Introduction
Acute lung injury (ALI), and its more severe
syndrome, acute respiratory distress syndrome (ARDS), is pulmonary
inflammation with devastating disorders, leading to diffuse
alveolar damage, which causes hypoxemia, pulmonary edema as well as
respiratory failure (1,2). The pediatric ALI incidence is high
in developed countries (3). ALI
is related to pediatric intensive care unit admissions, which
causes an increasing of pediatric intensive care unit deaths
(4). Previous studies for
children and adults exhibit strong relationship between positive
fluid balance and the worse outcomes, such as death, in patients
with respiratory failure and/or ALI (5,6).
In addition, a previous study indicated that the percentage of
fluid overload has a close relationship with worse oxygenation,
enhanced duration of mechanical ventilation, as well as increasing
of hospital length stay (7). In
children with ALI accordingly, only some of them were successfully
being treated to prevent fluid overload (8,9).
Thus, finding effective method and exploring the molecular
mechanism are necessary.
Inflammatory response is the common pathological
process, contributing to many diseases, which are regulated by
nuclear factor-κB (NF-κB) signaling pathway (10). As previously reported, TLR family
plays an important role in pathogen recognition and activation of
innate immunity (11). Once
activated by stimuli, TLRs family performs corresponding response
by stimulating a cascade of various distinct events. The TLRs in a
variety of formations exhibit different patterns for expression.
Accumulating evidence has showed that Toll-like receptor 4 (TLR4),
stimulated by lipopolysaccharide (LPS), can induce acute lung
injury by activating TLR4/NF-κB, associated with inflammation
response (12–14). Additionally, reactive oxygen
species (ROS) are linked to many diseases, such as diabetes, heart
injury, liver injury and renal dysfunction. The species of ROS
involve molecular oxygen, such as superoxide
(O2−) and hydrogen peroxide
(H2O2). Excessive O2−
and H2O2 produce signaling responses,
disrupting cellular processes and leading to tissue injury
(15).
Retinoids regulate vital biological processes,
including cellular proliferation, inflammation, apoptosis, and
differentiation (16–18). However, bioavailable and less
toxic synthetic retinoids, such as the atypical adamantyl retinoid
ST1926, have been developed and tested in clinical trials (19). For example, ST1926 showed
potential efficacy on solid tumors and hematological malignancies
with minimal side effects or toxicity (20). Further, oral ST1926 is
pharmacokinetically stable, bioavailable and pharmacologically
attainable in the plasma of patients (21,22). Although this compound has been
investigated in phase 1 clinical trials, it is still little known
in acute lung injury treatment, especially from inflammation and
ROS suppression. We explored the efficacy of ST1926 in acute lung
injury models in vivo and in vitro to investigate its
underlying pharmacological mechanisms, which may be of potential
value for acute lung injury treatment in children.
Materials and methods
Animal treatment
Sixty male, 6-week-old C57BL6 mice, weighing 20–22 g
were purchased from experimental animal center of Nanjing Medical
University of laboratory animal center. All the mice were carefully
maintained at room temperature on a 12:12 h light:dark cycle, with
free access to chow and water in the cages. This study was approved
by the Ethics Committee on Animal Research at the Department of
Pediatrics, Huai'an First People's Hospital, Nanjing Medical
University, Nanjing, China. The mice were divided into 4 groups:
the control group without LPS and ST1926 administration (Con,
n=15); LPS-induced group (15 mg/kg, n=15); 10 mg/kg ST1926-treated
group after LPS treatment (ST1926/L, n=15); 20 mg/kg ST1926-treated
group after LPS (ST1926/H, n=15) (23). After 7 days adaptation, mice from
LPS group were treated by intraperitoneal injection with 15 mg/kg
body weight LPS for 6 h. The control group was also administered
with the same volume of Hanks' buffer. Then, the mice were
administered with different concentrations of ST1926 via oral
gavage for 2 weeks. The suspension was mixed in high concentration
for use immediately after it was ready within 1 min through oral
gavage. ST1926, purchased from Sigma-Tau and Biogem (Ariano Irpino,
Italy) was dissolved in distilled water. Then, all the mice were
sacrificed. Eyeball blood was collected for the next investigation,
and the whole lung tissues were carefully harvested on 4°C glacial
table.
Cell culture
Mouse lung epithelial cells, MLE-12, and human
bronchial epithelial cells (NHBE) were obtained from Shanghai
Haoran Biological Technology Co., Ltd. (Shanghai, China) and both
cultured in Dulbecco's modified Eagle's medium (DMEM)/F12
containing 1% penicillin/streptomycin and 10% fetal bovine serum
(FBS). All cells were cultured in a humidified atmosphere with 5%
CO2 and 95% humidity at 37°C in an incubator. Cells were
treated with 100 ng/ml LPS with or without different concentrations
(4 µM, ST1926/L; 8 µM, ST1926/H) of ST1926 for 24
h.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT) assays
Lung epithelial cells (5×103) were
planted into a 96-well plate (Corning Inc., Corning, NY, USA) per
well. ST1926, from 0–8 µM, was added to the medium for 24 h.
The cells were then incubated at 37°C in an incubator, and the cell
viability was detected by the colorimetric MTT assay at 570 nm
following the manufacturer's instructions (KeyGen Biotech, Nanjing,
China).
Biochemical analysis
Levels of superoxide dismutase (SOD), Catalase (CAT)
and malondialdehyde (MDA) in serum and lung tissue samples were
determined by commercially available kits (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) according to the
manufacturer's instructions.
The serum tumor necrosis factor-α (TNF-α, MTA00B),
interleukin-1β (IL-1β, MLB00C) (both from R&D Systems,
Minneapolis, MN, USA), IL-8 (BYL02382; Beinglay, Wuhan, China),
IL-18 (DY122-05), IL-5 (M5000) (both from R&D Systems), IL-6
(BMS603; Bender MedSystems, Vienna, Austria), IL-12 (PK-EL-62216M;
Promocell, Heidelberg, Germany), IL-10 (M1000B; R&D Systems),
and IL-17 (BMS6001; Bender MedSystems) were measured according to
the manufacturer's protocol using an enzyme-linked immunosorbent
assay (ELISA) kit. For the determination of cytokine levels, blood
samples from the rats were obtained after sacrifice and stored at
−80°C until use.
H2O2 levels in lung tissue
were detected using a hydrogen peroxide assay kit (catalase assay
kit, S0051; Beyotime Institute of Biotechnology, Jiangsu, China)
following the manufacturer's instructions.
O2− of lung tissue samples was measured by
lucigenin chemiluminescence method. Briefly, lung tissue of mice
under different conditions were weighed and homogenized in a
homogenization buffer using Hepes and EDTA. After centrifugation,
an aliquot of the supernatant was incubated with 5 µM
lucigenin in Krebs-Hepes buffer. Light emission was measured with a
Tecan Infinite 200. Specificity for O2− was
evaluated by adding 350 U/ml SOD to the incubation medium. Protein
concentration was assessed with BCA Protein Quantitative analysis
kit (Thermo Fisher Scientific, Waltham, MA, USA).
Inflammatory cell counts of BALF
After treatment, eight mice in each group were
sacrificed and BALF was obtained by washing three times with 1 ml
of cold sterile phosphate-buffered saline (PBS) through a tracheal
cannula. BALF samples were centrifuged at 3,000 rpm for 10 min at
4°C, and the cell pellet was resuspended in PBS for the total cell
number assessment with a hemacytometer, and the cytospins were
prepared for other differential cell number through staining with
the method of Wright-Giemsa staining. Percentages of BALF
macrophages, neutrophils and lymphocytes were obtained by counting
leukocytes under light microscopy.
Measurement of NOx levels
Mouse lung tissue lysates were treated with cold
ethanol for 1 h at −20°C and then centrifuged at 20,000 × g to
remove proteins that can interfere with NO measurements. The
potassium iodide-acetic acid reagent was prepared fresh by
dissolving 0.05 g of potassium iodide in 7 ml of acetic acid. The
KI/AcOH mixture was added into a septum-sealed purge vessel and
bubbled with nitrogen gas. The gas stream was connected via a trap
containing 1 N NaOH to a Sievers 280i Nitric Oxide Analyzer (GE
Analytical Instruments, Boulder, CO, USA). The samples were
injected with a syringe through a silicone-Teflon septum. The data
were then analyzed via assessing the area under the curve of
chemiluminescence signal with the Liquid software (GE Analytical
Instruments). The resultant NOx value presents the total nitric
oxide and nitrite in pmols/mg protein.
Myeloperoxidase (MPO) activity
MPO activity in mouse lung tissue was determined
using MPO assay kit (BioVision, Milpitas, CA, USA) following the
manufacturer's instructions. Briefly, the MPO in the samples
catalyzes the production of NaClO from H2O2
and NaCl. Next, the NaClO reacts with exogenously added aminophenyl
fluorescein to generate fluorescein, which is assessed with a
fluorometer at 485 nm excitation and 525 nm emission. The relative
fluorescent units of each sample are converted into pmol of
fluorescein by a standard curve. The data are reported as pmol
fluorescein generated/min/mg of protein extract.
Immunohistochemical (IHC) analysis
The lung and liver tissue samples were fixed using
10% buffered formalin, imbedded in paraffin and then sliced into 4
µM thick sections. Following the hematoxylin and eosin
(H&E) staining, the pathological alterations of the lung tissue
samples were observed under a light microscope. Periodic acid-Shiff
(PAS) staining was performed using a standard protocol. The number
of PAS positive cells was quantified by inspection by a
single-blinded investigator. Individual slides were examined
independently. Lung tissue samples were IHC stained for the
analysis of the lung tissue after different treatments. The
sections were stained with TNF-α and IL-1β (Abcam, Cambridge, MA,
USA) overnight at 4°C, prior to incubation according to the
manufacturer's instructions, followed by horseradish peroxidase
conjugated anti-rabbit IgG (Dako, Glostrup, Denmark) as a secondary
antibody.
Fluorescence imaging
Lung tissue sections and cells were washed twice
with PBS and fixed with 3.7% (v/v) formaldehyde in PBS for 15 min.
Cells were permeabilised for 5 min with 0.1% Triton X-100, then
they blocked with 1% bovine serum albumin for 30 min. For TLR4 and
p-NF-κB staining in tissue and cell respectively, 50 µg/ml
mouse anti-p-NF-κB and TLR4 antibodies were employed at 4°C
overnight, followed by staining with 2 µg/ml Alexa Fluor
488-goat anti-mouse secondary antibodies at room temperature.
4′,6-Diamidino-2-phenylindole (DAPI; Sigma-Aldrich) were used.
Images were acquired by confocal laser scanning (Leica TCS SP5;
Leica, Heidelberg, Germany) by epifluorescence microscopy (Ningbo
Sunny Instruments Co., Ltd., Ningbo, China).
Real-time quantitative polymerase chain
reaction (RT-qPCR) and western blot assays
Total RNA was extracted from lung tissue samples and
cells by using TRI-reagent (Sigma-Aldrich) following the
manufacturer's instructions and treated with deoxyribonuclease I.
Then the mRNA was converted into cDNA for real-time PCR analysis.
Real-time PCR was carried out (35 cycles of 95°C for 20 sec, 54°C
for 30 sec, and 72°C for 30 sec). Fold alterations in mRNA levels
of the targeting gene relative to the endogenous cyclophilin
control were calculated. Briefly, the cycle threshold (=Ct) values
of each target gene were subtracted from the Ct values of the
housekeeping gene cyclophilin (ΔCt). Targeting gene ΔΔCt was
calculated as ΔCt of target gene minus ΔCt of control. The fold
change in mRNA expression was calculated as 2−ΔΔCt
following a previous study. The sequences used in this study are as
follows: forward IL-18, (5′-3′) TAA GGA TAC GGA CTA CGG CT and
reverse primers, (5′-3′) GTT GGT GGA GGT CTG AGT TTA; forward IL-6,
(5′-3′) ACT ACT TATC GTC GAG GTG CTA T and reverse primers, (5′-3′)
CGA GCT TGG AGT ACC ATG TTA CTT; forward TNF-α, (5′-3′) ATA GGA ACC
AGG GGC AGT T and reverse primers, (5′-3′) CTG CGT TCA GAT GAT TGA
TG; forward SOD1, (5′-3′) CTG GCC TGC TCT GCT GCT TGT and reverse
primers, (5′-3′) TGG TAG GTG CGG ACT GTG GT; forward SOD2, (5′-3′)
CCT TGC GTC ATT CTA TCC A and reverse primers, (5′-3′) GAA ACG CGC
CAG AAG GGT TA; forward TLR4, (5′-3′) CTG CTG CCT GCT TGC TGC GT
and reverse primers, (5′-3′) GTG GTG TGT GCG GTG TAG AC; forward
MyD88, (5′-3′) CAC TTC GCT GTC ATC TCC A and reverse primers,
(5′-3′) AGC ACA GAG CGT CAA GAG GT; forward CAT, (5′-3′) CTC TCT
GTG ACC GTA ACG CTG GT and reverse primers (5′-3′) TGC GTG TGG TGC
GTG ATG GAG ACC; forward haeme oxygenase-1 (HO-1), (5′-3′) CTA GCT
TCG TGC ACA GCT GCG T and reverse primers, (5′-3′) GTG ATG ACT GCG
CGT GGT GCT AGA AC; forward Nrf2, (5′-3′) CTC TCT GGA CCT TCC GTT
GCG CTG T and reverse primers, (5′-3′) GTT GTA GGT CAG TGT TCG AGG
ATC; forward TGF-β1, (5′-3′) AGG AAC TAC GCA GTG GGA T and reverse
primers, (5′-3′) CGT CTG TCA GAT GAA TGT TG; forward Foxp3, (5′-3′)
GCC TCT GGC TTG TCT GCT CTG and reverse primers (5′-3′) GGT ATG GAG
TGC GTG ATG TCT G; forward interferon γ (IFNγ), (5′-3′) GCG CTC ATC
TCC TCA TAC TAT and reverse primers, (5′-3′) GAA CCA GCC CGG AGA
CAG TTG A; forward granzyme B (GzmB), (5′-3′) CCT GAG TCT GAC TGT
CGC CAT GT and reverse primers, (5′-3′) GTG TCT GAG TAC TTG GAC
GTA; forward Tbx-21, (5′-3′) CAC GTG CTG CTA GCT GGT GCT CAT and
reverse primers, (5′-3′) GTC ATG TGC ATG ATA GAT AC; forward
glyceraldehyde 3-phosphate dehydrogenase (GAPDH), (5′-3′) CTA AGT
CGA ATG CAA ACA GTT CAG and reverse primers, (5′-3′) AAC ATA CCA
TCC ACG ACA CGC TC, which was used as the loading control.
