Introduction
Non-alcoholic fatty liver disease (NAFLD) is rapidly
becoming a serious global health concern due to its prevalence,
which has rapidly increased with the prevalence of obesity and type
2 diabetes (T2D) (1,2). NAFLD comprises a spectrum of liver
conditions. The dominant feature of NAFLD is the abundant
accumulation of hepatic triglycerides (TGs). Non-alcoholic
steatohepatitis (NASH), involving the presence of both lobular
inflammation and signs of hepatocellular damage, can develop in the
context of chronic NAFL (3,4).
Although the details mechanisms responsible for the disease remain
to be fully understood, NASH is considered a liver-related
complication, such as fibrosis, cirrhosis as well as hepatocellular
carcinoma (5). Importantly, to
date, there are no approved pharmacological therapeutic strategies
for NAFLD, and it is presently the second-leading etiology for
liver transplantation (6).
Carnosic acid (CA), known as a natural benzenediol
abietane diterpene detected in rosemary and common sage. CA is used
as a preservative or antioxidant in food and non-food products,
including toothpaste, mouthwash and chewing gum (7,8).
CA has been reported to have antitumor properties in various types
of cancer, such as adenoma formation, myeloid leukemia and skin
tumors by regulating cancer cell growth, apoptosis and oxidative
stress (9,10).
Myristoylated alanine-rich protein kinase C
substrate (MARCKS) is known as a major protein kinase C (PKC)
substrate in different cell types (11). MARCKS is bound to the plasma
membrane, and recent studies have indicated that this binding needs
both hydrophobic insertion of its myristate chain to the bilayer
and electrostatic interaction of its cluster of basic residues with
acidic lipids (12,13). The phosphorylation of MARCKS by
PKC leads to negative charges in the basic cluster, reducing its
electrostatic interaction with acidic lipids and leading to the
translocation of MARCKS from the membrane onto cytoplasm (14,15). Modulating MARCKS expression seems
to be mediated by different molecular mechanisms, which may utilize
PKC. MARCKS was highly related to α-smooth muscle actin (α-SMA), a
representative marker of fibrosis in many organs, such as the
liver, lungs and heart (16–18). Recent data emphasize the
importance of fibrosis, as it is a key factor during the natural
progression and development of NAFLD (19). However, the role of MARCKS in
high-fat (HF) diet-fed mice with NAFLD and the underlying
mechanisms have not yet been fully investigated.
The inflammatory response regulated by NLR family
pyrin domain containing 3 (NLRP3) and nuclear factor (NF)-κB has
been shown to be associated with NAFLD development through the
release of pro-inflammatory cytokines, such as interleukin (IL),
IL6 and IL-18 (20,21). Additionally, recent studies on
peroxisome proliferator-activated receptor (PPAR) transcription
factors found decreased hepatic PPARα expression accompanied by an
increased NASH severity (22–24). Furthermore, following
intervention, PPARα expression is then restored in patients whose
liver histology has improved, as with the expression of many of the
metabolic target genes of PPARα, including the gene that encodes
carnitine palmitoyltransferase-1 (CPT-1)α, a rate limiting enzyme
for mitochondrial β-oxidation (25,26).
In the present study, we investigated the molecular
mechanisms through which CA suppresses inflammation and lipogenesis
in mice with HF diet-induced NAFLD. We found that the effects of CA
are mediated via the regulation of MARCKS expression and related
signaling pathways.
Materials and methods
Animals and reagents
A total of 60 male C57BL/6 mice weighed 18–22 g
[used as wild-type (WT) mice] were purchased from the Experimental
Animal Center of Nanjing Medical University (Nanjing, China).
Thirty MARCKS-deficient mice were purchased from Jackson Laboratory
(Bar Harbor, ME, USA). All mice were provided with drinking water,
and housed in a controlled environment with a temperature of 22±2°C
and relative humidity of 60±10% under a 12-h light/dark cycle. This
study was approved by the Ethics Committee on Animal Research at
the Department of Otolaryngology-Head and Neck Surgery, Huai'an
Hospital Affiliated to Xuzhou Medical College, Huaian, China.
The 90 animals were divided into 6 groups (15 in
each group) as follows: i) wild-type mice fed the normal chow diet
(WT/Chow); ii) wild-type mice fed the HF diet (WT/HF); iii)
MARCKS-knockout mice fed the normal chow diet
(MARCKS−/−/Chow); iv) MARCKS-knockout mice fed the HF
diet (MARCKS−/−/HF); v) wild-type mice fed the HF diet
combined with 15 mg/kg CA (purity >98%; BioBioPha, yunnan,
China); and vi) wild-type mice fed the HF diet combined with 30
mg/kg CA. CA was dissolved in distilled water at 0.3 mg/ml and
administered to the mice with free access. Mice given distilled
water were used as controls (WT/Chow and
MARCKS−/−/Chow). CA administration was initiated when
the mice were fed with HF diets. Mice were administered a standard
diet containing most essential nutrients, such as vitamins A
(≥14,000 IU), D (≥1,500 IU), E (≥120 IU), K (≥5 mg), B1 (≥13 mg),
B2 (≥12 mg), B6 (≥12 mg), B12 (≥0.022 mg), biotin (≥0.2 mg) and
niacin (≥60 mg) per kg. During the period of study, the fodder was
changed to a HF diet (60 kcal% fat, D12492; Research Diets, USA)
until the mice were sacrificed for further analysis. At the end of
week 8, body weight was monitored and all experimental mice were
sacrificed after 12 h of fasting. Eyeball blood was collected, and
serum was obtained by centrifugation at 13,000 rpm for 15 min at
4°C and stored at −80°C for analysis. The whole liver tissues were
harvested and weighed on a 4°C glacial table, and were either
frozen in liquid nitrogen and kept at −80°C for analysis, or fixed
in 4% paraformaldehyde for histological analysis. All the methods
were carried out in accordance with the approved guidelines.
Oral glucose tolerance test (OGTT) and
insulin tolerance test (ITT)
At the end of the feeding period, OGTT and ITT were
conducted. The mice from the WT/Chow, WT/HF,
MARCKS−/−/HF and MARCKS−/−/Chow groups were
treated with 20% glucose dissolved in saline orally or injected
with insulin intraperitoneally (1 U/kg body weight). Tail-vein
blood was collected at 0, 30, 60, 90 and 120 min after glucose or
insulin treatment, and centrifuged at 4000 × g for 10 min at 4°C to
obtain serum for glucose assay.
Biochemical indicator evaluation
TG, total cholesterol (TC), aspartate transaminase
(AST), alanine transaminase (ALT), uric acid and non-esterified
fatty acid (NEFA) levels in serum or liver tissue obtained from the
WT/Chow, WT/HF, MARCKS−/−/HF and
MARCKS−/−/Chow groups of mice were determined using
biochemical kits (Nanjing Jiancheng Biotechnology, Nanjing, China)
following the manufaturer's instructions. TG, TC, AST, and ALT in
serum or liver from the Con, HF, CAL and CAH groups were measured
using biochemical kits.
Serum glucose and serum insulin
assays
Mice from the WT/Chow, WT/HF,
MARCKS−/−/HF, MARCKS−/−/Chow, CAL, and CAH
groups were fasted for 6 h, after which their blood was analyzed
for glucose measurement with a glucose meter (Bayer, Mishawaka, IN,
USA). For insulin analysis, the mice were fasted for 6 h and plasma
insulin levels were measured with an Ultra Sensitive Mouse insulin
enzyme-linked immunosorbent assay (ELISA) kit from Crystal Chem
(Downers Grove, IL, USA).
ELISA
The serum from 6 groups of mice was analyzed for the
levels of major inflammatory cytokines, such as TNF-α (#MTA00B),
IL-6 (#M6000B), IL-1β (#MLB00C), IL-18 (#7625), IL-2 (#M2000), IL-4
(#M4000B), IL-6 (#D6050), IL-12 (#M1270) and IFNγ (#MIF00), by
ELISA, following the manufacturer's instructions (R&D Systems,
Inc., Minneapolis, MN, USA). Color changes were determined at 450
nm.
Histopathological examination of liver
tissues
The tissue isolated from WT/Chow, WT/HF,
MARCKS−/−/HF and MARCKS−/−/Chow groups of
mice were fixed with 10% buffered formalin, imbedded in paraffin
and sliced into 4–5 µm thick sections. Following hematoxylin
and eosin (H&E) staining, the pathological changes of the liver
tissues were observed under a light microscope. Hepatic lipid
content in mice from WT/Chow, WT/HF, MARCKS−/−/HF,
MARCKS−/−/Chow, CAL, and CAH groups was determined using
5-µm-thick frozen sections stained with Oil Red O
(Sigma-Aldrich). Masson's trichrome staining was performed
according to the manufacturer's instructions (Diagnostic Biosystems
Inc., Pleasanton, CA, USA). In addition, some tissues also were
subjected to immunohistochemical (IHC) staining for the analysis of
MARCKS (except for MARCKS−/−/HF and
MARCKS−/−/Chow groups) and PPARα from each group of mice
expression. The sections were stained with MARCKS (ab51100; Abcam,
Shanghai, China) and PPARα (ab8934; Abcam). All histological
examinations were carried out according to standard procedures
reported previously (27).
Furthermore, immunofluorescence assays for PPARα for liver tissue
were performed according to the manufacturer's instructions.
Western blot analysis
The liver tumors were homogenized into 10% (wt/vol)
hypotonic buffer (pH 8.0, 1 mM EDTA, 5 µg/ml leupeptin, 25
mM Tris-HCl, 1 mM Pefabloc SC, 5 µg/ml soybean trypsin
inhibitor, 50 µg/ml aprotinin, 4 mM benzamidine) to yield a
homogenate. Primary antibodies were shown in Table I. The final supernatants from
cells and tumors were then obtained by centrifugation at 14,000 × g
for 20 min at 4°C. The protein concentration was determined using a
BCA protein assay kit (Thermo Fisher Scientific, Inc., Waltham, MA,
USA) with bovine serum albumin as a standard. Sample-loading buffer
was added, the mixture was boiled for 5 min. And the total protein
extract was used for western blot analysis. Total protein (40
µg) was loaded and proteins were separated using 10%
SDS-PAGE and electrophoretically transferred to the polyvinylidene
difluoride membranes (Millipore, Billerica, MA, USA). The membranes
were then blocked with 5% skim milk Tris-buffered saline with 0.1%
Tween 20 (TBST), washed, and then incubated with primary antibodies
overnight at 4°C. The membrane was then washed with TBST 3 times,
followed by incubation with a horseradish peroxidase
(HRP)-conjugated secondary antibody (1:2,500; KeyGen Biotech,
Nanjing, China) at room temperature for 2 h. Following another
round of washing with TBST, the membrane was then developed using
ECL (Thermo Fisher Scientific, Inc.), and exposed to Kodak X-ray
film (Eastman Kodak Company, Rochester, Ny, USA). Every protein
expression level was defined as grey value using ImageJ 1.38
software (National Institutes of Health, Bethesda, MD, USA) and
standardized to housekeeping gene of GAPDH and expressed as a fold
of control. All experiments were performed in triplicate and done
three times independently.
 | Table IPrimary antibodies used in western
blot analysis. |
Table I
Primary antibodies used in western
blot analysis.
| Primary
antibodies | Dilution ratio | Corporation |
|---|
| Rabbit anti-MARCKS
(ab51100) | 1:1,000 | Abcam |
| Rabbit anti-IL-1β
(ab9722) | 1:1,000 | Abcam |
| Rabbit anti-IL-18
(ab71495) | 1:1,000 | CST |
| Rabbit
anti-SREBP-1c (ab28481) | 1:1,000 | CST |
| Rabbit anti-FAS
(ab82419) | 1:1,000 | Abcam |
| Rabbit anti-NF-κB
(ab207297) | 1:1,000 | Abcam |
| Mouse anti-p-NF-κB
(3033) | 1:1,000 | CST |
| Rabbit
anti-caspase-1 (ab1872) | 1:1,000 | Abcam |
| Rabbit anti-PPARα
(ab8934) | 1:1,000 | Abcam |
| Rabbit anti-PI3K
(ab189403) | 1:1,000 | Abcam |
| Mouse anti-p-AKT
(9611) | 1:1,000 | CST |
| Rabbit anti-AKT
(ab182729) | 1:1,000 | Abcam |
| Rabbit anti-NLRP3
(ab214185) | 1:1,000 | Abcam |
| Mouse anti-ACCα
(sc-137104) | 1:200 | Santa Cruz |
| Mouse anti-SCD1
(sc-515844) | 1:200 | Santa Cruz |
| GAPDH
(sc-51631) | 1:200 | Santa Cruz |
Reverse transcription-quantitative PCR
(RT-qPCR)
For analysis of qPCR, was performed as previously
described (28). Total RNA was
extracted from liver tissues using TRI-Reagent (Sigma-Aldrich)
following the manufacturer's instructions and treated with
deoxyribonuclease I. First-strand cDNA was then synthesized and
amplified from 0.5 µg of total RNA using the ReverTra Ace
qPCR RT kit (Toyobo, Tokyo, Japan). Real-time PCR was carried out
for 35 cycles of 95°C for 20 sec, 54°C for 30 sec, and 72°C for 30
sec using SyBR Green Real-time PCR Master Mix (PE Applied
Biosystems, Foster City, CA, USA) in a total volume of 20
µl. Fold changes in mRNA levels of target gene relative to
the endogenous cyclophilin control were calculated. Briefly, the
cycle threshold (=Ct) values of each target gene were subtracted
from the Ct values of the housekeeping genecy clophilin (ΔCt).
