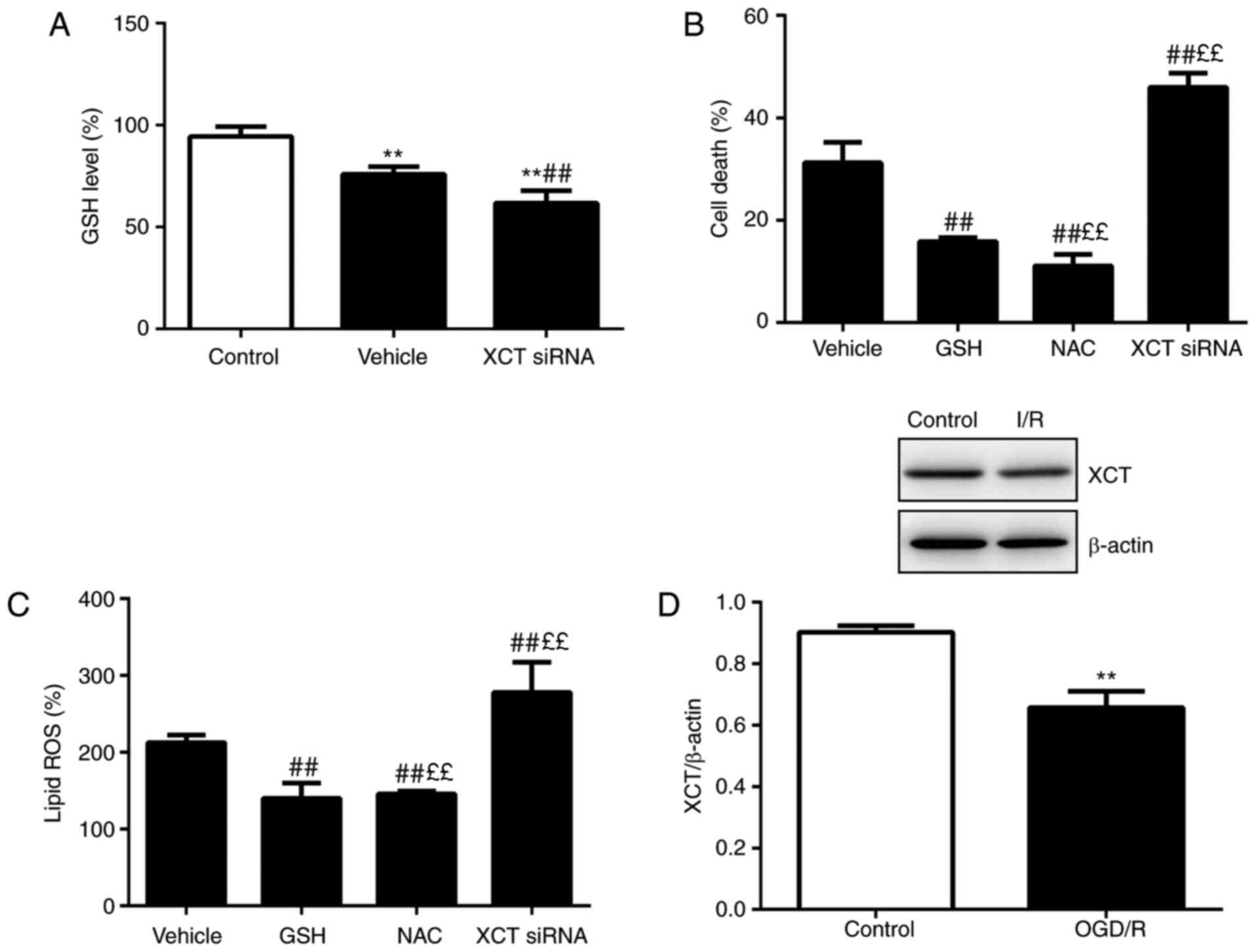 | Figure 4Lipid peroxidation results from GSH
depletion. (A) GSH levels were determined following treatment with
vehicle or XCT siRNA administration. (B) Cell death was analyzed
following treatment with vehicle, GSH (5 mM), NAC (5 nM) or XCT
siRNA for 24 h (C) Lipid ROS was assessed following treatment with
vehicle, GSH, NAC or XCT siRNA treatment for 12 h. (D) XCT
expression was assessed using western blotting. n=6.
**P<0.01 vs. control, ££P<0.01 vs. GSH
and ##P<0.01 vs. vehicle. GSH, glutathione; si, small
interfering; NAC, N-acetyl-cysteine; ROS, reactive oxygen species;
OGD/R, oxygen-glucose deprivation/reoxygenation. |
Overexpression of Fpn rescues
OGD/R-induced ferroptosis via suppression of iron accumulation
Increased iron uptake and reduced iron output
through iron metabolism proteins both lead to iron overload during
the ferroptotic cell death process (4,35).
First, it was determined which iron regulatory proteins were
expressed in Sertoli cells using immunocytochemistry and western
blotting. As presented in Fig.
5A, the following factors were expressed as normal in Sertoli
cells: Tf, which binds ferric iron (Fe3+); TFR1, which
imports iron into cells; DMT1, which mediates ferrous iron
(Fe2+) release into the cytoplasm; cytosolic ferritin,
which stores excess iron; and the membrane protein Fpn, which
mediates iron export (35). The
expression of Fpn was markedly reduced in the OGD/R group
(P<0.01), and no alterations were observed in the expression of
Tf, TFR1, DMT1 or ferritin. These results indicate that OGD/R may
reduce iron export to induce ferroptosis by decreasing Fpn
expression. Previous studies have revealed that Fpn mutation may
lead to iron overload and subsequent diseases accompanied by normal
or low transferrin expression (4,36).
To further demonstrate whether Fpn inhibits OGD/R-induced lipid ROS
and ferroptosis, Sertoli cells were transfected with the
Ferroportin-1 CRISPR Activation Plasmid, treated with the Fpn
activator ponasterone and subsequently analyzed for ROS and iron
levels. Cells transfected with the Ferroportin-1 CRISPR activation
plasmid or treated with ponasterone exhibited increased protein and
mRNA expression of Fpn and blocked accumulation of lipid ROS and
iron (P<0.01; Fig. 5). Cell
death was also significantly decreased (P<0.01; Fig. 5F). By contrast, knockdown of Fpn
with ferroportin-1 siRNA increased the OGD/R-induced lipid ROS
level and iron content, but not cell death (P<0.01; Fig. 5D–F). Taken together, these results
indicate that Fpn overexpression inhibited OGD/R-induced
ferroptosis in Sertoli cells by reducing the content of iron and
generation of lipid ROS and that the Fpn inducer ponasterone served
the same role.