In immunoblotting, lung tissue samples and
epithelial cells were homogenized into 10% (wt/vol) hypotonic
buffer (25 mM Tris-HCl, pH 8.0, 1 mM EDTA, 5 µg/ml
leupeptin, 1 mM Pefabloc SC, 50 µg/ml aprotinin, 5
µg/ml soybean trypsin inhibitor, 4 mM benzamidine) to yield
a homogenate. Then the final supernatants were obtained by
centrifugation at 12,000 rpm for 20 min. Protein concentration was
determined by BCA protein assay kit (Thermo Fisher Scientific) with
bovine serum albumin as a standard. Then, same amount of total
protein were clapped into 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) followed by immunoblotting using the
following antibodies: rabbit anti-p-NF-κB (1:1,000), NF-κB
(1:1,000) (both from Abcam), SOD1 (1:1,000), SOD2 (1:1,000), TLR4
(1:1,000), MyD88 (1:1,000) (all from Cell Signaling Technology),
p38 (1:1,000), p-p38 (1:1,000), extracellular receptor kinase 1/2
(ERK1/2) (1:1,000), p-ERK1/2 (1:1,000), inhibitor-κB kinase (IKKα)
(1:1,000), inhibitor-κB kinase-α (IκBα) (1:1,000), TGF-β1
(1:1,000), Foxp3 (1:1,000), IFNγ (1:1,000), CAT (1:1,000) (all from
Abcam), HO-1 (1:1,000; Cell Signaling Technology), Nrf2 (1:1,000;
Abcam), GzmB (1:1,000; Cell Signaling Technology), Tbx-21 (1:1,000;
Abcam), and GAPDH (1:500; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA). Western blot bands were observed using GE
Healthcare ECL western blotting analysis system and exposed to
Kodak X-ray film. Each protein expression level was defined as grey
value (version 1.4.2b, Mac OS X, ImageJ; National Institutes of
Health, Bethesda, MA, USA) and standardized to housekeeping genes
(GAPDH) and expressed as a fold of control.
Statistical analysis
Data are expressed as the means ± SD. The treated
tissues, cells, and corresponding controls were compared using
GraphPad PRI SM (version 6.0; GraphPad Software, La Jolla, CA,
USA). Significant differences between groups were considered at
p<0.05.
Results
ST1926 reduces inflammatory cell
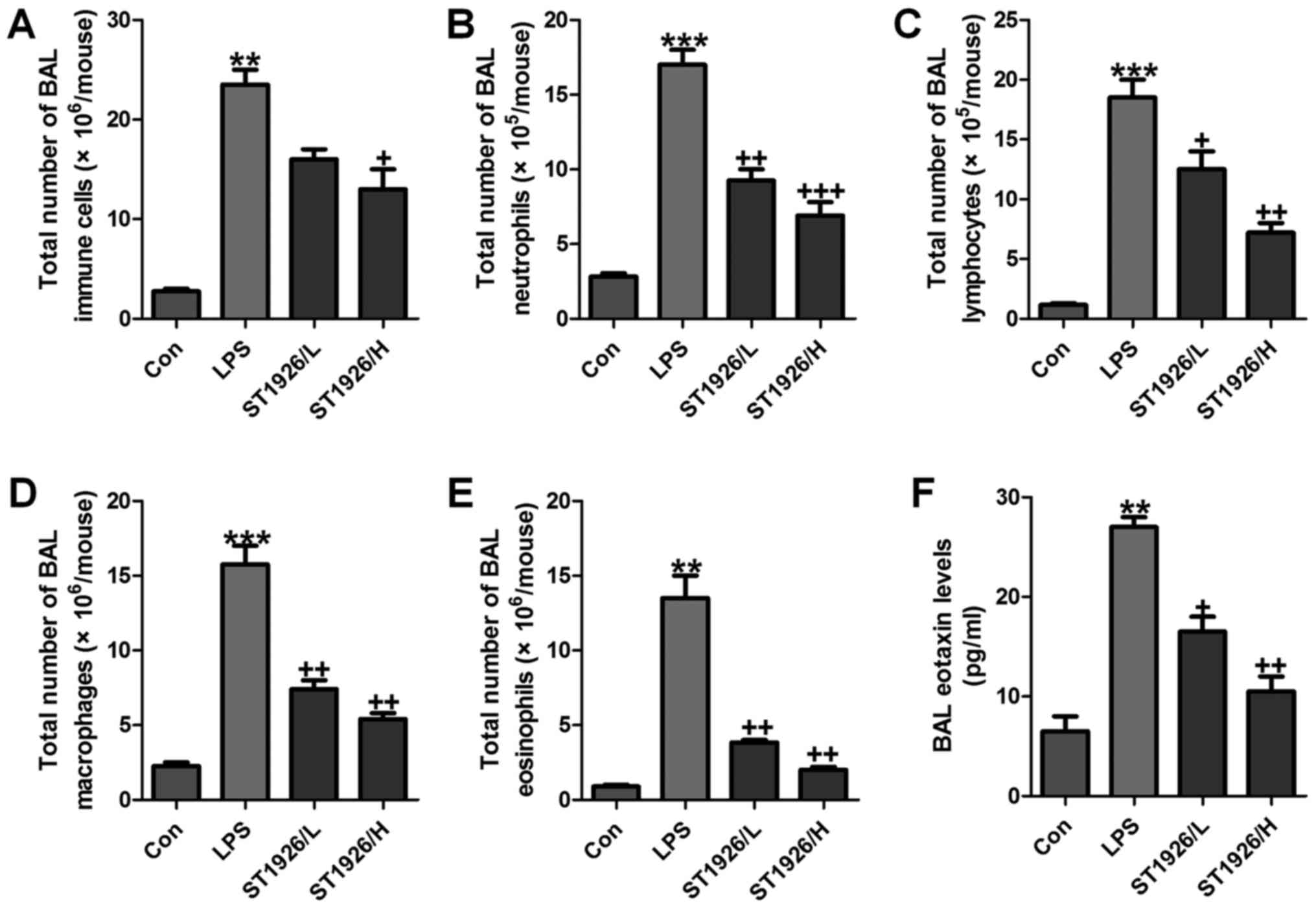
infiltrate in LPS-induced mice with acute lung injury
As an index reflecting airway inflammation, here the
role of ST1926 in inflammation regulation was investigated. First,
the bronchoalveolar lavages (BAL) was conducted and explored. As
shown in Fig. 1A, LPS treatment
resulted in an elevation of the total number of BAL, contributing
to an acceleration of neutrophils (Fig. 1B), lymphocytes (Fig. 1C), macrophages (Fig. 1D), and eosinophils (Fig. 1E). Of note, ST1926 significantly
reduced the total number of BAL, as well as neutrophils (Fig. 1B), lymphocytes (Fig. 1C), macrophages (Fig. 1D), and eosinophils (Fig. 1E), in a dose-dependent manner.
Additionally, in the lung tissue samples with LPS induction, BAL
eotaxin was apparently decreased, which was attenuated by ST1926
treatment (Fig. 1F).
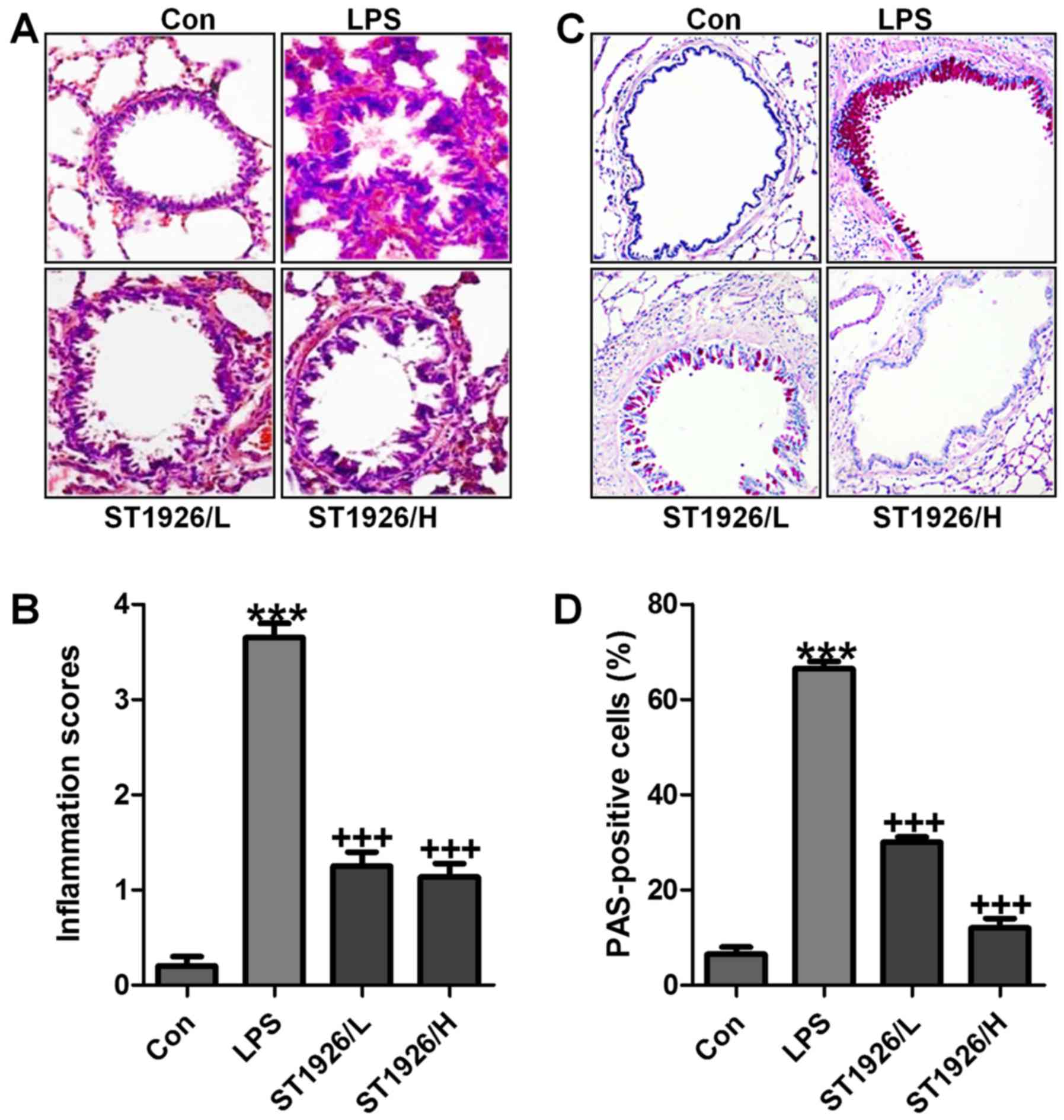
LPS-induced acute lung injury in mice is
ameliorated by ST1926 administration
LPS induction is well known to cause injury in
different organs, including the liver, renal injury and heart, even
the lung, as explored previously (24). Here, in order to assess whether
ST1926 has potential effects on airway inflammation in children,
LPS was used to induce acute lung injury. In this regard, H&E
staining was carried out to observe the injuries of lung tissue
specimens. Fig. 2A and B shows,
that after LPS treatment in mice, the lung tissue sample showed
higher inflammation score compared to the Con group, which was
comparable. After ST1926 treatment, the inflammatory response was
attenuated markedly. In addition, PAS analysis also exhibited that
the percent of PAS positive cells was notably upregulated by LPS
exposure, whereas reversed in ST1926 administration in a
dose-dependent manner (Fig. 2C and
D). The data above indicated that ST1926 exhibited protective
role against the LPS-induced acute lung injury in mice.