Target gene ΔΔCt was calculated as ΔCt of target gene minus ΔCt of
control. The fold change in mRNA expression was calculated as
2−ΔΔCt. The primers used in the study were showed in
Table II.
| Table IISequences of primers used for RT-qPCR
analysis. |
Table II
Sequences of primers used for RT-qPCR
analysis.
| Gene | Forward primers
(5′→3′) | Reverse primers
(5′→3′) |
|---|
| GAPDH |
CATTCAAGACCGGACAGAGG |
ACATACTCAGCACCAGCATCACC |
| MARCKS |
AGCACAAAGAGAGTGTCGC |
CGTCAGTCAGTGTGTATG |
| IL-1β G |
ACAGCAAAGTGATAGGCC |
CGTCGGCAATGTATGTGTTGG |
| IL-18 |
GCAGCAGGTGAGTGGGCAGT |
CTGTACGCCTGGTTCGCTCTGT |
| IL-2 |
CATGCTGGGGCCGTACAG | TTGTCCGACCTTTGGCAA
CT |
| IL-4 |
CAGAAGGAAGTTAGGCC |
CGTCGCAGTGGATGATGTG |
| IL-12 |
CTTCTCACTGTCGACTACCGC | GCGTC
TCCTGTGCATTCG |
| TNF-α |
GACTCTTCCTGGTCTTACCATATT |
CTGCTATTGCAAGGACCCAATT |
| IFN-γ |
GCAAGGACAAGATTCGATACT |
GCCAGACTACATGGAAATCTA |
qPCR analysis was performed as previously described
(23). Fold induction values were
calculated using the to 2−ΔΔCq method, where ΔCq
represents the differences in cycle threshold number between the
target gene and glyceraldehyde 3-phosphate dehydrogenase (GAPDH),
and ΔΔCq represents the relative change in the differences between
the control and treatment groups. The primers used in this study
are listed in Table II.
Statistical analysis
Data are expressed as the means ± SD. The treated
cells, tissues and corresponding controls were compared using
GraphPad PRI SM (version 6.0; GraphPad Software, Inc., La Jolla,
CA, USA) by a one-way ANOVA with Dunn's least significant
difference tests or Student's t-tests. Differences between groups
were considered significant at p<0.05.
Results
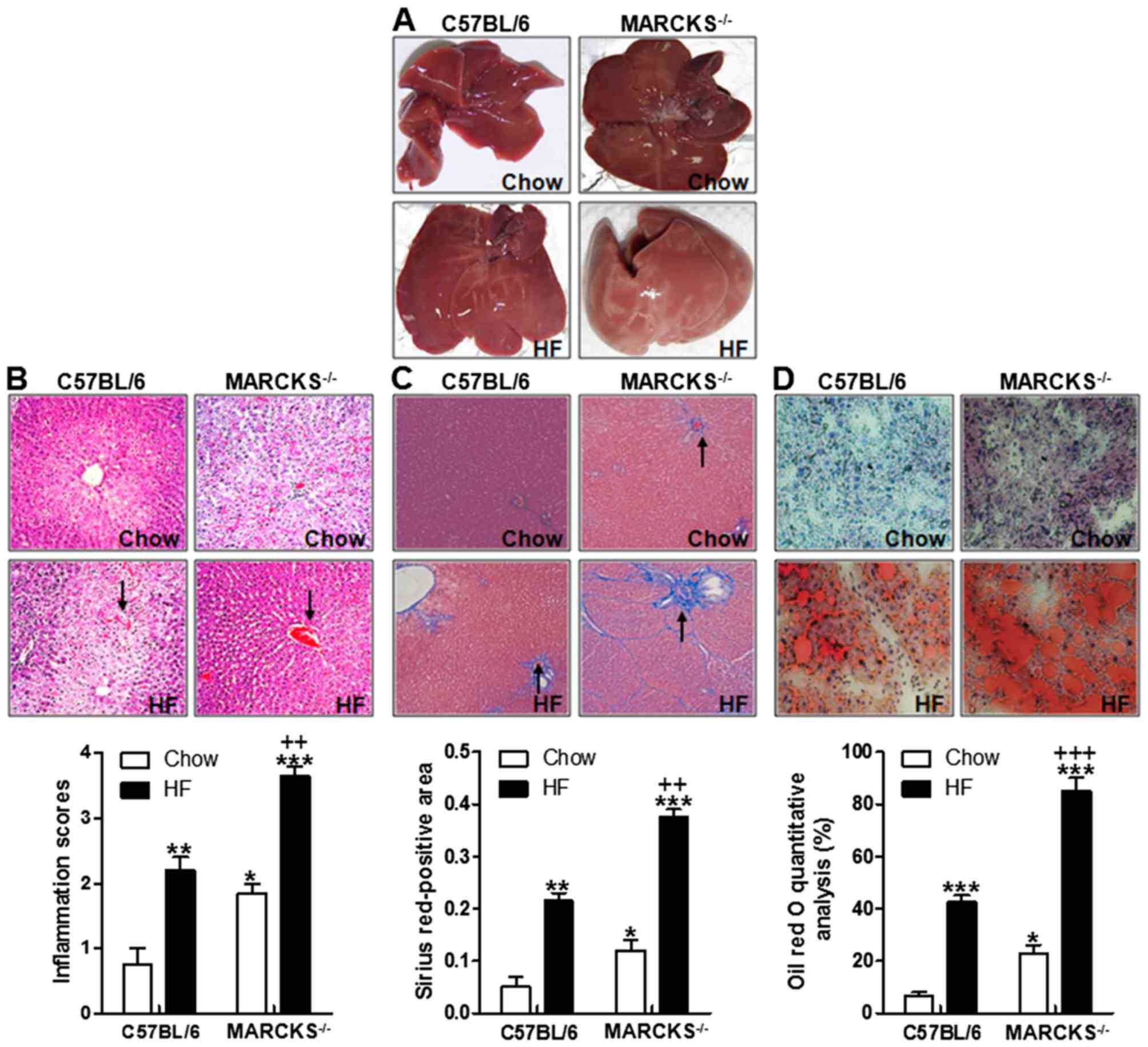
MARCKS deficiency accelerates liver
injury and lipid accumulation in response to being fed a HF
diet
At the end of the experimental period, there was a
striking difference in liver morphology in the Chow group compared
to the high-fat diet feeding mice (Fig. 1A). As shown in Fig. 1A, the mice with MARCKS exhibited
more severe liver injury. Significantly, the livers from the HF
diet-fed MARCKS-deficient mice were pale in color, suggesting the
evident accumulation of liver lipids in comparison to the HF
diet-fed wild-type mice. Based on the liver morphology, we
hypothesized that MARCKS might have a potential role in regulating
lipid accumulation in the liver. Additionally, the results of
H&E staining revealed that liver injury was induced in the HF
diet-fed wild-type mice compared with the mice fed the normal chow.
Importantly, the liver inflammation score was the highest in the HF
diet-fed MARCKS-deficient mice (Fig.
1B). These data indicated MARCKS may be of great importance in
preventing liver injury induced by HF. Similarly, Masson's
Trichrome staining indicated that liver fibrosis was mildly
increased in wild-type, but more robustly increased in
MARCKS−/− mice responding to HF diet (Fig. 1C). Indeed, the number of lipid
droplets were also higher in the HF diet-fed wild-type mice
compared with the normal chow-fed mice. However, an even higher
number of lipid droplets was observed in the livers of the
MARCKS-deficient mice fed the HF diet (Fig. 1D). These data indicated that
MARCKS deficiency promoted liver injury and liver lipid
accumulation in mice fed with HF diet.
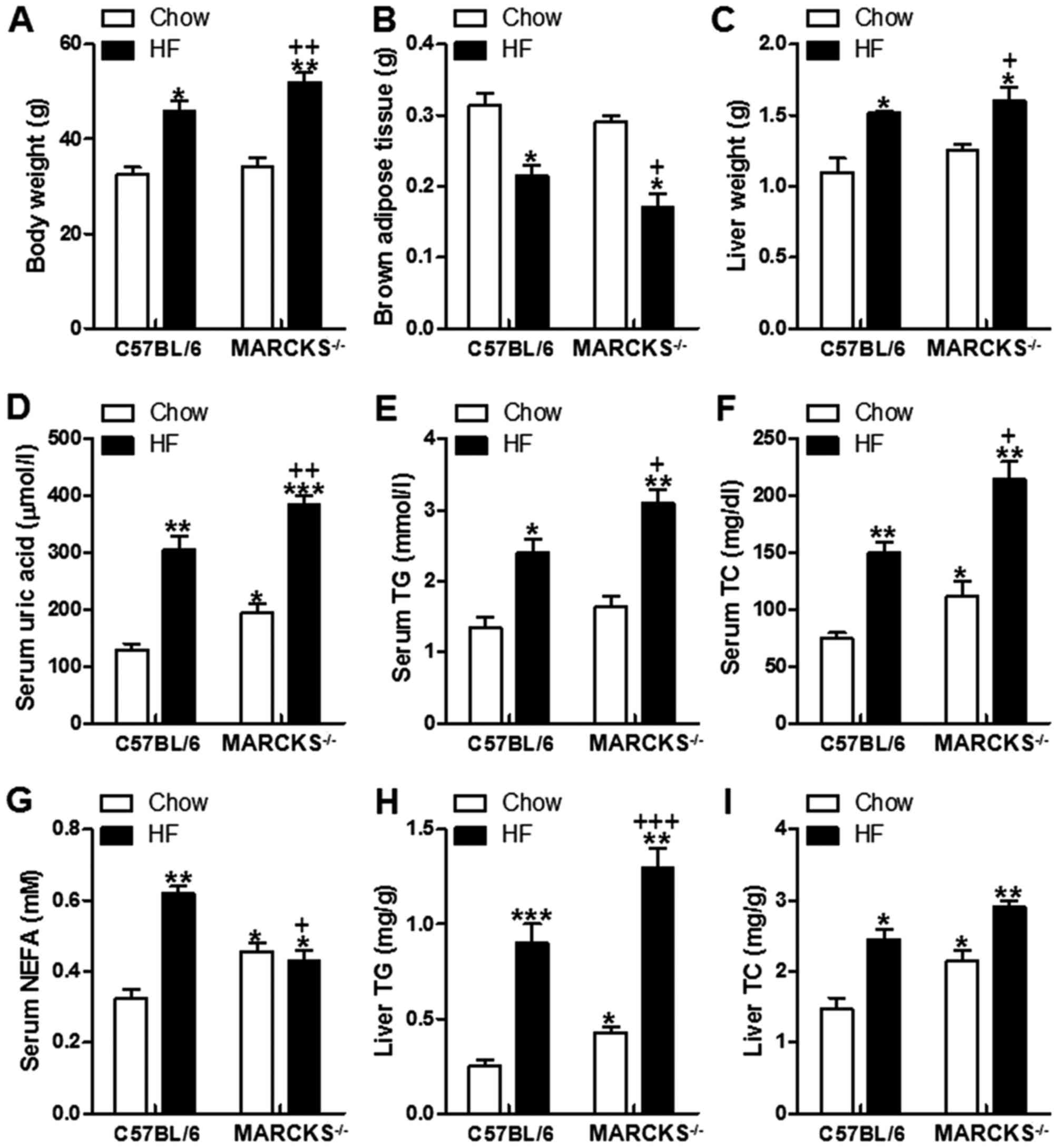
HF diet-induced liver lipid accumulation
is accelerated in MARCKS-deficient mice
We measured the body weight at the end of the
experimental period. As shown in Fig.
