Introduction
Fracture repair progresses through the following
stages: Hematoma formation, fibrocartilaginous callus formation,
bony callus formation and bone remodeling (1,2).
Fractures are often accompanied by ruptured blood vessels, thus
causing local hypoxia. In a hypoxic microenvironment, bone marrow
mesenchymal stem cells (BMSCs) differentiate into chondrocytes and
form an area of cartilage in the central region of the fracture.
Mature cartilage cells can secrete a large amount of cartilage
matrix factors, and new blood vessels then grow into the
cartilaginous callus along the matrix. Revascularization improves
the local oxygen supply, stimulating osteoblastic differentiation
of BMSCs and bone-lining cells in coordination with bone
morphogenetic proteins (BMPs), runt-related transcription factor 2
(RUNX2) and other cytokines, thus promoting endochondral bone
formation and matrix calcification, which subsequently form the
bony callus and ultimately lead to fracture healing (3–5).
Tissue hypoxia can induce angiogenesis following
fracture, and hypoxia-inducible factor (HIF) is the key protein
expressed in most tissues in response to localized low oxygen
tension (6). Under normoxic
conditions, HIF1α binds to the von Hippel-Lindau tumor suppressor
(pVHL) and is subsequently hydrolyzed. Conversely, this process is
suppressed under hypoxic conditions, and HIF1α forms dimers with
HIF1β, which then enter the nucleus and activate transcription of
HIF target genes (7–9). Vascular endothelial growth factor
(VEGF) is the most important HIF target gene (10,11). A significant reduction in the
formation of blood vessels and bone is observed in vascular
endothelial-cell-specific HIF1α-deficient mice. Significantly
increased metaphyseal vascular volume and bone formation have been
observed in pVHL knockout mice, whereas administration of a VEGF
receptor antagonist can offset angiogenesis caused by excessive
activation of HIF1α (10).
Reduced bone density, as well as decreased amounts of trabecular
bone and thinner cortical bone have been detected in VEGF knockout
mice. In vitro studies have demonstrated that osteoblasts in
VEGF knockout mice exhibit diminished capacity for differentiation
and maturation (12).
Parathyroid hormone (PTH) is an 84-amino acid
peptide hormone secreted by the parathyroid gland. Numerous studies
have demonstrated that intermittent low-dose injections of
recombinant human (rh)PTH can increase bone anabolism and promote
fracture healing (13,14). Conversely, trabecular bone volume
and osteoblast number are significantly reduced (15,16), and fracture healing is delayed in
PTH knockout (PTHKO) mice (17).
At present, the mechanisms by which PTH promotes fracture healing
remain to be elucidated; stimulation of angiogenesis may be one of
the mechanisms (18–20). A previous study indicated that the
effects of PTH on osteogenesis are predominantly mediated by
adenylate cyclase (AC)/cyclic adenosine monophosphate
(cAMP)/protein kinase A (PKA)/cAMP response element-binding protein
pathway (21). In addition, it
has been confirmed that the activated
cAMP/PKA/phosphorylated-serine/threonine protein kinase
(pAKT)/HIF1α/VEGF signaling pathway can promote angiogenesis
(22). Previous studies have
revealed that PTH can quickly promote the expression of VEGF in
osteoblasts (13,23). Therefore, the present study
hypothesized that PTH may affect fracture healing through its
effects on angiogenesis mediated by the PKA/pAKT/HIF1α/VEGF
pathway. In the present study, a PTHKO mouse fracture model was
constructed, and fracture healing and the expression of associated
angiogenic factors were measured, in order to investigate whether
endogenous PTH affects angiogenesis via the PKA/pAKT/HIF1α/VEGF
pathway, thus stimulating fracture healing.
Materials and methods
Reagents
Anti-mouse platelet endothelial cell adhesion
molecule (PECAM; 3528; 1:100) and GAPDH (5174/51332) were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA) and
osteocalcin (OCN; ab93876; 1:400) monoclonal antibodies were
purchased from Abcam (Cambridge, MA, USA). RUNX2 (ab76956; 1:600),
HIF1α (ab8366; 1:600), pVEGF receptor 2 (pVEGFR2; ab131241; 1:200),
proliferating cell nuclear antigen (PCNA; ab92552; 1:400), VEGF
(ab46154; 1:200), PKA (ab75991) and pAKT monoclonal antibodies
(ab81283) were purchased from Abcam (Cambridge, MA, USA). Type II
collagen (COL II; sc-7763; 1:200) primary antibody and all
secondary antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Streptavidin-biotin complex (SABC)
immunohistochemistry kit and DAB chromogenic kit were purchased
from Vector Laboratories, Inc. (Burlingame, CA, USA). Polymerase
chain reaction (PCR) kit (PCR kit Takara A5003) was provided by
Takara Bio, Inc. (Otsu, Japan). High glucose or low glucose
Dulbecco's modified Eagle's medium (DMEM), and fetal bovine serum
were purchased from HyClone; GE Healthcare (Logan, UT, USA).
Penicillin-streptomycin double-resistant solution was purchased
from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Dexamethasone, β-glycerophosphate and rhPTH (1–34)
were purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
PBS and total protein extraction kit [containing
radioimmuno-precipitation assay (RIPA) buffer and
phenylmethylsulfonyl fluoride) were provided by Beyotime Institute
of Biotechnology (Nantong, China). mRNA extraction kit was
purchased from Qiagen GmbH (Hilden, Germany). Alkaline phosphatase
(ALP) staining kit was provided by Nanjing Jiancheng Bioengineering
Institute (Nanjing, China). Osteogenic induction medium was
prepared by adding dexamethasone, ascorbic acid and
β-glycerophosphate to basal medium and serum at final
concentrations of 10 nmol/l, 50 µg/ml and 10 mmol/l,
respectively.
Construction and identification of PTHKO
mice
Briefly, the PTHKO mouse model was constructed by
inserting a neomycin resistance gene (NEO) into exon III of the
mouse PTH gene to replace the entire mature PTH coding sequence
(24). Parental knockout mice
were provided by McGill University (Montreal, QC, Canada), and were
housed in a specific pathogen-free (SPF) standard animal facility
under the following conditions: Humidity, 45–75%; temperature,
22–26°C; 12 h light/12 h dark cycle, in the specific-pathogen-free
animal facility at the Experimental Animal Center of Nanjing
Medical University (Nanjing, China); with ad libitum access
to food and water. Mouse genotypes and PTH gene knockout were
confirmed by PCR amplification of DNA fragments. Primer sequences
were as follows: PTH, forward 5′-GATGTGTGCAAACACCGTGGCTAA-3′,
reverse 5′-TCCAAAGTTTCATTACAGTAGAAG-3′; and NEO, forward
5′-TCTTGATTCCCACTTTGTGGTTCTA-3′, reverse primer sequence was the
same as the PTH forward primer. Annealing temperature was 55°C. For
experiments, 18 two-month-old PTHKO mice and 15 wild-type (WT)
littermates were randomly selected regardless of gender. Their
weigh was 20–30 g, WT were heavier than the PTHKO mice. The present
study was approved by the Ethics Committee of Nanjing Medical
University.
Establishment and analysis of the murine
fracture model
The murine fracture model was established as
follows: Mice were anesthetized with an intraperitoneal injection
of ketamine/xyla-zine. Subsequently, skin tissue around the knee
was disinfected and a 25G needle was inserted through the patellar
tendon into the femoral canal. A femoral fracture was created by
3-point bending using an Einhorn device, and was confirmed by X-ray
examination (Faxitron Bioptics, LLC, Tucson, AZ, USA). Each mouse
received daily subcutaneous injections of 0.5 mg/kg buprenorphine
for 3 consecutive days (25).
Fracture healing was assessed by X-ray radiography and callus
formation was examined using Skyscan 1176 high-resolution
micro-computerized tomography (micro-CT) (Bruker Corporation,
Billerica, MA, USA) at 1 and 2 weeks after establishment of the
fracture model.
