Introduction
Skin cancer, particularly non-melanoma skin cancers
(NMSCs), including basal cell carcinoma and squamous cell
carcinoma, is considered one of the most prevalent health concerns
worldwide. Approximately 132,000 cases of melanoma and 2–3 million
cases of NMSC are reported per annum globally (1,2).
Extensive exposure to sunlight has been closely associated with
skin cancer (3), and the
formation of skin cancer has been reported to be associated with
one or numerous signaling pathways (4). Various coordinated biological
alterations, including but not limited to epidermal hyperplasia,
inflammation, oxidative stress and proliferation, are essential
events associated with skin tumor progression (4,5).
Juglanin is a natural compound extracted from crude
Polygonum aviculare, which has been reported to exert
inhibitory activity against the inflammatory response, as well as
cancer growth (6–8). It has previously been suggested that
juglanin inhibits apoptosis and the inflammatory response through
Toll-like receptor 4-modulated mitogen-activated protein kinase
(MAPK)/nuclear factor (NF)-κB and Janus kinase 2/signal transducer
and activator of transcription 3 signaling pathways, respectively,
in rats with hepatitis (9). In
addition, juglanin has been revealed to decrease the levels of
reactive oxygen species in senescent cells induced by adriamycin
treatment (10). Furthermore,
juglanin may exert inhibitory effects against
lipopolysaccharide-induced cytokine production in RAW 264.7
macrophages, including interleukin (IL)-1β, tumor necrosis factor
(TNF)-α and IL-6 (11). However,
to the best of our knowledge, the role of juglanin in autophagy and
apoptosis in UVB-induced skin injury remains to be elucidated.
Furthermore, it remains to be determined as to whether juglanin
suppresses the growth of human skin cancer.
A previous study indicated that NF-κB may serve an
important role in the maintenance of skin homeostasis, as well as
the regulation of various types of cell proliferation, resistance
to apoptosis and survival (12).
Various signaling pathways have been reported to have a crucial
role in the response of cells to ultraviolet B (UVB) irradiation,
including phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)
signaling. In addition, the MAPK family subgroups, c-Jun N-terminal
kinase (JNK) and p38, have been reported to serve an important role
in cell apoptosis and proliferation (13,14). UVB induces potentially lethal
injuries to cellular DNA, thus initiating cellular recovery
mechanisms, including activation of DNA damage response pathways
and apoptosis (15). However,
some DNA-injured cells could induce apoptosis, destruct
differentiation control and even lead to the formation of cancerous
cells in the end (16).
Therefore, exploring novel therapeutic strategies for apoptotic
induction, and the suppression of cell proliferation and
inflammation in skin cancer is necessary.
The present study aimed to explore the effects of
various doses of juglanin on UVB-induced mouse skin tumor formation
and promotion in vivo. In vitro, B16F10 cells were
used as a model of melanoma to determine the molecular mechanisms
underlying the proapoptotic effects of juglanin on UVB-irradiated
melanoma cells. The results demonstrated that juglanin promoted
cell death of UVB-irradiated B16F10 cells, thus indicating that
juglanin may be considered a potential compound to prevent against
UVB-irradiated skin damage and skin cancer.
Materials and methods
Animals and treatment
A total of 40 SKH-1 hairless mice (age, 5–6 weeks;
weight, ~20 g) were initially obtained from Jackson Laboratory (Bar
Harbor, ME, USA), and were maintained in our facility with free
access to water and food, under a 12-h light/dark cycle, with 35%
humidity. The mice were divided into four groups: The control (Con)
group (without any UVB irradiation and juglanin treatment), the
model (Mod) group (UVB irradiation), and the 10 and 20 mg/kg
juglanin-treated groups, which were treated with juglanin after UVB
irradiation. All experimental procedures were carried out in
accordance with the Guide for the Care and Use of Laboratory
Animals, and, before the animal experiments were carried out, the
procedures were approved by the Research Ethical Committee of
Southern Medical University (Guangdong, China).
The UV lamp (UVA-340; SolarSystems & Solutions,
LLC, Clifford, PA, USA) used to irradiate mice in the present study
simulates sunlight in the critical short wavelength region, from
365 nm to the solar cut-off of 295 nm, with a peak emission at 340
nm. The radiant dose was quantified, at the appropriate distance,
using a UVB Daavlin Flex Control Integrating Dosimeter (Daavlin,
Bryan, OH, USA) and was further confirmed using an external
radiometer (X-96 Irradiance Meter; Daavlin) before and after each
irradiation session. An electrical fan was used to avoid excessive
heating. As for the acute radiation experiments, mice were exposed
to UV radiation at a dose of 600 mJ/cm2 in groups and
were euthanized 24 or 72 h after irradiation. For skin
carcinogenesis experiments, the animals were exposed twice a week
for 10 weeks on Mondays and Wednesdays to solar-simulated UV
radiation in clear bedding-free cages. During the process, the
animals were consecutively administered various doses (10 and 20
mg/kg) of juglanin by gavage. Finally, the dorsal skin was
harvested, and computer-assisted image analysis, using a Zeiss
KS300 (version 3.0; München, Germany,) was used to observe vessel
size. Some of the fresh skin tissue samples were immediately frozen
in liquid N2 and stored at −80°C for subsequent
experiments.
Histopathological analysis
Fresh tissue samples were fixed in formalin for 48 h
at room temperature. Then, the formalin-fixed, paraffin-embedded
mouse skin samples were processed and were then stained with
hematoxylin and eosin, based on a routine protocol (17). The stained tissue sections
(5-μm thick) were then observed under a light microscope and
digital micrographs of slides were captured for analysis.
Immunohistochemical analysis
For immunohistochemistry, fresh tissue samples were
fixed in formalin for 48 h at room temperature. Subsequently,
tissue samples were embedded in paraffin and cut into sections
(5-μm thick) using a microtome. The sections were then fixed
onto slides. Endogenous peroxidase was blocked with 3%
H2O2, and non-specific proteins were blocked
with 10% goat serum for 30 min. The samples were then incubated
with antibodies against KI67, TNF-α and proliferating cell nuclear
antigen (PCNA) (1:100 dilution) in 5% horse serum (Life
Technologies, Grand Island, NY, USA) with PBS at 4°C overnight.
Sections were then incubated with diluted streptavidin-peroxidase
horseradish peroxidase (1:200; cat. no. 6721; Abcam, Cambridge, MA,
USA) for 30 min at room temperature using a Foxp3 staining kit
(eBioscience, San Diego, CA, USA), according to the manufacturer's
protocol. Finally, sections were stained with hematoxylin for 3
min, and were mounted and assessed using a phase-contrast
microscope (DP70; Olympus, Tokyo, Japan).