2A, body weight was increased in the wild-type mice fed the HF
diet compared with those fed the normal chow. MARCKS deficiency
increased the body weight of the HF diet-fed mice even further
(Fig. 2A). The brown adipose
tissue weight was decreased in the HF diet-fed mice, and this was
further decreased in the MARCKS-deficient mice fed with HF
(Fig. 2B). In addition, liver
weight was increased in the wild-type mice in response to the HF
diet, and an even greater increase was observed in the
MARCKS-deficient mice fed the HF diet (Fig. 2C). Similarly, serum uric acid was
highly increased in wild-type mice induced by HF. In the
MARCKS-deficient− mice, the uric acid levels were even
higher than those of the wild-type mice fed the HF diet (Fig. 2D). The HF diet-fed wild-type mice
also exhibited significantly higher serum levels of TGs, TC and
NEFA than the chow diet-fed wild-type mice. The mice with MARCKS
deficiency fed the HF diet exhibited even higher levels of serum
TGs, TC and NEFA (Fig. 2E–G).
Consistent with the serum alterations, the hepatic TG and TC
contents were significantly higher in the HF diet-fed wild-type
mice compared with the chow diet-fed wild-type mice. The
MARCKS-deficient mice fed the HF diet exhibited even higher TG and
TC (Fig. 2H and I). These results
indicated that MARCKS deficiency intensified the HF diet-induced
accumulation of TGs and TC.
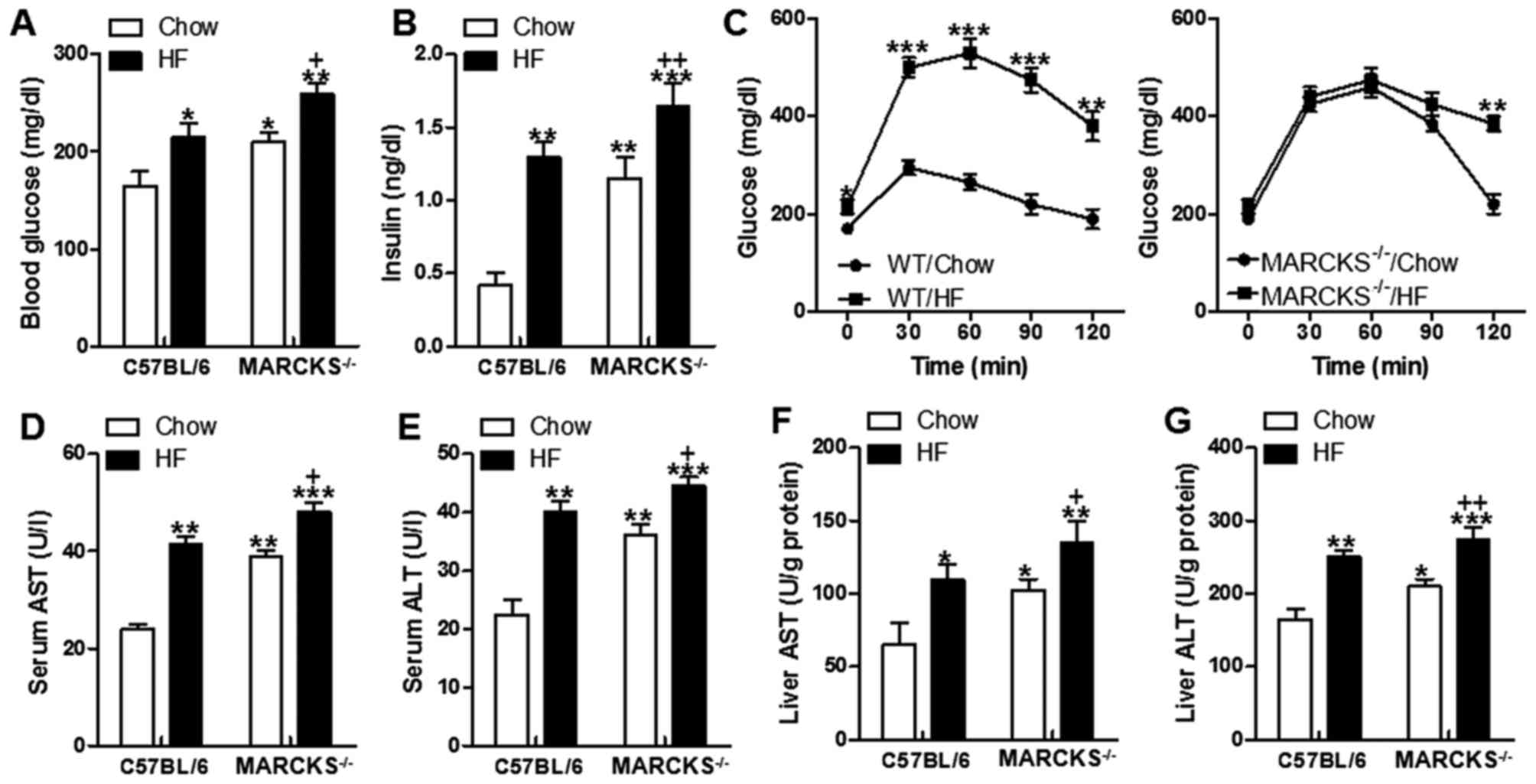
MARCKS deficiency disrupts glucose
homeostasis, and promotes insulin resistance and liver injury
We then determined whether MARCKS deficiency is is
involved in glucose and insulin resistance in HF diet-fed mice. The
fasting blood glucose levels and serum insulin levels were
increased in HF-fed mice, which was robustly upregulated in
responding to MARCKS deficiency (Fig.
3A and B). We then performed a glucose tolerance test.
Consistent with their elevated fasting glucose and insulin
concentrations on the chow diet, MARCKS−/− mice were
more glucose intolerant than the mice on chow diet (Fig. 3C). The mice exhibited increased
AST and ALT in serum as well as the increased concentrations of
hepatic AST and ALT after HF diet (Fig. 3D–G). The MARCKS-deficient mice had
even higher AST and ALT levels in serum and in the liver tissues,
particularly those fed the HF diet (Fig. 3D–G).
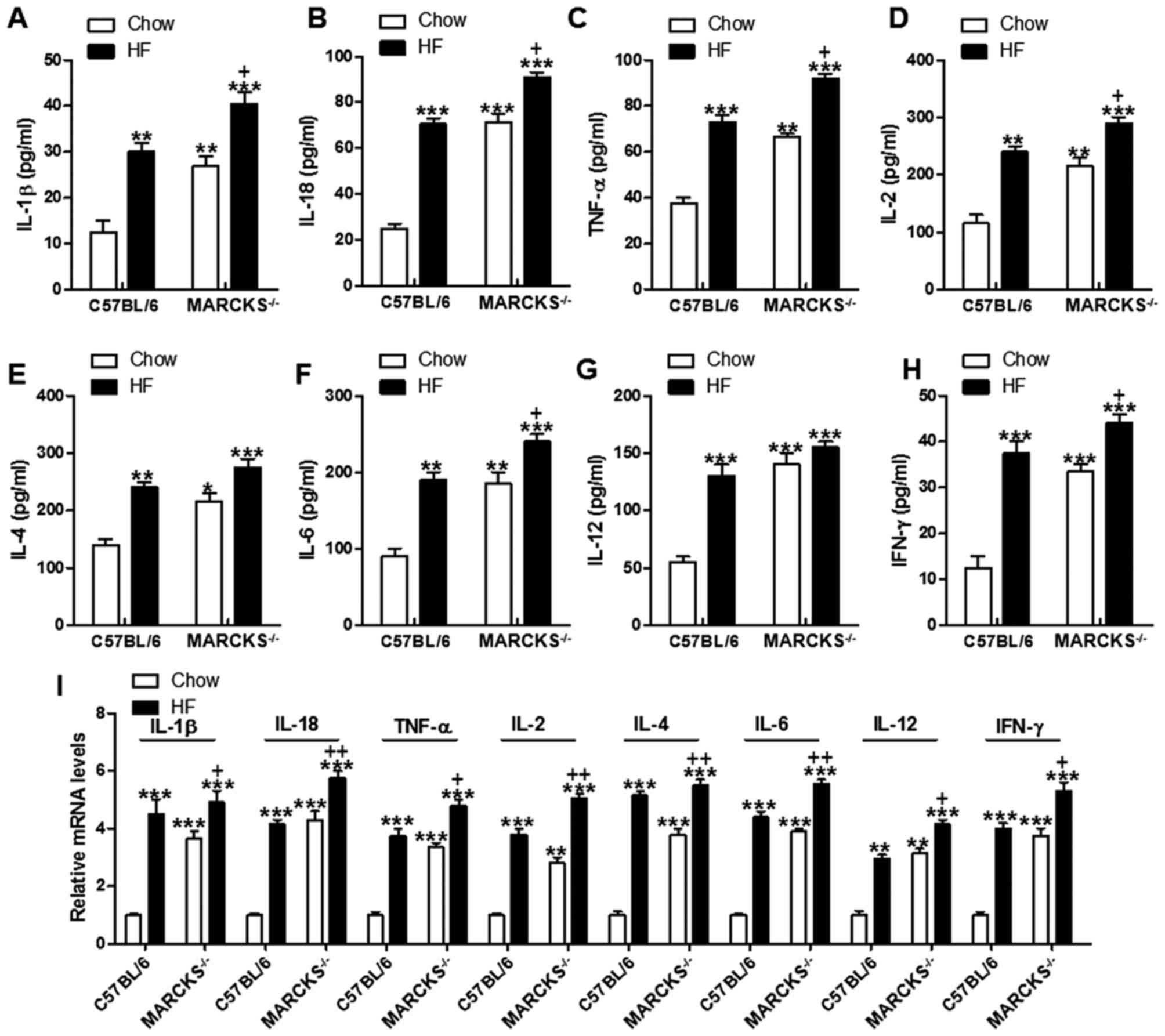
MARCKS deficiency induces
pro-inflammatory cytokine secretion in vivo
HF diet-induced liver injury is always associated
with inflammation (29). Thus, in
this regard, we evaluated pro-inflammatory cytokine expression. The
data indicated that the expression levels of IL-1β, IL-18, TNF-α,
IL-2, IL-4, IL-6, IL-12 and IFN-γ in serum were markedly increased
in mice fed the HF diet compared to those fed the normal chow diet.
The MARCKS-deficient mice fed the HF diet exhibited even further
upregulated levels of these pro-inflammatory cytokines (Fig. 4A–H). To further confirm these
results, we also measured the pro-inflammatory cytokine levels in
liver tissue samples by RT-qPCR. The results revealed that the
hepatic mRNA levels of IL-1β, IL-18, TNF-α, IL-2, IL-4, IL-6, IL-12
and IFN-γ were elevated in the HF diet-fed mice, and MARCKS
deficiency further stimulated these genes expressions (Fig. 4I). These data indicated that the
presence of MARCKS greatly attenuated inflammation in liver of mice
fed with HF diet.