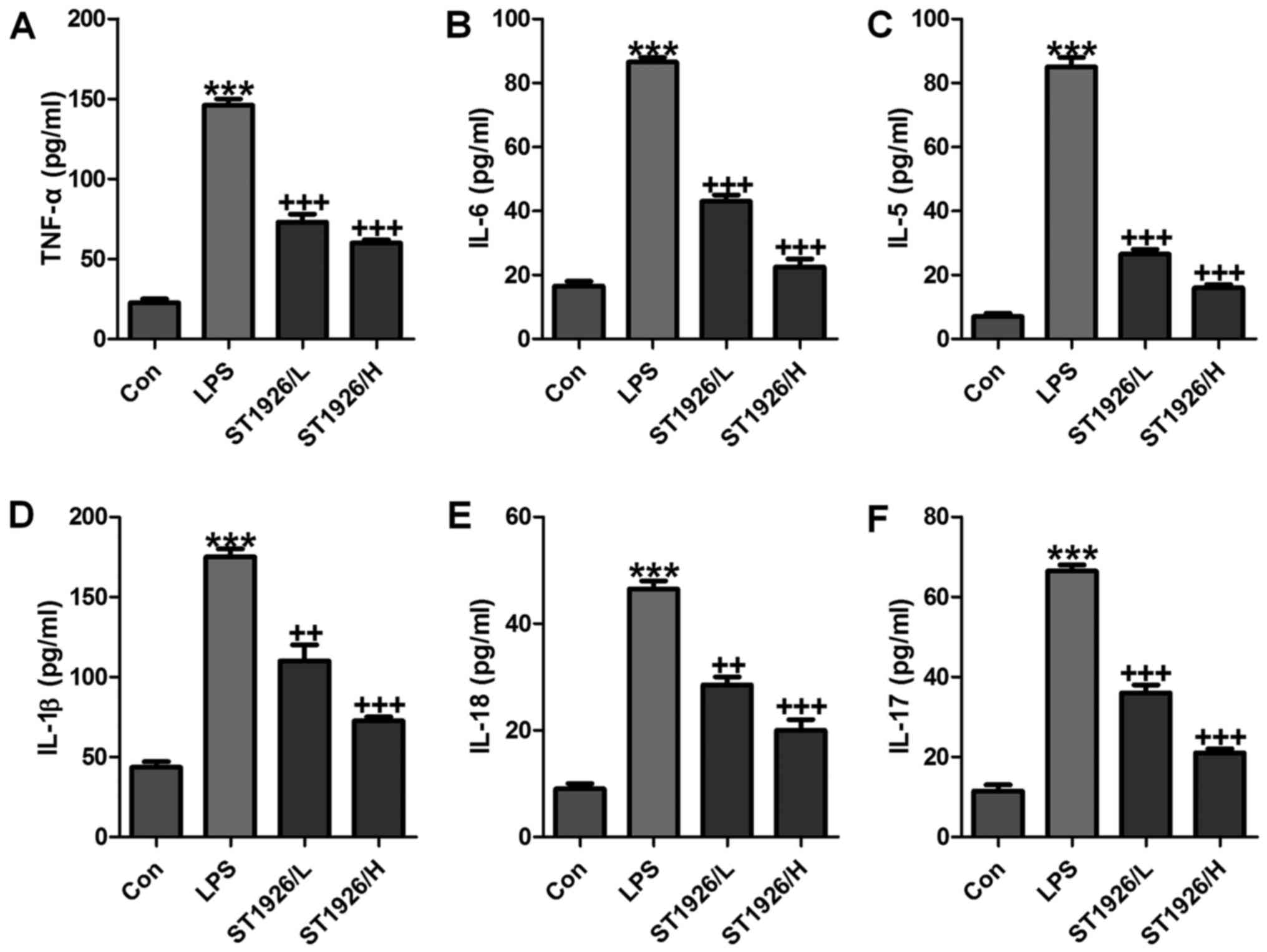
In order to further confirm that ST1926 could
attenuate LPS-caused lung injury via inflammation improvement,
pro-inflammatory cytokines level in serum were measured. As shown
in Fig. 3, we found that
pro-inflammatory cytokines of TNF-α (Fig. 3A), IL-6 (Fig. 3B), IL-5 (Fig. 3C), IL-1β (Fig. 3D), IL-18 (Fig. 3E) and IL-17 (Fig. 3F) in serum were considerably
stimulated in mice with LPS exposure. In agreement with the results
as mentioned above, ST1926 displayed suppressive role in the
inflammatory cytokine secretion, suggesting that ST1926, at least
partly, improved LPS-induced injury from inflammation
inhibition.
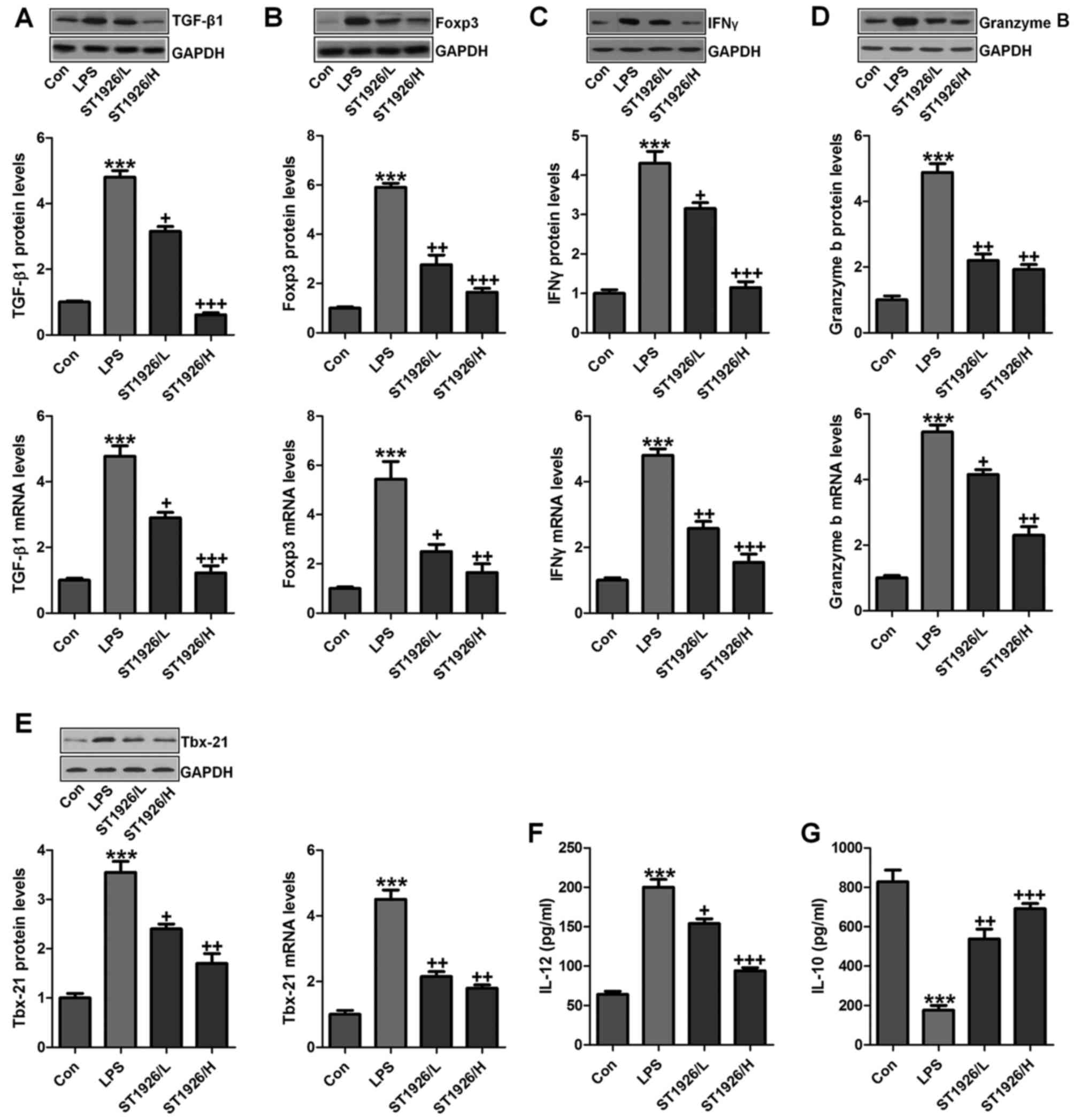
TGF-β1 is one of the most studied cytokines that
play an essential role in induction and development of acute
injury, contributing to the inflammatory cell recruitment (25). GzmB is a serine protease with
intracellular and extracellular activities capable of regulating
inflammation through cytokine processing and apoptosis of effector
cells (26). We tested the
hypothesis that TGF-β1 and GzmB will be changed in LPS treatment,
and ST1926 may have potential to reverse it. IL-10 secretion is the
characteristic activity of cells in regulating inflammatory disease
and a potent anti-inflammatory molecule, thereby suppressing the
action of many pro-inflammatory and pro-fibrotic molecules. TBX21
is an important transcription factor of adaptive immunity (27–29). As shown in Fig. 4A, we found that TGF-β1 was highly
upregulated in LPS treatment, which was reduced due to ST1926
administration from the protein and gene levels, indicating the
inflammatory cell accumulation. Foxp3, as another factor of T cell
differentiation for lung injury regulation, was also found to be
increased in LPS group. Notably, ST1926 significantly decreased its
expression in a dose-dependent manner (Fig. 4B). INFγ and GzmB are also reported
to be highly expressed in acute lung injury (30). Similarly, INFγ, GzmB and Tbx21
protein and gene levels were apparently enhanced by LPS treatment,
which were reduced by ST1926 administration through western
blotting and RT-qPCR assays (Fig.
4C–E). In the end, IL-12 and IL-10 protein levels in serum of
mice were calculated. IL-12, a proinflammatory cytokine, was
upregulated by LPS. ST1926 showed suppressive role in IL-12
expression. In contrast, IL-10, as an important anti-inflammatory
cytokine, was found to be reduced by LPS, which was reversed in
ST1926 treatment (Fig. 4F and G).
Taken together, the data above indicated that ST1926, at least
partly had an inhibitory role in regulating inflammatory
response.
ST1926-ameliorated inflammation response
in mice induced by LPS is dependent on NF-κB signaling pathway
As mentioned above, inflammation response was
observed. Thus, here we attempted to explore how ST1926 altered
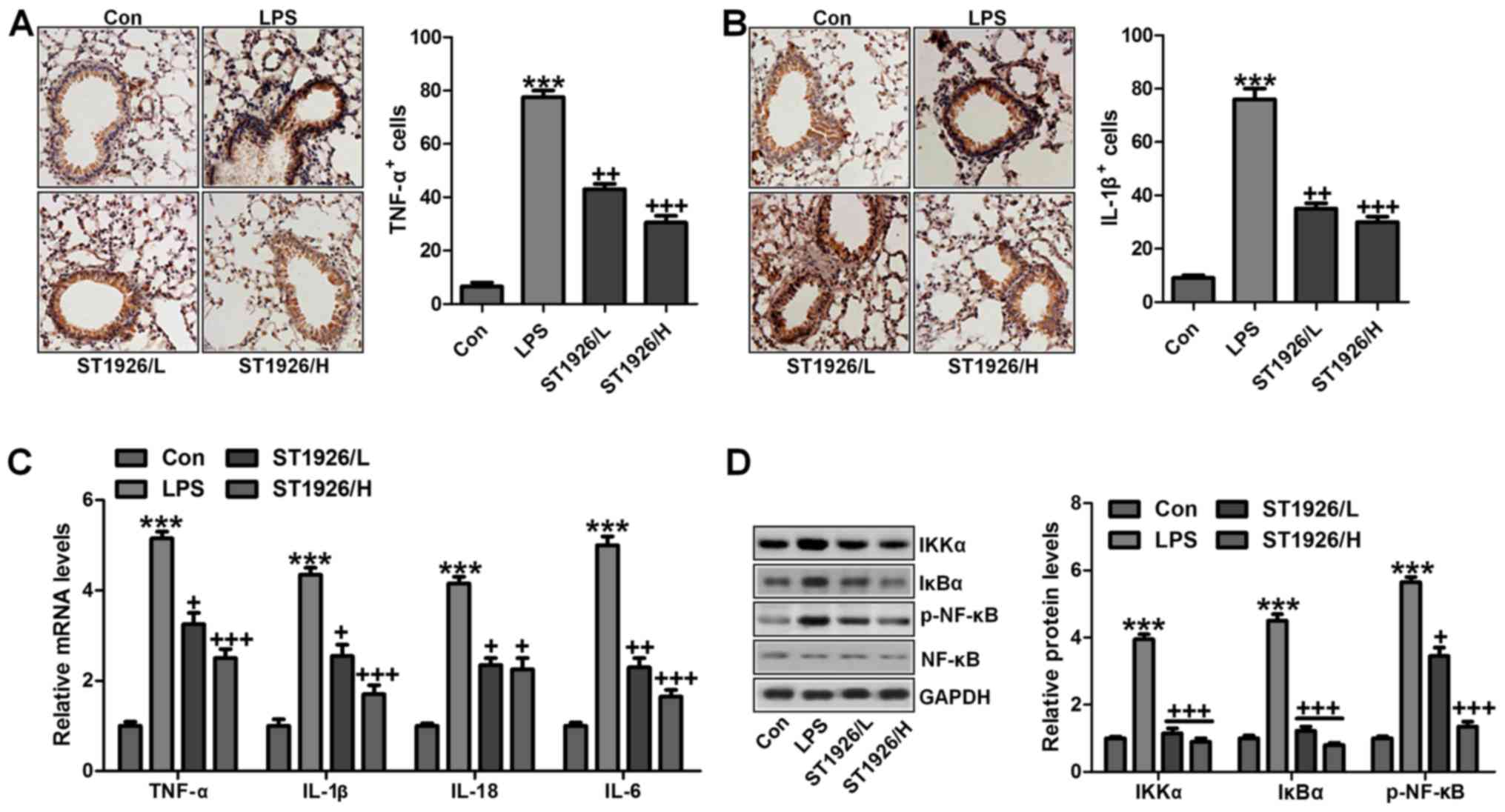
LPS-induced acute lung injury in mice. First, as shown in Fig. 5A and B, we further confirmed that
pro-inflammatory cytokines of TNF-α and IL-1β were significantly
stimulated through IHC analysis. ST1926 displayed attenuated role
in controlling pro-inflammatory cytokine release. Furthermore,
RT-qPCR analysis was carried out to confirm that ST1926 indeed
suppressed LPS-caused inflammation response in the lung tissue of
mice. As shown in Fig. 5C, TNF-α,
IL-18, IL-6 and IL-1β were enhanced for LPS treatment, which was
reduced by ST1926 administration in a dose-dependent manner. NF-κB
signaling pathway is well known in modulating inflammatory response
and controlling pro-inflammatory cytokine secretion, associated
with IKKα and IκBα activation (31). As shown in Fig. 5D, western blot analysis indicated
that IKKα activation was significantly improved by LPS treatment,
resulting in IκBα expression and NF-κB phosphorylation eventually.