Cell culture and treatment
B16F10 murine melanoma cells and Hs68 human foreskin
fibroblast cells were purchased from American Type Culture
Collection (Manassas, VA, USA). The cells were maintained in a
monolayer culture in Dulbecco's modified Eagle's medium (DMEM;
Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine
serum (FBS; Gibco), 0.012% penicillin G, 0.027% streptomycin,
0.022% sodium pyruvate and 0.26% sodium bicarbonate at 37°C in an
atmosphere containing 95% air and 5% CO2. Juglanin stock
solution was prepared in dimethyl sulfoxide (DMSO) and was diluted
to the desired final concentration (10 and 20 μM) in culture
medium 24 h prior to use. The final concentration of DMSO did not
exceed 0.1% (v/v).
Cell viability analysis
The antiproliferative role of juglanin in B16F10 and
human Hs68 cells was determined using the MTS Cell Proliferation
Colorimetric Assay kit (Biovision, Inc., Milpitas, CA, USA).
Briefly, cells were plated in 96-well plates at a density of
5×103 cells/well. After 12 h, the cells were treated
with various concentrations of juglanin (0–30 μM) and/or UVB
(5 mJ/cm2) for 24 h. Subsequently, fresh MTS and PMS
mixture was added and incubated for 2–4 h at 37°C according to the
manufacturer's protocol. A MR7000 microplate reader (Dynatech
Laboratories Inc., Chantilly, VA, USA) was used to determine the
absorbance at 500 nm. In addition, half maximal inhibitory
concentration values were determined. Data are presented as the
mean of five replicates; each experiment was conducted in
triplicate.
Flow cytometric analysis
Cancer cell apoptosis was calculated via flow
cytometry. Cells were harvested and washed three times with PBS,
after which they were stained with Annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI) in binding buffer.
Apoptosis was assessed using a FACSCalibur flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA) after 15 min incubation at
room temperature in the dark. Fluorescence was detected at an
excitation wavelength of 480 nm via FL-1 (530 nm) and FL-2 filters
(585 nm). The number of apoptotic cells was then analyzed using
Win-MDI software (version 2.9; http://facs.scripps.edu/wm29w98.exe).
Fluorescence imaging
Tissue samples were washed twice with PBS and fixed
with 3.7% (v/v) formaldehyde in PBS for 15 min. Cells were
permeabilized for 5 min with 0.1% Triton X-100. For
immunofluorescence, phosphorylated (p)-NF-κB and 50 μg/ml
mouse anti-caspase-3 antibodies were employed, followed by
incubation with 2 μg/ml Alexa Fluor 488-goat anti-mouse
secondary antibodies. DAPI (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) was also used to stain cells. Images were acquired by
confocal laser scanning using epifluorescence microscopy (Nikon
TE2000-E; Shanghai Sunny Biotech Co., Ltd., Shanghai, China).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissue samples using
the mirVana miRNA Isolation kit (Ambion; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) according to the manufacturer's protocol.
Subsequently, cDNA was synthesized from total RNA using the TaqMan
miRNA RT kit (Applied Biosystems; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. qPCR was conducted using
the Applied Biosystems 7500 Sequence Detection system with iQ™
SYBR-Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
containing 5 ng cDNA and 10 pM each primer. Amplification of
pre-denaturated products was conducted at 94°C for 60 sec; followed
by 45 cycles at 95°C for 30 sec, 58°C for 30 sec and 72°C for 30
sec; followed by 95°C for 10 sec, 65°C for 45 sec, and 40°C for 60
sec. Data were normalized to the geometric mean of the housekeeping
gene GAPDH, and relative mRNA expression levels were calculated
using the 2−ΔΔCq method (18). Fold changes in the mRNA levels of
target genes relative to the endogenous GAPDH control were
calculated. Briefly, the quantification cycle (Cq) values of each
target gene were subtracted from the Cq values of the housekeeping
gene GAPDH (ΔCq). The target gene ΔΔCq was calculated as ΔCq of
target gene - ΔCq of control gene. The fold change in mRNA
expression was evaluated as 2−ΔΔCq. The primer sequences
used in the present study were as follows: IL-1β, sense (5′-3′) GTG
AGG AGA AGA TGG GTA G, antisense (5′-3′) AGA CCT AGG GAA GAA CCA
AT; IL-18, sense (5′-3′) AGG TAA CCT ACA AGA CGT GG, antisense
(5′-3′) TTA CCA GAT CGG TGA GAG AT; and GAPDH, sense (5′-3′) ATC
AAC ACG AGG CTA GCA GG and antisense (5′-3′) CAT CAT ACA CGC ACC
ACA GTC AC.
Western blotting
Melanoma cells were homogenized in 10% (wt/vol)
hypotonic buffer [25 mM Tris-HCl (pH 8.0), 1 mM EDTA, 5
μg/ml leupeptin, 1 mM Pefabloc SC, 50 μg/ml
aprotinin, 5 μg/ml soybean trypsin inhibitor and 4 mM
benzamidine] to yield a homogenate. The final supernatants were
obtained by centrifugation at 10,800 × g for 20 min at 4°C. Protein
concentration was determined using the bicinchoninic acid protein
assay kit (Thermo Fisher Scientific, Inc.) with bovine serum
albumin as a standard. Total protein extracts were used for western
blotting. Equal amounts of total protein (40 μg) were loaded
and proteins were separated using 10% SDS-PAGE and
electrophoretically transferred to the polyvinylidene difluoride
membranes (Millipore, Billerica, MA, USA). The membranes were then
blocked with 5% skim milk Tris buffered saline with 0.1% Tween 20
(TBST), washed, and then incubated with primary antibodies
overnight at 4°C. The membrane was then washed with TBST for three
times, followed by incubation with a horseradish peroxidase
(HRP)-conjugated secondary antibody (1:2,500; cat. no. 6721; Abcam)
at room temperature for 2 h. Following another round of washing
with TBST, the membrane was then developed using ECL (Thermo Fisher
Scientific, Inc.), and exposed to Kodak X-ray film (Kodak,
Rochester, NY, USA). Protein expression levels were defined as grey
value using ImageJ (version 1.4.2b, Mac OS X; National Institutes
of Health, Bethesda, MD, USA) and standardized to the housekeeping
gene (GAPDH) and expressed as a fold of control. All experiments
were performed in triplicate and done three times independently.