Cell culture
For BMSCs culture, mice were sacrificed and
immediately immersed in 75% alcohol for 10 min. Bilateral femurs
were then removed under sterile conditions, and the marrow cavity
was repeatedly flushed using serum-free low glucose DMEM to obtain
a single-cell suspension. Cells were collected by centrifugation at
1,200 × g/min for 5 min, then resuspended, inoculated into culture
flasks, and incubated in a humidified incubator in an atmosphere
containing 5% CO2 at 37°C. The 3rd generation of BMSCs
was induced to differentiate into osteoblasts. Briefly, BMSCs from
WT (+/+) and PTHKO (−/−) mice (5×106/ml) were inoculated
into 6-well plates and into culture plates containing slides;
induction medium containing exogenous PTH (final concentration,
10−7 mol/l) was added to the cells once they reached 80%
confluence. Induction medium was replaced with ordinary culture
medium after 6 h. The cells were treated with PTH for 6 h every 48
h. Cells were collected 1 and 2 weeks after induction, and protein
and RNA were extracted using specific kits according to the
manufacturers' protocols. ALP staining of fixed cells was performed
using an ALP staining kit according to the manufacturer's
protocol.
Coculture of human umbilical vein
endothelial cells (HUVECs) and BMSCs
BMSCs were cultured as aforementioned (for 2 weeks)
and allowed to reach 80% confluence, after which they were treated
with induction medium, and HUVECS were cocultured with the lower
layer of BMSCs for 1 week. Dishes were observed on day 7 after the
start of coculture. Cell nuclei were stained with hematoxylin and
images were captured.
Immunohistochemical examination
Fracture calluses (captured 1 and 2 weeks after
femur fracture) were fixed in 10% formalin for 12–24 h at room
temperature (20–25°C), decalcified in 5% EDTA-2Na for 4–5 weeks,
dehydrated through a graded ethanol series, cleared in xylene,
embedded in paraffin wax and cut into 5-µm sections.
Histological observations were performed after hematoxylin and
eosin (H&E) staining, cartilage morphology was observed using
alcian blue staining, and differences in osteoblasts were examined
by ALP staining. Immunohistochemical staining, followed by SABC and
DAB color development were conducted according to the
manufacturer's protocols. Images of sections were captured using a
light microscope; four fields of each section were randomly
selected, and the positive expression rate and gray values of
stained areas were measured under the same light intensity to
semi-quantify protein expression levels using Image-Pro-Plus 6.0
(Media Cybernetics, Inc., Rockville, MD, USA).
Western blot analysis
Cells were lysed with RIPA lysis buffer, total
protein was extracted and concentration was measured using the
Bradford method. Proteins (10 µg) were separated by 12%
SDS-PAGE. Following SDS-PAGE, protein samples were transferred to
polyvinylidene fluoride (PVDF) membranes (EMD Millipore, Billerica,
MA, USA). PVDF membranes were then incubated with primary
antibodies (OCN, 1 µg/ml; RUNX2, 2 µg/ml; VEGF, 1
µg/ml; pVEGFR2, 1 µg/ml; HIF1α, 1 µg/mL; PKA,
0.5 µg/ml; AKT, 1 µg/ml; pAKT, 1 µg/ml)
overnight at 4°C, followed by incubation with horseradish
peroxidase-conjugated secondary antibody for 2 h at 37°C. Enhanced
chemiluminescence (Beyotime Institute of Biotechnology) was used to
visualize the bands, and blots were analyzed using Quantity One
software (version 4.62; Bio-Rad Laboratories Inc., Hercules, CA,
USA).
Reverse transcription-quantitative
(RT-qPCR)
Total RNA was extracted from adherent cells using a
kit according to the manufacturer's protocol. Concentration and
purity of mRNA were determined using an ultraviolet spectrometer.
RT was performed using the Superscript™ first-strand synthesis
system (Invitrogen; Thermo Fisher Scientific, Inc.) in RT reaction
buffer (Takara Bio, Inc.) according to Takara PCR kit protocol. The
resulting cDNA was quantified using a dual hybridization probe
system (forward primer, 5-GATGTCTGCAAACACCGTGGCTAA-3, reverse
primer, 5-TCCAAAGTTTCATTACAGTAGAAG-3; Neo,
5-TCTTGATTCCCACTTTGTGGTTCTA-3; Roche Diagnostics) and a semi-qPCR
procedure according to the manufacturer's protocol (Takara Bio,
Inc.). The PCR conditions were as follows: 1 cycle at 95°C for 4
min, 30 cycles at 95°C for 30 sec, 30 cycles at 55°C for 30 sec, 30
cycles at 72°C for 30 sec, 1 cycle at 72°C for 10 min and then 1
cycle at 4°C. The thermocycling conditions were as follows:
degeneration at 95°C for 4 min; extension at 72°C; renaturation at
55°C.
Statistical analysis
All experimental data are presented as the means ±
standard deviation, and were processed using SPSS 13.0 software
(SPSS, Inc., Chicago, IL, USA). All experiments were repeated
independently three times. Differences among groups were analyzed
by independent-samples t-test or single-factor analysis of
variance. IHC data were analysed by ANOVA followed by Boferroni's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference (26).
Results
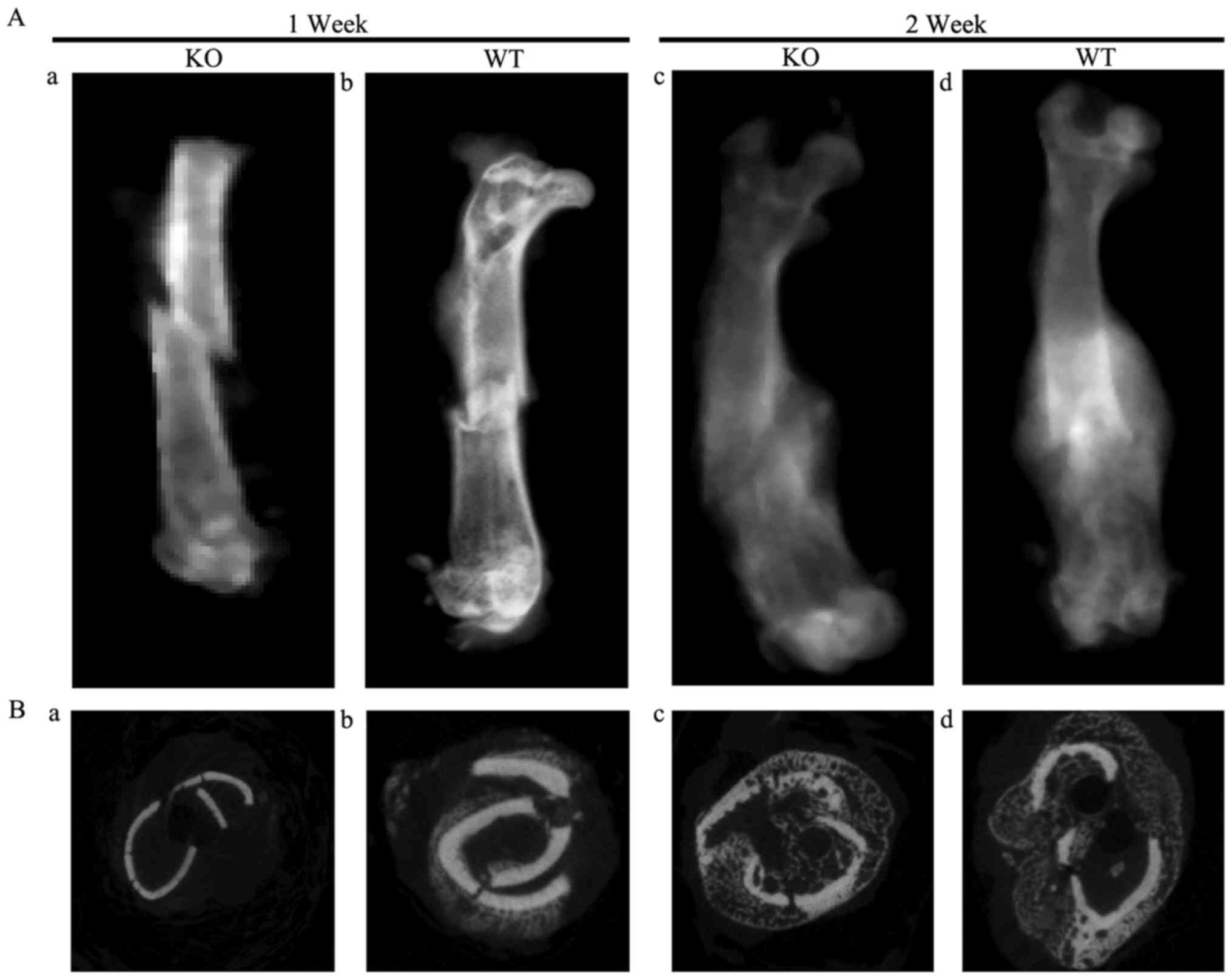
Delayed fracture healing in PTHKO
mice
X-ray examination demonstrated that fracture lines
were clearly visible and a small region of calcification was
observed in the surrounding area close to fractures in WT and PTHKO
mice 1 week after fracture. However, the calcified region in the
PTHKO mice was smaller than that in the WT mice (Fig. 1Aa and b). Furthermore, fracture
lines were less distinct in WT mice 2 weeks after fracture, but
remained clearly visible in PTHKO mice. PTHKO mice exhibited
relatively smaller calluses compared with WT mice (Fig. 1Ac and d).