Interestingly, ST1926 treatment could reverse NF-κB activation
through inhibition of IKKα and IκBα expression, impeding
inflammatory response. Taken together, ST1926 suppressed
LPS-induced acute lung injury by inactivating NF-κB/IκBα.
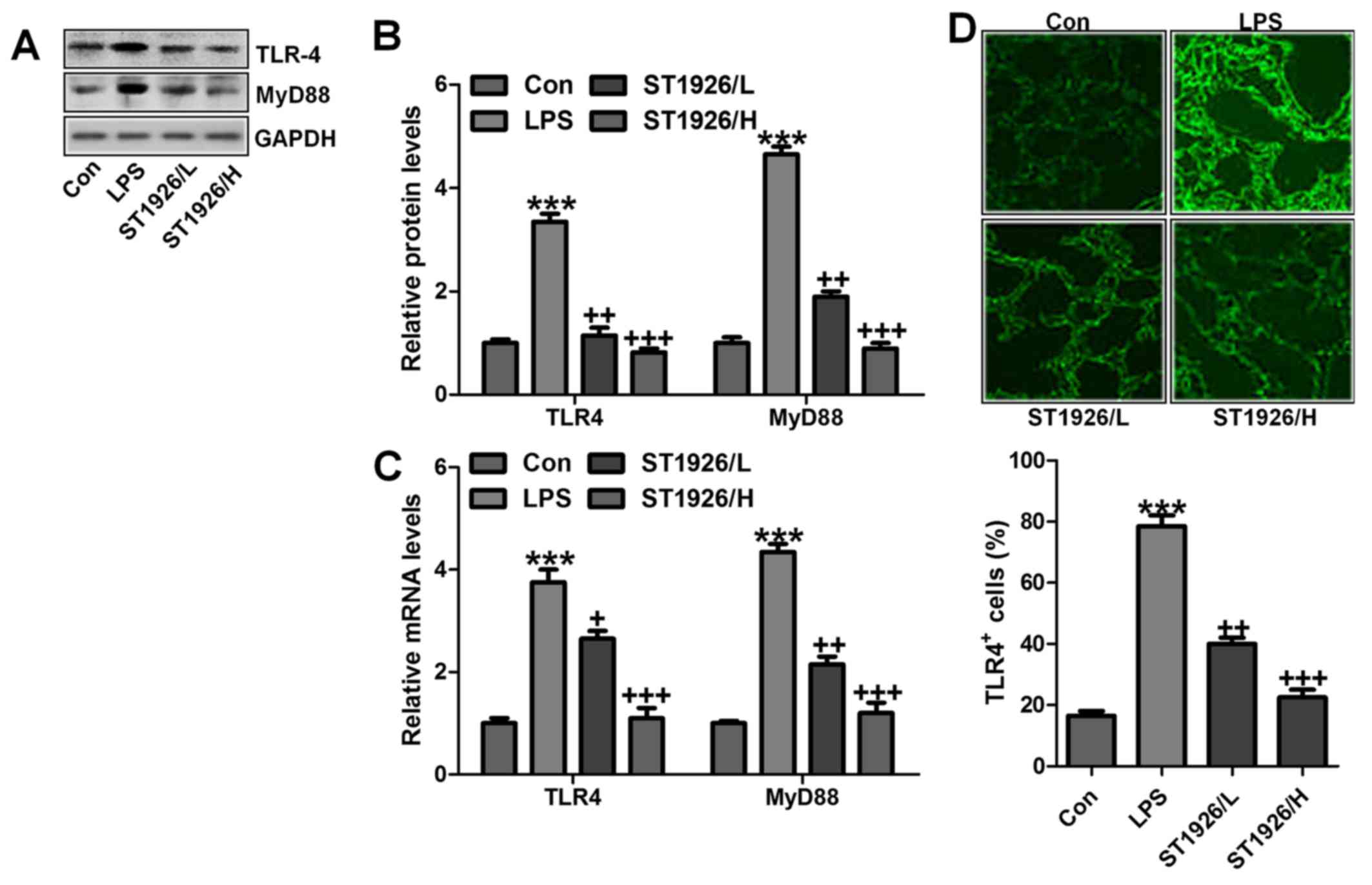
ST1926 suppresses TLR4/MyD88 signal
pathway to inactivate NF-κB activity
TLR4/MyD88 signaling pathway has been well explored
in inflammation response through NF-κB signaling pathway regulation
(32). Thus, here we attempted to
explore if TLR4/MyD88 was involved in NF-κB activation. As shown in
Fig. 6A and B, TLR4 and MyD88
were found to be upregulated in LPS-treated group, which was in
line with NF-κB alteration. ST1926 showed inhibitory role in TLR4
and MyD88 activation on protein levels. Furthermore, RT-qPCR
analysis also indicated that LPS induced high gene expression of
TLR4 and MyD88, and reversed TLR4 and MtD88 gene levels were
observed (Fig. 6C).
Immunofluorescent assay was performed to evidence that TLR4 was
activated in LPS-treated group, which was reduced for ST1926
administration (Fig. 6D). The
data indicated that ST1926 attenuated inflammation response induced
by LPS relying on TLR4 signaling pathway suppression.
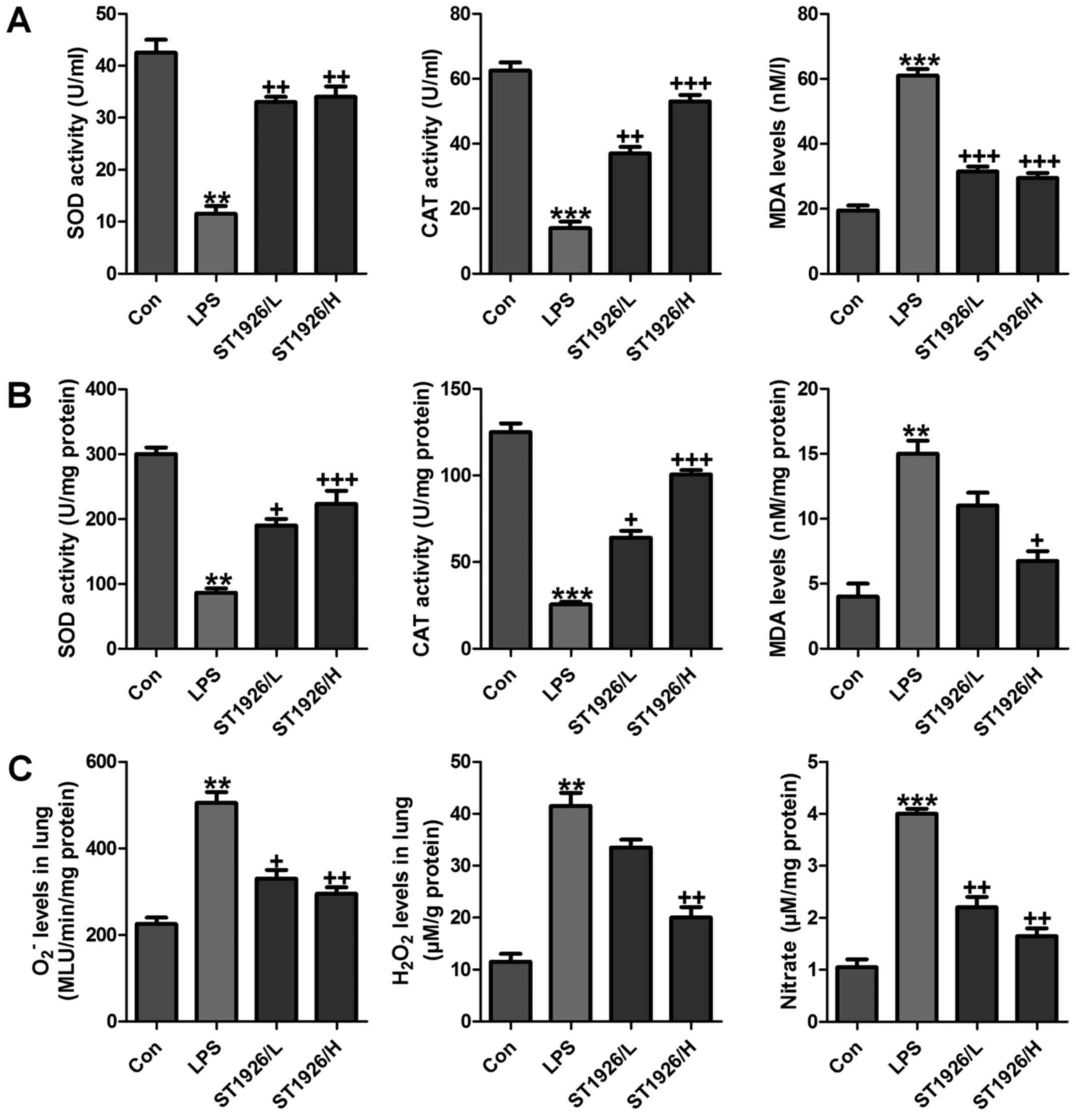
ST1926 impeded oxidative stress in mice
with acute lung injury induced by LPS
Oxidative stress is a leading cause in various
diseases, including a variety of cancers, and acute liver and renal
injury (33). Also, in acute lung
injury oxidative stress was also reported (34). Thus, we proposed that ST1926 may
perform its role in LPS-induced acute lung injury by inhibiting ROS
generation. In order to prove our hypothesis, first in serum and
lung tissue samples, SOD and CAT activity was investigated after
LPS treatment, activity of SOD and CAT was highly reduced, while
MDA was discovered with higher levels in LPS-treated group both in
serum and in lung tissue samples (Fig. 7A and B). However, ST1926
administration significantly upregulated SOD and CAT activity,
while the MDA levels were reduced. In addition, oxidants of
O2− and H2O2 as well as
nitrate levels in the LPS-induced lung tissue samples were found to
be higher than that in the control group (Fig. 7C). Also, ST1926 showed inhibitory
role in expression of these oxidants, which may be another property
of ST1926 to attenuate LPS-induced acute lung injury.
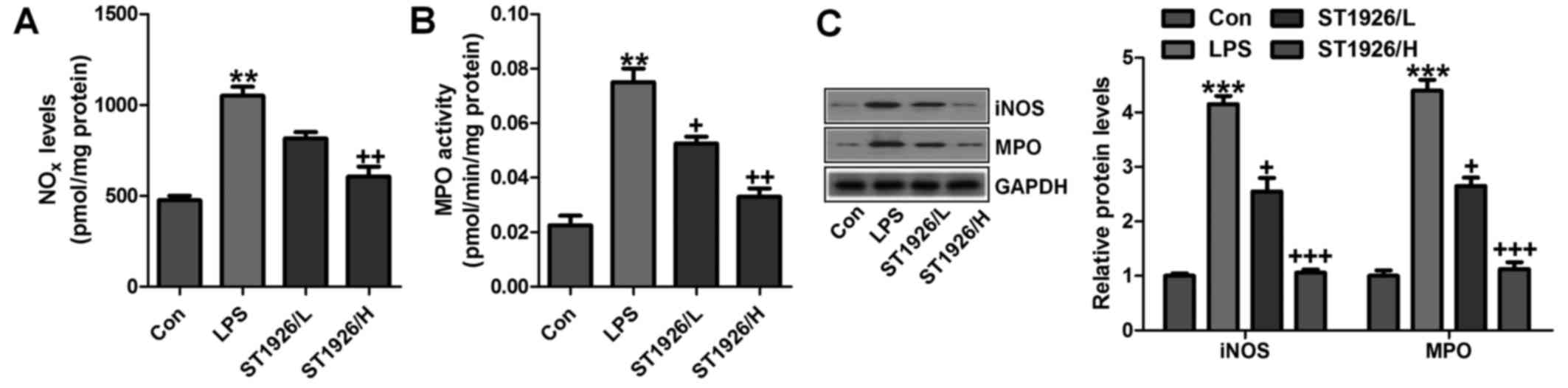
In addition, NOx, iNOS and MPO levels were assessed
to indicate the extent of lung injury. As shown in Fig. 8A, LPS enhanced NO levels in the
tissue samples, which could be reversed by ST1926 administration.
Using MPO activity, we found that LPS induced neutrophil
infiltration in the lungs of LPS-treated mice, and ST1926 reduced
MPO activity (Fig. 8B). MPO
activity in pulmonary parenchyma, reflecting the activation of
neutrophils, was largely upregulated in acute lung injury
condition. NO is produced by the oxidation of l-arginine (l-arg) to
l-citrulline and NO, which is catalyzed by NOS. Thus, iNOS is
generally considered to be the major contributor to the
pathogenesis of acute lung injury (35). In our study, we found that iNOS
protein expression levels were highly induced for LPS treatment,
which were reversed by ST1926 administration (Fig. 8C). Further, protein levels of MPO,
in line with the data above, were promoted in LPS-treated group.