The primary polyclonal antibodies used were as follows: rabbit
anti-GAPDH (1:500; cat. no. sc-293335; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), p38 (1:1,000; cat. no. 8690; Cell Signaling
Technology, Inc., Danvers, MA, USA), p-p38 (1:1,000; cat. no. 4511,
Cell Signaling Technology, Inc.), p-JNK (1:1,000; cat. no. 4668,
Cell Signaling Technology, Inc.), JNK (1:1,000; cat. no. 9252, Cell
Signaling Technology, Inc.), TNF-α (1:1,000; cat. no. ab1793;
Abcam), IL-18 (1:1,000; cat. no. ab71495; Abcam), IL-1β (1:1,000;
cat. no. ab9722; Abcam), p-NF-κB (1:1,000; cat. no. ab86299;
Abcam), NF-κB (1:1,000; cat. no. ab207297; Abcam), PI3K (1:1,000;
cat. no. ab86714; Abcam), p-AKT (1:1,000; cat. no. 81283; Abcam),
AKT (1:1,000; cat. no. ab8805; Abcam), p-mammalian target of
rapamycin (mTOR; 1:1,000; cat. no. ab109268; Abcam), mTOR (1:1,000;
cat. no. ab2732; Abcam), poly (ADP-ribose) polymerase (PARP;
1:1,000; cat. no. ab13907; Abcam), cyclin-dependent kinase (CDK1;
1:1,000; cat. no. ab18; Abcam), cyclin D (1:1,000; cat. no.
ab134175; Abcam), p53 (1:1,000; cat. no. ab26; Abcam), p21
(1:1,000; cat. no. ab109199; Abcam), p27 (1:1,000; cat. no.
ab62364; Abcam), caspase-8 (1:1,000; cat. no. ab25901; Abcam),
caspase-3 (1:1,000; cat. no. ab52293; Abcam) and PCNA (1:1,000;
cat. no. ab29; Abcam).
Statistical analysis
Data are presented as the means ± standard deviation
(SD). Experimental groups were compared using GraphPad PRISM
(version 6.0; GraphPad Software, Inc., La Jolla, CA, USA) by
one-way analysis of variance with Dunn's least significant
difference tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
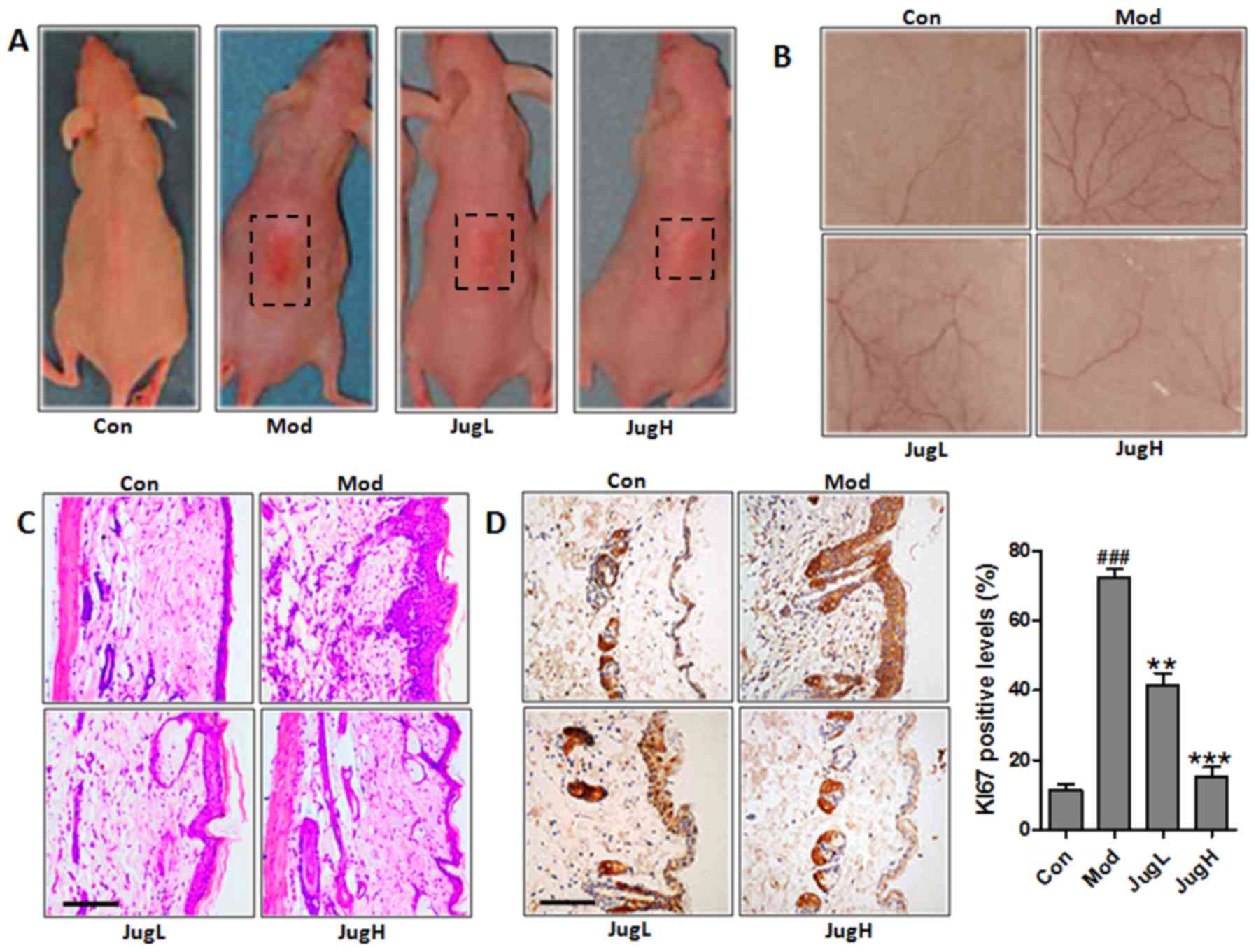
Juglanin ameliorates UVB-induced skin
carcinogenesis
UVB exposure has been known to stimulate hyperplasia
or infiltration of inflammatory cells into animal skin (19). Therefore, the present study
explored the role of juglanin in UVB-induced skin carcinogenesis.
Notably, 72 h after exposure to solar-simulated UV radiation, when
erythema development was most obvious, the skin of the
juglanin-treated mice exhibited no apparent erythema compared with
the mice in the Mod group (Fig.
1A). Having established the effects of juglanin on UVB-induced
skin injury, the present study explored the effects of 600
mJ/cm2 solar-simulated UV radiation on normal mice;
juglanin exerted dose-dependent alterations in the appearance of
skin vasculature 24 h after exposure (Fig. 1B). UVB exposure led to upregulated
epidermal thickness in the Mod group, as well as the infiltration
of inflammatory cells compared with in the control mice (Fig. 1C). Following UVB exposure,
administration of juglanin (10 and 20 mg/kg to mice) led to the
suppression of epidermal hyperplasia and inflammatory cell
infiltration compared with in the Mod group (Fig. 1C). KI67 expression was assessed in
the skin of UVB-induced mice administered juglanin. In mice in the
Mod group, the expression levels of KI67 were markedly higher
compared with in the control mice. However, juglanin treatment
significantly downregulated KI67 expression in the skin of
UVB-exposed mice compared with in the Mod group (Fig. 1D). Taken together, these results
suggested that juglanin exerts potential effects on the
amelioration of UVB-stimulated skin carcinogenesis in animals.