Coronal micro-CT views and three-dimensional surface
reconstruction analysis of fracture lines revealed no differences
between bone volume (BV), total volume (TV), bone surface (BS),
BS/TV and trabecular number (Tb.N) in PTHKO mice compared with WT
mice at 1 week after fracture. However, PTHKO mice had
significantly lower BV/TV ratio, reduced trabecular thickness
(Tb.Th), larger trabecular spacing (Tb.Sp) and increased porosity
compared with WT mice (Fig. 1Ba and
b). A total of 2 weeks after fracture, PTHKO mice exhibited
significantly lower BV, TV, BV/TV, BS/TV, Tb.N and Tb.Th, and
substantially increased Tb.Sp and porosity compared with in WT
mice, thus indicating that PTHKO mice displayed reduced
ossification, poor trabecular development and cancellous bone
during fracture healing (Fig. 1Bc and
d).
H&E staining of tissue sections demonstrated
that PTHKO mice developed a significantly smaller cartilaginous
callus compared with WT mice (Fig.
2Aa and b); even though callus formation was detected in both
groups 1 week after fracture. WT mice exhibited normal endochondral
bone formation 2 weeks after fracture (Fig. 2Ad). Osteoid was formed in and
surrounding the cartilage expansion area and cartilage area was
reduced (Fig. 2Ad). Conversely,
endochondral bone formation was observed only in regions adjacent
to the cartilage expansion area in PTHKO mice; chondrocytes were
arranged in an abnormal manner, and cartilage areas were
significantly larger than those in WT mice (Fig. 2Ac). The results of
cartilage-specific alcian blue staining were similar to those
detected by H&E staining, but cartilaginous callus is more
significant (Fig. 2B). COL II is
secreted by chondrocytes and is an important component of the
cartilage matrix (27).
Immunohistochemical staining revealed that PTHKO mice exhibited
impaired ability to secrete COL II in addition to a diminished
capacity for endochondral bone formation. WT mice exhibited strong
positive staining for COL II 1 week after fracture, whereas COL II
expression was identified only in the central region of the
cartilage expansion area in PTHKO mice (Fig. 2Ca and b). Although the expression
levels of COL II were slightly increased in PTHKO mice, it remained
lower than in WT mice 2 weeks after fracture (Fig. 2Cc and d).
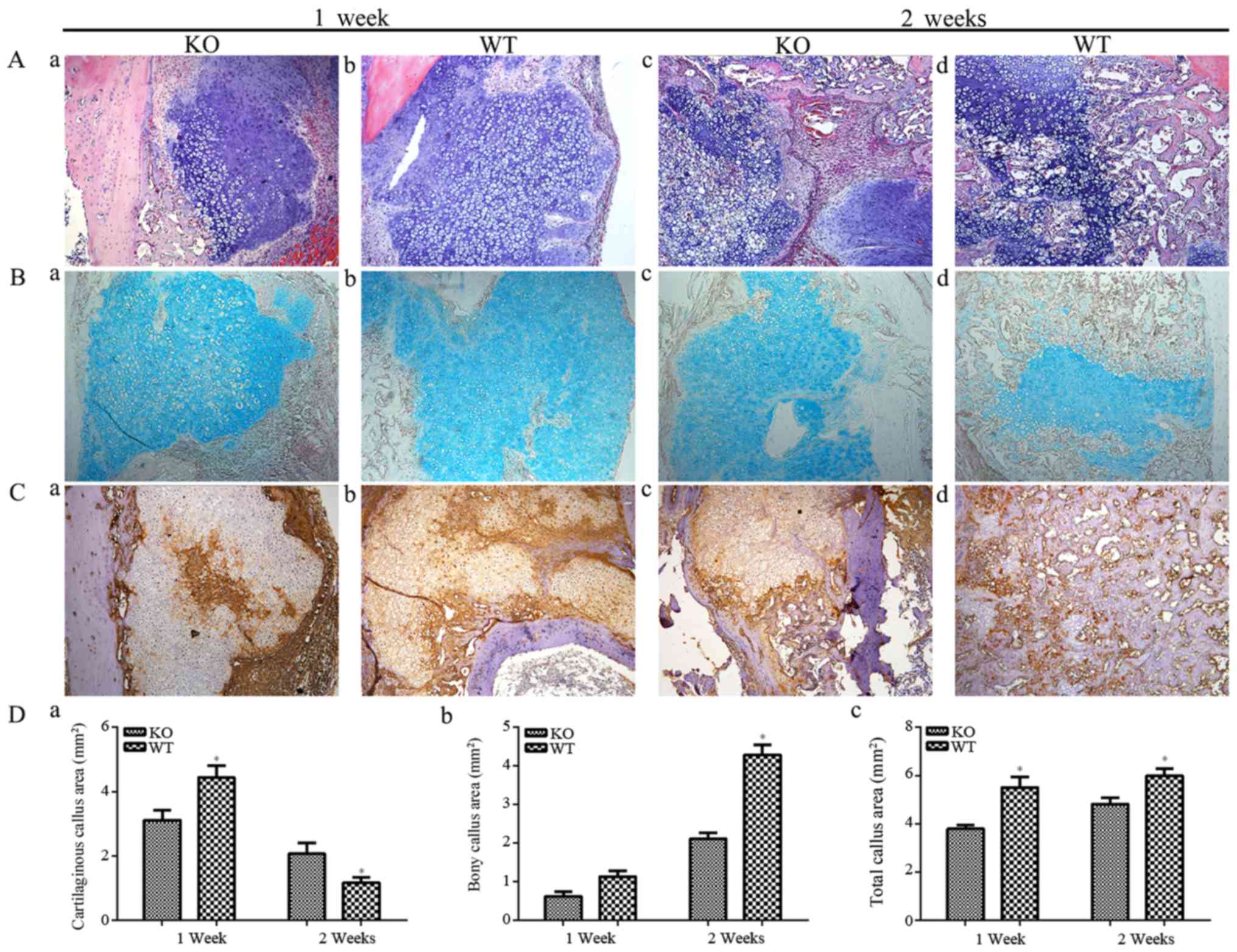 | Figure 2(A) H&E staining (magnification,
×100) of fractures. (a) In PTHKO mice, fractures developed a
smaller cartilaginous callus compared with in (b) WT mice 1 week
after fracture. (c) In PTHKO mice at 2 weeks after fracture, little
endochondral bone formation and a large cartilage area were
detected. (d) In WT mice at 2 weeks after fracture, normal
endochondral bone formation and decreased cartilage area were
detected. (B) Alcian blue staining (magnification, ×100) of
fractures. Light blue area shows cartilaginous callus, which was
consistent with the results of H&E staining. (C)
Immunohistochemical staining of COL II in fractures (magnification,
×100) in (a) PTHKO and (b) WT mice 1 week after fracture. WT mice
exhibited strong positive staining for COL II; however, COL II
expression was detected only in the central region of the cartilage
expansion area in PTHKO mice. Immunohistochemical staining of COL
II in (c) PTHKO and (d) WT mice 2 weeks after fracture. COL II
expression was slightly increased in PTHKO mice, but remained lower
than that in WT mice. (D) Statistical analysis of (a) cartilaginous
callus area, (b) bony callus area and (c) total callus area.
*P<0.05. PTH, parathroid hormone; PTHKO, PTH
knockout; WT, wild-type. |
Defective bone formation in PTHKO
mice
Expression of OCN, an indicator of osteoblast
activity, was examined to further confirm alterations in
endochondral bone formation in PTHKO mice (Fig. 3A). Immunohistochemical staining
detected higher levels of OCN expression in WT mice 1 week after
fracture (Fig. 3Ab), whereas OCN
expression was much lower and was observed primarily around the
edge of the callus in PTHKO mice (Fig. 3Aa). In addition, PTHKO mice
exhibited significantly lower OCN expression compared with in WT
mice at 2 weeks; however, expression levels were increased in WT
and PTHKO mice 2 weeks after fracture (Fig. 3Ac and d).