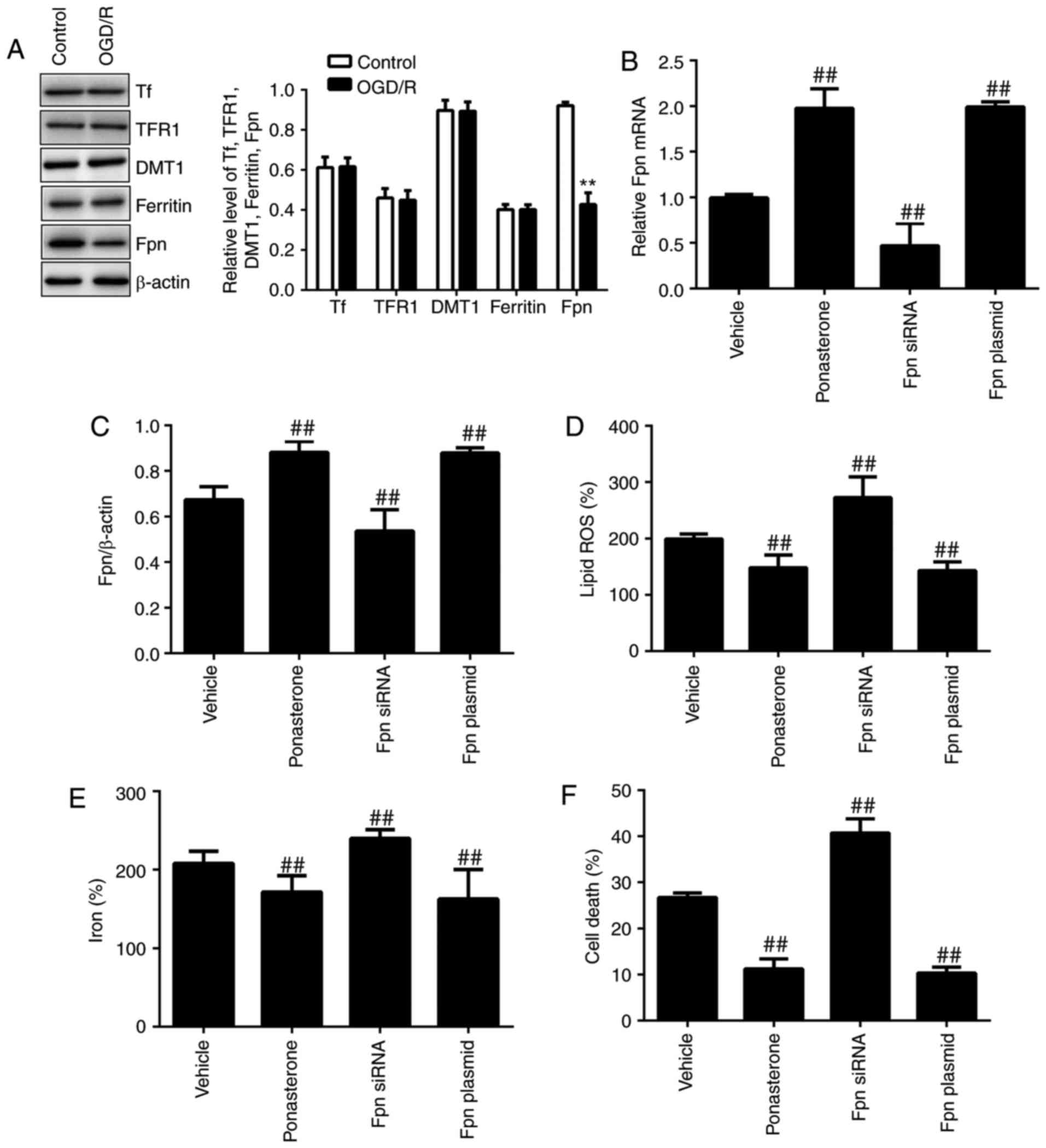 | Figure 5Overexpression of Fpn rescues
OGD/R-induced ferroptosis via suppression of iron accumulation. (A)
Protein levels of Tf, TFR1, DMT1, ferritin and Fpn were analyzed
using western blotting. TM4 cells were incubated with vector,
ponasterone (10 µM), Ferroportin-1 siRNA or Ferroportin-1
CRISPR Activation Plasmid. (B) Fpn mRNA expression, (C) Fpn protein
expression, (D) lipid ROS levels, (E) iron levels and (F) cell
death were assessed. **P<0.01 vs. control, and
##P<0.01 vs. vehicle. Fpn, ferroportin; OGD/R,
oxygen-glucose deprivation/reoxygenation; TF, transferrin; TFR1,
transferrin receptor 1/cluster of differentiation; DMT1, divalent
metal transporter 1/natural resistance-associated macrophage
proteins; si, small interfering; ROS, reactive oxygen species. |
GPX4 is inactivated following OGD/R
injury via GSH depletion
Considering that GSH depletion is crucial for
OGD/R-induced ROS generation, it was examined how OGD/R-induced GSH
depletion may lead to ferroptosis in Sertoli cells. The cells were
treated with the GSH synthesis inhibitor BSO and antioxidant
inhibitors, including the superoxide dismutase (SOD) inhibitor DETC
and the catalase inhibitor ATZ, and subsequently, examined for cell
death and GSH levels. The results revealed that inhibitors induced
cell death and that the GSH content was not significantly reduced
by these antioxidant inhibitors compared with BSO (Fig. 6A and B). The results indicated
that altered downstream GSH depletion was likely responsible for
the induction of ferroptotic cell death. It was considered that
GPX4 inactivation due to GSH depletion induces ferroptosis by
accumulating lipid ROS from lipid peroxidation. Previous studies
have demonstrated that GPX4 regulates ferroptosis via indirect or
direct targeting of lipid peroxidation and iron metabolism
(4). Furthermore, inactivation of
GPX4 triggers ferroptotic cell death in renal tubular epithelial
cells via lipid peroxidation (24,37,38). To determine whether GPX4
expression is decreased in the cell model of OGD/R-induced
testicular damage, the total activity of GPXs was examined as well
as GPX4 expression using Western blotting. Notable decreases in
GPXs activity and GPX4 expression were observed (P<0.01;
Fig. 6C and D). Furthermore,
treatment with GSH or NAC revealed that these molecules may
activate GPXs (P<0.01; Fig.
6C). These results further indicate that GSH depletion
inactivates GPXs with low GPX4 protein expression, resulting in the
production of lipid ROS.