MARCKS deficiency impairs PI3K/AKT and
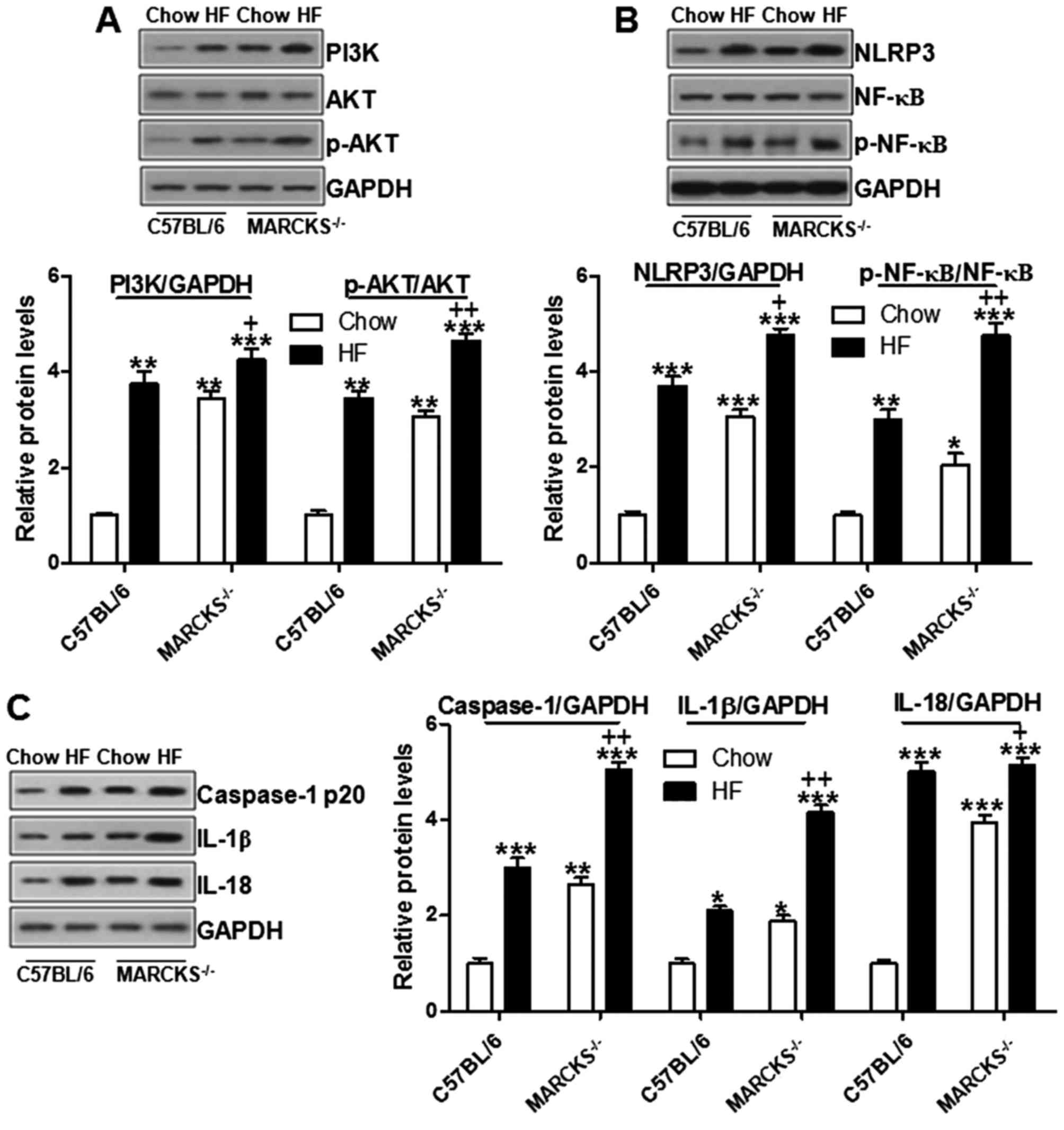
NLRP3 inflammasome signaling in vivo
As previously demonstrated, the PI3K/AKT signaling
pathway is regulated by MARCKS (30). Hence, we attempted to explore
whether MARCKS could regulate NAFLD in mice induced by high fat
diet through regulating PI3K/AKT. Western blot analysis was
performed to examine the levels of PI3K/AKT. As shown in Fig. 5A, we found that the PI3K/AKT
signaling pathway was upregulated in the wild-type mice fed the HF
diet. MARCKS deficiency further stimulated PI3K and phosphorylated
AKT. AKT has a close association with the NLRP3-regulated NF-κB
signaling pathway (31). As shown
in Fig. 5B, NLRP3 expression was
upregulated in the HF-fed wild-type mice and in the
MARCKS-deficient mice. Subsequently, NF-κB was activated through
caspase-1 stimulation in the HF diet-fed wild-type mice, and this
was further enhanced by MARCKS deficiency (Fig. 5B and C). NF-κB activation is
responsible for the release of pro-inflammatory cytokines,
including IL-1β and IL-18 (32).
In this study, both IL-1β and IL-18 were highly expressed in the HF
diet-fed mice, particularly in the MARCKS-deficient mice (Fig. 5C). These data indicated that
MARCKS, consistent with previous results (33), may play a potential role in
suppressing the inflammatory response via the PI3K/AKT and NLRP3
inflammasome signaling pathways.
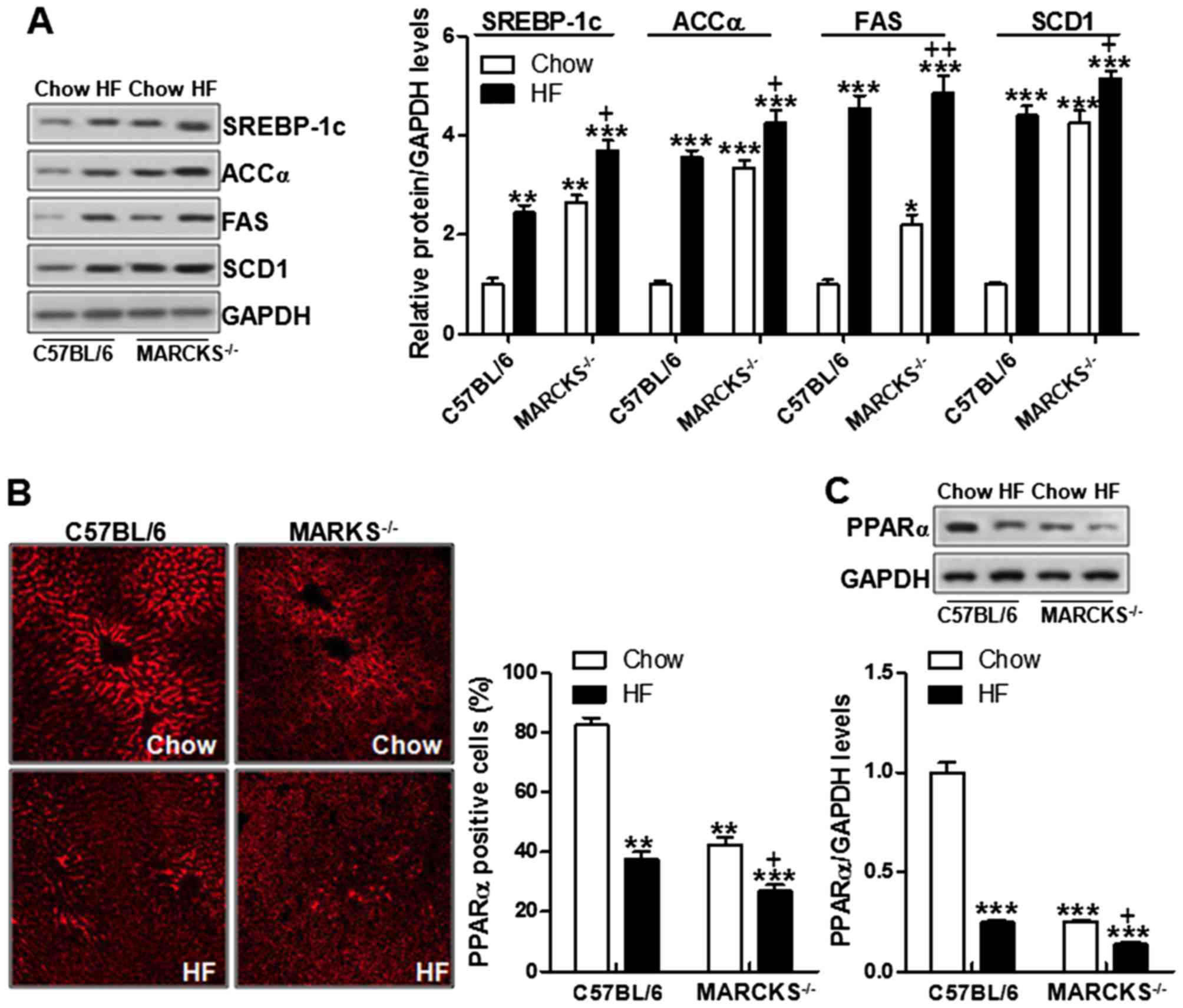
MARCKS deficiency enhances
lipogenesis-related signaling pathway activation
NAFLD development has a close association with
lipogenesis-related proteins. SREBP-1c, ACCα, FAS and SCD1 have
been well known to play a crucial role in lipid generation
(34–36). We thus then attempted to
investigate the mechanisms through which MARCKS regulates lipid
metabolism via these proteins. As shown in Fig. 6A, SREBP-1c was highly expressed in
HF diet fed mice, something that was consistent with a previous
report (37). Similarly, the
levels of ACCα, FAS and SCD1 were also elevated in the mice fed the
HF diet. Of note, MARCKS deficiency markedly increased these
proteins levels even further (Fig.
6A). PPARα is known as an important factor regulating lipid
metabolism (38). In this study,
we found that PPARα was expressed at low levels in the HF-fed mice.
MARCKS deficiency even further downregulated PPARα levels in the HF
diet-fed mice (Fig. 6B).
Consistently, the results of western blot analysis further proved
the PPARα alterations in the MARCKS-deficient mice fed the HF diet
(Fig. 6C). These data suggested
that MARCKS may be associated with lipid metabolism in HF diet-fed
mice, which influenced NAFLD development.
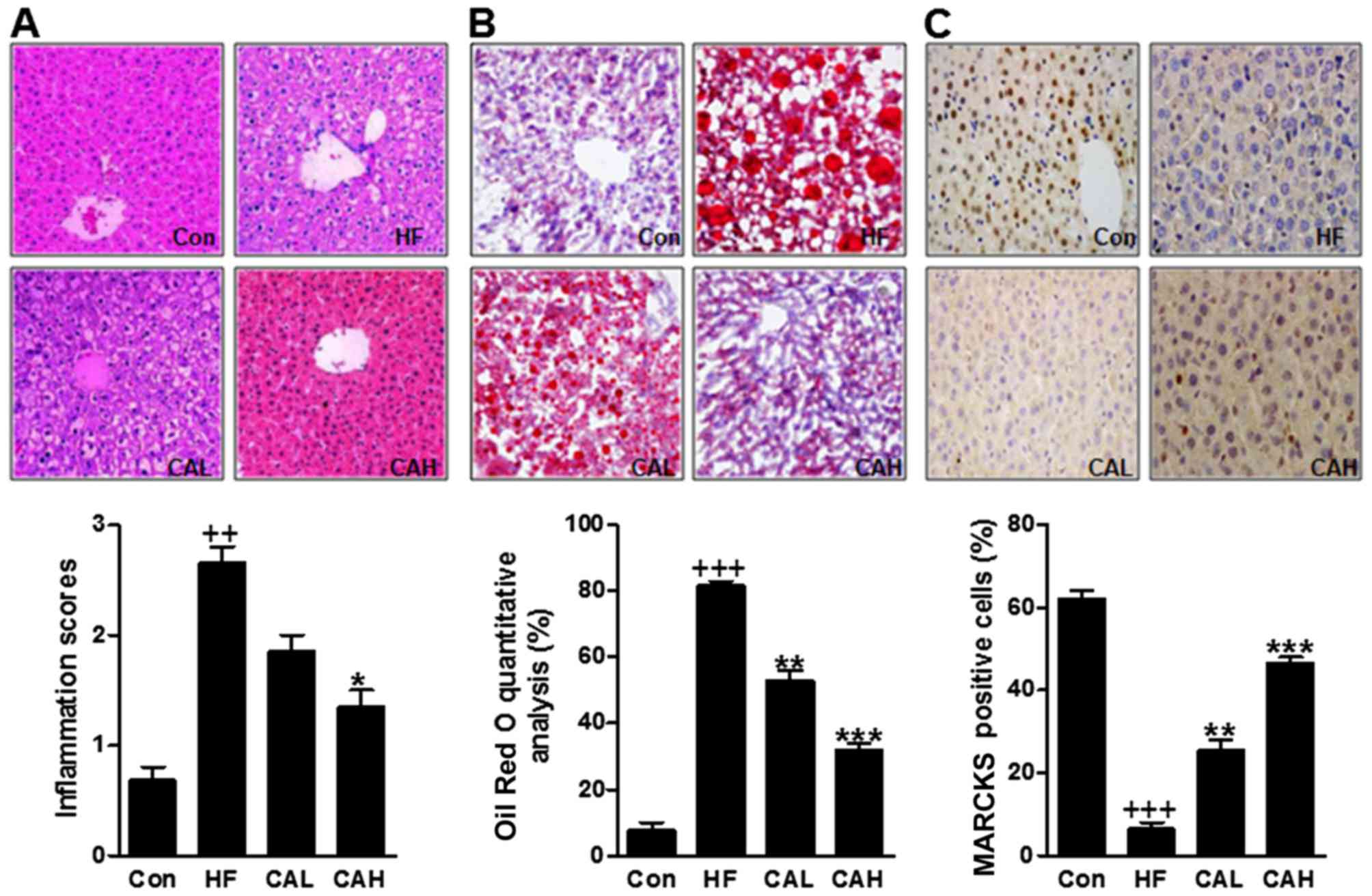
CA treatment attenuates liver injury in
the livers of mice fed the HF diet
We then attempted to reveal how CA affects NAFLD
induced by a HF diet in mice. At the end of the treatment period,
the H&E staining results showed that the higher inflammatory
score in HF-treated mouse, the livers were much larger. By
contrast, a lower inflammatory score was observed in the livers of
mice treated with CA (Fig. 7A).