The expression levels were downregulated for ST1926 through western
blot analysis (Fig. 8C). The data
above further indicated that LPS induced acute lung injury due to
NOx, MPO and iNOS acceleration, which was in line with previous
studies (36,37). ST1926, at least partly, could
ameliorate LPS-induced acute lung injury by downregulating the
activity of these factors.
p38/ERK1/2 signaling pathway is inhibited
by ST1926 administration in LPS-induced mice
The data above indicated that ST1926 suppressed
oxidants levels, which is related to upregulated anti-oxidants to
improve lung injury. Thus, here we attempted to further explore the
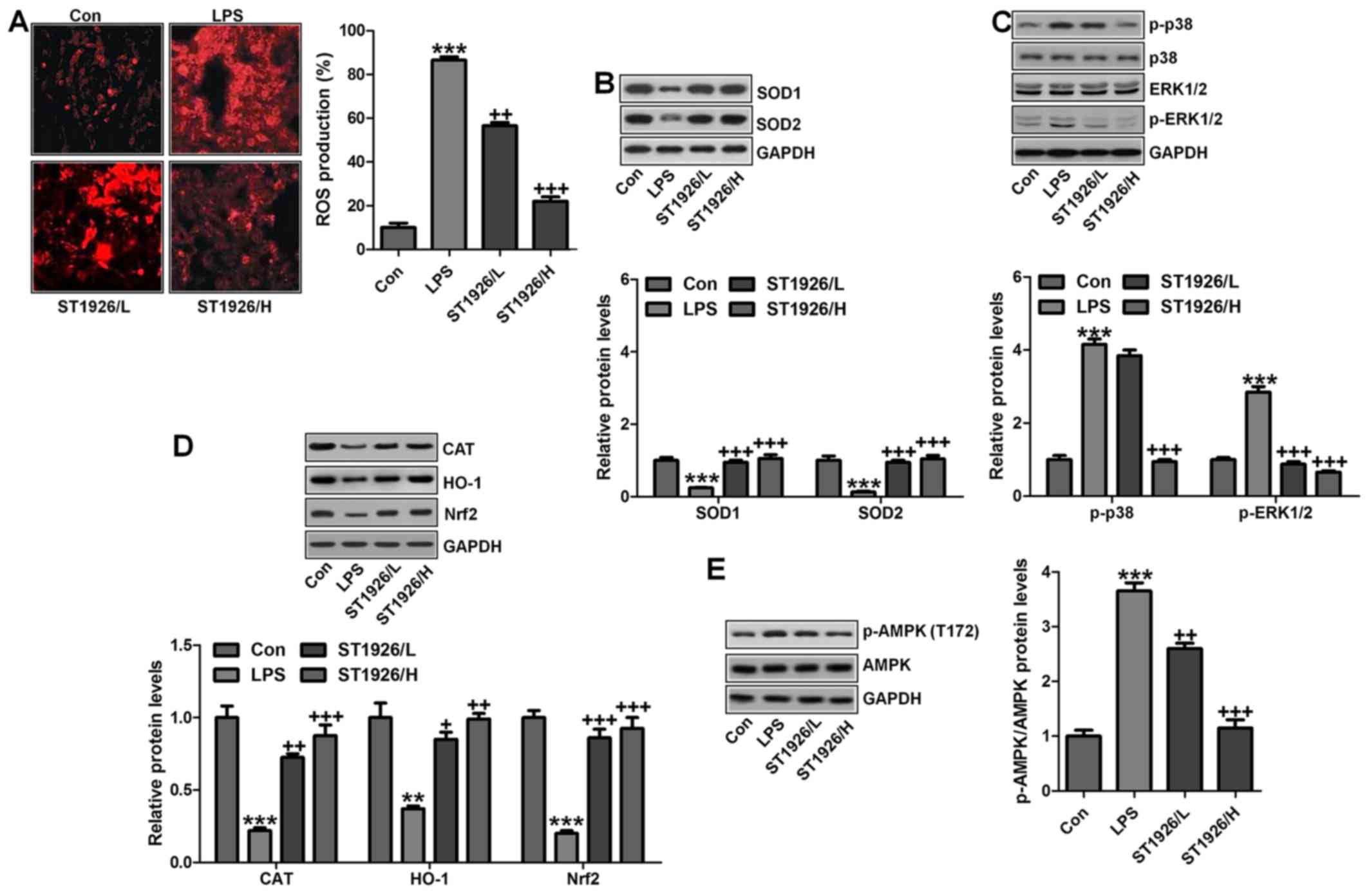
possible molecular mechanism regarding ROS. As shown in Fig. 9A, ROS production was highly
elevated in LPS-treated group, which was reversed by ST1926
administration. Consistently, anti-oxidants of SOD1 and SOD2 were
also upregulated by ST1926 treatment in LPS-induced mice with acute
lung injury (Fig. 9B). Finally,
p38 and ERK1/2 signaling pathway was investigated to explore
whether they were involved in ROS production, mediated by ST1926 in
LPS-treated lung tissue samples. As reported previously, p38 and
ERK1/2 are of great importance in ROS generation (38). Similarly, in our study, we found
that phosphorylated p38 and ERK1/2 were markedly enhanced by LPS
treatment, which was reduced by ST1926 administration (Fig. 9C). The data above indicated that
ST1926 could reduce ROS production, which was related to p38/ERK1/2
signaling pathway inactivation.
Moreover, this study indicated that in the
LPS-induced lung injury, CAT, HO-1 and Nrf2 levels were
downregulated in LPS treatment. In contrast,
O2− and H2O2 were
upregulated. Of note, ST1926 suppressed ROS production by enhancing
anti-oxidant expression while reducing oxidants (Fig. 9D). AMP-activated protein kinase
(AMPK) is a serine/threonine protein kinase that serves as an
energy sensor in the regulation of cellular metabolism.
Additionally, AMPK is reported to be closely associated with ROS
production (39). Here, in our
study, we found that AMPK phosphorylated levels were significantly
reduced for LPS, and ST1926 augmented AMPK phosphorylation of
Thr172, indicating that AMPK phosphorylation induced by ST1926 may
be a key point to modulate ROS generation (Fig. 9E).
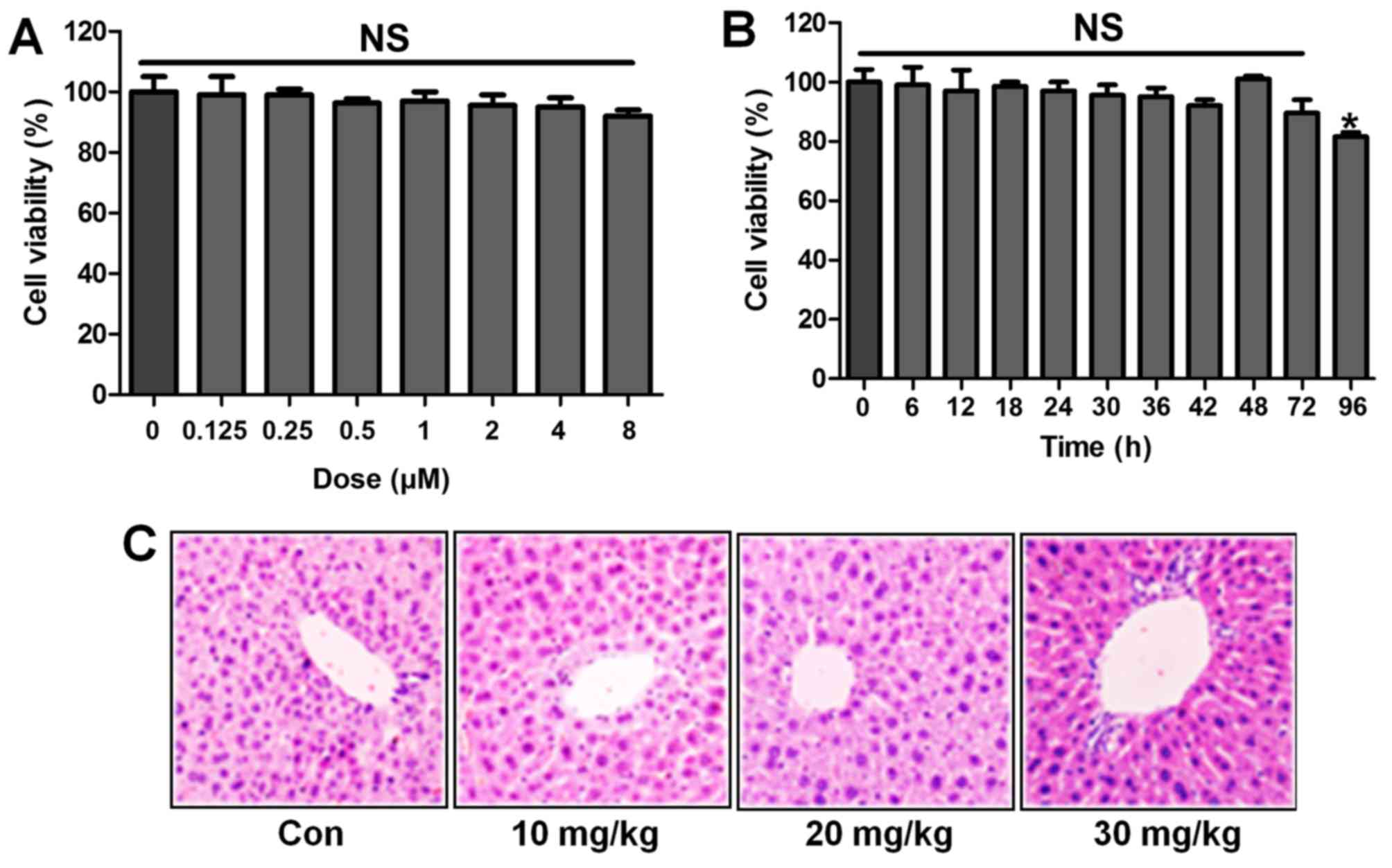
ST1926 shows no toxicity in vitro and in
vivo
Following the results above, we attempted to explore
if ST1926 was effective in LPS-induced acute lung injury in
vitro by the use of lung epithelial cells. As shown in Fig. 10A, the results indicated that
with the increasing of ST1926, even at the highest level, no
significant difference was observed on cells. In Fig. 10B, the cell viability showed no
significant difference at 8 µM ST1926 treatment for
different times until 72 h. In addition, H&E staining suggested
that ST1926 at different concentrations exhibited no toxic role in
the liver of mice without LPS treatments (Fig. 10C). The data above indicated that
ST1926 could ameliorate acute lung injury without any toxicity.
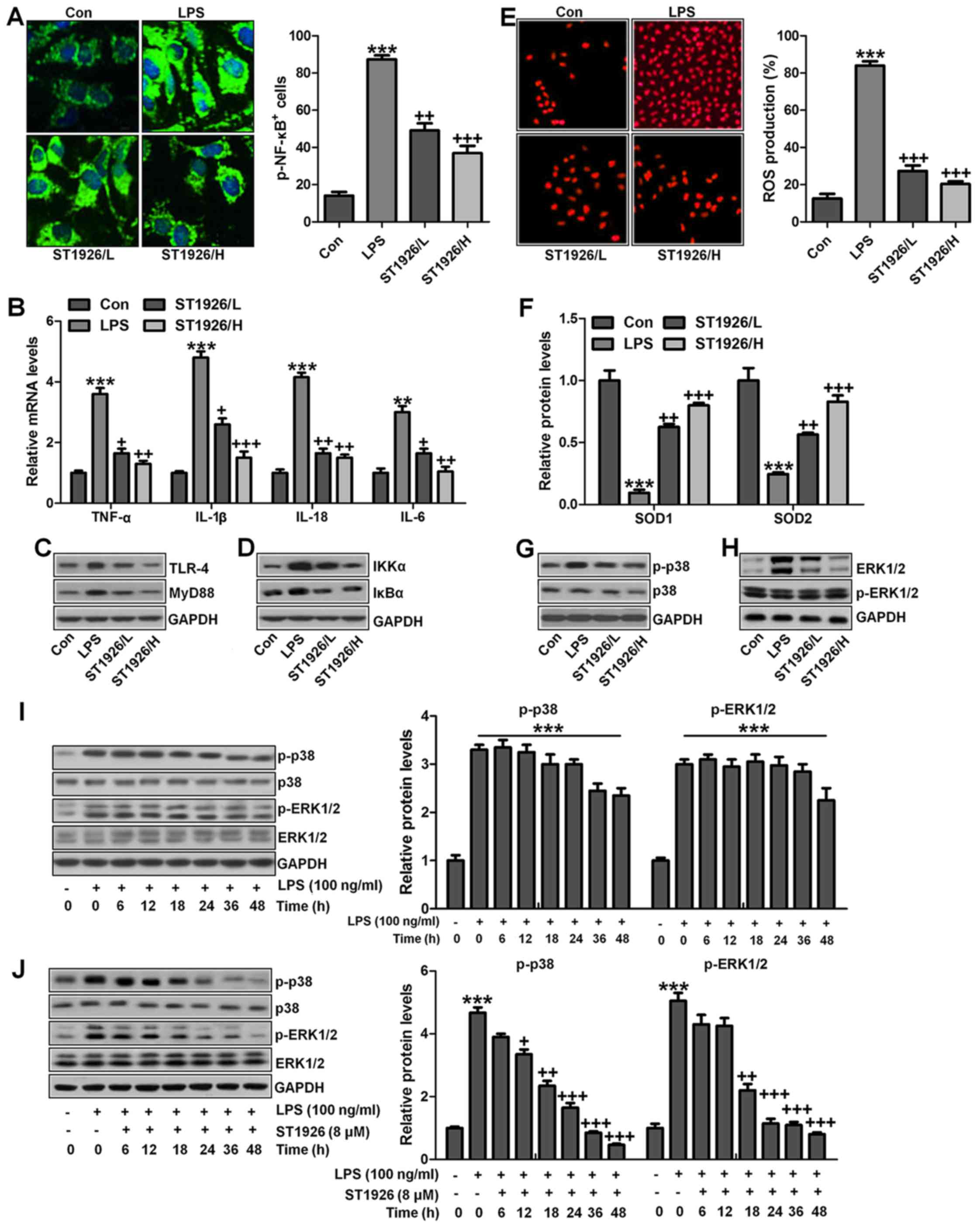
ST1926 suppresses inflammation response
and ROS production in lung epithelia cells in vitro
We confirmed that ST1926 showed no toxicity in cells
in vitro. Thus, here, we attempted to explore if ST1926
could perform its role in vitro. As shown in Fig. 11A, p-NF-κB was found to be
upregulated in LPS-induced lung epithelial cells through
immunofluorescence. ST1926 at 4 and 8 µM significantly
reduced NF-κB phosphorylated levels. In line with the results in
vivo, pro-inflammatory cytokines of TNF-α, IL-1β, IL-18 and
IL-6 were apparently upregulated for LPS induction and ST1926
downregulated the inflammatory cytokine release induced by LPS
(Fig. 11B). TLR4/MyD88 and
IKKα/IκBα signaling pathways were activated by LPS induction, which
were inactivated by ST1926 administration (Fig. 11C and D). Additionally,
LPS-induced ROS production was decreased in ST1926-treated groups,
accompanied by high level of SOD1 and SOD2 (Fig. 11E and F). Further, phosphorylated
p38 and ERK1/2 improved by LPS treatment. Significantly, ST1926
administration could inactivate p38 and ERK1/2 activity,
ameliorating ROS production (Fig.