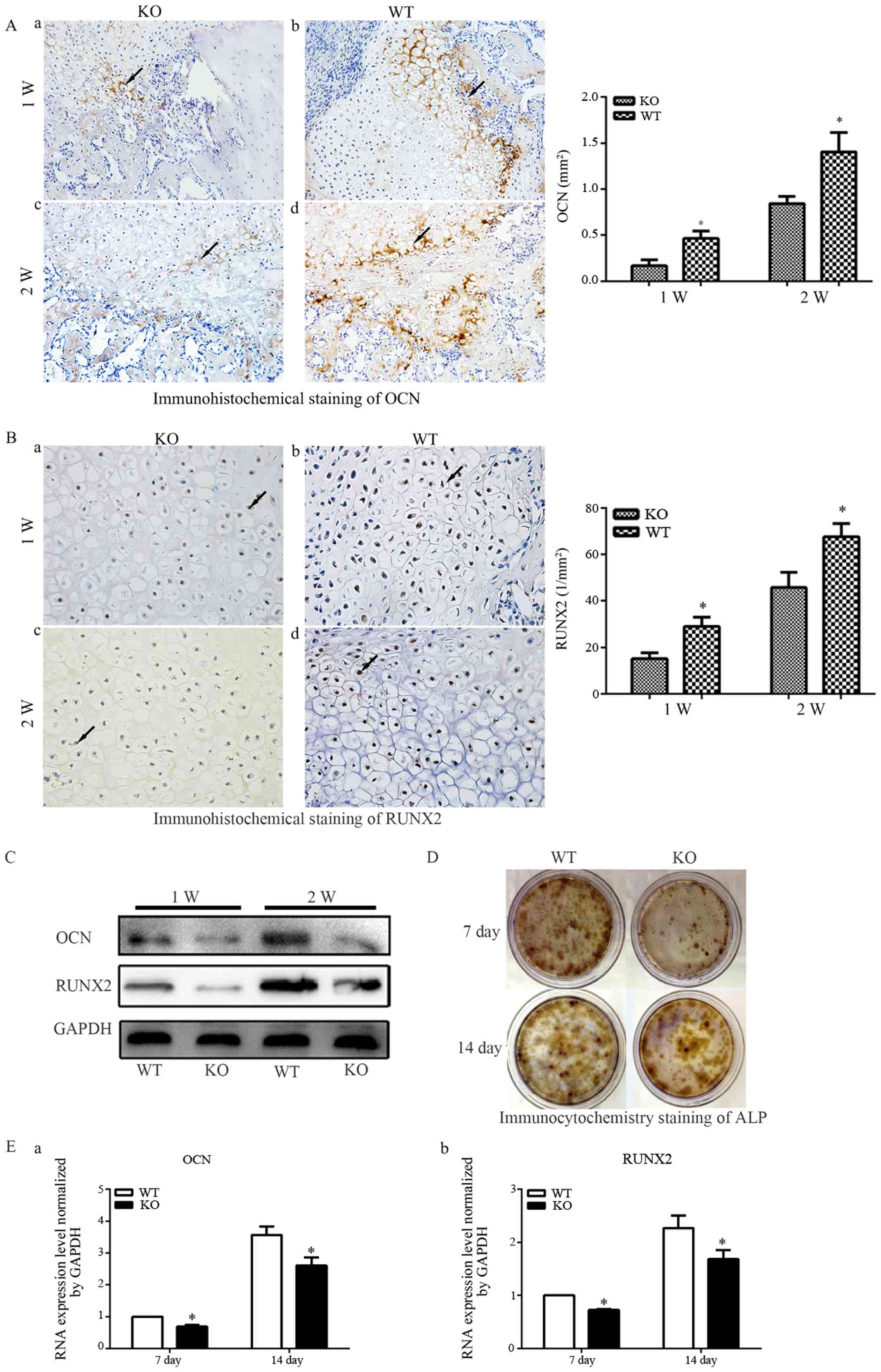 | Figure 3(A) Immunohistochemical staining of
OCN (magnification, ×200) in fractures of (a) PTHKO and (b) WT mice
1 week after fracture. OCN expression was significantly lower in
PTHKO mice compared with in WT mice and was primarily distributed
around the edge of the callus. Immunohistochemical staining of OCN
in fractures of (c) PTHKO and (d) WT mice 2 weeks after fracture.
OCN expression was significantly reduced in PTHKO mice compared
with in WT mice; however, it was increased in both groups compared
with after 1 week. (B) Immunohistochemical staining of RUNX2
(magnification, ×400) in fractures of (a) PTHKO and (b) WT mice 1
week after fracture. RUNX2 expression was significantly lower in
PTHKO mice compared with in WT mice, although it was relatively
strong in the area surrounding the cartilaginous callus in both
groups. Immunohistochemical staining of RUNX2 in fractures of (c)
PTHKO and (d) WT mice 2 weeks after fracture. RUNX2 was strongly
expressed in WT mice, whereas RUNX2 expression remained lower in
PTHKO mice compared with in WT mice, and was mainly concentrated in
the outer periphery of the cartilaginous callus. Black arrows
indicate positive areas. (C) Callus proteins, including OCN and
RUNX2, were detected using western blot analysis. (D) ALP
immunocytochemistry. Reduced ALP staining intensity was detected in
PTHKO mice compared with in WT mice. (E) Reverse
transcription-quantiative polymrease chain reaction analysis of
mRNA expression of (a) OCN and (b) RUNX2 in BMSC-derived
osteoblasts; the mRNA expression levels were significantly lower in
PTHKO mice compared with in WT mice 1 and 2 weeks after induction.
*P<0.05. ALP, alkaline phosphatase; BMSC, bone marrow
mesenchymal stem cells; OCN, osteocalcin; PTH, parathroid hormone;
PTHKO, PTH knockout; RUNX2, runt-related transcription factor 2;
WT, wild-type. |
RUNX2 is an important regulatory factor that
promotes bone formation (28).
Immunohistochemical examination detected relatively strong RUNX2
expression in the area surrounding the cartilage callus 1 week
after fracture; however, expression was significantly reduced in
PTHKO mice compared with in WT mice (Fig. 3Ba and b). WT mice exhibited strong
positive staining for RUNX2 2 weeks after fracture, whereas RUNX2
expression remained lower in PTHKO mice compared with in WT mice,
and was concentrated in areas adjacent to the cartilage callus
(Fig. 3Bc and d).
Callus-specific proteins, including OCN and RUNX2,
were semi-quantified by western blot analysis. The results were
consistent with those of immunohistochemical staining (Fig. 3C).
BMSCs were isolated from mouse femoral bone marrow,
and osteoblastic differentiation was induced with dexamethasone,
ascorbic acid and β-glycerophosphate, in order to investigate
whether lack of endogenous PTH affects differentiation of
mesenchymal stem cells into osteoblasts. Immunocytochemical
analysis revealed markedly lower levels of ALP in PTHKO mice
compared with in WT mice, thus indicating a diminished capacity of
BMSCs for osteoblastic differentiation in PTHKO mice (Fig. 3D). Total RNA was extracted 7 and
14 days after cell culture, and osteogenic indicators were
quantified by RT-qPCR. The results indicated that the mRNA
expression levels of osteoblast-specific proteins, including RUNX2
and OCN, were significantly lower in PTHKO mice compared with in WT
mice (Fig. 3Ea and b).
Reduced angiogenesis in PTHKO mice
Fracture healing involves the remodeling of blood
vessels (3), and endochondral
bone formation is closely associated with angiogenesis. The present
study hypothesized that defective endochondral bone formation in
PTHKO mice may be associated with decreased angiogenic capacity.
Immunohistochemical detection of the vascular specific marker PECAM
confirmed this hypothesis. The area of angiogenesis around the edge
of the callus in WT mice was significantly larger than that in
PTHKO mice (Fig. 4Aa and b) 1
week after fracture. In addition, a large number of blood vessels
were observed in the cartilage callus in WT mice 2 weeks after
fracture, whereas PTHKO mice exhibited far fewer blood vessels,
which were mainly located around the cartilage callus (Fig. 4Ac and d).