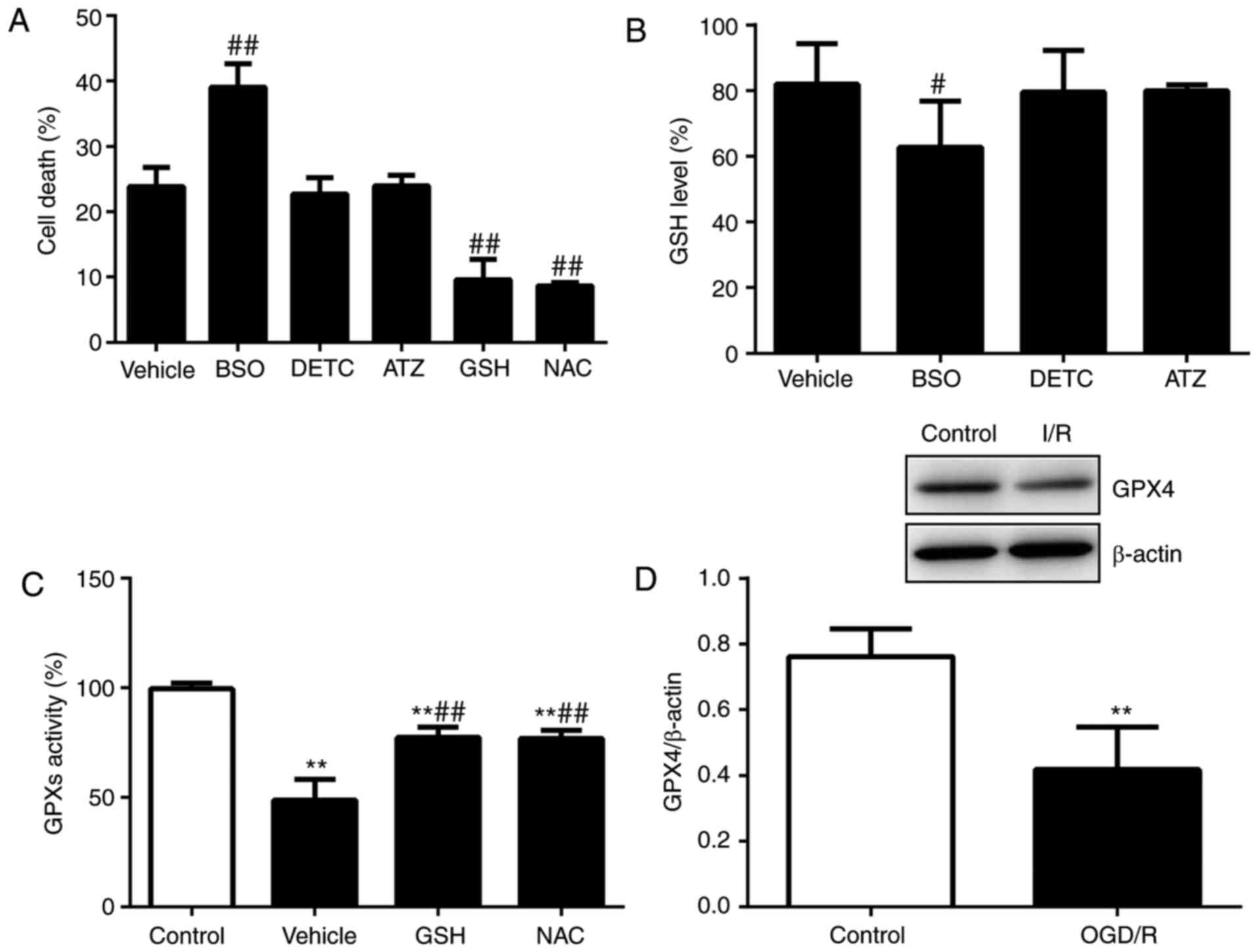 | Figure 6GPX4 is inactivated following
testicular OGD/R injury via GSH depletion. (A) Cell death was
assessed following treatment with vector, BSO (50 nM), DETC (10 nM)
or ATZ (10 µM) for 24 h. (B) GSH levels were analyzed
following treatment with vector, BSO (50 nM), DETC (10 nM), ATZ (10
µM), GSH (5 mM) or NAC (5 nM) for 12 h. (C) Activities of
GPXs were examined following treatment with vehicle, GSH or NAC for
6 h. (D) GPX4 protein expression was analyzed using western
blotting. n=6. **P<0.01 vs. control,
#P<0.05 and ##P<0.01 vs. vehicle. GSH,
glutathione; GPX4, GSH-dependent peroxidase 4; OGD/R,
oxygen-glucose deprivation/reoxygenation; BSO,
buthionine-sulfoximine; DETC, diethyldithiocarbamate; ATZ,
3-amino-1,2,4-triazole. |
Activation of GPX4 blocks OGD/R-induced
ferroptosis via reducing lipid ROS
GPX4 has been described as the major regulator of
erastin-induced cancer cell ferroptosis and OGD/R-induced
ferroptotic renal tubular epithelial cell death (24,37,39–41). A previous study demonstrated that
GPX4 is a prerequisite for sperm development (42). It was therefore investigated
whether GPX4 regulates OGD/R-induced ferroptotic cell death in
Sertoli cells. The results revealed that knocking down GPX4 with
GPX-4 siRNA reduced the mRNA and protein levels of GPX4 and
increased cell death as well as lipid ROS compared with a vector
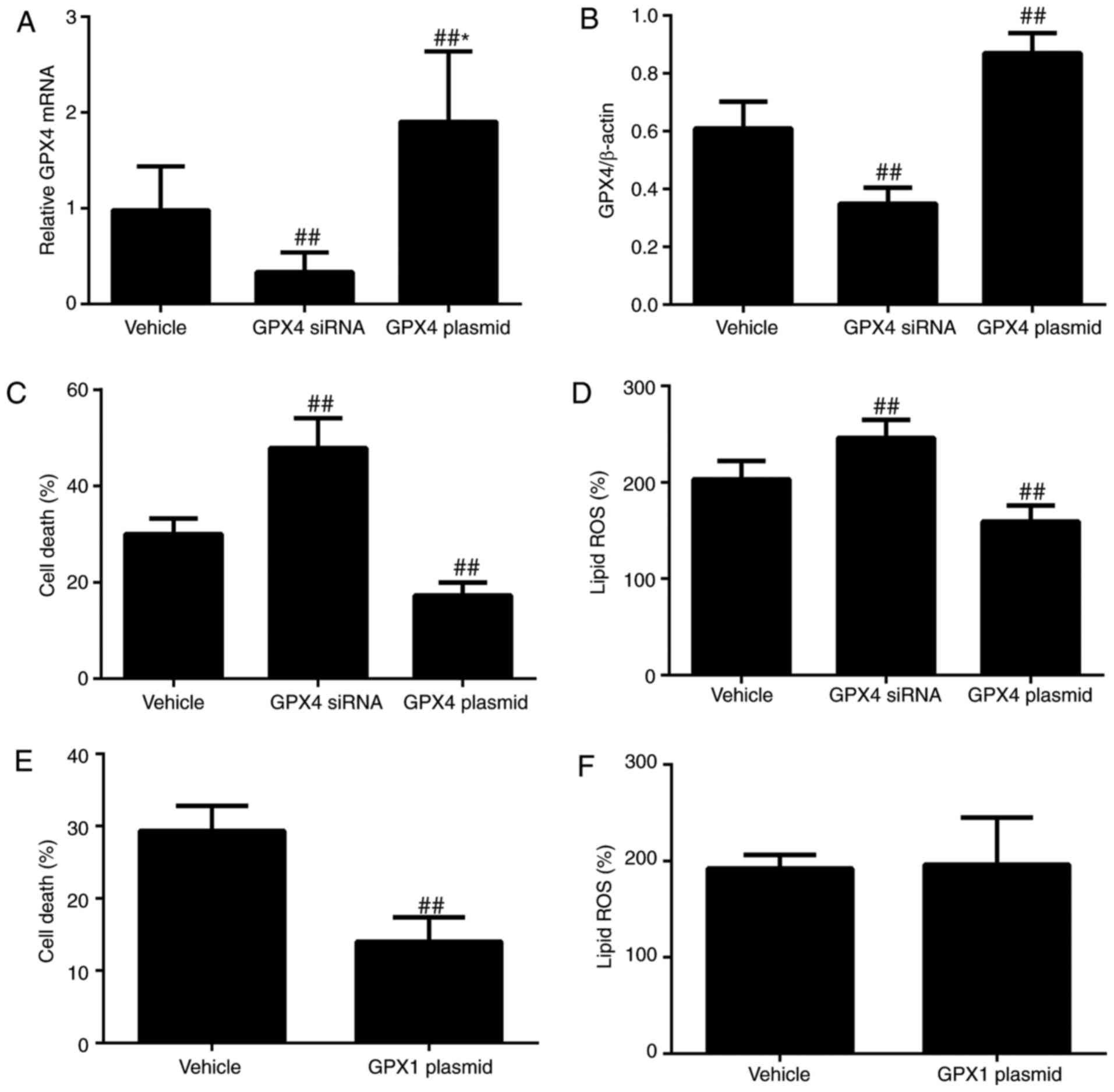
(P<0.01; Fig. 7A–D). GPX4
expression was subsequently upregulated in cells using the GPx-4
CRISPR Activation Plasmid and it was observed that GPX4 mRNA and
protein levels were increased and OGD/R-induced ferroptosis and
lipid ROS were decreased (P<0.01; Fig. 7A–D). The role of GPX1, a soluble
hydroperoxide reducer, was also examined. GPX1 overexpression