Juglanin suppresses UVB-induced p38/JNK
and PI3K/AKT activity
Phosphorylation of p38 and JNK is an indicator of
cellular stress following exposure to UV radiation (20). Therefore, the phosphorylated
levels of p38 and JNK were investigated in the present study. The
results suggested that p38 and JNK phosphorylation levels were
increased in mice following UV exposure compared with in the
control group (Fig. 2A). However,
juglanin treatment significantly reduced the increased expression
of p-p38 and p-JNK compared with in the Mod group (Fig. 2A). In addition, the PI3K/AKT
signaling pathway has been reported to be involved in UV
exposure-induced skin cancer (21). In the present study, PI3K was
activated in the Mod group, whereas juglanin reduced PI3K
expression. In addition, UV-induced p-AKT expression was
downregulated by juglanin in a dose-dependent manner (Fig. 2B). mTOR is regulated by the
PI3K/AKT signaling pathway, and is closely associated with cancer
progression (22). Finally, the
present study demonstrated that upregulated mTOR phosphorylation
was induced by UV, and was downregulated in the juglanin-treated
groups (Fig. 2B). These findings
indicated that p38/JNK and PI3K/AKT may be involved in the
inhibitory effects of juglanin on skin cancer.
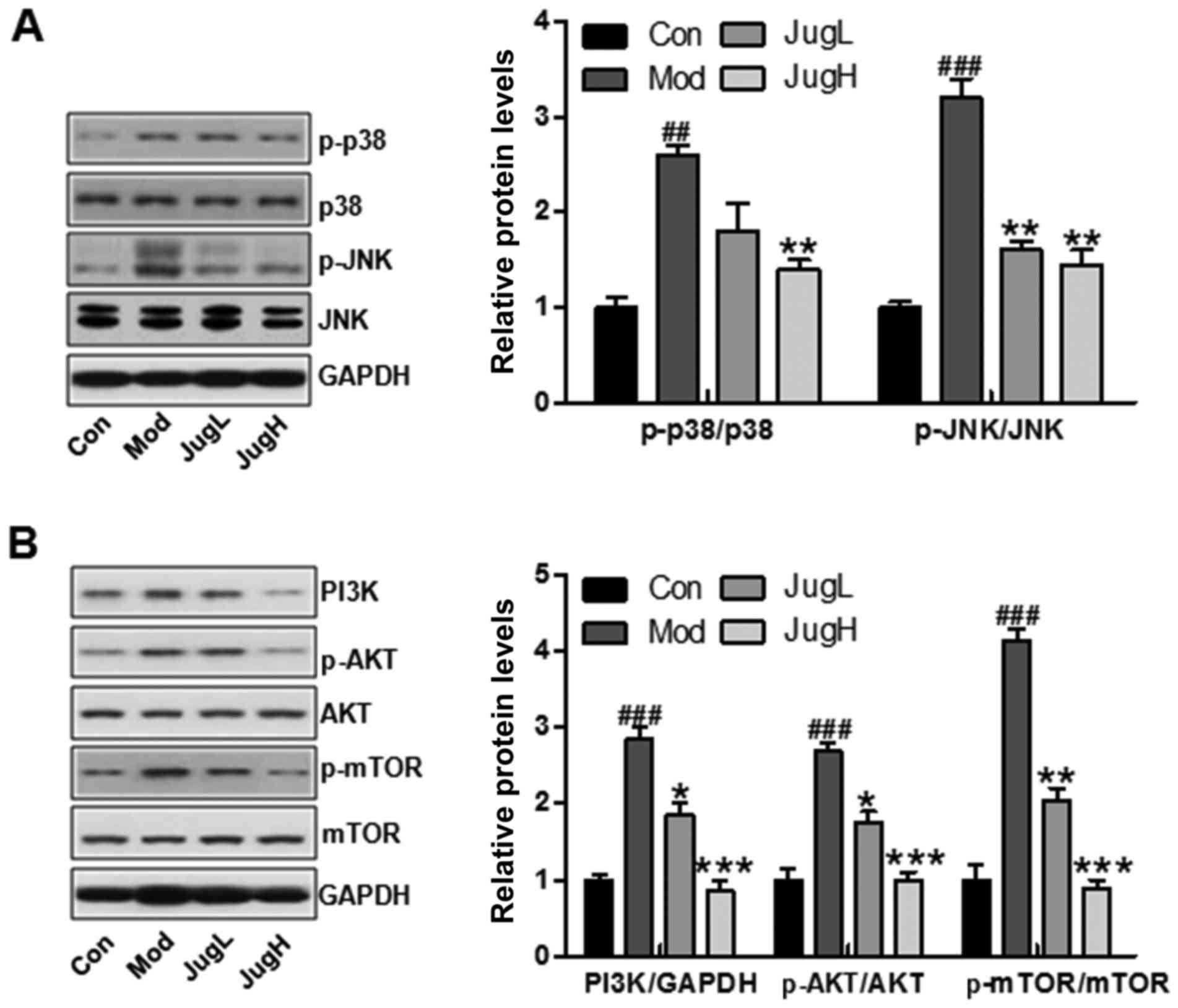 | Figure 2Effects of juglanin on UVB-induced
p38/JNK and PI3K/AKT signaling activity. Western blotting was
conducted and relative protein expression levels of (A) p-p38 and
p-JNK, and (B) PI3K, p-AKT and p-mTOR were determined. Data are
presented as the means ± standard deviation of three independent
experiments. ##P<0.01 and ###P<0.001
compared with the Con group; *P<0.05,
**P<0.01 and ***P<0.001 compared with
the Mod group. AKT, protein kinase B; Con, control; JNK, c-Jun
N-terminal kinase; JugH, high juglanin; JugL, low juglanin; Mod,
model; mTOR, mammalian target of rapamycin; p-, phosphorylated;
PI3K, phosphoinositide 3-kinase; UVB, ultraviolet B. |
Juglanin inhibits UVB-induced
inflammation
Skin inflammation has been reported to serve a role
in UVB exposure-induced cancer progression, as determined by the
enhancement of proinflammatory cytokines, including IL-1β, TNF-α
and IL-18 (23). IL-1β is
associated with the hemostatic function of normal skin in animals,
but is also involved in various pathophysiological situations,
including inflammation, when generated excessively. IL-1β can be
secreted from infiltrated macrophages, mast cells and keratinocytes
following exposure to UV (24).
TNF-α enhances the local inflammatory response in the epidermis and
possesses an essential role in photodamage (25). Furthermore, IL-6 has an essential
role in the acute phase response during acute inflammation, and
production of IL-6 is increased in the skin following UV exposure
(26). The expression levels of
proinflammatory cytokines were detected in the skin from
UVB-exposed mice treated with juglanin. In UVB-exposed mice, the
protein expression levels of TNF-α, IL-1β and IL-6 were
significantly upregulated compared with in the control group.