11G and H). Following, the cells were exposed to 100 ng/ml LPS
for different times, ranging from 0 to 48 h. Next, western blot
analysis was used to calculate p38 and ERK1/2 phosphorylation. As
shown in Fig. 11I, we found that
LPS treatment activated p38 and ERK1/2 levels, and with the
increase in treatment time, the phosphorylated form was sustained
until 48 h, indicating that ROS may be activated by LPS exposure.
As shown in Fig. 11J, we found
that LPS significantly upregulated p38 and ERK1/2 phosphorylation,
which were reduced for ST1926 in a time-dependent manner. After
ST1926 treatment for 12 and 18 h, significant difference was
observed in p38 and ERK1/2 activation, respectively. Furthermore,
as shown in Fig. 11K, the NF-κB
levels in the nucleus were found to be upregulated for LPS
treatment, which was in line with p-NF-κB alteration in cytoplasm
to enhance pro-inflammatory transcription, contributing to
pro-inflammatory cytokines secretion. Of note, ST1926 showed
significant role in reducing NF-κB in the nucleus, subsequently
downregulating pro-inflammatory cytokine release. In addition, the
whole cell IκBα levels were upregulated due to LPS treatment, while
p-IκBα was down-regulated. ST1926 could promote IκBα and p-IκBα
levels considerably (Fig. 11L).
Thus, we supposed that ST1926 could ameliorate inflammation
response by reducing NF-κB translocation into the nucleus. The
above data indicated that ST1926 indeed attenuated LPS-induced lung
injury in vitro via inhibiting inflammation and ROS
production.
ST1926 inhibits inflammation response and
ROS generation in human bronchial epithelial cells
In this regard, normal human bronchial epithelial
cells (NHBE) were included to further explore how ST1926 regulated
LPS-induced damage in lung cells. As shown in Fig. 12A, cell viability was
investigated with increasing concentrations of ST1926, no
significant difference was observed among various groups. Further,
8 µM ST1926 was administered to cells for different times as
indicated, ranging from 0 to 90 h. Then, MTT analysis was carried
out to investigate the cell viability, which showed no significant
change among different groups (Fig.
12B). Inflammatory response and ROS have been revealed to be
induced by LPS. The phosphorylated NF-κB and ROS generation was
dramatically upregulated by LPS induction (Fig. 12C and D), which were
downregulated in ST1926-treated groups, accompanied with the
reduced pro-inflammatory cytokines mRNA levels through RT-qPCR
analysis (Fig. 12E).
Additionally, anti-oxidants of SOD1, SOD2, CAT, HO-1 and Nrf2 were
reduced in LPS treatment, contributing to ROS generation, while
ST1926 enhanced these anti-oxidants with apparent difference in
comparison to the LPS group (Fig.
12F). Next, TLR4/MyD88 signaling pathway was calculated, which
was highly activated for LPS, leading to IKKα expression. ST1926
showed inhibitory role in TLR4, MyD88 and IKKα expression induced
by LPS (Fig. 12G). NF-κB levels
in the nucleus were increased by LPS, while decreased due to
ST1926. Accordingly, IκBα and p-IκBα were also augmented in LPS
single treatment group, in line with the results in MLE-12 cells
mentioned above. ST1926 could reduce IκBα and phosphorylated IκBα
expression levels (Fig. 12H).
Next, NHBE cells were exposed to LPS for different times as
indicated to explore the p-p38 and p-ERK1/2 expression levels.
Fig. 12J shows that p38 and
ERK1/2 were activated due to LPS treatment, indicating ROS
generation. With the increase of treatment time, the
phosphorylation of p38 and ERK1/2 was maintained in high levels.
Finally, p38 and ERK1/2 protein levels were assessed. As shown in
Fig. 12I, phophorylated p38 and
ERK1/2 were highly expressed in LPS-treated group, which were
reduced by ST1926. In NHBE cells, we found similar results that
ST1926 could reduce LPS-induced p38 and ERK1/2 phosphorylation
beginning from 12 h with significant difference (Fig. 12K). Thus, the data above
indicated that ST1926 could reduce LPS-induced p38 and ERK1/2
phosphorylation from 12 or 18 h in a time-dependent manner.
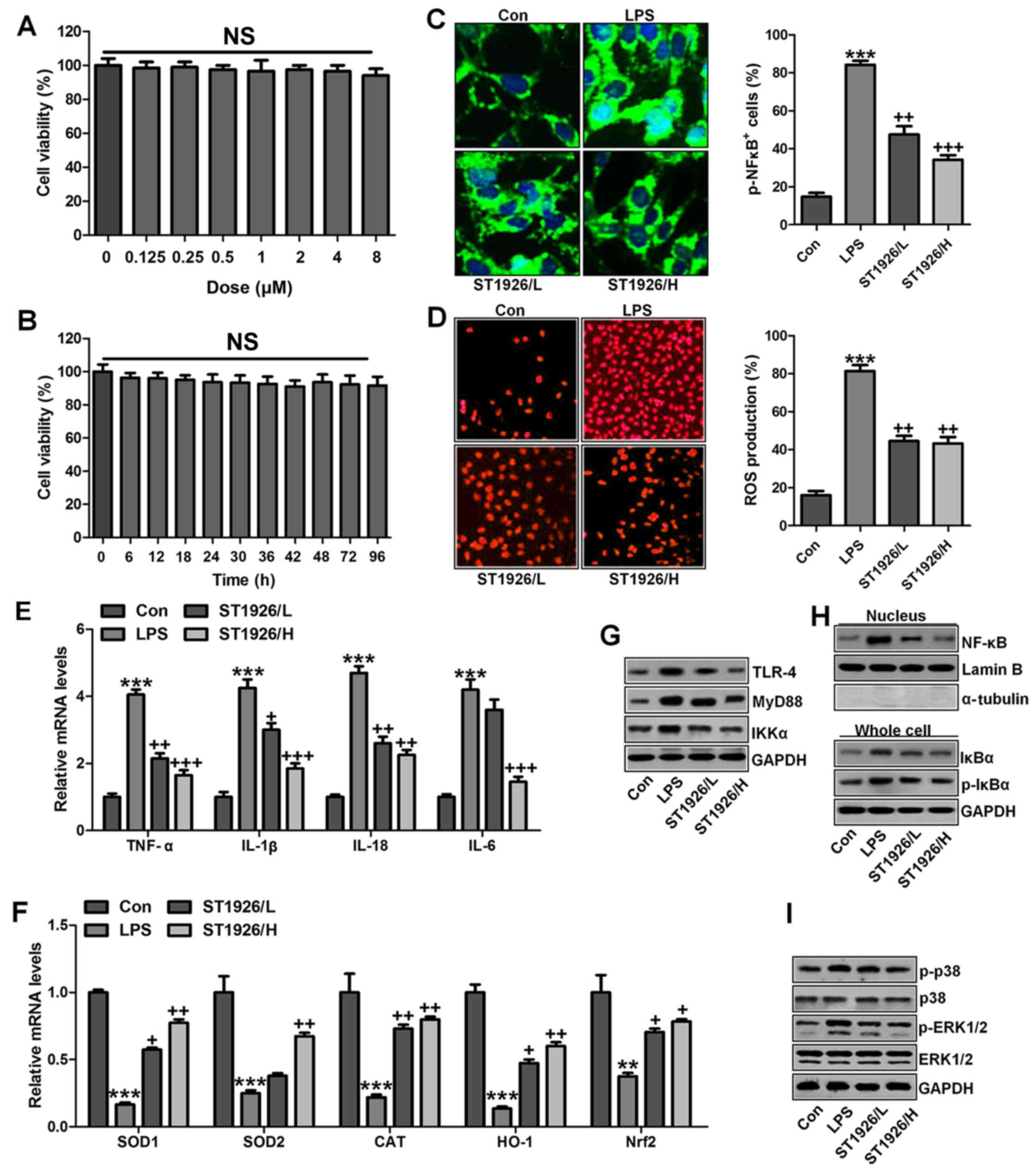 | Figure 12ST1926 inhibits inflammation response
and reactive oxygen species (ROS) generation in human bronchial
epithelial cells. (A) Human bronchial epithelial cells were treated
with different concentrations of ST1926 at different concentrations
for 24 h. Then, the cell viability was calculated through MTT
analysis. (B) Human bronchial epithelial cells were treated with 8
µM ST1926 for different time, followed by MTT assays. (C)
The phosphorylated nuclear factor-κB (NF-κB) levels were measured
by immunofluorescent analysis. (D) ROS generation was evaluated and
the quantification was displayed. (E) RT-qPCR assays were performed
to determine tumor necrosis factor-α (TNF-α), interleukin-1β
(IL-1β), IL-18 and IL-6 gene levels in cells under different
treatment. (F) SOD1, SOD2, CAT, haeme oxygenase-1 (HO-1) and Nrf2
mRNA levels were calculated through RT-qPCR assays. (G) Toll-like
receptor 4 (TLR4), MyD88 and inhibitor-κB kinase (IKKα) protein
expression levels were measured by western blot analysis. (H)
Western blotting was carried out to investigate nuclear NF-κB
levels. The cytoplasm p-inhibitor-κB kinase-α (IκBα) and IκBα
levels were assessed through western blot analysis. (I)
Phosphorylated p38, and (H) extracellular receptor kinase 1/2
(ERK1/2) was determined by western blot analysis. (J) NHBE cells
were exposed to 100 ng/ml lipopolysaccharide (LPS) for different
times as indicated, followed by western blot analysis. (K) ST1926
reduced p38 and ERK1/2 phosphorylation in human bronchial
epithelial cells after LPS treatment with or without ST1926
administration from 0 to 48 h. Then, western blot assays were used
to explore p38 and ERK1/2 activation. The data are presented as
mean ± SD (n=8). *p<0.05 and **p<0.001
vs. the control (Con); +p<0.05,
++p<0.01 and +++p<0.001 vs. LPS-induced
mice (LPS). |
Discussion
Acute lung injury (ALI) is a severe disease
syndrome, which consists of hypoxemic respiratory failure
accompanied by bilateral pulmonary infiltrates, and has been
considered as a serious threat for people, especially children with
relatively weak immune system (1,2,40,41). Furthermore, as the acute
respiratory distress syndrome, the formation and development of ALI
is linked to high morbidity and mortality (42). According to previous studies, many
therapeutic strategies were investigated and used to prevent acute
lung injury in children (43).
However, more effective approach and the possible molecular
mechanism is still needed to be explored.
Natural retinoids such as all-trans retinoic acid
are currently used as important therapeutic agents in human
diseases, mainly acute promyelocytic leukemia (44,45). Synthetic retinoid of ST1926 has
been developed to overcome resistance and attenuate side effects.
ST1926 has the same crucial structural and pharmacological groups
as CD437, the lipophilic adamantyl moiety, and the carboxylic
function. However, ST1926 was designed to be with a styrene moiety
replacing the naphthalene ring of CD437, leading to a
pharmacokinetically stable, highly orally bioavailable, as well as
pharmacologically attainable in plasma of patients at the
micromolar concentrations (21,22). ST1926 has been suggested to induce
apoptosis in tumor cells, which was related to anti-apoptotic Bcl-2
protein suppression (46). Bcl-2
is known to be upregulated in NF-κB activation (47). Thus, we supposed that ST1926 may
have a possible effect on NF-κB suppression. NF-κB is well known to
modulate inflammation response. Hence, ST1926 was chosen in our
study to explore if it could be used in treating lung injury,
providing a new therapeutic strategy for acute lung injury. In this
study, ST1926 showed downregulated role in inflammation
infiltration, suggesting that ST1926 may have a potential effect on
controlling inflammation response in acute lung injury related to
the immune system.
As we mentioned above, inflammation response is well
known to play an essential role in various disease development,
including cancers, diabetes, as well as lung injury in different
forms (48). Neutrophils and
macrophages were the main inflammatory cells in acute lung injury.
They infiltrated into the lung tissues, releasing enzymes and
phagocytizing the pathogen. These inflammatory cells were the
fundamental source of inflammatory mediators in vivo. In
LPS-induced inflammation, neutrophils and macrophages were
activated (49). After
activation, neutrophils and macrophages were recruited to the
inflammation site (50).