partially inhibited OGD/R-induced cell death without affecting
lipid ROS generation (Fig. 7E and
F), indicating that GPX1 may not scavenge additional ROS
species. Taken together, these data indicate that GPX4 is
inactivated following testicular OGD/R injury via GSH depletion and
blocks OGD/R-induced ferroptosis by reducing lipid ROS.
Inactivation of p38 MAPK prevents
OGD/R-induced ferroptosis
Previous studies have reported that MAPK signaling
is associated with erastin-induced ferroptosis in cancer cells and
OGD/R-induced testicular injury (4,43,44). However, whether the MAPK signaling
pathway serves a role in OGD/R-induced ferroptosis in Sertoli cells
remains unclear. Three major MAPKs, p38 MAPK, ERK1/2 and JNK, which
are involved in I/R-induced ferroptosis, were investigated. OGD/R
promoted the phosphorylation of p38, but not JNK and ERK1/2
(P<0.01; Fig. 8A). In
addition, NAC prominently depressed OGD/R-induced p38 MAPK
phosphorylation (P<0.01; Fig.
8B). Cells were treated with the p38 activation inhibitor
SB203580, JNK phosphorylation inhibitor SP600125 or ERK1/2 upstream
activator inhibitor SCH772984 for 1 h and subsequently examined.
OGD/R-induced cell death was blocked by SB203580 (P<0.01;
Fig. 8C), indicating that p38
MAPK may be associated with OGD/R-induced ferroptosis in Sertoli
cells. By contrast, SP600125 and SCH772984 had no effect on
OGD/R-induced cell death (Fig.
8C). The OGD/R-induced p38 phosphorylation was notably
inhibited by SB203580 (P<0.01; Fig. 8B). Furthermore, knocking down a
major isoform of p38 using p38α siRNA reversed OGD/R-induced
ferroptosis in Sertoli cells (P<0.01; Fig. 8C). Collectively, these results
suggest that p38 MAPK participates in OGD/R-induced ferroptotic
cell death.