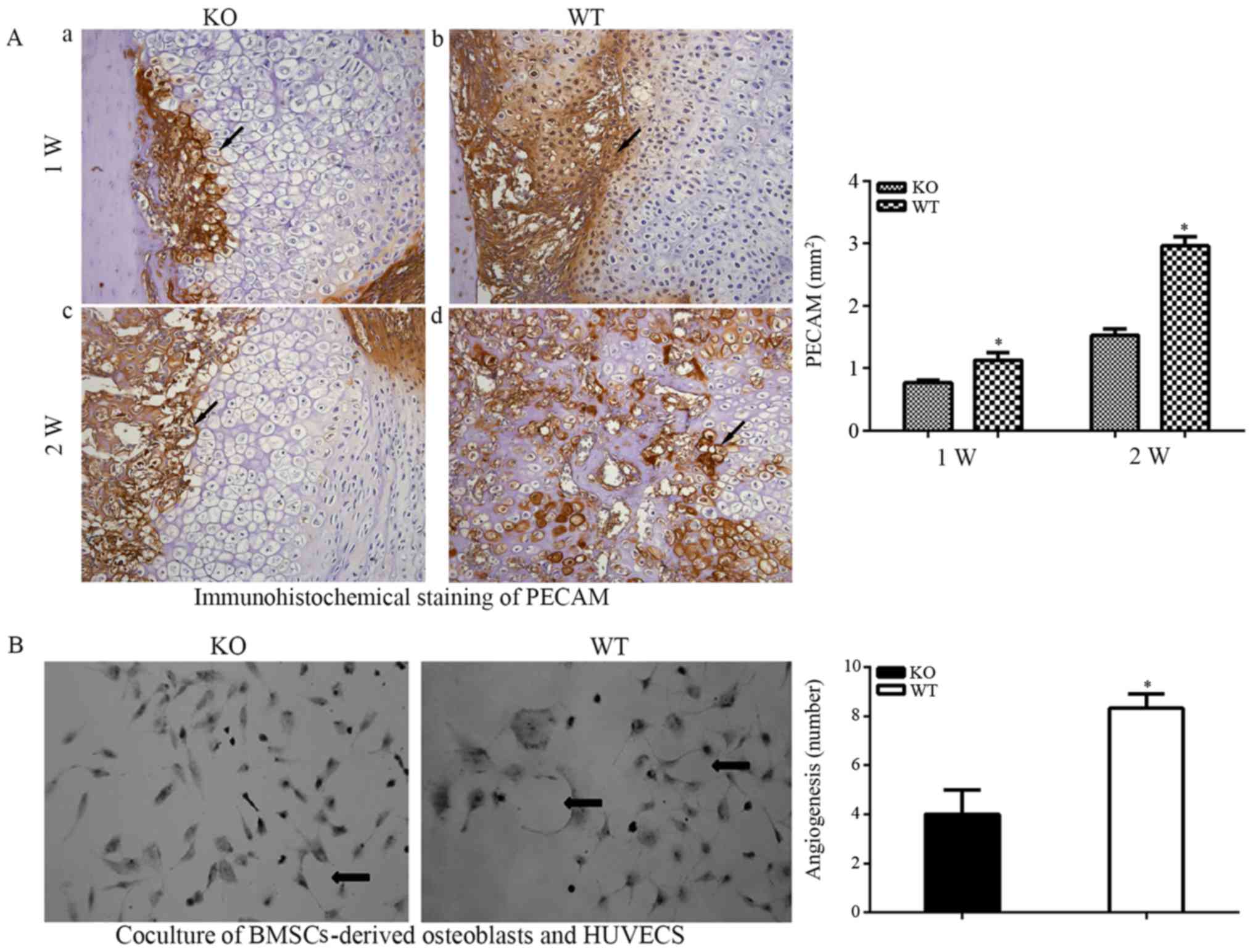 | Figure 4(A) Immunohistochemical staining of
PECAM (magnification, ×200) in fractures. (a) In PTHKO and (b) WT
mice 1 week after fracture, the area of angiogenesis was
significantly smaller in PTHKO mice compared with in WT mice. (c)
In PTHKO and (d) WT mice 2 weeks after fracture, a large number of
blood vessels were observed in the cartilaginous callus in WT mice,
whereas fewer blood vessels were detected in PTHKO mice and were
mainly located abound the cartilaginous callus. Black arrows
indicate positive areas. (B) BMSCs-derived osteoblasts were
cocultured with HUVECs for 2 weeks. HUVECs cocultured with PTHKO
BMSCs-derived osteoblasts displayed a significantly reduced
capacity for aggregation and cross-linking. Black arrows indicate
cross-linked HUVECs. *P<0.05. BMSC, bone marrow
mesenchymal stem cells; HUVECs, human umbilical vein endothelial
cells; PECAM, platelet endothelial cell adhesion molecule; PTH,
parathroid hormone; PTHKO, PTH knockout; WT, wild-type. |
BMSC-derived osteoblasts and HUVECs were cocultured
for 2 weeks, in order to confirm the adverse effects of the
impaired osteoblastic differentiation capacity of BMSCs and their
decreased angiogenic activity. The results indicated that HUVECs
cocultured with BMSCs from PTHKO mice exhibited significantly
reduced capacity for aggregation and cross-linking (Fig. 4B).
Various growth factors are required for angiogenesis
in the process of fracture healing, and VEGF is particularly
important (29,30). Immunohistochemical analysis
demonstrated that the expression of VEGF-A was significantly
increased in WT mice compared with in PTHKO mice (Fig. 5Aa and b) 1 week after fracture.
VEGF-A expression remained higher than that in WT mice 2 weeks
after fracture (Fig. 5Ac and d)
even though it was slightly increased in WT and PTHKO mice compared
with at 1 week. The receptor tyrosine kinase VEGFR2 is
phosphorylated and activated by binding to extracellular VEGF-A and
mediates almost all known VEGF-A-induced cellular responses
(31). Immunohistochemical
analysis detected pVEGFR2 in a large number of callus cells in WT
mice 1 week after fracture (Fig.
5Bb), whereas far fewer positive cells were detected in PTHKO
mice (Fig. 5Ba). In addition,
PTHKO mice possessed significantly fewer pVEGFR2-positive cells in
the cartilage callus compared with in WT mice 2 weeks after
fracture (Fig. 5Bc and d). HIF1α
expression within the callus was examined to determine the
association between HIF1α and VEGF (30). Whereas strong cytoplasmic and
endonulcear HIF1α expression was observed in WT mice 1 week after
fracture, HIF1α expression was much weaker in PTHKO mice (Fig. 5Ca and b). HIF1α expression
increased more slowly in PTHKO mice compared with in WT mice;
however, stronger expression was detected in both mice groups 2
weeks after fracture (Fig. 5Cc and
d). In addition, the protein expression levels of VEGF, pVEGFR2
and HIF1α were determined by western blot analysis. The results
were similar to those of immunohistochemical staining (Fig. 5D).