Furthermore, Oil Red O staining results showed that the lipid
droplets in livers of mice with HF diet were much larger. By
contrast, fewer lipid droplets were observed in the livers of mice
treated with CA in a dose-dependent manner (Fig. 5B). As mentioned above, MARCKS has
a potential role in regulating HF diet-induced NAFLD. Thus, we
performed immunohistochemical assays and the results indicated that
a a high number of MARCKS-positive cells was observed in the
control group; however, only a few MARCKS-positive cells were
observed in the HF diet-fed mice. This number was increased by CA
treatment (Fig. 7C). These data
indicated that CA prevented liver injury and increased MARCKS
expression in the livers of mice fed the HF diet.
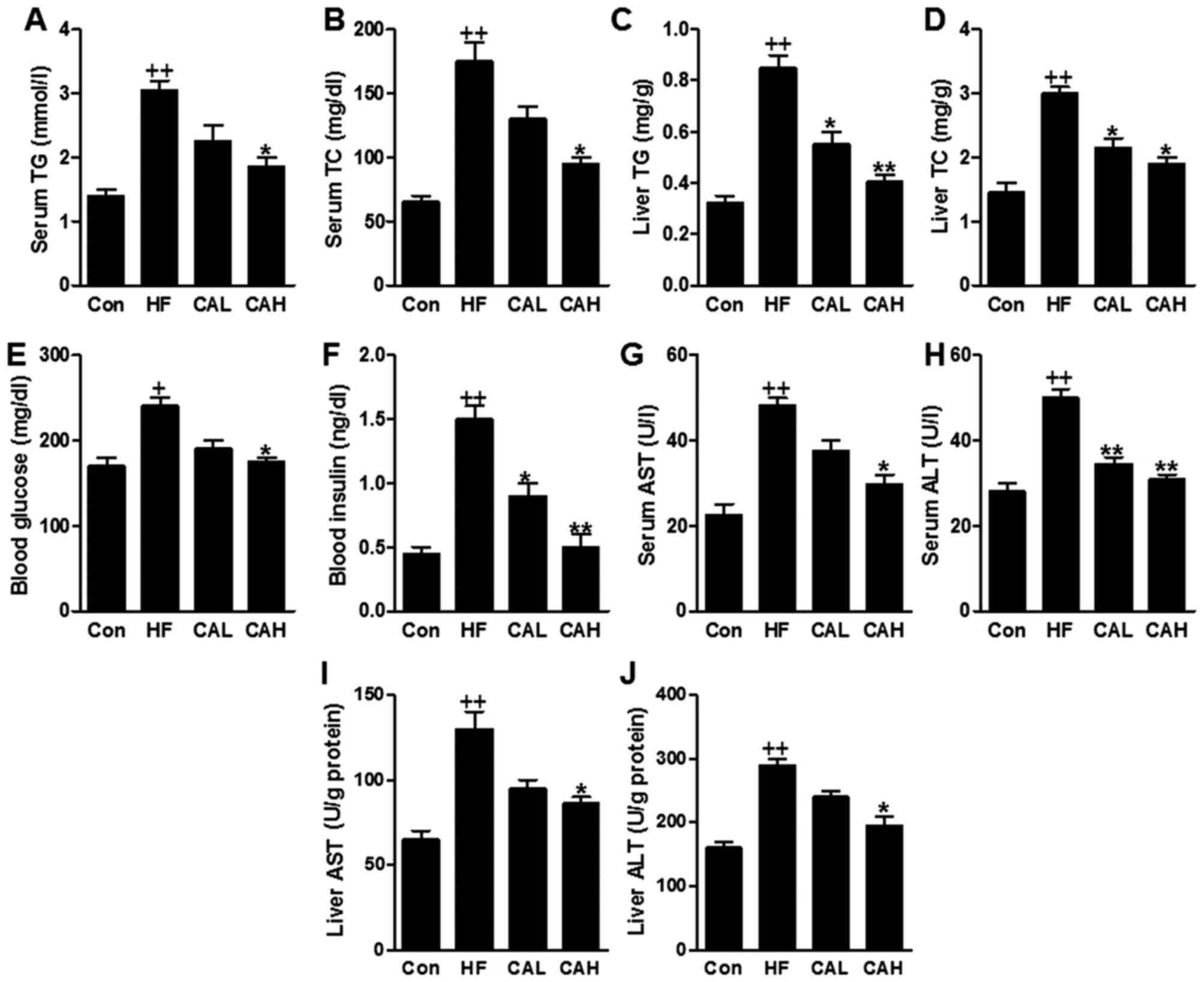
HF diet-induced liver lipid accumulation
is attenuated by CA in mice
The TG and TC levels in serum were markedly
increased following feeding on the HF diet, consistent with the
results mentioned above; however, these levels were reduced in the
HF diet-fed mice treated with CA (Fig. 8A and B). In addition, TG and TC
contents in liver induced by HF diet were significantly decreased
for CA administration in a dose-dependent manner (Fig. 8C and D). We then examined whether
treatment with CA improves glucose and insulin tolerance in the
HF-fed mice. The fasting blood glucose levels and the plasma
insulin levels were decreased following treatment with CA (Fig. 8E and F). Additionally, the serum
AST and ALT levels were upregulated in the HF diet-fed mice, and
were downregulated following treatment with CA (Fig. 8G and H). Similarly, the hepatic
AST and ALT contents were also increased in HF groups compared to
that in the Con group, which were decreased in HF diet-fed mice
treated with CA (Fig. 8I and J).
These results clearly indicated that CA treatment suppressed lipid
accumulation by reducing TG and TC in serum and in liver, improving
liver injury in mice administered with high fat diet.
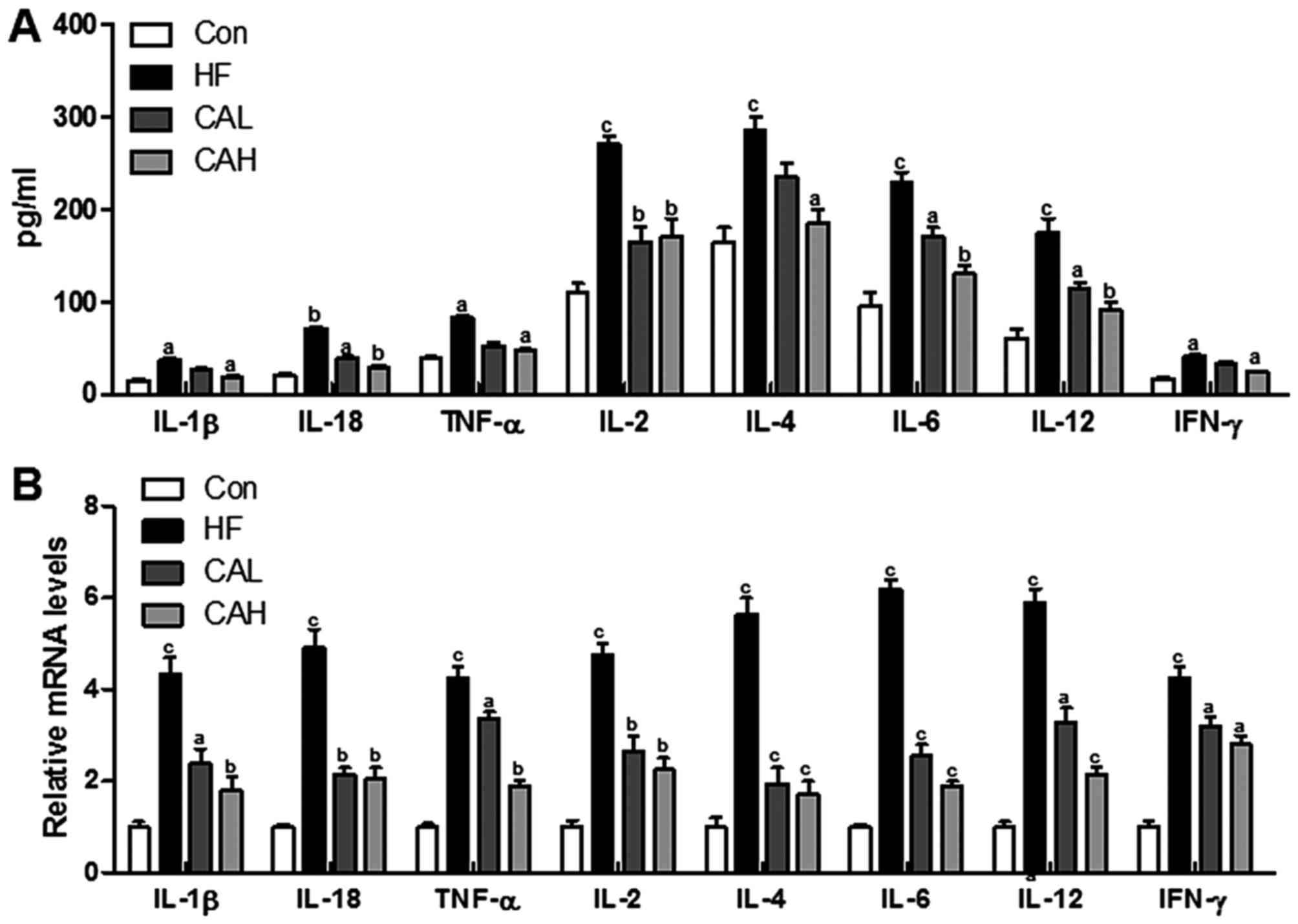
CA reduces pro-inflammatory cytokine
expression in mice fed a HF diet
Accelerated NAFLD is often associated with the
increased production of inflammatory cytokines (39). Therefore, in this study, we
examined whether CA represses the productions of inflammatory
cytokines. To examine this, we evaluated the expression levels of
the inflammatory cytokines, IL-1β, IL-18, TNF-α, IL-2, IL-4, IL-6,
IL-12 and IFN-γ, in serum. The data indicated that the expression
levels of IL-1β, IL-18, TNF-α, IL-2, IL-4, IL-6, IL-12 and IFN-γ,
which had been increased by the HF diet, were markedly decreased by
CA in the mice fed the HF diet (Fig.