Lymphocytes have drawn increased attention, and there is
accumulating evidence indicating that lymphocytes play important
roles in the development of acute lung injury. It has been
suggested that lymphocytes contribute to the progression of
autoimmune and inflammatory diseases (51). In our study, we found that LPS
upregulated lymphocytes, neutrophils and macrophages, contributing
to inflammation response, which could be reversed by ST1926
administration. Additionally, pro-inflammatory cytokines, including
TNF-α, IL-1β, IL-18, IL-6 and IL-17, are main factors causing
inflammatory response, and their activation is a key to accelerate
disease progression (52). In
this study, we found that LPS induced higher expression of these
pro-inflammatory cytokines, was in line with a previous report in
acute lung injury caused by LPS (53,54). However, ST1926 showed
significantly suppressive role in controlling the cytokine
secretion, inhibiting inflammation. Inflammatory cytokine release
could be stimulated by NF-κB phosphorylation, which is dependent on
IKKα and IκBα signaling pathway activation (55). Consistently, in our present study,
we found that IKKα and IκBα were upregulated in LPS treatment in
vivo and in vitro, subsequently contributing to NF-κB
phosphorylation and pro-inflammatory cytokines secretion
eventually, which may be a main cause, leading to lung injury in
animal models and cells. Furthermore, IκBα and NF-κB forms a
complex, inhibiting NF-κB translocation into nuclear and
suppressing pro-inflammatory cytokines release. In contrast,
phosphorylated IκBα abolished the IκBα/NF-κB complex, promoting
NF-κB translocation into nuleus and causing inflammatory response
(56,57). In line with a previous study, we
found that NF-κB levels in nucleus were promoted in LPS treatment,
consistent to cytoplasmic p-NF-κB alteration in, leading to
inflammation response. Obviously, ST1926 had a significant role in
reducing nuclear NF-κB, subsequently suppressing pro-inflammatory
cytokine secretion. Additionally, the cytoplasm IκBα levels were
increased for LPS treatment, while p-IκBα was decreased. ST1926
downregulated IκBα and p-IκBα levels. Thus, ST1926 could improve
inflammation response by preventing NF-κB translocation into the
nucleus. Toll-like receptor-4 (TLR-4) could identify pathogenic
microorganisms in a natural immune system, bind the specific ligand
and generate corresponding inflammation following identification
(58,59). TLR-4 exogenous ligands include
LPS. Here, we also found that TLR4/MyD88 signaling pathway was
highly activated by LPS induction, which was consistent with NF-κB
activity. ST1926 apparently downregulated TLR4 and MyD88 expression
levels after LPS induction, revealing that TLR4 was also involved
in ST1926-improved lung injury.
Oxidative stress is widely involved in progression
of many diseases (60). SOD and
CAT are two typical anti-oxidants, representing oxidative stress
level (61). SOD is well known as
an essential anti-oxidase that converses O2−
into O2 and H2O2 (62). CAT could catalyze
H2O2 into H2O and O2,
reducing ROS generation (63).
Additionally, phase II detoxifying enzymes such as HO-1 is also
known as an effective antioxidant enzyme (64). HO-1, encoded by HMOX1 gene, can
change haeme into the strong pro-oxidant biliverdin, which is
subsequently transformed into bilirubin, a potent antioxidant
(65). Nuclear factor erythroid 2
related factor 2 (Nrf2), a key regulator of antioxidant defense
system, protects cells against oxidative stress (66). Downregulated SOD, CAT, HO-1 and
Nrf2 for LPS were found to be upregulated after ST1926 treatment.
In contrast, LPS-induced higher levels of MDA and ROS species of
O2− and H2O2 were
decreased in ST1926-treated groups. Also, according to previous
studies, an inverse correlation was observed between anti-oxidant
mRNA and ROS levels. With the increase of anti-oxidant expression
via Nrf-2/HO-1 pathway, the ROS generation was accordingly reduced,
proved by downregulated O2, H2O2,
NO, MPO and iNOS levels. In this study, similar results were
observed, displaying an inverse correlation between anti-oxidant
and ROS levels (61,63,64). In addition, p38/ERK1/2 signaling
pathway has a close relationship with ROS generation (67,68). Following previous studies,
p38/ERK1/2 signaling pathway is activated, contributing to
oxidative stress progression in different injuries (69). Similarly, here we found that LPS
induction caused higher ROS production. Interestingly, ST1926
displayed significant inhibitory role in controlling ROS
production. AMP-activated protein kinase (AMPK) is a
serine/threonine protein kinase that serves as an energy sensor in
the regulation of cellular metabolism. Additionally, AMPK is
reported to be closely associated with ROS production (70). AMPK is activated by changes in the
AMP:ATP ratio that occur in response to energetic stress, and
activation requires the phosphorylation of Thr172. Promotion of
AMPK decreased ROS generation to improve injuries (71). AMPK phosphorylated levels were
significantly reduced by LPS, and ST1926 augmented AMPK
phosphorylation of Thr172, indicating ROS suppression by ST1926.
Collectively, our data indicated that ST1926 was associated with
LPS-induced acute lung injury progression via TLR4/NF-κB and
AMPK/p38/ERK1/2 signaling suppression.
In conclusion, the results above showed that
LPS-induced acute lung injury in mice was developed from
inflammation activation and ROS generation in lung tissue samples
and epithelial cells through TLR4/NF-κB and p38/ERK1/2 pathways.
This study elucidated the possible mechanism by which ST1926
attenuated acute lung injury with little toxicity, which may be a
potential therapy for acute lung injury for children.
Acknowledgments
Not applicable.
Notes
[1]
Funding
No funding was received.
[2] Availability
of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
ZD designed the project and wrote the paper. YY
performed the lab experiment and data analysis. Both authors read
and approved the final manuscript.
[4] Ethics
approval and consent to participate
This study was approved by the Ethics Committee on
Animal Research at the Department of Pediatrics, Huai'an First
People's Hospital, Nanjing Medical University, Nanjing, China.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Pediatric Acute Lung Injury Consensus
Conference Group: Pediatric acute respiratory distress syndrome:
Consensus recommendations from the Pediatric Acute Lung Injury
Consensus Conference. Pediatr Crit Care Med. 16:428–439. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rettig JS, Smallwood CD, Walsh BK,
Rimensberger PC, Bachman TE, Bollen CW, Duval EL, Gebistorf F,
Markhorst DG, Tinnevelt M, et al: High-frequency oscillatory
ventilation in pediatric acute lung injury: A multicenter
international experience. Crit Care Med. 43:2660–2667. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thomas NJ, Jouvet P and Willson D: Acute
lung injury in children - kids really aren't just 'little adults'.
Pediatr Crit Care Med. 14:429–432. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
López-Fernández Y, Azagra AM, de la Oliva
P, Modesto V, Sánchez JI, Parrilla J, Arroyo MJ, Reyes SB,
Pons-Ódena M, López-Herce J, et al: Pediatric Acute Lung Injury
Epidemiology and Natural History (PED-ALIEN) Network: Pediatric
acute lung injury epidemiology and natural history study: Incidence
and outcome of the acute respiratory distress syndrome in children.
Crit Care Med. 40:3238–3245. 2012. View Article : Google Scholar
|
|
5
|
Zhang B, Liu ZY, Li YY, Luo Y, Liu ML,
Dong HY, Wang YX, Liu Y, Zhao PT, Jin FG, et al: Antiinflammatory
effects of matrine in LPS-induced acute lung injury in mice. Eur J
Pharm Sci. 44:573–579. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Santschi M, Randolph AG, Rimensberger PC
and Jouvet P; Pediatric Acute Lung Injury Mechanical Ventilation
Investigators; Pediatric Acute Lung Injury and Sepsis Investigators
Network; European Society of Pediatric and Neonatal Intensive Care:
Mechanical ventilation strategies in children with acute lung
injury: A survey on stated practice pattern. Pediatr Crit Care Med.
14:e332–e337. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hernu R, Wallet F, Thiollière F, Martin O,
Richard JC, Schmitt Z, Wallon G, Delannoy B, Rimmelé T, Démaret C,
et al: An attempt to validate the modification of the
American-European consensus definition of acute lung injury/acute
respiratory distress syndrome by the Berlin definition in a
university hospital. Intensive Care Med. 39:2161–2170. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yehya N, Servaes S and Thomas NJ:
Characterizing degree of lung injury in pediatric acute respiratory
distress syndrome. Crit Care Med. 43:937–946. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Valentine SL, Sapru A, Higgerson RA,
Spinella PC, Flori HR, Graham DA, Brett M, Convery M, Christie LM,
Karamessinis L, et al: Pediatric Acute Lung Injury and Sepsis
Investigator's (PALISI) Network; Acute Respiratory Distress
Syndrome Clinical Research Network (ARDSNet): Fluid balance in
critically ill children with acute lung injury. Crit Care Med.
40:2883–2889. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar
|
|
11
|
Fu Y, Liu B, Zhang N, Liu Z, Liang D, Li
F, Cao Y, Feng X, Zhang X and Yang Z: Magnolol inhibits
lipopolysaccharide-induced inflammatory response by interfering
with TLR4 mediated NF-κB and MAPKs signaling pathways. J
Ethnopharmacol. 145:193–199. 2013. View Article : Google Scholar
|
|
12
|
Wang Y, Tu Q, Yan W, Xiao D, Zeng Z,
Ouyang Y, Huang L, Cai J, Zeng X, Chen YJ, et al: CXC195 suppresses
proliferation and inflammatory response in LPS-induced human
hepatocellular carcinoma cells via regulating
TLR4-MyD88-TAK1-mediated NF-κB and MAPK pathway. Biochem Biophys
Res Commun. 456:373–379. 2015. View Article : Google Scholar
|
|
13
|
Capiralla H, Vingtdeux V, Zhao H,
Sankowski R, Al-Abed Y, Davies P and Marambaud P: Resveratrol
mitigates lipopolysaccharide- and Aβ-mediated microglial
inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. J
Neurochem. 120:461–472. 2012. View Article : Google Scholar
|
|
14
|
Wang W, Xia T and Yu X: Wogonin suppresses
inflammatory response and maintains intestinal barrier function via
TLR4-MyD88-TAK1-mediated NF-κB pathway in vitro. Inflamm Res.
64:423–431. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Valavanidis A, Vlachogianni T, Fiotakis K
and Loridas S: Pulmonary oxidative stress, inflammation and cancer:
Respirable particulate matter, fibrous dusts and ozone as major
causes of lung carcinogenesis through reactive oxygen species
mechanisms. Int J Environ Res Public Health. 10:3886–3907. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fratelli M, Fisher JN, Paroni G, Di
Francesco AM, Pierri F, Pisano C, Godl K, Marx S, Tebbe A, Valli C,
et al: New insights into the molecular mechanisms underlying
sensitivity/resistance to the atypical retinoid ST1926 in acute
myeloid leukaemia cells: The role of histone H2A.Z, cAMP-dependent
protein kinase A and the proteasome. Eur J Cancer. 49:1491–1500.
2013. View Article : Google Scholar
|
|
17
|
El Hajj H, Khalil B, Ghandour B, Nasr R,
Shahine S, Ghantous A, Abdel-Samad R, Sinjab A, Hasegawa H, Jabbour
M, et al: Preclinical efficacy of the synthetic retinoid ST1926 for
treating adult T-cell leukemia/lymphoma. Blood. 124:2072–2080.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Di Francesco AM, Cusano G, Franzese O,
Orienti I, Falconi M, Vesci L and Riccardi R: Resistance to the
atypical retinoid ST1926 in SK-N-AS cells selected the subline
rAS-ST with enhanced sensitivity to ATRA mediated by not
conventional mechanisms: DNA damage, G2 accumulation and late
telomerase inhibition. Toxicol In Vitro. 29:1628–1638. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zuco V, Benedetti V, De Cesare M and
Zunino F: Sensitization of ovarian carcinoma cells to the atypical
retinoid ST1926 by the histone deacetylase inhibitor, RC307:
Enhanced DNA damage response. Int J Cancer. 126:1246–1255.
2010.
|
|
20
|
Bernasconi E, Gaudio E, Kwee I, Rinaldi A,
Cascione L, Tarantelli C, Mensah AA, Stathis A, Zucca E, Vesci L,
et al: The novel atypical retinoid ST5589 downregulates Aurora
Kinase A and has anti-tumour activity in lymphoma pre-clinical
models. Br J Haematol. 171:378–386. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Basma H, Ghayad SE, Rammal G, Mancinelli
A, Harajly M, Ghamloush F, Dweik L, El-Eit R, Zalzali H, Rabeh W,
et al: The synthetic retinoid ST1926 as a novel therapeutic agent
in rhabdomyosarcoma. Int J Cancer. 138:1528–1537. 2016. View Article : Google Scholar
|
|
22
|
Sala F, Zucchetti M, Bagnati R, D'Incalci
M, Pace S, Capocasa F and Marangon E: Development and validation of
a liquid chromatography-tandem mass spectrometry method for the
determination of ST1926, a novel oral antitumor agent, adamantyl
retinoid derivative, in plasma of patients in a phase I study. J
Chromatogr B Analyt Technol Biomed Life Sci. 877:3118–3126. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pisano C, Vesci L, Foderà R, Ferrara FF,
Rossi C, De Cesare M, Zuco V, Pratesi G, Supino R and Zunino F:
Antitumor activity of the combination of synthetic retinoid ST1926
and cisplatin in ovarian carcinoma models. Ann Oncol. 18:1500–1505.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
González-Terán B, Cortés JR, Manieri E,
Matesanz N, Verdugo Á, Rodríguez ME, González-Rodríguez Á, Valverde
ÁM, Martín P, Davis RJ, et al: Eukaryotic elongation factor 2
controls TNF-α translation in LPS-induced hepatitis. J Clin Invest.