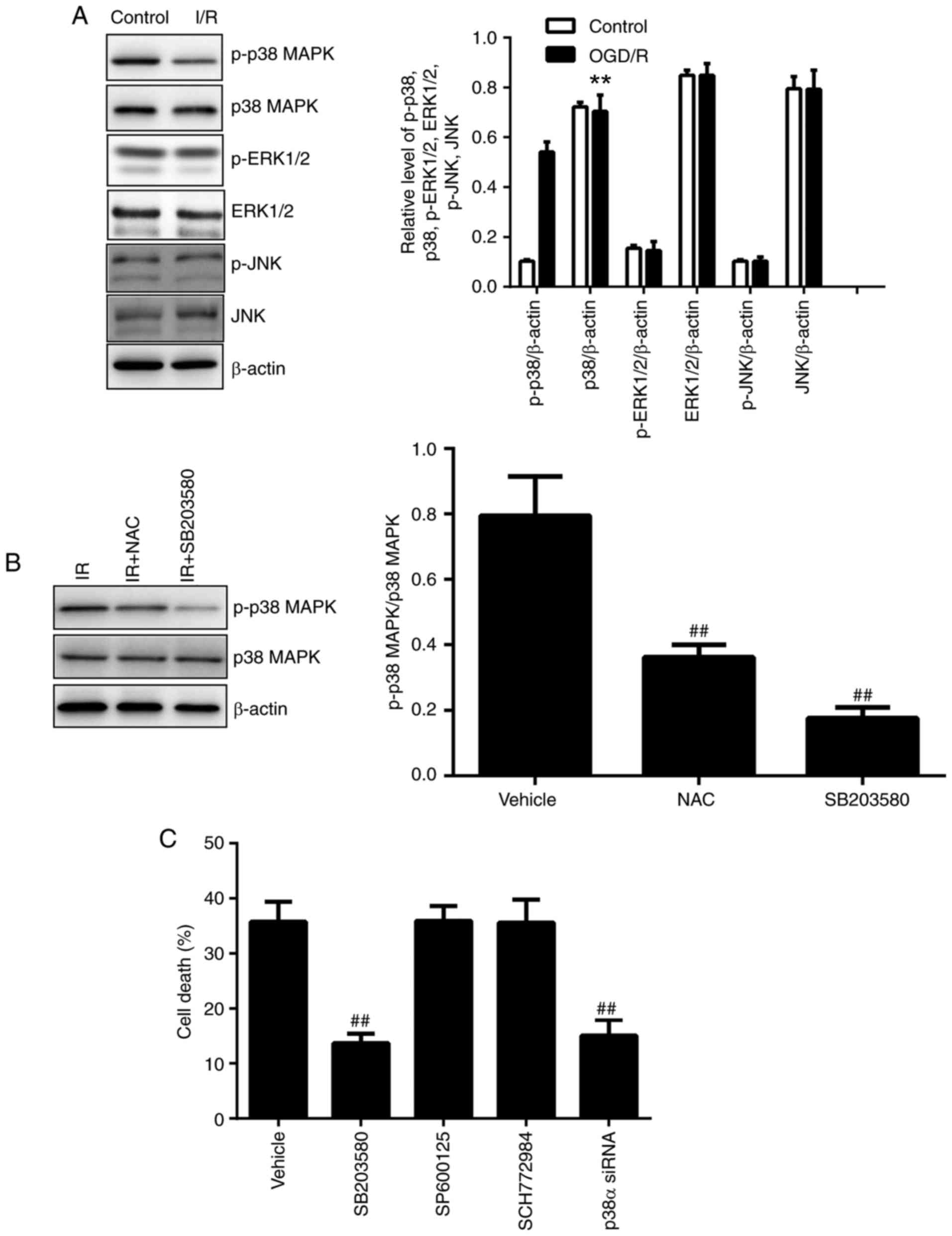 | Figure 8Effect of p38 MAPK on OGD/R-induced
ferroptosis in TM4 cells. (A) p38 MAPK, p-p38 MAPK, ERK1/2,
p-ERK1/2, JNK and p-JNK were analyzed using western blotting. (B)
Comparative levels of p-p38 MAPK/p38 MAPK were detected following
treatment with vehicle, NAC or SB203580 using western blotting. (C)
Cell death was assessed following treatment with vehicle, SB203580,
SP600125, SCH772984 or p38α siRNA administration. n=6.
**P<0.01 vs. control and ##P<0.01 vs.
vehicle MAPK, mitogen-activated protein kinase; OGD/R,
oxygen-glucose deprivation/reoxygenation; p, phosphorylated; ERK,
extracellular-regulated kinase; JNK, c-Jun N-terminal kinase; si,
small interfering. |
Discussion
The loss of spermatogenic cells contributes to the
poor therapeutic effect of acute testicular I/R injury,
highlighting the medical need for new clinical treatment strategies
to prevent spermatogenesis arrest. In I/R injury, testicular
tissues, including germ cells and Sertoli cells, exhibit high
levels of lipid ROS and antioxidant adaptation mechanisms, which
renders these tissues susceptible to anti-lipid
peroxidation-targeting treatments (2,44,45). In the present study, the type of
cell death induced by OGD/R injury and the potential regulating
mechanisms of cell death in TM4 cells was assessed using a Sertoli
cell line in vitro. The results provide evidence that lipid
peroxidation production induced by OGD/R stress leads to the death
of Sertoli cells. The main form of cell death following testicular
I/R injury was demonstrated to be ferroptosis, resulting from iron
accumulation and GSH depletion. Overexpression of Fpn, activation
of GPX4 and inactivation of p38MAPK blocked OGD/R-induced
ferroptosis in TM4 cells.
Oxidative stress and lipid peroxidation are
considered to be fatal contributors to the pathophysiological
process of testicular I/R injury (2). In the testes, lipid peroxidation
results in oxidative damage and cytotoxicity to germ cells and
Sertoli cells, inducing massive apoptosis-mediated germ cell death
(2,19,29,46–48). Considering the yet-uncharacterized
role of I/R injury-induced lipid peroxidation in Sertoli cell
death, the modality of TM4 cell death was investigated. The results
confirmed that the pathological process of OGD/R-induced cell
injury is similar to that of clinical tissue damage resulting from
organic I/R injury; cellular I/R injury is also mediated by lipid
ROS generation (49–51). It has previously been demonstrated
that OGD/R increases lipid peroxidation, intracellular iron levels
and cell death in human neuroblastoma cell lines and that cell
death is not inhibited by inhibitors of apoptosis, necrosis or
autophagy (52). In the present
study, it was observed that OGD/R promotes cell death, lipid ROS
generation and iron accumulation in TM4 cells without activating
caspase 3. These findings are consistent with those previously
reported (52). Iron has been
demonstrated to mediate lipid peroxidation-induced ferroptotic cell
death by supplying lipid ROS, which may be reduced by antioxidants
and ferroptosis inhibitors (4).
In the present study, it was observed that the antioxidant NAC, a
ROS scavenger, reduced lipid ROS levels via inhibiting lipid
peroxidation to prevent OGD/R-induced ferroptosis. The effect of
ferroptosis has been estimated in various diseases using the
small-molecule ferroptosis inhibitor, Fer-1, which acts as a lipid
ROS scavenger and suppresses ferroptosis but not apoptosis,
necrosis or autophagy (4,23,25,53). In the present study, treatment
with Fer-1 significantly depressed OGD/R-induced cell death and
lipid ROS generation, indicating that lipid peroxidation may be
associated with OGD/R-induced cell death in TM4 cells. Testicular
OGD/R injury shares some characteristics with classical I/R-induced
damage, including apoptosis-mediated germ cell death and lipid ROS
generation, concomitant with persisting Sertoli cell populations
and Leydig cell functions (54,55). The results of the present study
indicate that OGD/R injury induces ferroptosis in TM4 cells and
that lipid ROS generation contributes to the pathogenesis of I/R
injury. In vitro studies have demonstrated that
liproxstatin-1, a specific inhibitor of ferroptosis that prevents
cell death-induced ferroptosis revulsants, alleviates acute renal
failure induced by Gpx4 depletion by restraining ferroptotic cell
death, whilst also increasing the survival of hepatocytes by
blocking lipid peroxidation (6).