9A). As can be seen in Fig.
9B, CA suppressed pro-inflammatory cytokines release through
RT-qPCR analysis (Fig. 9B). These
data indicated that treatment with CA markedly attenuated
inflammation in mice fed a HF diet.
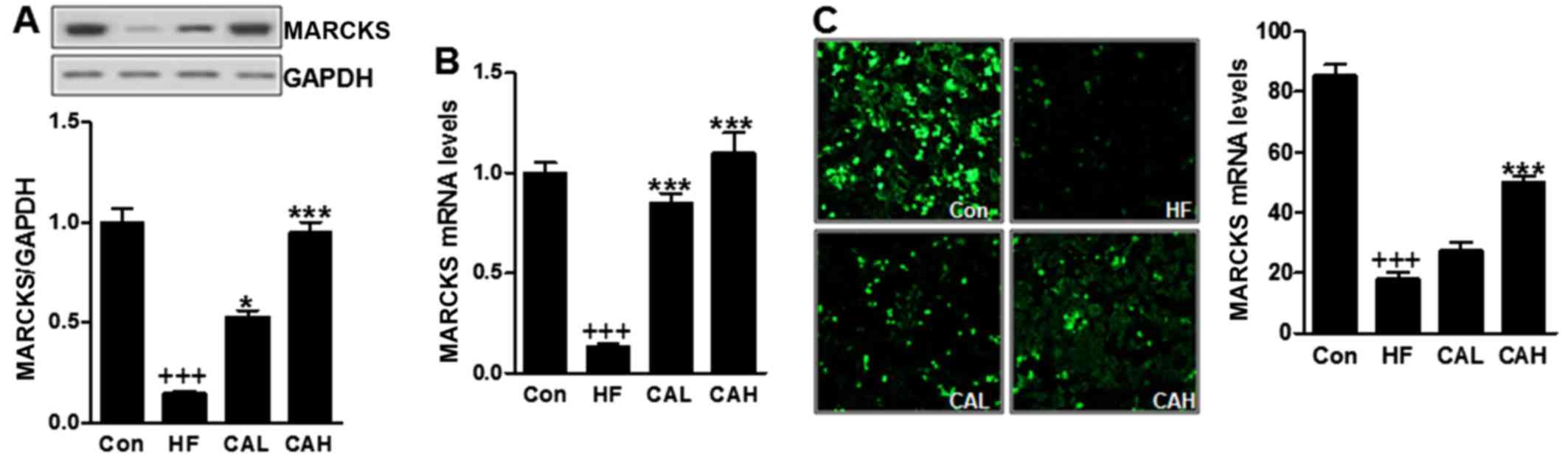
CA increases MARCKS expression in mice
fed a HF diet
To elucidate the precise mechanisms responsible for
the effects of CA in HF diet fed-mice and explore whether it can
target MARCKS to attenuate NAFLD, we examined MARCKS expressed
levels. The results revealed that the MARCKS levels were affected
by the administration of CA in the HF diet-fed mice. MARCKS protein
levels, which had been decreased by the HF diet, were markedly
increased by CA (Fig. 10A). The
mRNA level of MARCKS was also decreased in the HF diet-fed mice,
and this reduction was reversed significantly following treatment
with CA (Fig. 10B). To further
examine this, we performed immunostaining using an antibody against
MARCKS. Our results revealed that a great number of MARCKS-positive
cells was observed in the control group; however, only a few
MARCKS-positive cells were observed in the HF diet-fed mice, which
were stimulated in mice with HF diet (Fig. 10C). These data indicated that CA
attenuates NAFLD in mice fed a HF diet by regulating MARCKS
expression.
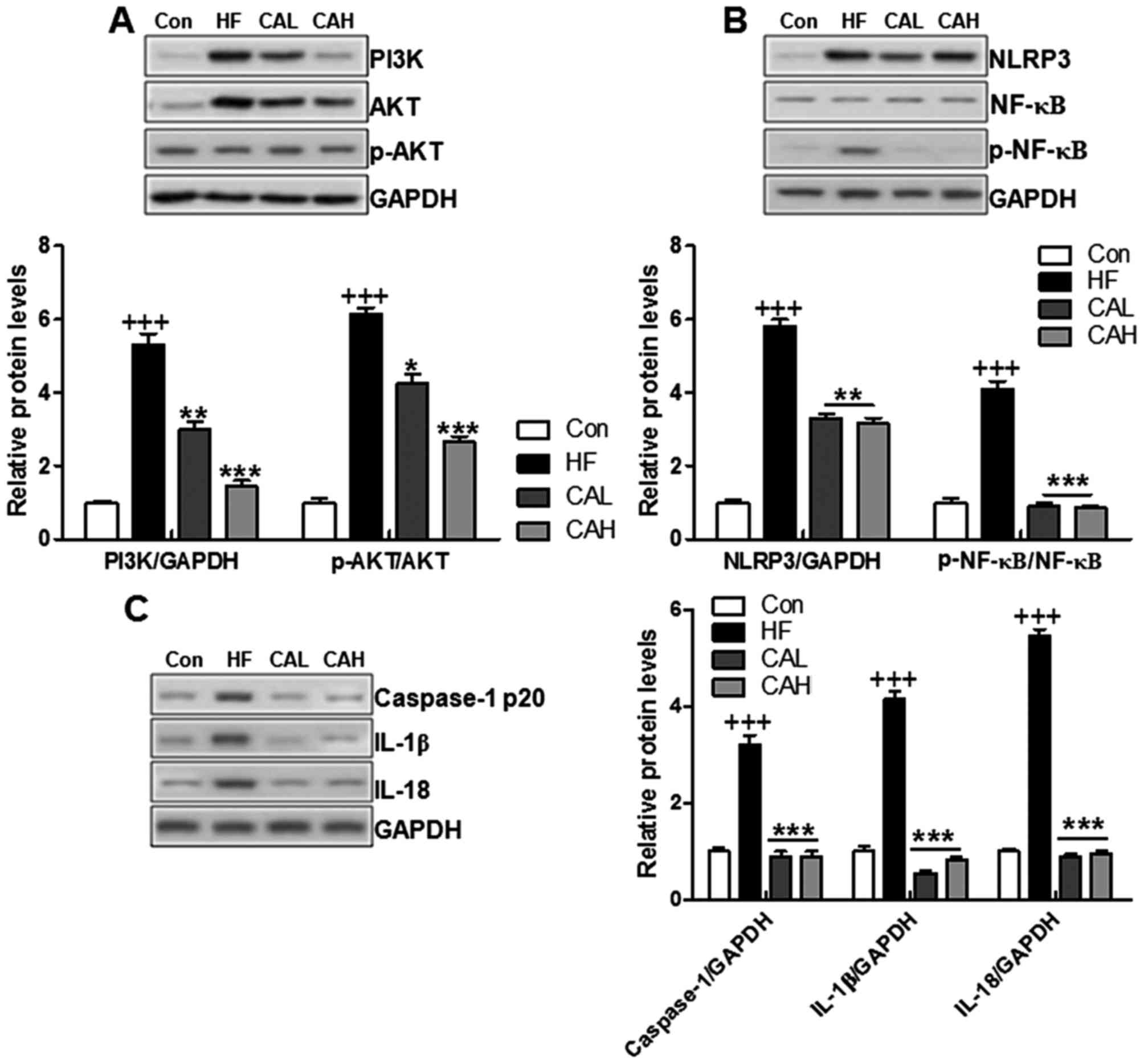
CA protects mice against HF diet-induced
liver injury by suppressing PI3K/AKT and NLRP3 inflammasome
signaling pathway activation
PI3K/AKT is a potential target of MARCKS in
attenuating NAFLD based on the results mentioned above. A recent
study demonstrated that CA treatment attenuated the inflammatory
response by regulating the NF-κB signaling pathway (40). To confirm this conclusion, we
measured the levels of PI3K/AKT in the livers of mice fed a HF diet
and treated with CA. We found that the upregulation of PI3K and
p-AKT were reduced by carnosic acid in liver of mice with HF diet
induction (Fig. 11A). Increased
PI3K/AKT was also a pivotal factor for inducing NF-κB activation
(41). Therefore, we examined
whether NLRP3 and NF-κB expression in the liver was altered by CA.
The overexpression of NLRP3 and the phophorylated levels of NF-κB
in the HF diet-fed mice were reduced by CA treatment, whereas the
levels of total NF-κB were not affected by CA (Fig. 11B). Caspase-1 was stimulated in
HF diet fed mice, which was down-regulated due to carnosic acid
administration, leading to NF-κB inactivity and pro-inflammatory
cytokines suppression, including IL-1β and IL-18 (Fig. 11C). These data indicated that
PI3K/AKT and NLRP3/NF-κB in the liver may be involved in the
inhibitory effects of CA against the inflammatory response in mice
with NAFLD induced by a HF diet.
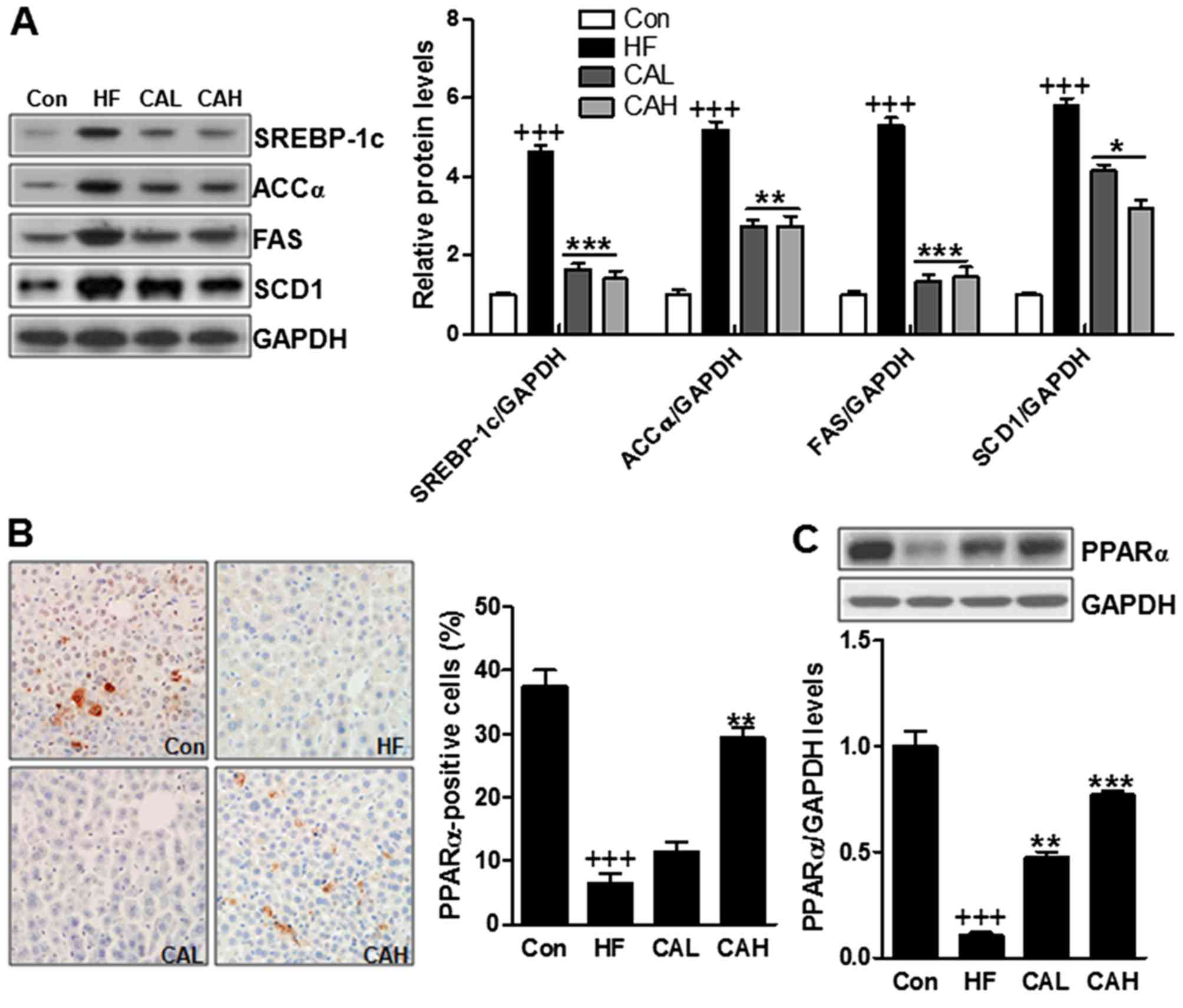
CA inhibits lipogenesis-related signaling
pathway activation
In the end of the treatment period, we investigated
whether lipogenesis is affected by CA. To examine this, we measured
the expression levels of several key transcriptional factors in
lipogenesis, including SREBP-1c, ACCα, FAS and SCD1. The
overexpression of SREBP-1c, ACCα, FAS and SCD1, induced by the HF
diet, was significantly attenuated by CA (Fig. 12A). Moreover, we also examined
the PPARα levels by immunohistochemical assays, and the results
revealed that PPARα expression was downregulated in the HF diet-fed
mice, and that this effect was reversed by CA (Fig. 12B). The mRNA level of PPARα was
also decreased in the mice fed the HF diet, and similar to the data
mentioned above, CA increased PPARα mRNA expression (Fig. 12C). These results clearly
indicated that CA treatment suppressed lipogenesis in HF diet
induced animals with NAFLD.