123:164–178. 2013. View Article : Google Scholar
|
|
25
|
Akbarshahi H, Sam A, Chen C, Rosendahl AH
and Andersson R: Early activation of pulmonary TGF-β1/Smad2
signaling in mice with acute pancreatitis-associated acute lung
injury. Mediators Inflamm. 2014:1480292014. View Article : Google Scholar
|
|
26
|
Hirota JA, Hiebert PR, Gold M, Wu D,
Graydon C, Smith JA, Ask K, McNagny K, Granville DJ and Knight DA:
Granzyme B deficiency exacerbates lung inflammation in mice after
acute lung injury. Am J Respir Cell Mol Biol. 49:453–462. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nieves W, Hung LY, Oniskey TK, Boon L,
Foretz M, Viollet B and Herbert DR: Myeloid-restricted AMPKα1
promotes host immunity and protects against IL-12/23p40-dependent
lung injury during hookworm infection. J Immunol. 196:4632–4640.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Glista-Baker EE, Taylor AJ, Sayers BC,
Thompson EA and Bonner JC: Nickel nanoparticles cause exaggerated
lung and airway remodeling in mice lacking the T-box transcription
factor, TBX21 (T-bet). Part Fibre Toxicol. 11:72014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Do-Umehara HC, Chen C, Urich D, Zhou L,
Qiu J, Jang S, Zander A, Baker MA, Eilers M, Sporn PH, et al:
Suppression of inflammation and acute lung injury by Miz1 via
repression of C/EBP-δ. Nat Immunol. 14:461–469. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kuchroo VK, Wu C, Pot C, Apetoh L and
Anderson AC: Methods for modulating immune responses during chronic
immune conditions by targeting metallothioneins. US Patent
Application WO 2014172606 A1. Filed April 18, 2014; issued October
23, 2014.
|
|
31
|
Wang Y, Wang H, Zhang W, Shao C, Xu P, Shi
CH, Shi JG, Li YM, Fu Q, Xue W, et al: Genistein sensitizes bladder
cancer cells to HCPT treatment in vitro and in vivo via
ATM/NF-κB/IKK pathway-induced apoptosis. PLoS One. 8:e501752013.
View Article : Google Scholar
|
|
32
|
Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X,
Chen F, Wang CS, Feng H and Lin JK: Curcumin attenuates acute
inflammatory injury by inhibiting the TLR4/MyD88/NF-κB signaling
pathway in experimental traumatic brain injury. J
Neuroinflammation. 11:592014. View Article : Google Scholar
|
|
33
|
Arslan F, Lai RC, Smeets MB, Akeroyd L,
Choo A, Aguor EN, Timmers L, van Rijen HV, Doevendans PA,
Pasterkamp G, et al: Mesenchymal stem cell-derived exosomes
increase ATP levels, decrease oxidative stress and activate
PI3K/Akt pathway to enhance myocardial viability and prevent
adverse remodeling after myocardial ischemia/reperfusion injury.
Stem Cell Res. 10:301–312. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
de Lima FM, Albertini R, Dantas Y,
Maia-Filho AL, Santana Cde L, Castro-Faria-Neto HC, França C,
Villaverde AB and Aimbire F: Low-level laser therapy restores the
oxidative stress balance in acute lung injury induced by gut
ischemia and reperfusion. Photochem Photobiol. 89:179–188. 2013.
View Article : Google Scholar
|
|
35
|
Golden T, Crabtree M, Channon K and Gow
AJ: Uncoupled inducible nitric oxide synthase influences macrophage
polarization in acute lung injury. Am J Respir Crit Care Med.
193:A2651. 2016.
|
|
36
|
Tsai CL, Lin YC, Wang HM and Chou TC:
Baicalein, an active component of Scutellaria baicalensis, protects
against lipopolysaccharide-induced acute lung injury in rats. J
Ethnopharmacol. 153:197–206. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Barut F, Ozacmak VH, Turan I,
Sayan-Ozacmak H and Aktunc E: Reduction of acute lung injury by
administration of spironolactone after intestinal ischemia and
reperfusion in rats. Clin Invest Med. 39:E15–E24. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Koinzer S, Reinecke K, Herdegen T, Roider
J and Klettner A: Oxidative stress induces biphasic ERK1/2
activation in the RPE with distinct effects on cell survival at
early and late activation. Curr Eye Res. 40:853–857. 2015.
View Article : Google Scholar
|
|
39
|
Cheng PW, Ho WY, Su YT, Lu PJ, Chen BZ,
Cheng WH, Lu WH, Sun GC, Yeh TC, Hsiao M, et al: Resveratrol
decreases fructose-induced oxidative stress, mediated by NADPH
oxidase via an AMPK-dependent mechanism. Br J Pharmacol.
171:2739–2750. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Acute Respiratory Distress Syndrome
Network; Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT
and Wheeler A: Ventilation with lower tidal volumes as compared
with traditional tidal volumes for acute lung injury and the acute
respiratory distress syndrome. N Engl J Med. 342:1301–1308. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Caudrillier A, Kessenbrock K, Gilliss BM,
Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z and Looney MR:
Platelets induce neutrophil extracellular traps in
transfusion-related acute lung injury. J Clin Invest.
122:2661–2671. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li YW, Zhang Y, Zhang L, Li X, Yu JB,
Zhang HT, Tan BB, Jiang LH, Wang YX, Liang Y, et al: Protective
effect of tea polyphenols on renal ischemia/reperfusion injury via
suppressing the activation of TLR4/NF-κB p65 signal pathway. Gene.
542:46–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lieberman L, Petraszko T, Yi QL, Hannach B
and Skeate R: Transfusion-related lung injury in children: A case
series and review of the literature. Transfusion. 54:57–64. 2014.
View Article : Google Scholar
|
|
44
|
Niu H, Chacko J, Hadwiger G and Welch JS:
Absence of natural intracellular retinoids in mouse bone marrow
cells and implications for PML-RARA transformation. Blood Cancer J.
5:e2842015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Morriss-Kay G, Ward S and Sokolova N: The
role of retinoids in normal development. In: Use of Mechanistic
Information in Risk Assessment: Proceedings of the 1993 EUROTOX
Congress Meeting Held; Uppsala, Sweden. June 30-July 3, 1993;
Springer Science and Business Media; 2012, 16. pp. 112
|
|
46
|
Di Francesco AM, Cusano G, Franzese O,
Orienti I, Falconi M, Vesci L and Riccardi R: Resistance to the
atypical retinoid ST1926 in SK-N-AS cells selected the subline
rAS-ST with enhanced sensitivity to ATRA mediated by not
conventional mechanisms: DNA damage, G2 accumulation and late
telomerase inhibition. Toxicol In Vitro. 29:1628–1638. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chiron D, Dousset C, Brosseau C, Touzeau
C, Maïga S, Moreau P, Pellat-Deceunynck C, Le Gouill S and Amiot M:
Biological rational for sequential targeting of Bruton tyrosine
kinase and Bcl-2 to overcome CD40-induced ABT-199 resistance in
mantle cell lymphoma. Oncotarget. 6:8750–8759. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cani PD, Bibiloni R, Knauf C, Waget A,
Neyrinck AM, Delzenne NM and Burcelin R: Changes in gut microbiota
control metabolic endotoxemia-induced inflammation in high-fat
diet-induced obesity and diabetes in mice. Diabetes. 57:1470–1481.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hidalgo MA, Romero A, Figueroa J, Cortés
P, Concha II, Hancke JL and Burgos RA: Andrographolide interferes
with binding of nuclear factor-kappaB to DNA in HL-60-derived
neutrophilic cells. Br J Pharmacol. 144:680–686. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lee L, Kelher MR, Moore EE, Banerjee A and
Silliman CC: Hypertonic saline inhibits arachidonic acid priming of
the human neutrophil oxidase. J Surg Res. 174:24–28. 2012.
View Article : Google Scholar
|
|
51
|
Noack M and Miossec P: Th17 and regulatory
T cell balance in autoimmune and inflammatory diseases. Autoimmun
Rev. 13:668–677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pasarica M, Sereda OR, Redman LM, Albarado
DC, Hymel DT, Roan LE, Rood JC, Burk DH and Smith SR: Reduced
adipose tissue oxygenation in human obesity: Evidence for
rarefaction, macrophage chemotaxis, and inflammation without an
angiogenic response. Diabetes. 58:718–725. 2009. View Article : Google Scholar :
|
|
53
|
Xu CQ, Liu BJ, Wu JF, Xu YC, Duan XH, Cao
YX and Dong JC: Icariin attenuates LPS-induced acute inflammatory
responses: Involvement of PI3K/Akt and NF-kappaB signaling pathway.
Eur J Pharmacol. 642:146–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Schwartz EA, Zhang WY, Karnik SK, Borwege
S, Anand VR, Laine PS, Su Y and Reaven PD: Nutrient modification of
the innate immune response: A novel mechanism by which saturated
fatty acids greatly amplify monocyte inflammation. Arterioscler
Thromb Vasc Biol. 30:802–808. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jia Z, Nallasamy P, Liu D, Shah H, Li JZ,
Chitrakar R, Si H, McCormick J, Zhu H, Zhen W, et al: Luteolin
protects against vascular inflammation in mice and
TNF-alpha-induced monocyte adhesion to endothelial cells via
suppressing IκBα/NF-κB signaling pathway. J Nutr Biochem.
26:293–302. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Manna P, Ghosh M, Ghosh J, Das J and Sil
PC: Contribution of nano-copper particles to in vivo liver
dysfunction and cellular damage: Role of IκBα/NF-κB, MAPKs and
mitochondrial signal. Nanotoxicology. 6:1–21. 2012. View Article : Google Scholar
|
|
58
|
Kawai T and Akira S: Signaling to
NF-kappaB by Toll-like receptors. Trends Mol Med. 13:460–469. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Baker RG, Hayden MS and Ghosh S: NF-κB,
inflammation, and metabolic disease. Cell Metab. 13:11–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chouchani ET, Pell VR, Gaude E,
Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN,
Smith AC, et al: Ischaemic accumulation of succinate controls
reperfusion injury through mitochondrial ROS. Nature. 515:431–435.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lipovsky A, Nitzan Y, Gedanken A and
Lubart R: Antifungal activity of ZnO nanoparticles - the role of
ROS mediated cell injury. Nanotechnology. 22:1051012011. View Article : Google Scholar
|
|
62
|
Beladi-Mousavi SS, Hajibabaei K, Tamadon
MR and Rafieian-Kopaei M: Relationship between free radicals and
risk of kidney diseases; the role of antioxidants and their
reaction mechanisms. Ann Res Antioxid. 1:e022016.
|
|
63
|
Sousa RHV, Carvalho FEL, Ribeiro CW,
Passaia G, Cunha JR, Lima-Melo Y, Margis-Pinheiro M and Silveira
JA: Peroxisomal APX knockdown triggers antioxidant mechanisms
favourable for coping with high photorespiratory
H2O2 induced by CAT deficiency in rice. Plant
Cell Environ. 38:499–513. 2015. View Article : Google Scholar
|
|
64
|
Kusunoki C, Yang L, Yoshizaki T, Nakagawa
F, Ishikado A, Kondo M, Morino K, Sekine O, Ugi S, Nishio Y, et al:
Omega-3 polyunsaturated fatty acid has an anti-oxidant effect via
the Nrf-2/HO-1 pathway in 3T3-L1 adipocytes. Biochem Biophys Res
Commun. 430:225–230. 2013. View Article : Google Scholar
|
|
65
|
Kah J, Volz T, Lütgehetmann M and Dandri
M: Hemoxygenase-1 and its downstream product Biliverdin suppress
HCV replication and provides hepatoprotective effects in humanized
uPA/SCID mice. Zeitschrift für Gastroenterologie. 52:5–17. 2014.
View Article : Google Scholar
|
|
66
|
Staitieh BS, Ding L, Neveu WA, Spearman P,
Guidot DM and Fan X: HIV-1 decreases Nrf2/ARE activity and
phagocytic function in alveolar macrophages. J Leukoc Biol. May
26–2017.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Sun WH, Liu F, Chen Y and Zhu YC: Hydrogen
sulfide decreases the levels of ROS by inhibiting mitochondrial
complex IV and increasing SOD activities in cardiomyocytes under
ischemia/reperfusion. Biochem Biophys Res Commun. 421:164–169.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lan A, Liao X, Mo L, Yang C, Yang Z, Wang
X, Hu F, Chen P, Feng J, Zheng D, et al: Hydrogen sulfide protects
against chemical hypoxia-induced injury by inhibiting ROS-activated
ERK1/2 and p38MAPK signaling pathways in PC12 cells. PLoS One.
6:e259212011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Choi DC, Lee JY, Lim EJ, Baik HH, Oh TH
and Yune TY: Inhibition of ROS-induced p38MAPK and ERK activation
in microglia by acupuncture relieves neuropathic pain after spinal
cord injury in rats. Exp Neurol. 236:268–282. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Cheng PW, Ho WY, Su YT, Lu PJ, Chen BZ,
Cheng WH, Lu WH, Sun GC, Yeh TC, Hsiao M, et al: Resveratrol
decreases fructose-induced oxidative stress, mediated by NADPH
oxidase via an AMPK-dependent mechanism. Br J Pharmacol.
171:2739–2750. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Cheng PW, Lee HC, Lu PJ, Chen HH, Lai CC,
Sun GC, Yeh TC, Hsiao M, Lin YT, Liu CP, et al: Resveratrol
inhibition of Rac1-derived reactive oxygen species by AMPK
decreases blood pressure in a fructose-induced rat model of
hypertension. Sci Rep. 6:253422016. View Article : Google Scholar : PubMed/NCBI
|