Conversely, treatment with juglanin significantly reduced the
protein expression levels of IL-1β, TNF-α and IL-6 in the skin of
UVB-exposed mice compared with in the Mod group (Fig. 3A). NF-κB is an essential regulator
that contributes to the release of proinflammatory cytokines
(27). The present study
demonstrated that NF-κB was activated in mice exposed to UV, which
was downregulated by juglanin administration (Fig. 3A). As shown in Fig. 3B, RT-qPCR was used to confirm that
UV-induced activation of IL-1β and IL-6 was downregulated by
juglanin administration (Fig.
3B). In addition, TNF-α was stimulated in the Mod group,
whereas juglanin treatment could reverse this UV-induced
overexpression, as determined by immunohistochemical analysis
(Fig. 3C). Finally,
immunofluorescence analysis indicated that NF-κB was activated in
the Mod group, whereas juglanin administration was able to reduce
p-NF-κB activation (Fig. 3D).
Taken together, these data indicated that juglanin may ameliorate
skin carcinogenesis via the suppression of inflammation.
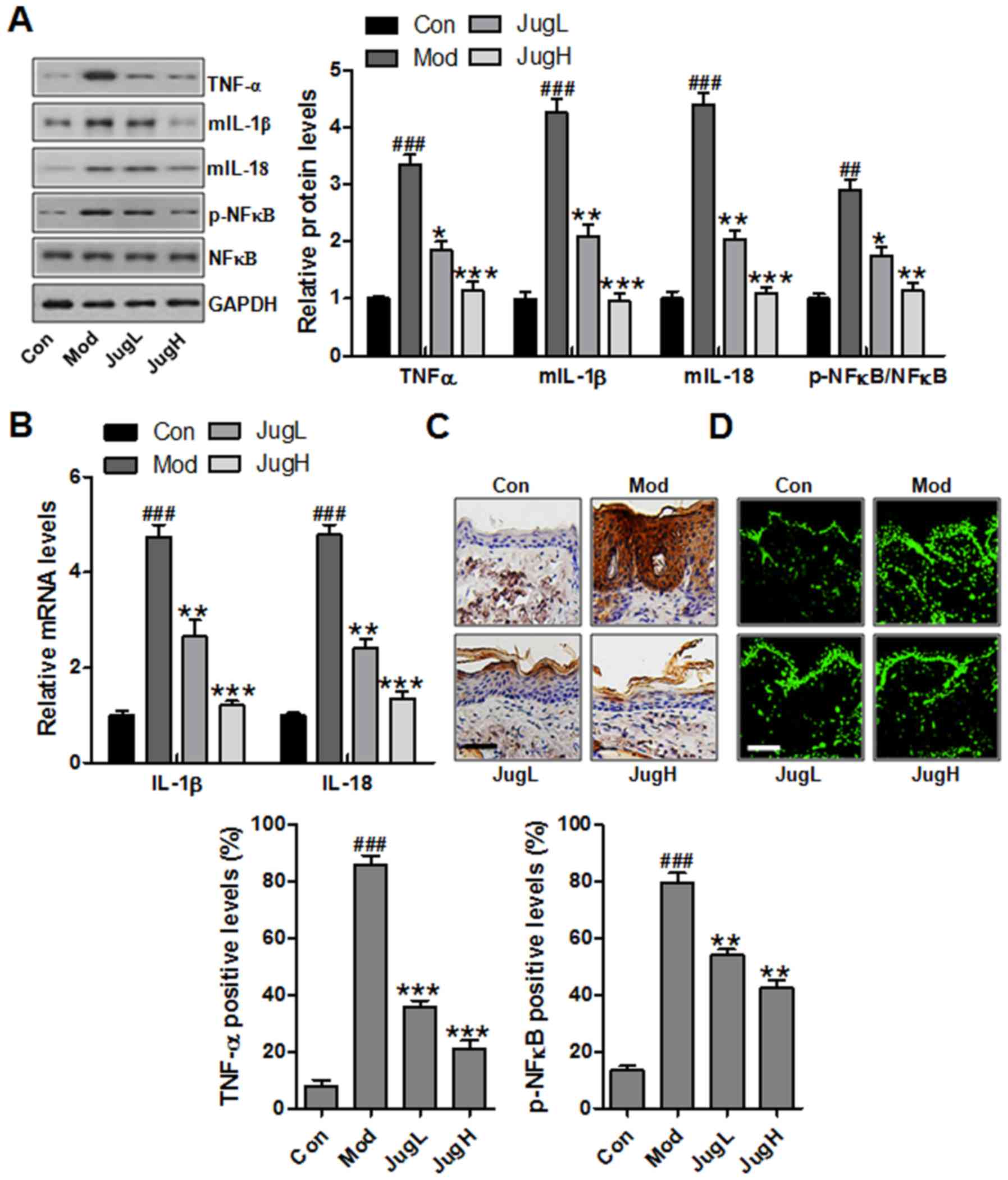 | Figure 3Effects of juglanin on UVB-induced
inflammation. (A) Western blotting was conducted and relative
expression levels of the following inflammation-associated
proteins, TNF-α, IL-1β, IL-18 and p-NF-κB, were determined. (B)
Reverse transcription-quantitative polymerase chain reaction
analysis was performed to determine the mRNA expression levels of
inflammation-associated genes, IL-1β and IL-18. (C)
Immunohistochemical analysis of TNF-α was conducted on 5-μm
sections. Representative photomicrographs from each group were
shown (scale bar, 100 μm). (D) Immunofluorescence analysis
of p-NF-κB was conducted on tissue sections. Representative
photomicrographs from each group were shown (scale bar, 100
μm). Data are presented as the means ± standard deviation of
three independent experiments. ###P<0.001 compared
with the Con group; **P<0.01 and
***P<0.001 compared with the Mod group. Con, control;
IL, interleukin; JugH, high juglanin; JugL, low juglanin; Mod,
model; NF-κB, nuclear factor-κB; p-, phosphorylated; TNF-α, tumor
necrosis factor-α; UVB, ultraviolet B. |
Role of juglanin in UVB-induced cell
proliferation
UVB induces cell proliferation by initiating cell
cycle-associated signaling pathway activation. Therefore, the
present study explored the role of juglanin in the proliferative
potential of epidermal cells following exposure to UVB. As shown in
Fig. 4A, exposure of mice to UVB
led to upregulated expression of cyclin D1, CDK1 and PCNA compared
with in the control group. Conversely, juglanin treatment decreased
the expression levels of these proteins. UVB-induced skin damage is
associated with the ability of cells to assess or reverse DNA
damage, regulating the cell cycle and inducing apoptosis if
necessary. In UVB-exposed skin, DNA damage leads to p53 expression,
as well as its downstream target proteins p27 and p21 (28). As presented in Fig. 4B, UVB exposure downregulated p53,
p27 and p21 protein expression compared with in the control group.