In the present study, it was confirmed that liproxstatin-1
ameliorates ferroptotic cell death and OGD/R-induced lipid ROS
generation in TM4 cells. These results suggest that Sertoli cells
are susceptible to OGD/R-induced ferroptosis, confirming the role
of lipid peroxidation in and highlighting the effect of ferroptosis
inhibitors and antioxidants in improving testicular I/R-induced
cell loss.
Previous studies have provided evidence that
ferroptosis serves a major role in I/R-induced tissue and cell
damage, and that targeting ferroptotic pathways may improve
protection against I/R injury (4–9,21).
Ferroptosis is a non-apoptotic form of regulated cell death
triggered by inactivation of the lipid repair enzyme GPX4 and
subsequent lipid peroxidation (4,37,39,40). Lipid peroxidation has routinely
been described as a key factor that may lead to spermatogenesis
disturbance by damaging Sertoli cells in testicular I/R injury
(16,56). Iron-dependent lipid ROS-mediated
I/R-induced renal cell ferroptosis and ROS generation-mediated
OGD/R-induced Sertoli cell dysfunction have also been reported
(4,16,17,22–24,28,57). The essential role of the GSH/GPX4
axis in impeding lipid peroxidation-induced acute renal impairment
and associated ferroptotic cell death has been described (6). In the present study, similar
findings were obtained in the mouse testicular Sertoli cell line,
TM4. Reduced GSH serves a role in antioxidant defense, and the
GSH-GPX4 interaction is critical for the regulation of redox
(22). Depletion of GSH, a basal
cofactor of GPXs, impairs GPX4 function, leading to cellular redox
state changes and ferroptosis (4,22).
Previous studies have reported that inactivation of GPX4 or
degradation of GPX4 induces ferroptosis in vitro (58,59). Knocking out GPX4 in mice results
in tissue damage that is associated with the induction of
ferroptosis (6). GPX4 has also
been demonstrated to protect cells from apoptosis (37,38) and necroptosis (39), suggesting that it serves a
protective role in programmed cell death. In the present study,
OGD/R-mediated ferroptosis resistance was achieved via upregulation
of the GSH-GPX4 pathway. Similarly, knockdown of GPX4-induced
ferroptosis and lipid ROS generation, as well as treatment with GSH
or NAC, significantly inhibited cell death in TM4 cells. These
results suggest that by reducing lipid ROS levels, GPX4 prevents
cell death by actively depressing ferroptosis in TM4 cells
Ferroptosis is characterized by the accumulation of
lipid ROS derived from iron overload and may be prevented using an
iron chelator; this indicates that iron-mediated lipid ROS
generation regulates ferroptosis in addition to GSH/GPX4-dependent
lipid peroxidation (4,6,59)
Excess iron increases lipid ROS through the Fenton reaction,
thereby leading to cell death (4,59).
Previous studies have verified that, in germ cells, the lethal
lipid peroxidation reaction is accelerated by intracellular iron,
whereas other divalent cationic metals have no effect (60–62). In the present study, it was
observed that I/R stress induced Sertoli cell ferroptosis via the
accumulation of iron and lipid peroxidation products. This is
consistent with a previous study in which it was demonstrated that
iron overload injured Sertoli cells in mice (34,63). Iron metabolism signaling
associated with the absorption, transport, storage, utilization and
export of iron is essential for ferroptosis (4,6).
In the present study, OGD/R injury-induced cell death was depressed
by the iron chelator DFO or genetic inhibition of cellular iron
exclusion, suggesting that intracellular iron overload is a key
factor leading to ferroptosis. The role of Sertoli cells in iron
acquisition of developing spermatocytes has also been documented
(64). In testicular seminiferous
tubules, iron circulation through Fpn is reduced in Sertoli cells
during iron overload and developing germ cells are protected from
iron excess (64–66). Recent studies have reported that
Tf is essential for ferroptosis and may be vital in I/R-induced
heart injury (5). However, no
alterations in the expression of Tf and ferritin were observed in
the present study. The discrepancy between these findings may
reflect differences in the unique iron metabolism process in
Sertoli cells. These results suggest that iron regulation through
Fpn is altered in TM4 cells and may be a target for testicular I/R
injury treatment.
The molecular mechanism underlying OGD/R-induced
ferroptosis in Sertoli cell has not yet been explored. The MAPK
signaling pathway has been implicated in OGD/R-induced ferroptosis,
in addition to iron metabolism and lipid peroxidation (4,6,59).
Previous studies have demonstrated that lipid ROS activates the p38
MAPK and JNK pathways and subsequently triggers ferroptosis
(4,58). In the present study, it was
confirmed that OGD/R injury activates p38 MAPK, but not ERK1/2 and
JNK in TM4 cells. A p38 MAPK inhibitor (SB203580) was also
demonstrated to prevent OGD/R-induced cell ferroptosis, similar to
the antioxidant NAC, confirming the effect of p38 MAPK in
ferroptosis. In addition, siRNA experiments against p38 MAPK were
performed to further confirm the role of p38 MAPK in OGD/R-induced
ferroptosis in TM4 cells. Transfection with p38 siRNA reduced
OGD/R-induced cell death. Furthermore, OGD/R-induced activation of
p38 MAPK was depressed by NAC. Collectively, these results suggest
that activation of the p38 MAPK signaling pathway contributes to
ferroptosis in TM4 cells.
The result s of the present study demonstrate that
depressing lipid ROS generation, overexpressing Fpn, activating
GPX4, or inactivating p38 MAPK may prevent OGD/R-induced
ferroptosis in Sertoli cells. Accordingly, these results may be
used as a clinical basis for the development of new efficacious
treatments for testicular I/R-induced cell loss and male
infertility.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant no. 81373787) and the Hebei
Provincial Natural Science Fund (grant no. H2013201139).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Dokmeci D: Testicular torsion, oxidative
stress and the role of antioxidant therapy. Folia Med (Plovdiv).