Discussion
NAFLD is the hepatic manifestation of obesity and
insulin resistance (1,2,42).
The progression involves the development of liver steatosis,
increased inflammation and hepatocyte injury, and eventually the
deposition of extracellular matrix proteins, leading to liver
fibrosis and cirrhosis. The clinical features of NAFLD include
obesity, insulin resistance and dyslipidemia. The consumption of a
HF diet is an important factor in the increasing occurrence of
these metabolic disorders, primarily NAFLD (43). MARCKS is an intrinsically unfolded
protein with a conserved cationic effector domain, regulating the
crosstalk between several signaling transduction pathways. MARCKS
has been shown to be involved in the regulation of cell adhesion
and migration, of exocytosis, endocytosisand phagocytosis, as well
as brain development (44,45).
MARCKS has been investigated in cancer progression and LPS-induced
disease from PI3K/AKT and inflammation-related signaling pathways
(46). However, the effects of
MARCKS on NAFLD development have not been fully determined. Thus,
in this present study, we attempted to examine the role of MARCKS
in HF diet-induced NAFLD in mice and to elucidate the underlying
mechanisms. In addition, we examined the effects of CA, which is an
essential antioxidant compound in Rosmarinus officinalis L.,
on liver cancer. CA has anticancer effects on colon cancer, acute
myeloid leukemia and skin cancer through anti-inflammatory,
antioxidant and antimicrobial properties. However, the molecular
mechanisms responsible for NAFLD remain poorly understood. In the
present study, the consumption of a HF diet decreased the
expression of MARCKS in mouse livers, suggesting a role of MARCKS
in modulating the pathogenesis of NAFLD. However, the genetic
deletion of MARCKS increased hepatic steatosis, the inflammatory
response, lipid accumulation and ALT/AST levels in the liver in
response to a HF diet. Taken together, these data suggest that the
inhibition of MARCKS exacerbates the fibrotic response to a HF
diet.
First of all, we used MARCKS-deficient mice to
construct a model of HF diet-induced NAFLD model in order to
investigate the effects of MARCKS in NAFLD. Insulin resistance or
impaired insulin signaling is a main contributor to the disruption
of glucose metabolism and plays a pivotal role in pathogenesis of
T2DM (47,48). Reciprocally, the improvement or
restoration of insulin signaling is an effective treatment for
restoring abnormal glucose metabolism in T2DM (49). In this study, we found that MARCKS
deficiency increased glucose levels in serum and accelerated
insulin resistance in HF diet-fed mice. Thus, we hypothesized that
MARCKS may play a potential role in regulating insulin resistance
in mice with NAFLD induced by a HF diet. Of note, CA administration
not only improved MARCKS expressed levels from mRNA and protein
levels, but also improved insulin sensitivity from down-regulating
glucose levels in blood. In the insulin signal pathway, Akt is a
central regulator for insulin signal transduction and accumulating
evidences revealed that many chemicals activating Akt signal have
potential role in improving insulin signal transduction (50,51). Previous data have revealed that
MARCKS has a close association with AKT activity in cancer
(30). In this study, PI3K/AKT
signaling pathway activity was increased in mice deficient in
MARCKS, suggesting that MARCKS plays a role in suppressing AKT
activity in HF diet-fed mice with NAFLD. Thus, the administration
of CA improved glucose homeostasis in the HF diet-fed mice with
NAFLD. Also, carnosic acid showed inhibitory role in mediating AKT
activity, which might be related to the suppressive effects of
MARCKS on AKT activity.
The reasons for ameliorated insulin signal
transduction in liver tissues are probably due to the
downregulation of circulating pro-inflammatory cytokines (52). The inflammatory response in the
livers of mice with NAFLD is widely considered as a key factor for
inducing systemic insulin resistance (53). Pro-inflammatory cytokines,
including TNFα, IL-18, IL-2, IL-4, IL-1β and IL-6, disrupt the
insulin sensitivity of adipocytes and liver (54). In this study, demonstrated that
the expression levels of pro-inflammatory cytokines in serum and
liver tissues were greatly increased in MARCKS-deficient mice the
HF diet. In the HF diet-fed mice, the levels of these
pro-inflammatory cytokines were suppressed by CA. In agreement with
our results, it has been shown that CA inhibits the inflammation
through reducing the release of pro-inflammatory cytokines
(55). NF-κB signaling-related
factors are involved in inflammatory responses (56). The key components of the
functional NLRP3 inflammasome include NLRP3 and caspase-1. The
activation of the NLRP3 inflammasome increases the maturation and
release of pro-inflammatory cytokines, such as IL-1β and pro-IL-18
potently (57). In this study, we
found that MARCKS deficiency stimulated NLRP3 expression, leading
to caspase-1 activation and causing pro-inflammatory cytokine
release through NF-κB phosphorylation. Here, the secretion of
pro-inflammatory cytokines in liver of mice with HF diet are
greatly suppressed by CA.
In addition to inflammation, lipid accumulation was
also affected by MARCKS and CA. Our results revealed a marked
enhancement of TG and TC accumulation in the MARCKS-deficient mice
fed the HF diet. MARCKS deficiency led to lipid accumulation in HF
diet fed mice, indicating that MARCKS, at least partly, could
influence lipid metabolism in mice with NAFLD. In addition, a
marked reduction in the levels of TGs and TC in mice treated with
CA was observed in our study, suggesting that CA has a
lipid-regulating effect. SREBP-1c is a member of the family of
SREBP membrane-bound transcription factors, activating the
transcription of lipogenic genes, which contain the sterol
regulatory elements in their promoter regions, including FAS and
SCD1. The expression of two genes in particular, FAS and SCD1, is
closely related to the levels of the de novo fatty acid
synthesis and to cellular lipid accumulation ultimately (58). Some lines of evidence have
indicated that the induction of SREBP-1c expression in the liver
aids in the enhancement of hepatic TG concentrations, causing the
progression of NAFLD (59).
Consistently, in our study, SREBP-1c, FAS and SCD1 were upregulated
in mice fed a HF diet, and this effect was enhanced in the
MARCKS-deficient mice, leading to a high expression of FAS and
SCD1, indicating that MARCKS is involved in lipid metabolism in
mice with NAFLD. However, the specific underlying mechanisms
require further investigation. PPARα is a major transcriptional
regulator of lipid metabolism, which is involved in fatty acid
β-oxidation (60). This study
demonstrated that PPARα expression was downregulated by MARCKS
deficiency in HF diet-fed mice. By contrast, PPARα was upregulated
due to CA administration in mice with fed the HF diet. The present
study showed that carnosic acid reduced lipogenesis in HF
diet-induced mice with NAFLD, which might be linked to MARCKS
upregulation.
In conclusion, the findings of the present study
reveal a protective effect of MARCKS of MARCKS on mice treated with
a HF diet. Our data indicate that MARCKS may protect the liver from
the lipid accumulation, inflammation, fibrosis in the context of
NAFLD and the damaged glucose homeostasis. In addition, our results
revealed that CA possesses the ability to improve high fat
diet-induced NAFLD in mice through reducing the lipogenesis and
inflammation in liver. Therefore, our data indicate that CA may be
used as a potential therapeutic agent in the treatment of
NAFLD-related metabolic diseases, including insulin resistance and
lipid abnormal metabolism.
Acknowledgments
Not applicable
References
|
1
|
Duvnjak M, Lerotić I, Barsić N, Tomasić V,
Virović Jukić L and Velagić V: Pathogenesis and management issues
for non-alcoholic fatty liver disease. World J Gastroenterol.
13:4539–4550. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Angulo P: Nonalcoholic fatty liver
disease. N Engl J Med. 346:1221–1231. 2002. View Article : Google Scholar
|
|
3
|
Postic C and Girard J: The role of the
lipogenic pathway in the development of hepatic steatosis. Diabetes
Metab. 34:643–648. 2008. View Article : Google Scholar
|
|
4
|
Yeh MM and Brunt EM: Pathology of
nonalcoholic fatty liver disease. Am J Clin Pathol. 128:837–847.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Estep JM, Baranova A, Hossain N, Elariny
H, Ankrah K, Afendy A, Chandhoke V and Younossi ZM: Expression of
cytokine signaling genes in morbidly obese patients with
nonalcoholic steatohepatitis and hepatic fibrosis. Obes Surg.
19:617–624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lam B and Younossi ZM: Treatment options
for nonalcoholic fatty liver disease. Therap Adv Gastroenterol.
3:121–137. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang MT, Ho CT, Wang ZY, Ferraro T, Lou
YR, Stauber K, Ma W, Georgiadis C, Laskin JD and Conney AH:
Inhibition of skin tumorigenesis by rosemary and its constituents
carnosol and ursolic acid. Cancer Res. 54:701–708. 1994.PubMed/NCBI
|
|
8
|
Shi B, Wang LF, Meng WS, Chen L and Meng
ZL: Carnosic acid and fisetin combination therapy enhances
inhibition of lung cancer through apoptosis induction. Int J Oncol.
50:2123–2135. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moran AE, Carothers AM, Weyant MJ, Redston
M and Bertagnolli MM: Carnosol inhibits beta-catenin tyrosine
phosphorylation and prevents adenoma formation in the
C57BL/6J/Min/+ (Min/+) mouse. Cancer Res. 65:1097–1104.
2005.PubMed/NCBI
|
|
10
|
Sharabani H, Izumchenko E, Wang Q, Kreinin
R, Steiner M, Barvish Z, et al: Cooperative antitumor effects of
vitamin D3 derivatives and rosemary preparations in a mouse model
of myeloid leukemia. Int J Cancer. 118:3012–3021. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jarboe JS, Anderson JC, Duarte CW, Mehta
T, Nowsheen S, Hicks PH, Whitley AC, Rohrbach TD, McCubrey RO, Chiu
S, et al: MARCKS regulates growth and radiation sensitivity and is
a novel prognostic factor for glioma. Clin Cancer Res.
18:3030–3041. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brooks G, Brooks SF and Goss MW: MARCKS
functions as a novel growth suppressor in cells of melanocyte
origin. Carcinogenesis. 17:683–689. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bickeböller M, Tagscherer KE, Kloor M,
Jansen L, Chang-Claude J, Brenner H, Hoffmeister M, Toth C,
Schirmacher P, Roth W, et al: Functional characterization of the
tumor-suppressor MARCKS in colorectal cancer and its association
with survival. Oncogene. 34:1150–1159. 2015. View Article : Google Scholar
|
|
14
|
Kim J, Shishido T, Jiang X, Aderem A and
McLaughlin S: Phosphorylation, high ionic strength, and calmodulin
reverse the binding of MARCKS to phospholipid vesicles. J Bio Chem.