In addition, juglanin administration to UVB-induced mice promoted
p53, p27 and p21 expression. Immunohistochemical analysis indicated
that PCNA was upregulated in UV-induced mice, whereas juglanin
administration was able to reduce PCNA expression; this finding was
in accordance with the results of western blotting (Fig. 4C).
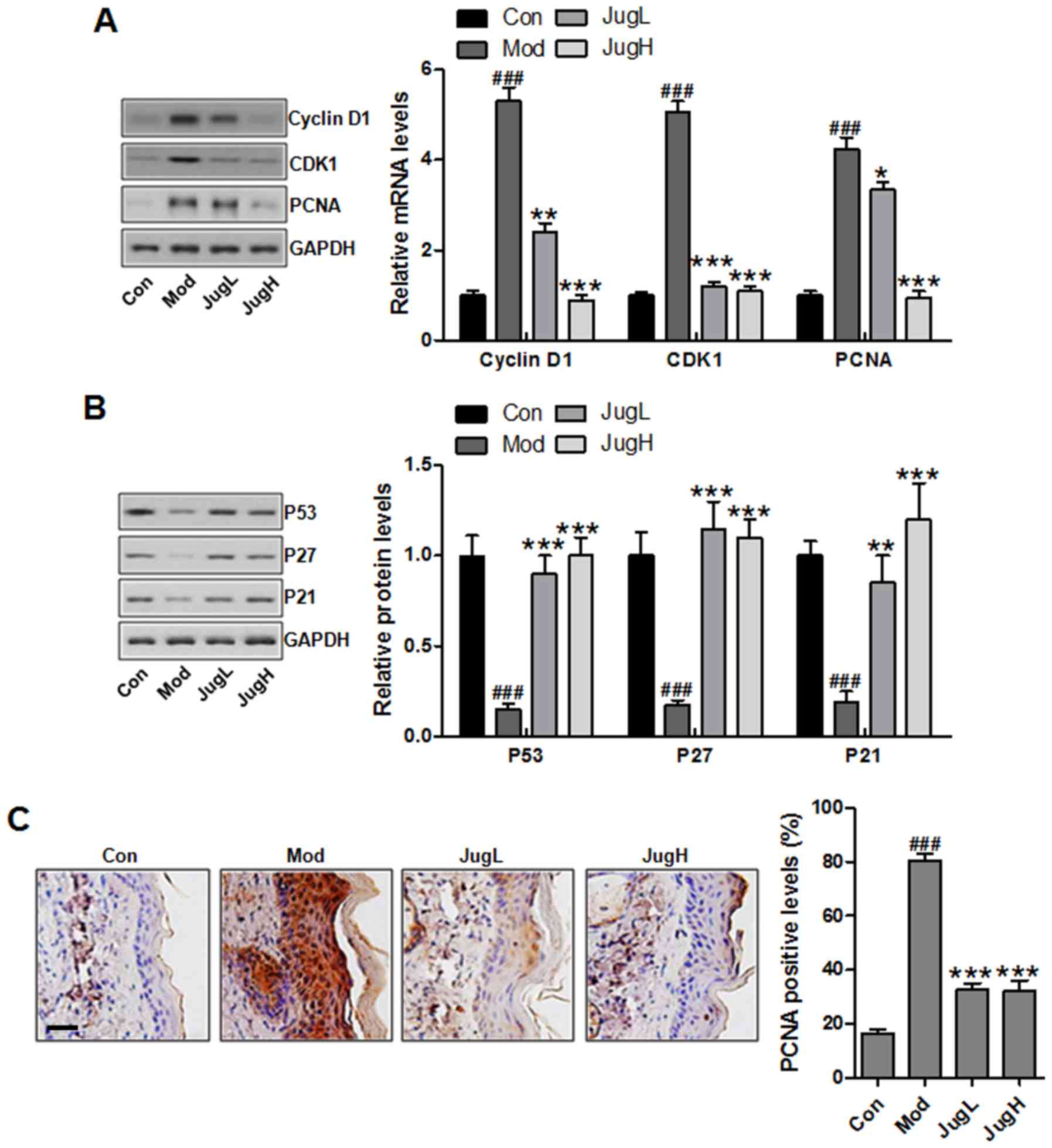 | Figure 4Effects of juglanin on UVB-induced
cell proliferation markers. (A) Western blotting was conducted and
relative protein expression levels of cyclin D1, CDK1 and PCNA were
determined. Equal protein loading was confirmed by stripping the
immunoblot and reprobing it for GAPDH. (B) Western blotting was
conducted and relative protein expression levels of p53, p27 and
p21 were determined. (C) Immunohistochemical analysis of PCNA was
conducted on 5-μm sections. Representative photomicrographs
from each group were shown (scale bar, 100 μm). Data are
presented as the means ± standard deviation of three independent
experiments. ###P<0.001 compared with the Con group;
**P<0.01 and ***P<0.001 compared with
the Mod group. CDK1, cyclin-dependent kinase 1; Con, control; JugH,
high juglanin; JugL, low juglanin; Mod, model; PCNA, proliferating
cell nuclear antigen; UVB, ultraviolet B. |
Juglanin induces caspase activation and
PARP-1 cleavage
As aforementioned, apoptosis may be associated with
UV-induced skin cancer progression. In the present study, the
results of an immunofluorescence assay suggested that caspase-3 was
suppressed following UV induction in mice (Fig. 5A). Notably, juglanin significantly
enhanced caspase-3 levels; caspase-3 is an essential regulator that
leads to apoptosis (29).
Furthermore, western blotting indicated that PARP, caspase-8 and -3
cleavage was enhanced in UV-exposed mice treated with juglanin
(Fig. 5B).