48:16–21. 2006.
|
|
2
|
Shimizu S, Tsounapi P, Dimitriadis F,
Higashi Y, Shimizu T and Saito M: Testicular torsion-detorsion and
potential therapeutic treatments: A possible role for ischemic
postconditioning. Int J Urol. 23:454–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Turner TT, Bang HJ and Lysiak JL: The
molecular pathology of experimental testicular torsion suggests
adjunct therapy to surgical repair. J Urol. 172:2574–2578. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Skouta R, Dixon SJ, Wang J, Dunn DE, Orman
M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A and
Stockwell BR: Ferrostatins inhibit oxidative lipid damage and cell
death in diverse disease models. J Am Chem Soc. 136:4551–4556.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Linkermann A, Skouta R, Himmerkus N, Mulay
SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz
PS, et al: Synchronized renal tubular cell death involves
ferroptosis. Proc Natl Acad Sci USA. 111:16836–16841. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lorincz T, Jemnitz K, Kardon T, Mandl J
and Szarka A: Ferroptosis is involved in acetaminophen induced cell
death. Pathol Oncol Res. 21:1115–1121. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Uyeturk U, Terzi EH, Gucuk A, Kemahli E,
Ozturk H and Tosun M: Prevention of torsion-induced testicular
injury by Rhodiola rosea. Urology. 82:254.e1–6. 2013. View Article : Google Scholar
|
|
11
|
Chen SR and Liu YX: Regulation of
spermatogonial stem cell self-renewal and spermatocyte meiosis by
Sertoli cell signaling. Reproduction. 149:R159–R167. 2015.
View Article : Google Scholar
|
|
12
|
Cheng CY and Mruk DD: The biology of
spermatogenesis: The past, present and future. Philos Trans R Soc
Lond B Biol Sci. 365:1459–1463. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Griswold MD: The central role of Sertoli
cells in spermatogenesis. Semin Cell Dev Biol. 9:411–416. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mruk DD and Cheng CY: Sertoli-Sertoli and
Sertoli-germ cell interactions and their significance in germ cell
movement in the seminiferous epithelium during spermatogenesis.
Endocr Rev. 25:747–806. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tanwar PS, Kaneko-Tarui T, Zhang L, Rani
P, Taketo MM and Teixeira J: Constitutive WNT/beta-catenin
signaling in murine Sertoli cells disrupts their differentiation
and ability to support spermatogenesis. Biol Reprod. 82:422–432.
2010. View Article : Google Scholar :
|
|
16
|
Dokmeci D, Kanter M, Inan M, Aydogdu N,
Basaran UN, Yalcin O and Turan FN: Protective effects of ibuprofen
on testicular torsion/detorsion-induced ischemia/reperfusion injury
in rats. Arch Toxicol. 81:655–663. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moon C, Kim JS, Jang H, Lee HJ, Kim SH,
Kang SS, Bae CS, Kim JC, Kim S, Lee Y and Shin T: Activation of
Akt/protein kinase B and extracellular signal-regulated kinase in
rats with acute experimental testicular torsion. J Vet Med Sci.
70:337–341. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kanter M: Protective effects of Ginkgo
biloba (EGb 761) on testicular torsion/detorsion-induced
ischemia-reperfusion injury in rats. Exp Mol Pathol. 91:708–713.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim HJ, Lee JW, Hwang BR, Lee YA, Kim JI,
Cho YJ, Jhun HJ and Han JS: Protective effect of pterostilbene on
testicular ischemia/reperfusion injury in rats. J Pediatr Surg.
51:1192–1196. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Conrad M and Sato H: The oxidative
stress-inducible cystine/glutamate antiporter, system x (c) (−):
Cystine supplier and beyond. Amino Acids. 42:231–246. 2012.
View Article : Google Scholar
|
|
21
|
Garg JP and Vucic D: Targeting cell death
pathways for therapeutic intervention in kidney diseases. Semin
Nephrol. 36:153–161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang WS and Stockwell BR: Ferroptosis:
Death by Lipid Peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar :
|
|
23
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hofmans S, Vanden Berghe T, Devisscher L,
Hassannia B, Lyssens S, Joossens J, Van Der Veken P, Vandenabeele P
and Augustyns K: Novel ferroptosis inhibitors with improved potency
and ADME properties. J Med Chem. 59:2041–2053. 2016. View Article : Google Scholar
|
|
25
|
Cao JY and Dixon SJ: Mechanisms of
ferroptosis. Cell Mol Life Sci. 73:2195–2209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Galluzzi L, Bravo-San Pedro JM, Kepp O and
Kroemer G: Regulated cell death and adaptive stress responses. Cell
Mol Life Sci. 73:2405–2410. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li W, Wu ZQ, Zhao J, Guo SJ, Li Z, Feng X,
Ma L, Zhang JS, Liu XP and Zhang YQ: Transient protection from
heat-stress induced apoptotic stimulation by metastasis-associated
protein 1 in pachytene spermatocytes. PLoS One. 6:e260132011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang S, Zeng Y, Qu J, Luo Y, Wang X and
Li W: Endogenous EGF maintains Sertoli germ cell anchoring junction
integrity and is required for early recovery from acute testicular
ischemia/reperfusion injury. Reproduction. 145:177–189. 2013.
View Article : Google Scholar
|
|
29
|
Erol B, Bozlu M, Hanci V, Tokgoz H, Bektas
S and Mungan G: Coenzyme Q10 treatment reduces lipid peroxidation,
inducible and endothelial nitric oxide synthases, and germ
cell-specific apoptosis in a rat model of testicular
ischemia/reperfusion injury. Fertil Steril. 93:280–282. 2010.
View Article : Google Scholar
|
|
30
|
Hotchkiss RS, Strasser A, McDunn JE and
Swanson PE: Cell death. N Engl J Med. 361:1570–1583. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Linkermann A: Nonapoptotic cell death in
acute kidney injury and transplantation. Kidney Int. 89:46–57.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Carlson BA, Tobe R, Yefremova E, Tsuji PA,
Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL and Conrad M:
Glutathione peroxidase 4 and vitamin E cooperatively prevent
hepatocellular degeneration. Redox Biol. 9:22–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar
|
|
34
|
Li L, Yu Z, Miao G, Hui-juan W and Fu-lu
G: Iron overload injures Sertoli cells of mouse. Basic Clin Med.
36:321–326. 2016.
|
|
35
|
Bogdan AR, Miyazawa M, Hashimoto K and
Tsuji Y: Regulators of Iron Homeostasis: New players in metabolism,
cell death, and disease. Trends Biochem Sci. 41:274–286. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Y, Zhang S, Wang X, Guo W, Wang L,
Zhang D, Yuan L, Zhang Z, Xu Y and Liu S: Disordered signaling
governing ferroportin transcription favors breast cancer growth.
Cell Signal. 27:168–176. 2015. View Article : Google Scholar
|
|
37
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar :
|
|
38
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dächert J, Schoeneberger H, Rohde K and
Fulda S: RSL3 and Erastin differentially regulate redox signaling
to promote Smac mimetic-induced cell death. Oncotarget.