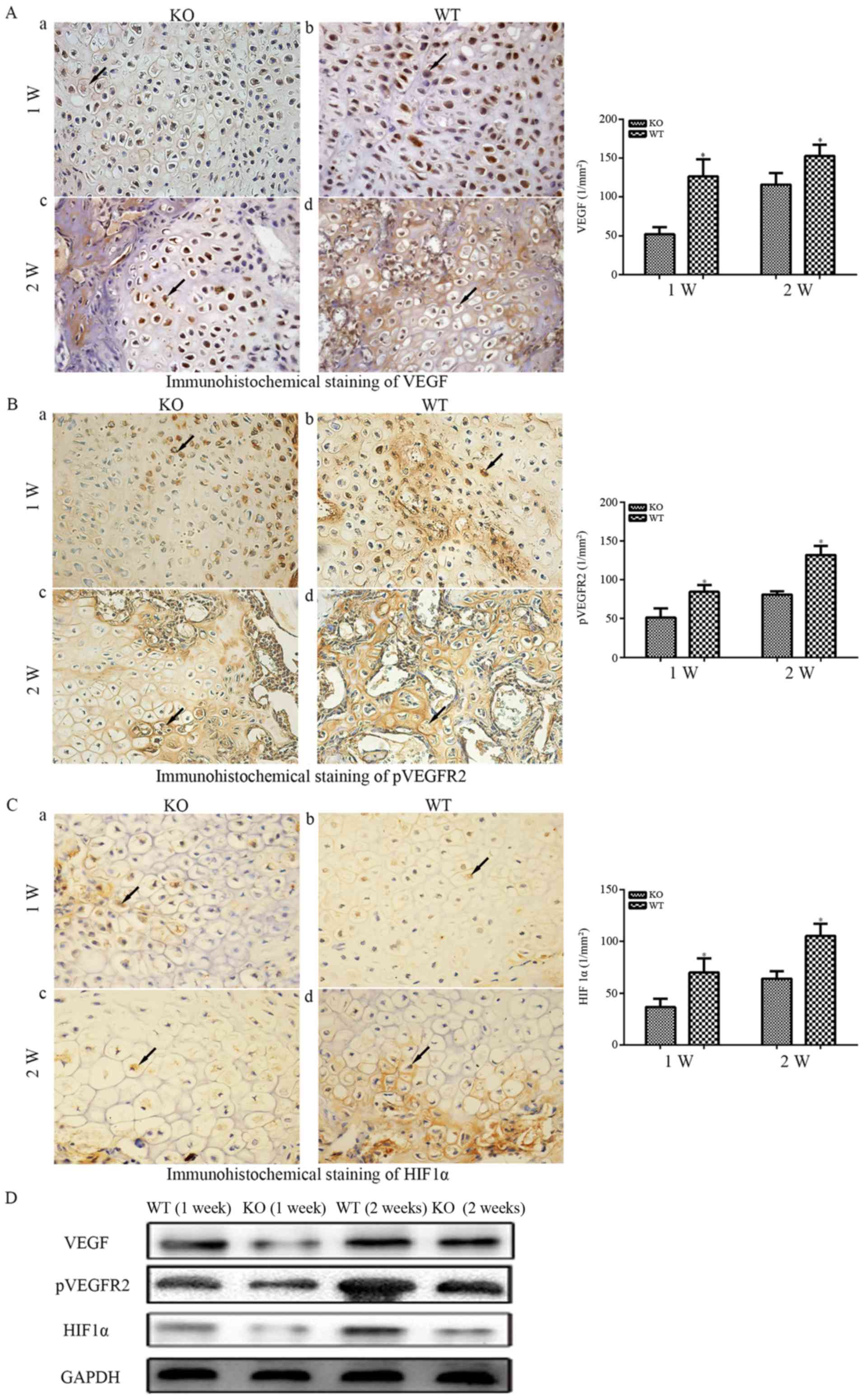 | Figure 5(A) Immunohistochemical staining of
VEGF-A (magnification, ×400) in fractures. (a) In PTHKO and (b) WT
mice 1 week after fracture, VEGF-A expression was significantly
reduced in PTHKO mice compared with in WT mice. (c) In PTHKO and
(d) WT mice 2 weeks after fracture, VEGF-A expression was increased
in PTHKO mice, but remained significantly lower than that in WT
mice. (B) Immunohistochemical staining of pVEGFR2 (magnification,
×400) in fractures. (a) In PTHKO and (b) WT mice 1 week after
fracture, a significantly smaller number of pVEGFR2-positive cells
was detected in the cartilaginous callus in PTHKO mice compared
with in WT mice. (c) In PTHKO and (d) WT mice 2 weeks after
fracture, a large number of pVEGFR2-positive cells was observed in
the cartilaginous callus in WT mice, whereas a much lower level of
angiogenesis was detected in PTHKO mice. (C) Immunohistochemical
staining for HIF1α (magnification, ×400) in fractures. (a) In PTHKO
and (b) WT mice 1 week after fracture, the expression levels of
cytoplasmic HIF1α were significantly lower in PTHKO mice. (c) In
PTHKO and (d) WT mice 2 weeks after fracture, HIF1α expression was
increased in both groups; however, the expression remained lower in
PTHKO mice compared with in WT mice. Black arrows indicate positive
areas. (D) Protein expression levels of VEGF, pVEGFR2 and HIF1α
were detected by western blot analysis. HIF1α, hypoxia inducible
factor-1α; PTH, parathroid hormone; PTHKO, PTH knockout; pVEGFR,
phosphorylated-VEGF receptor 2; VEGF, vascular endothelial growth
factor; WT, wild-type. |
Downregulation of cell proliferation and
the PKA/pAKT/HIF1α/VEGF signaling pathway in PTHKO mice
It has previously been reported that exogenous PTH
can reduce DNA damage caused by oxidative stress (32); however, the relationship between
endogenous PTH and the local oxygen environment, as well as its
association with cell proliferation, remain to be elucidated. Since
endogenous PTH can affect angiogenesis and consequently affect the
oxygen supply during fracture healing, it may also influence cell
proliferation. PCNA was measured in PTHKO and WT mice, and rapid
cell proliferation was observed in both groups 1 week after
fracture. However, the PCNA-positive rate was significantly reduced
in PTHKO mice compared with in WT mice (Fig. 6Aa and b). Cell proliferation was
reduced in both groups 2 weeks after fracture, and the
PCNA-positive rate in PTHKO mice remained lower than that in WT
mice (Fig. 6Ac and d), thus
suggesting that lack of endogenous PTH may cause reduced callus
formation by suppressing cell proliferation.
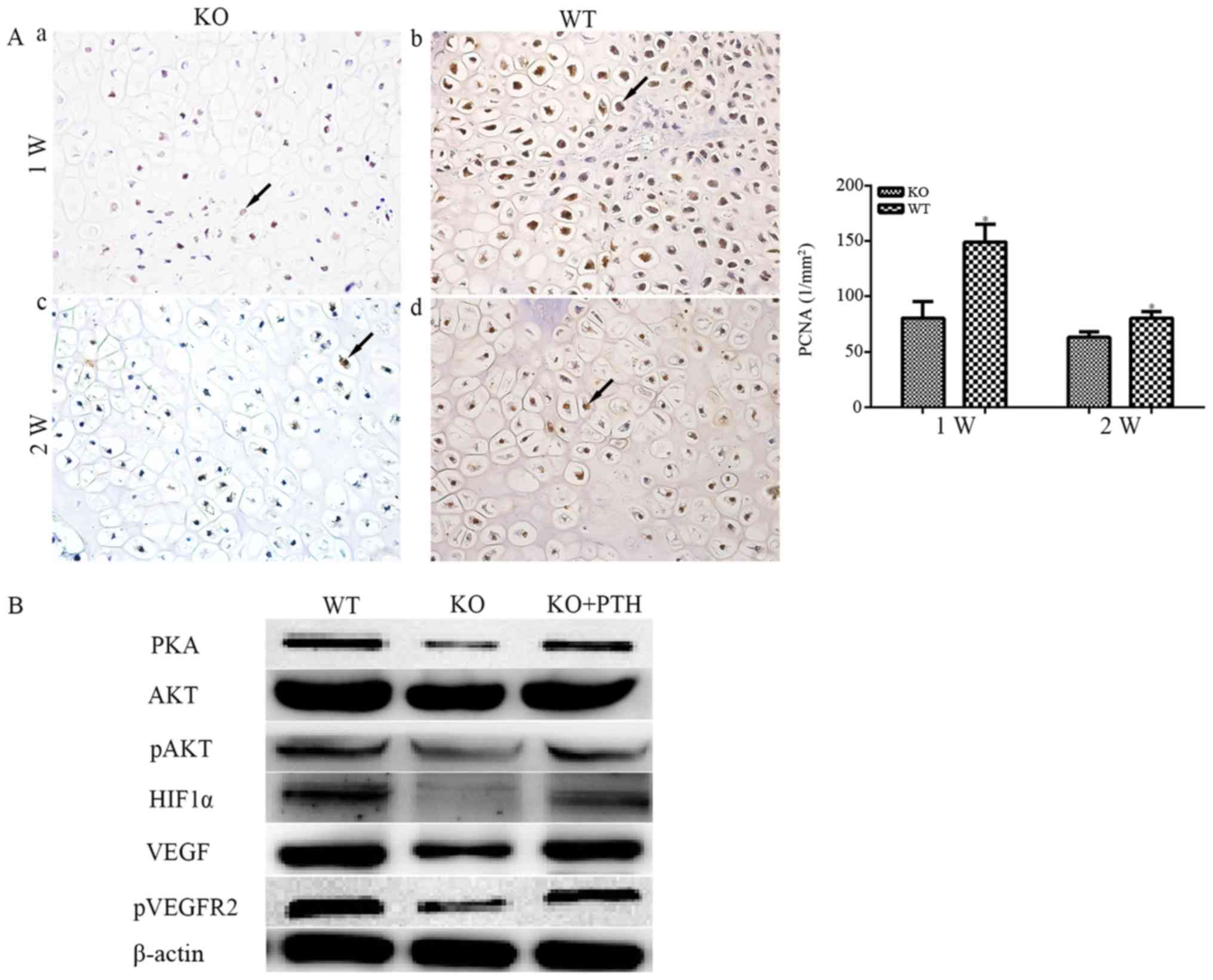 | Figure 6(A) Immunohistochemical staining of
PCNA (magnification, ×400) in fractures. (a) In PTHKO and (b) WT
mice 1 week after fracture, rapid cell proliferation was observed,
whereas PTHKO mice exhibited a significant reduction in
PCNA-positive rate compared with in WT mice. (c) In PTHKO and (d)
WT mice 2 weeks after fracture, cell proliferation in WT mice
remained faster than that in PTHKO mice; however, it slowed down in
both groups. Black arrows indicate positive areas.