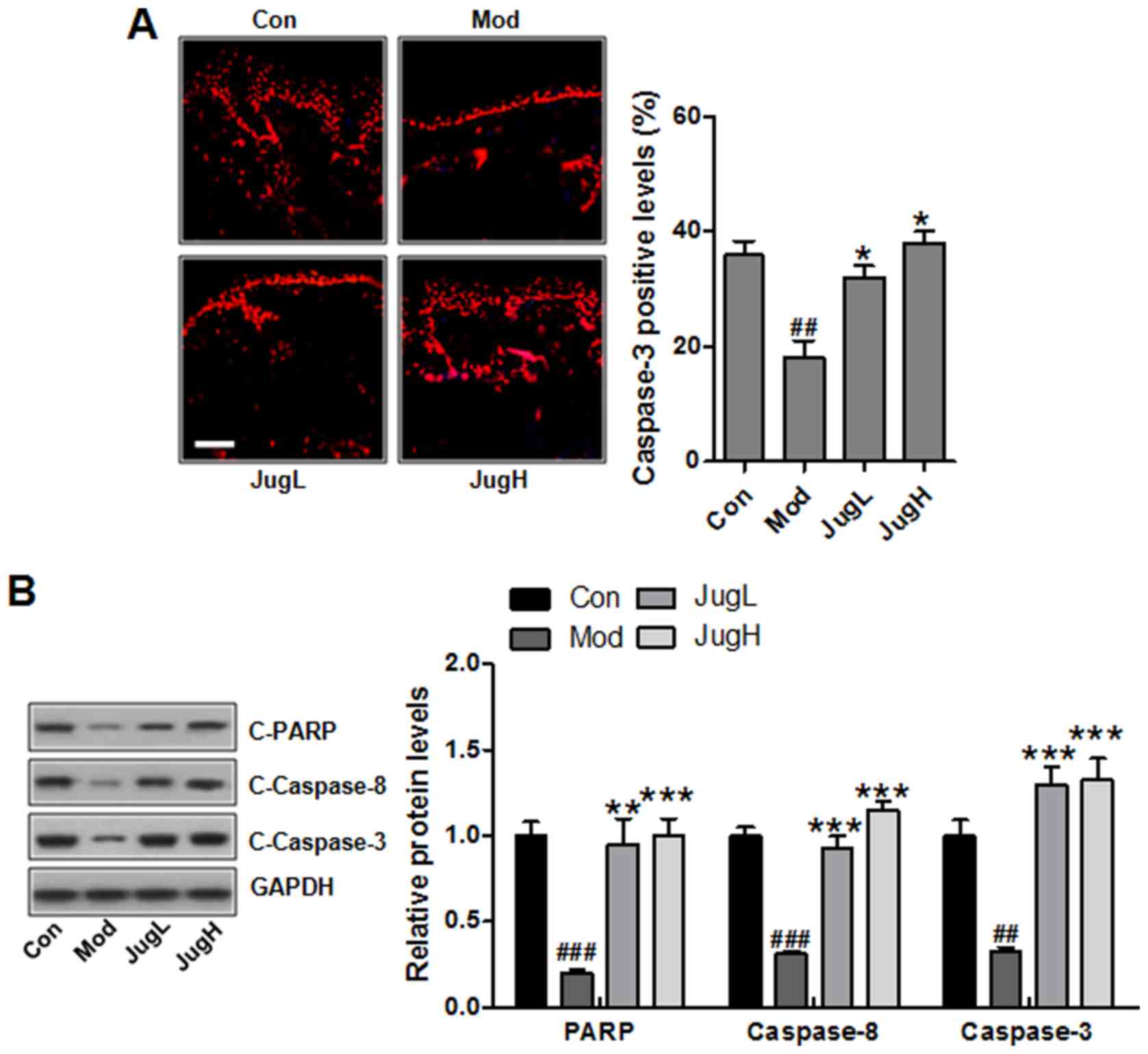 | Figure 5Juglanin induces caspase activation
and PARP-1 cleavage. (A) Immunofluorescence analysis of caspase-3
was conducted on tissue sections. Representative photomicrographs
from each group were shown (scale bar, 100 μm). (B) Western
blotting was conducted and relative protein expression levels of
C-caspase-3, C-caspase-8 and C-PARP were determined. Equal protein
loading was confirmed by stripping the immunoblot and reprobing it
for GAPDH. Data are presented as the means ± standard deviation of
three independent experiments. ##P<0.01 and
###P<0.001 compared with the Con group;
*P<0.05, **P<0.01 and
***P<0.001 compared with the Mod group. C-, cleaved;
Con, control; JugH, high juglanin; JugL, low juglanin; Mod, model;
PARP, poly (ADP-ribose) polymerase; UVB, ultraviolet B. |
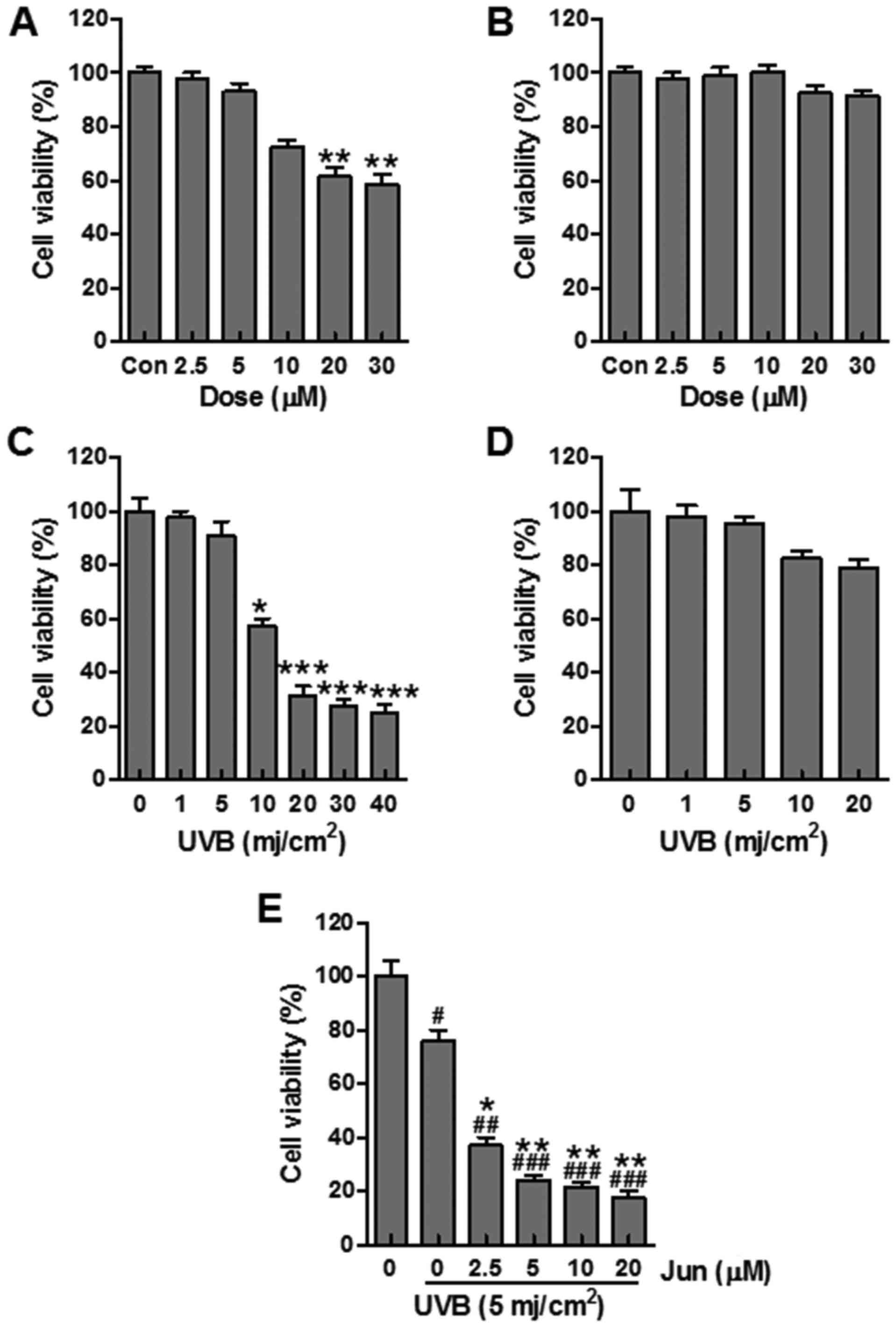
Juglanin promotes UVB-induced cell
death
In order to further examine the effects of juglanin
on skin cancer, B16F10 cells were exposed to UVB (1–40
mJ/cm2) and the cell viability was determined at 24 h
post-UVB irradiation. In addition, B16F10 cells were administered
juglanin (2.5–30 μM) and cell viability was investigated.