7:63779–63792. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Conrad M and Friedmann Angeli JP:
Glutathione peroxidase 4 (Gpx4) and ferroptosis: What's so special
about it? Mol Cell Oncol. 2:e9950472015. View Article : Google Scholar
|
|
41
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schneider M, Förster H, Boersma A, Seiler
A, Wehnes H, Sinowatz F, Neumüller C, Deutsch MJ, Walch A, Hrabe de
Angelis M, et al: Mitochondrial glutathione peroxidase 4 disruption
causes male infertility. FASEB J. 23:3233–3242. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Minutoli L, Antonuccio P, Polito F, Bitto
A, Fiumara T, Squadrito F, Nicotina PA, Arena S, Marini H, Romeo C
and Altavilla D: Involvement of mitogen-activated protein kinases
(MAPKs) during testicular ischemia-reperfusion injury in nuclear
factor-kappaB knock-out mice. Life Sci. 81:413–422. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Antonuccio P, Minutoli L, Romeo C,
Nicòtina PA, Bitto A, Arena S, Altavilla D, Zuccarello B, Polito F
and Squadrito F: Lipid peroxidation activates mitogen-activated
protein kinases in testicular ischemia-reperfusion injury. J Urol.
176:1666–1672. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yagmurdur H, Ayyildiz A, Karaguzel E, Ogus
E, Surer H, Caydere M, Nuhoglu B and Germiyanoglu C: The preventive
effects of thiopental and propofol on testicular
ischemia-reperfusion injury. Acta Anaesthesiol Scand. 50:1238–1243.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee JW, Kim JI, Lee YA, Lee DH, Song CS,
Cho YJ and Han JS: Inhaled hydrogen gas therapy for prevention of
testicular ischemia/reperfusion injury in rats. J Pediatr Surg.
47:736–742. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Al-Maghrebi M, Kehinde EO and Anim JT:
Long term testicular ischemia-reperfusion injury-induced apoptosis:
Involvement of survivin down-regulation. Biochem Biophys Res
Commun. 395:342–347. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ergur BU, Kiray M, Pekcetin C, Bagriyanik
HA and Erbil G: Protective effect of erythropoietin pretreatment in
testicular ischemia-reperfusion injury in rats. J Pediatr Surg.
43:722–728. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hong S, Kwon J, Kim DW, Lee HJ, Lee D and
Mar W: Mulberrofuran G protects ischemic injury-induced cell death
via inhibition of NOX4-mediated ROS generation and ER stress.
Phytother Res. 31:321–329. 2017. View Article : Google Scholar
|
|
50
|
Xu Q, Deng F, Xing Z, Wu Z, Cen B, Xu S,
Zhao Z, Nepomuceno R, Bhuiyan MI, Sun D, et al: Long non-coding RNA
C2dat1 regulates CaMKIIdelta expression to promote neuronal
survival through the NF-kappaB signaling pathway following cerebral
ischemia. Cell Death Dis. 7:e21732016. View Article : Google Scholar
|
|
51
|
Park H, Seol GH, Ryu S and Choi IY:
Neuroprotective effects of (-)-linalool against oxygen-glucose
deprivation-induced neuronal injury. Arch Pharm Res. 39:555–564.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang YT, Li FM, Guo YZ, Jiang LR, Ma J,
Ke Y and Qian ZM: (Z)-ligustilide increases ferroportin1 expression
and ferritin content in ischemic SH-SY5Y cells. Eur J Pharmacol.
792:48–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Martin-Sanchez D, Ruiz-Andres O, Poveda J,
Carrasco S, Cannata-Ortiz P, Sanchez-Niño MD, Ruiz Ortega M, Egido
J, Linkermann A, Ortiz A and Sanz AB: Ferroptosis, but Not
Necroptosis, is important in nephrotoxic folic acid-induced AKI. J
Am Soc Nephrol. 28:218–229. 2017. View Article : Google Scholar
|
|
54
|
Turner RM: Moving to the beat: A review of
mammalian sperm motility regulation. Reprod Fertil Dev. 18:25–38.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Namazi H: Decreasing the expression of
LFA-1 and ICAM-1 as the major mechanism for the protective effect
of hyperbaric oxygen on ischemia-reperfusion injury. Microsurgery.
28:3002008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yao C, Li G, Qian Y, Cai M, Yin H, Xiao L,
Tang W, Guo F and Shi B: Protection of pentoxifylline against
testis injury induced by intermittent hypobaric hypoxia. Oxid Med
Cell Longev. 2016:34068022016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dokucu AI, Ozturk H, Ozturk H, Tuncer MC
and Yilmaz F: The effects of molsidomine on hypoxia inducible
factor alpha and Sonic hedgehog in testicular ischemia/reperfusion
injury in rats. Int Urol Nephrol. 41:101–108. 2009. View Article : Google Scholar
|
|
58
|
Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze
MT, Zeh HJ, Kang R and Tang D: The ferroptosis inducer erastin
enhances sensitivity of acute myeloid leukemia cells to
chemotherapeutic agents. Mol Cell Oncol. 2:e10545492015. View Article : Google Scholar
|
|
59
|
Shimada K, Skouta R, Kaplan A, Yang WS,
Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ and
Stockwell BR: Global survey of cell death mechanisms reveals
metabolic regulation of ferroptosis. Nat Chem Biol. 12:497–503.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Barber AA: Lipid peroxidation in rat
tissue homogenates: Interaction of iron and ascorbic acid as the
normal catalytic mechanism. Lipids. 1:146–151. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
El-Seweidy MM, Hashem RM, Abo-El-matty DM
and Mohamed RH: Frequent inadequate supply of micronutrients in
fast food induces oxidative stress and inflammation in testicular
tissues of weanling rats. J Pharm Pharmacol. 60:1237–1242. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sundarraj K, Manickam V, Raghunath A,
Periyasamy M, Viswanathan MP and Perumal E: Repeated exposure to
iron oxide nanoparticles causes testicular toxicity in mice.
Environ Toxicol. 32:594–608. 2017. View Article : Google Scholar
|
|
63
|
Doreswamy K and Muralidhara: Genotoxic
consequences associated with oxidative damage in testis of mice
subjected to iron intoxication. Toxicology. 206:169–178. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Leichtmann-Bardoogo Y, Cohen LA, Weiss A,
Marohn B, Schubert S, Meinhardt A and Meyron-Holtz EG:
Compartmentalization and regulation of iron metabolism proteins
protect male germ cells from iron overload. Am J Physiol Endocrinol
Metab. 302:E1519–1530. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wauben-Penris PJ, Veldscholte J, van der
Ende A and van der Donk HA: The release of iron by Sertoli cells in
culture. Biol Reprod. 38:1105–1113. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sylvester SR and Griswold MD: The
testicular iron shuttle: A 'nurse' function of the Sertoli cells. J
Androl. 15:381–385. 1994.PubMed/NCBI
|