*P<0.05. (B) Western blot analysis detected reduced
expression of PKA/pAKT/HIF1α/VEGF, and a lower level of pVEGFR2 in
PTHKO BMSC-derived osteoblasts 2 weeks after induction. Addition of
exogenous PTH in the culture medium partially reversed
downregulation of the PKA/pAKT/HIF1α/VEGF pathway. AKT,
serine/threonine protein kinase; BMSC, bone marrow mesenchymal stem
cell; HIF1α, hypoxia inducible factor-1α; pAKT, phosphorylated-AKT;
PCNA, proliferating cell nuclear antigen; PKA, protein kinase A;
PTH, parathroid hormone; PTHKO, PTH knockout; pVEGFR2, p-VEGFR2;
VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2;
WT, wild-type. |
Previous studies have confirmed that exogenous PTH
can promote VEGF secretion from osteoblasts (13,26). BMSCs were isolated from mouse
femoral bone marrow, and osteoblastic differentiation was induced
with dexamethasone, ascorbic acid and β-glycerophosphate to
investigate whether lack of endogenous PTH affects the VEGF
pathway. Western blot analysis revealed that the expression of
proteins involved in the PKA/pAKT/HIF1α signaling pathway were
reduced 2 weeks after induction. In addition, a reduction in VEGF
and pVEGFR2 expression was observed. Conversely, administration of
exogenous PTH to cells from PTHKO mice partially reversed
downregulation of the PKA/pAKT/HIF1α/VEGF pathway (Fig. 6B).
Discussion
In the present study, PTHKO mice displayed delayed
fracture healing alongside formation of a smaller callus, defective
endochondral bone formation and reduced osteoblast activity.
Angiogenic markers were measured, due to the close association
between angiogenesis and endochondral bone formation (27). Decreased expression of VEGF and
the oxygen sensor HIF1α was observed in PTHKO mice, whereas reduced
angiogenesis was associated with endochondral bone formation. In
vitro experiments confirmed that lack of endogenous PTH
inhibited osteoblastic differentiation of mesenchymal stem cells
and downregulated signal transduction of the PKA/pAKT/HIF1α/VEGF
pathway. In addition, the aggregation and crosslinking of
endothelial cells was reduced when cocultured with BMSCs from PTHKO
mice. Furthermore, in vitro experiments demonstrated that
exogenous PTH partially reversed reduced signal transduction of the
PKA/pAKT/HIF1α/VEGF pathway.
The regeneration of blood vessels is crucial for
callus remodeling during fracture healing (3). In the early stage of fracture
healing, low oxygen levels induce a large number of differentiated
chondrocytes to form a cartilage callus. As mature chondrocytes
begin to secrete cartilage matrix factors, blood vessels grow into
the cartilage callus along the matrix, resulting in increased
levels of oxygen, which induce osteoblastic differentiation and
gradual mineralization of the matrix, thus resulting in the
generation of a large amount of woven bone (3–5).
Therefore, angiogenesis is closely associated with endochondral
bone formation. PTH has an anabolic effect on bone; however, its
association with angiogenesis remains unclear. The present study
demonstrated that lack of endogenous PTH may cause a reduction in
angiogenesis and defects in endochondral bone formation, ultimately
leading to delayed fracture healing.
Hypoxia is a major regulatory factor during
angiogenesis. The HIF signaling pathway is the key pathway by which
most body tissues react to local partial pressures of oxygen, and
HIF is simultaneously influenced by various hormones, as well as
nerve activity (6,22). Li et al reported that the
osteogenic effects of PTH are predominantly mediated by the
AC-cAMP-PKA pathway (21). Park
et al also indicated that, in tumor cells, activation of the
β-adrenergic receptor promotes angiogenesis by upregulating VEGF
expression via the cAMP/PKA/phosphatidylinositol
3-kinase/Akt/mammalian target of rapamycin/p70S6 kinase/HIF1α/VEGF
pathway (22). The present study
confirmed that the expression levels of all proteins involved in
the PKA/pAKT/HIF1α pathway were reduced in PTHKO mouse BMSC-derived
osteoblasts, whereas exogenous PTH partially increased their
expression. These findings suggested that PTH may affect the
expression of VEGF via the PKA/pAKT/HIF1α pathway and thereby
influence angiogenesis.
At present, VEGF is the most well-studied angiogenic
factor in the mammalian skeletal system. As the most important
HIF1α target gene following its translocation into the nucleus,
VEGF is considered the crucial link between angiogenesis and
osteogenesis (9,10,12,27). Numerous studies have demonstrated
that increased VEGF expression in osteoblasts may be induced by
various stimuli (20,23), and that VEGF can increase the
activities of endothelial cells and promote angiogenesis (33,34). In addition, it has been reported
that VEGF can act directly on osteogenic progenitor cells, thus
enhancing their recruitment and differentiation, and promoting
fracture healing (33,35). Deletion of the endogenous VEGF
gene in mice leads to impaired BMSC-derived osteoblast functions;
this finding confirms that VEGF may directly affect
differentiation, maturation and functionality of osteoblasts
(12). The present study revealed
that the expression levels of VEGF were reduced, and that
osteoblast activity and angiogenesis were inhibited in PTHKO mice.
However, further studies are required to elucidate whether VEGF
directly affects the functions of osteoblasts or acts through its
effects on angiogenesis. The present in vitro experiments
detected lower VEGF expression in PTHKO mouse BMSC-derived
osteoblasts compared with in those from WT mice. In addition,
osteoblasts from PTHKO mice exhibited a diminished ability to
promote aggregation and cross-linking of endothelial cells in a
coculture experiment. These findings also indicated that these
cells exhibited reduced ability to promote angiogenesis. Therefore,
it may be hypothesized that it is more likely that endogenous PTH
affects fracture healing indirectly by regulating angiogenesis via
the PKA/pAKT/HIF1α/VEGF pathway.
Mature cartilage cells can continuously secrete
cartilage matrix factors, which provide a scaffold for vascular
invasion into the callus (3–5).
Previous studies have suggested that mature osteoclasts can produce
heparanase during the fracture healing process, which degrades
proteoglycans containing sulfated heparin in the extracellular
matrix and results in the release of heparin-associated growth
factors, including VEGF and basic fibroblast growth factor
(36,37). These factors can increase
angiogenesis, and regulate the activities of osteoblasts and
osteoclasts. The present study demonstrated that the expression of
matrix proteins, including COL II, OCN and ALP were decreased, and
VEGF expression was also reduced, thus suggesting that the lack of
endogenous PTH may induce a simultaneous decrease in bone matrix
and angiogenesis. However, further research is required to clarify
the interaction between these two processes.
In conclusion, the present study suggested that the
lack of endogenous PTH may decrease expression of VEGF in BMSCs or
osteoblasts via downregulation of the PKA/pAKT/HIF1α/VEGF pathway,
which causes reduced angiogenesis and thereby affects endochondral
bone formation, ultimately leading to delayed fracture healing.
Acknowledgments
The present study was supported by the National
Natural and Science Foundation of China (grant nos. 81472080 and
81520108018).
References
|
1
|
Gerstenfeld LC, Cullinane DM, Barnes GL,
Graves DT and Einhorn TA: Fracture healing as a post-natal
developmental process: Molecular, spatial, and temporal aspects of
its regulation. J Cell Biochem. 88:873–884. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsiridis E, Upadhyay N and Giannoudis P:
Molecular aspects of fracture healing: Which are the important
molecules? Injury. 38(Suppl 1): S11–S25. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Geris L, Gerisch A, Sloten JV, Weiner R
and Oosterwyck HV: Angiogenesis in bone fracture healing: A
bioregulatory model. J Theor Biol. 251:137–158. 2008. View Article : Google Scholar
|
|
4
|
Taguchi K, Ogawa R, Migita M, Hanawa H,
Ito H and Orimo H: The role of bone marrow-derived cells in bone
fracture repair in a green fluorescent protein chimeric mouse
model. Biochem Biophys Res Commun. 331:31–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khosla S, Westendorf JJ and Oursler MJ:
Building bone to reverse osteoporosis and repair fractures. The
Journal of Clinical lnvestigation. 118:421–428.