269:28214–28219. 1994.
|
|
15
|
Lippoldt J, Händel C, Dietrich U and Käs
JA: Dynamic membrane structure induces temporal pattern formation.
BBA-Biomembranes. 1838:2380–2390. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rombouts K, Lottini B, Caligiuri A, Liotta
A, Mello T, Carloni V, Marra F and Pinzani M: MARCKS is a
downstream effector in platelet-derived growth factor-induced cell
motility in activated human hepatic stellate cells. Exp Cell Res.
314:1444–1454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu D, Makkar G, Strickland DK, Blanpied
TA, Stumpo DJ, Blackshear PJ, Sarkar R and Monahan TS:
Myristoylated alanine-rich protein kinase substrate (MARCKS)
regulates small GTPase Rac1 and Cdc42 activity and is a critical
mediator of vascular smooth muscle cell migration in intimal
hyperplasia formation. J Am Heart Assoc. 4:e0022552015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao Y and Davis HW: Thrombin-induced
phosphorylation of the myristoylated alanine-rich C kinase
substrate (MARCKS) protein in bovine pulmonary artery endothelial
cells. J Cell Physiol. 169:350–357. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Campbell JS, Hughes SD, Gilbertson DG,
Palmer TE, Holdren MS, Haran AC, Odell MM, Bauer RL, Ren HP, Haugen
HS, et al: Platelet-derived growth factor C induces liver fibrosis,
steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci USA.
102:3389–3394. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Diehl AM, Li ZP, Lin HZ and Yang SQ:
Cytokines and the pathogenesis of non-alcoholic steatohepatitis.
Gut. 54:303–306. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abiru S, Migita K, Maeda Y, Daikoku M, Ito
M, Ohata K, Nagaoka S, Matsumoto T, Takii Y, Kusumoto K, et al:
Serum cytokine and soluble cytokine receptor levels in patients
with non-alcoholic steatohepatitis. Liver Int. 26:39–45. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Neuschwander-Tetri BA, Brunt EM, Wehmeier
KR, Oliver D and Bacon BR: Improved nonalcoholic steatohepatitis
after 48 weeks of treatment with the PPAR-γ ligand rosiglitazone.
Hepatology. 38:1008–1017. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ratziu V, Harrison SA, Francque SM,
Bedossa P, Serfaty L, Gomez MR, et al: An international, phase 2
randomized controlled trial of the dual PPAR α-δ agonist GFT505 in
adult patients with NASH. Hepatology. 62:262A–263A. 2015.
|
|
24
|
Abitbol JL, Broqua P and Junien JL:
Metabolic effects and good tolerance of IVA337 a Pan-PPAR agonist
in diabetic patients warrant further investigation in NASH. J
Hepatol. 64:S1892016. View Article : Google Scholar
|
|
25
|
Pettinelli P, Del Pozo T, Araya J, Rodrigo
R, Araya AV, Smok G, Csendes A, Gutierrez L, Rojas J, Korn O, et
al: Enhancement in liver SREBP-1c/PPAR-α ratio and steatosis in
obese patients: correlations with insulin resistance and n-3
long-chain polyunsaturated fatty acid depletion. Biochim Biophys
Acta. 1792:1080–1086. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bajaj M, Suraamornkul S, Hardies LJ, Glass
L, Musi N and DeFronzo RA: Effects of peroxisome
proliferator-activated receptor (PPAR)-α and PPAR-γ agonists on
glucose and lipid metabolism in patients with type 2 diabetes
mellitus. Diabetologia. 50:1723–1731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Acikgoz Y, Can B, Bek K, Acikgoz A, Ozkaya
O, Genç G and Sarikaya S: The effect of simvastatin and
erythropoietin on renal fibrosis in rats with unilateral ureteral
obstruction. Ren Fail. 36:252–257. 2014. View Article : Google Scholar
|
|
28
|
Mohamed JS, Lopez MA and Boriek AM:
Mechanical stretch upregulates microRNA-26a and induces human
airway smooth muscle hypertrophy by suppressing glycogen synthase
kinase-3β. J Biol Chem. 285:29336–29347. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baker RG, Hayden MS and Ghosh S: NF-κB,
inflammation, and metabolic disease. Cell Metab. 13:11–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen CH, Statt S, Chiu CL, Thai P, Arif M,
Adler KB and Wu R: Targeting myristoylated alanine-rich C kinase
substrate phosphorylation site domain in lung cancer. Mechanisms
and therapeutic implications. Am J Resp Crit Care. 190:1127–1138.
2014. View Article : Google Scholar
|
|
31
|
Dos Santos S, Delattre AI, De Longueville
F, Bult H and Raes M: Gene expression profiling of LPS-stimulated
murine macrophages and role of the NF-κB and PI3K/mTOR signaling
pathways. Ann Ny Acad Sci. 1096:70–77. 2007. View Article : Google Scholar
|
|
32
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tatsumi S, Mabuchi T, Katano T, Matsumura
S, Abe T, Hidaka H, Suzuki M, Sasaki Y, Minami T and Ito S:
Involvement of Rho-kinase in inflammatory and neuropathic pain
through phosphorylation of myristoylated alanine-rich C-kinase
substrate (MARCKS). Neuroscience. 131:491–498. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Raghow R, Yellaturu C, Deng X, Park EA and
Elam MB: SREBPs: the crossroads of physiological and pathological
lipid homeostasis. Trends Endocrinol Metab. 19:65–73. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Blankenship K, Gilley A, Piekarski A,
Orlowski S, Greene E, Bottje W, Anthony N and Dridi S: Differential
expression of feeding-related hypothalamic neuropeptides in the
first generation of quails divergently selected for low or high
feed efficiency. Neuropeptides. 58:31–40. 2016. View Article : Google Scholar
|
|
36
|
Chen H, Zhang L, Li X, Li X, Sun G, Yuan
X, Lei L, Liu J, Yin L, Deng Q, et al: Adiponectin activates the
AMPK signaling pathway to regulate lipid metabolism in bovine
hepatocytes. J Steroid Biochem. 138:445–454. 2013. View Article : Google Scholar
|
|
37
|
Dossi CG, Tapia GS, Espinosa A, Videla LA
and D'Espessailles A: Reversal of high-fat diet-induced hepatic
steatosis by n-3 LCPUFA: role of PPAR-α and SREBP-1c. J Nutr
Biochem. 25:977–984. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pawlak M, Lefebvre P and Staels B:
Molecular mechanism of PPARα action and its impact on lipid
metabolism, inflammation and fibrosis in non-alcoholic fatty liver
disease. J Hepatol. 62:720–733. 2015. View Article : Google Scholar
|
|
39
|
du Plessis J, Korf H, van Pelt J,
Windmolders P, Vander Elst I, Verrijken A, Hubens G, Van Gaal L,
Cassiman D, Nevens F, et al: Pro-inflammatory cytokines but not
endotoxin-related parameters associate with disease severity in
patients with NAFLD. PloS One. 11:e01660482016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lin CY, Chen JH, Fu RH and Tsai CW:
Induction of Pi form of glutathione S-transferase by carnosic acid
is mediated through PI3K/Akt/NF-κB pathway and protects against
neurotoxicity. Chem Res Ttoxicol. 27:1958–1966. 2014. View Article : Google Scholar
|
|
41
|
Zhao M, Zhou A, Xu L and Zhang X: The role
of TLR4-mediated PTEN/PI3K/AKT/NF-κB signaling pathway in
neuroinflammation in hippocampal neurons. Neuroscience. 269:93–101.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Boppidi H and Daram SR: Nonalcoholic fatty
liver disease: hepatic manifestation of obesity and the metabolic
syndrome. Postgrad Med. 120:E01–E07. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nakamura A and Terauchi Y: Lessons from
mouse models of high-fat diet-induced NAFLD. Int J Mol Sci.
14:21240–21257. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Orecchia A, Mettouchi A, Uva P, Simon GC,
Arcelli D, Avitabile S, Ragone G, Meneguzzi G, Pfenninger KH,
Zambruno G and Failla CM: Endothelial cell adhesion to soluble
vascular endothelial growth factor receptor-1 triggers a cell
dynamic and angiogenic phenotype. FASEB J. 28:692–704. 2014.
View Article : Google Scholar
|
|
45
|
Dorris E, O'Neill A, Hanrahan K, Treacy A
and Watson RW: MARCKS promotes invasion and is associated with
biochemical recurrence in prostate cancer. Oncotarget. 8:720212017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee SM, Suk K and Lee WH: Myristoylated
alanine-rich C kinase substrate (MARCKS) regulates the expression
of proinflammatory cytokines in macrophages through activation of
p38/JNK MAPK and NF-κB. Cell Immunol. 296:115–121. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zheng S, Hoos L, Cook J, Tetzloff G, Davis
H Jr, van Heek M and Hwa JJ: Ezetimibe improves high fat and
cholesterol diet-induced non-alcoholic fatty liver disease in mice.
Eur J Pharmacol. 584:118–124. 2008. View Article : Google Scholar
|
|
48
|
Matono T, Koda M, Tokunaga S, Kato J,
Sugihara T, Ueki M and Murawaki Y: Therapeutic effects of ezetimibe
for non-alcoholic steatohepatitis in fatty liver shionogi-ob/ob
mice. Hepatol Res. 41:1240–1248. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kishino Y, Tanaka Y, Ikeda T, Yamamoto K,
Ogawa H, Iwatani Y and Kamisako T: Ezetimibe increases hepatic iron
levels in mice fed a high-fat diet. J Pharmacol Exp Ther.
345:483–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Choi K and Kim YB: Molecular mechanism of
insulin resistance in obesity and type 2 diabetes. Korean J Intern
Med. 25:119–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Mackenzie RW and Elliott BT: Akt/PKB
activation and insulin signaling: A novel insulin signaling pathway
in the treatment of type 2 diabetes. Diabetes Metab Syndr Obes.
7:55–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bagul PK, Middela H, Matapally S, Padiya
R, Bastia T, Madhusudana K, Reddy BR, Chakravarty S and Banerjee
SK: Attenuation of insulin resistance, metabolic syndrome and
hepatic oxidative stress by resveratrol in fructose-fed rats.
Pharmacol Res. 66:260–268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chandrashekaran V, Dattaroy D, Das S,
Alhasson F, Seth R, Carson J, Berger F, Zielnoka J, Kalyanaraman B
and Chatterjee S: The receptor for advanced glycation end product
(RAGE) binding to HMGB1 and subsequent NADPH oxidase activation
mediates ectopic intestinal inflammation in NAFLD. Free Radical
Biol Med. 100:S372016. View Article : Google Scholar
|
|
54
|
Calle MC and Fernandez ML: Inflammation
and type 2 diabetes. Diabetes Metab. 38:183–191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hadad N and Levy R: The synergistic
anti-inflammatory effects of lycopene, lutein, β-carotene, and
carnosic acid combinations via redox-based inhibition of NF-κB
signaling. Free Radical Biol Med. 53:1381–1391. 2012. View Article : Google Scholar
|
|
56
|
Segovia J, Sabbah A, Mgbemena V, Tsai SY,
Chang TH, Berton MT, Morris IR, Allen IC, Ting JP and Bose S:
TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux
activates NLRP3/ASC inflammasome during respiratory syncytial virus
infection. PLOs One. 7:e296952012. View Article : Google Scholar
|
|
57
|
Vandanmagsar B, Youm YH, Ravussin A,
Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM and Dixit
VD: The NLRP3 inflammasome instigates obesity-induced inflammation
and insulin resistance. Nat Med. 17:179–188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fu S, Yang L, Li P, Hofmann O, Dicker L,
Hide W, Lin X, Watkins SM, Ivanov AR and Hotamisligil GS: Aberrant
lipid metabolism disrupts calcium homeostasis causing liver
endoplasmic reticulum stress in obesity. Nature. 473:528–531. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ahmed MH and Byrne CD: Modulation of
sterol regulatory element binding proteins (SREBPs) as potential
treatments for non-alcoholic fatty liver disease (NAFLD). Drug
Discov Today. 12:740–747. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Reddy JK: Peroxisomal β-oxidation, PPARα,
and steatohepatitis. Am J Physiol-Gastr L. 281:G1333–G1339.
2001.
|