2008.10.1172/JCI33612. View
Article : Google Scholar
|
|
6
|
Wan C, Gilbert SR, Wang Y, Cao X, Shen X,
Ramaswamy G, Jacobsen KA, Alaql ZS, Eberhardt AW, Gerstenfeld LC,
et al: Activation of the hypoxia-inducible factor-1alpha pathway
accelerates bone regeneration. Proc Natl Acad Sci USA. 105:686–691.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ivan M, Kondo K, Yang H, Kim W, Valiando
J, Ohh M, Salic A, Asara JM, Lane WS and Kaelin WG Jr: HIFalpha
targeted for VHL-mediated destruction by proline hydroxylation:
Implications for O2 sensing. Science. 292:464–468. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jaakkola P, Mole DR, Tian YM, Wilson MI,
Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji
M, Schofield CJ, et al: Targeting of HIF-alpha to the von
Hippel-Lindau ubiquitylation complex by O2-regulated prolyl
hydroxylation. Science. 292:468–472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kallio PJ, Okamoto K, O'Brien S, Carrero
P, Makino Y, Tanaka H and Poellinger L: Signal transduction in
hypoxic cells: Inducible nuclear translocation and recruitment of
the CBP/p300 coactivator by the hypoxia-inducible factor-1alpha.
EMBO J. 17:6573–6586. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kusumbe AP, Ramasamy SK and Adams RH:
Coupling of angiogenesis and osteogenesis by a specific vessel
subtype in bone. Nature. 507:323–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brahimi-Horn MC and Pouysségur J:
Harnessing the hypoxia-inducible factor in cancer and ischemic
disease. Biochem Pharmacol. 73:450–457. 2007. View Article : Google Scholar
|
|
12
|
Liu Y, Berendsen AD, Jia S, Lotinun S,
Baron R, Ferrara N and Olsen BR: Intracellular VEGF regulates the
balance between osteoblast and adipocyte differentiation. J Clin
Invest. 122:3101–3113. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gensure RC, Gardella TJ and Jüppner H:
Parathyroid hormone and parathyroid hormone-related peptide, and
their receptors. Biochem Biophys Res Commun. 328:666–678. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barnes GL, Kakar S, Vora S, Morgan EF,
Gerstenfeld LC and Einhorn TA: Stimulation of fracture-healing with
systemic intermittent parathyroid hormone treatment. J Bone Joint
Surg Am. 90(Suppl 1): 120–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miao D, He B, Karaplis AC and Goltzman D:
Parathyroid hormone is essential for normal fetal bone formation. J
Clin Invest. 109:1173–1182. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xue Y, Karaplis AC, Hendy GN, Goltzman D
and Miao D: Genetic models show that parathyroid hormone and
1,25-dihydroxyvitamin D3 play distinct and synergistic roles in
postnatal mineral ion homeostasis and skeletal development. Hum Mol
Genet. 14:1515–1528. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ren Y, Liu B, Feng Y, Shu L, Cao X,
Karaplis A, Goltzman D and Miao D: Endogenous PTH deficiency
impairs fracture healing and impedes the fracture-healing efficacy
of exogenous PTH(1-34). PLoS One. 6:e230602011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jilka RL, O'Brien CA, Bartell SM,
Weinstein RS and Manolagas SC: Continuous elevation of PTH
increases the number of osteoblasts via both osteoclast-dependent
and -independent mechanisms. J Bone Miner Res. 25:2427–2437. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rhee Y, Park SY, Kim YM, Lee S and Lim SK:
Angiogenesis inhibitor attenuates parathyroid hormone-induced
anabolic effect. Biomed Pharmacother. 63:63–68. 2009. View Article : Google Scholar
|
|
20
|
Prisby R, Guignandon A, Vanden-Bossche A,
Mac-Way F, Linossier MT, Thomas M, Laroche N, Malaval L, Langer M,
Peter ZA, et al: Intermittent PTH(1-84) is osteoanabolic but not
osteoangiogenic and relocates bone marrow blood vessels closer to
bone-forming sites. J Bone Miner Res. 26:2583–2596. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li X, Liu H, Qin L, Tamasi J, Bergenstock
M, Shapses S, Feyen JH, Notterman DA and Partridge NC:
Determination of dual effects of parathyroid hormone on skeletal
gene expression in vivo by microarray and network analysis. J Biol
Chem. 282:33086–33097. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park SY, Kang JH, Jeong KJ, Lee J, Han JW,
Choi WS, Kim YK, Kang J, Park CG and Lee HY: Norepinephrine induces
VEGF expression and angiogenesis by a hypoxia-inducible factor-1α
protein-dependent mechanism. Int J Cancer. 128:2306–2316. 2011.
View Article : Google Scholar
|
|
23
|
Esbrit P, Alvarez-Arroyo MV, De Miguel F,
Martin O, Martinez ME and Caramelo C: C-terminal parathyroid
hormone-related protein increases vascular endothelial growth
factor in human osteoblastic cells. J Am Soc Nephrol. 11:1085–1092.
2000.PubMed/NCBI
|
|
24
|
Miao D, He B, Lanske B, Bai XY, Tong XK,
Hendy GN, Goltzman D and Karaplis AC: Skeletal abnormalities in
Pth-null mice are influenced by dietary calcium. Endocrinology.
145:2046–2053. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marturano JE, Cleveland BC, Byrne MA,
O'Connell SL, Wixted JJ and Billiar KL: An improved murine femur
fracture device for bone healing studies. J Biomech. 41:1222–1228.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Menon P, Yin G, Smolock EM, Zuscik MJ, Yan
C and Berk BC: GPCR kinase 2 interacting protein 1 (GIT1) regulates
osteoclast function and bone mass. J Cell Physiol. 225:777–785.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yin G, Sheu TJ, Menon P, Pang J, Ho HC,
Shi S, Xie C, Smolock E, Yan C, Zuscik MJ, et al: Impaired
angiogenesis during fracture healing in GPCR kinase 2 interacting
protein-1 (GIT1) knock out mice. PLoS One. 9:e891272014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Krishnan V, Moore TL, Ma YL, Helvering LM,
Frolik CA, Valasek KM, Ducy P and Geiser AG: Parathyroid hormone
bone anabolic action requires Cbfa1/Runx2-dependent signaling. Mol
Endocrinol. 17:423–435. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tischer E, Mitchell R, Hartman T, Silva M,
Gospodarowicz D, Fiddes JC and Abraham JA: The human gene for
vascular endothelial growth factor. Multiple protein forms are
encoded through alternative exon splicing. J Biol Chem.
266:11947–11954. 1991.PubMed/NCBI
|
|
31
|
Holmes K, Roberts OL, Thomas AM and Cross
MJ: Vascular endothelial growth factor receptor-2: Structure,
function, intracellular signalling and therapeutic inhibition. Cell
Signal. 19:2003–2012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schnoke M, Midura SB and Midura RJ:
Parathyroid hormone suppresses osteoblast apoptosis by augmenting
DNA repair. Bone. 45:590–602. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mayr-Wohlfart U, Waltenberger J, Hausser
H, Kessler S, Günther KP, Dehio C, Puhl W and Brenner RE: Vascular
endothelial growth factor stimulates chemotactic migration of
primary human osteoblasts. Bone. 30:472–477. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Street J, Bao M, deGuzman L, Bunting S,
Peale FV Jr, Ferrara N, Steinmetz H, Hoeffel J, Cleland JL,
Daugherty A, et al: Vascular endothelial growth factor stimulates
bone repair by promoting angiogenesis and bone turnover. Proc Natl
Acad Sci USA. 99:9656–9661. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Keramaris NC, Calori GM, Nikolaou VS,
Schemitsch EH and Giannoudis PV: Fracture vascularity and bone
healing: A systematic review of the role of VEGF. Injury. 39(Suppl
2): S45–S57. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Collin-Osdoby P, Rothe L, Bekker S,
Anderson F, Huang Y and Osdoby P: Basic fibroblast growth factor
stimulates osteoclast recruitment, development, and bone pit
resorption in association with angiogenesis in vivo on the chick
chorioallantoic membrane and activates isolated avian osteoclast
resorption in vitro. J Bone Miner Res. 17:1859–1871. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Saijo M, Kitazawa R, Nakajima M, Kurosaka
M, Maeda S and Kitazawa S: Heparanase mRNA expression during
fracture repair in mice. Histochem Cell Biol. 120:493–503. 2003.
View Article : Google Scholar : PubMed/NCBI
|