Juglanin (2.5–10 μM) did not induce B16F10 cell death,
whereas 20 and 30 μM juglanin significantly reduced cell
viability (Fig. 6A). To determine
the specificity of the proapoptotic effects of juglanin, Hs68
fibroblast cells were treated with juglanin and cell viability was
analyzed. Notably, juglanin induced no significant cell death in
Hs68 cells (Fig. 6B). In
addition, the present study indicated that UVB exposure led to a
significant downregulation in the viability of B16F10 cells
(Fig. 6C). Upon UVB irradiation
at 10 mJ/cm2, the viability of B16F10 cells was
decreased by ~40% compared with in unirradiated control cells.
However, 10 mJ/cm2 UVB induced no apparent cell death of
normal Hs68 human fibroblast cells, and was therefore selected for
further experiments (Fig. 6D).
The present study investigated whether juglanin enhanced cell death
of UVB-irradiated B16F10 cells. The results indicated that
treatment of UVB-irradiated B16F10 cells with 2.5, 5, 10 and 20
μM juglanin increased the percentage of cell death (Fig. 6E). These results indicated that
the effectiveness of juglanin to induce cell death was, at least
partly, contributed to specific cell type and may rely on the
concentration of administration.
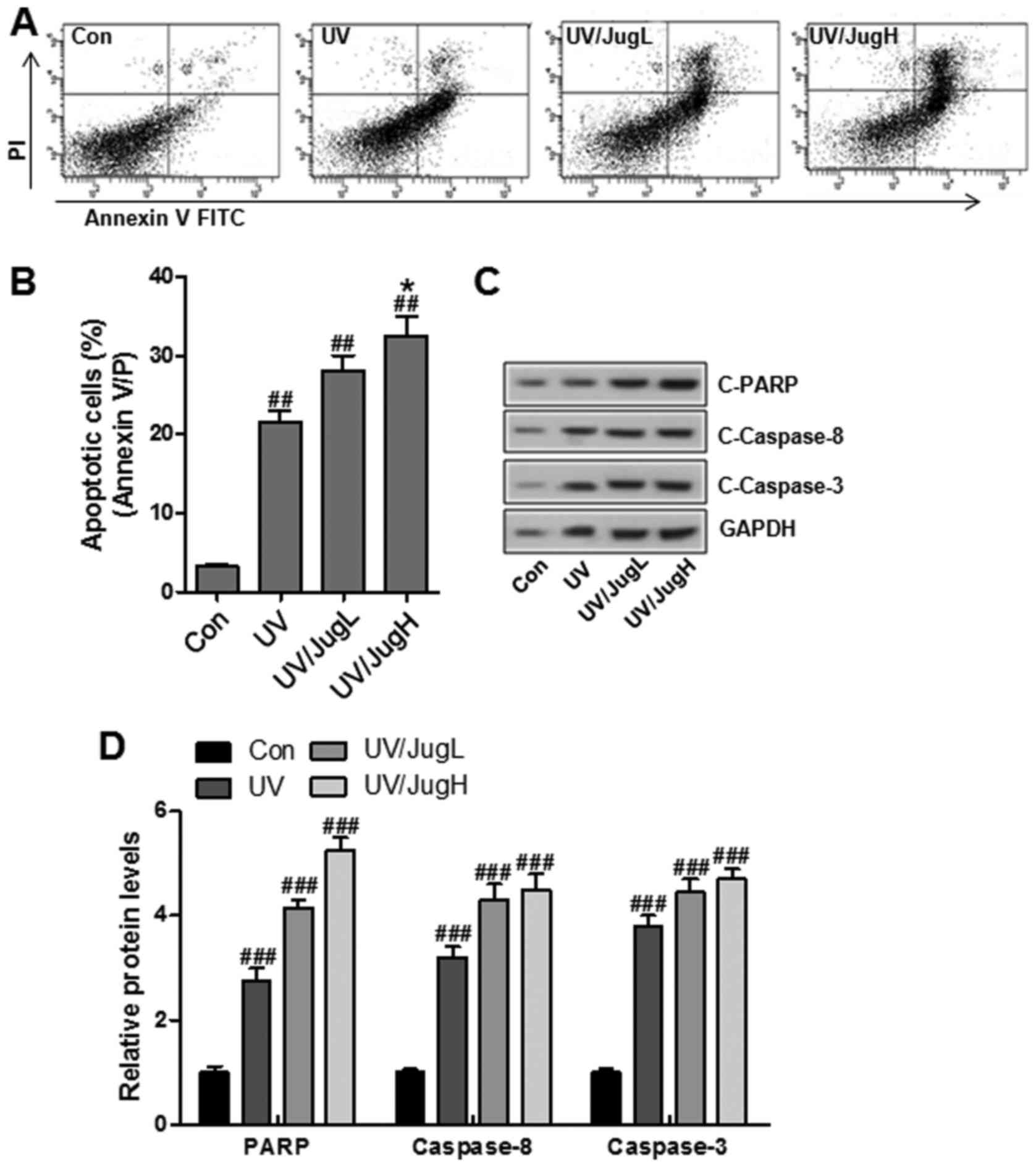
UVB and juglanin cotreatment enhances
apoptotic cell death and induces caspase activation and PARP-1
cleavage
To determine if the reduction in cell viability was
caused by apoptosis, Annexin V/PI double staining was used to
detect apoptosis in control and treated cells. Consistent with the
previous results, juglanin led to a substantial increase in
UVB-induced apoptosis (Fig. 7A
and B). In addition, western blotting suggested that UVB and
juglanin cotreatment promoted caspase activation and PARP cleavage,
which are hallmark features of apoptosis (Fig. 7C and D).
Effects of juglanin on major protein
regulators of p38/JNK and cell proliferation-associated signals in
UVB-irradiated B16F10 cells
The MAPK pathway, which is activated in virtually
all melanomas, regulates cell metastasis, proliferation and
survival (13). The present study
explored the effects of juglanin on p38 and JNK in UVB-irradiated
B16F10 cells via western blotting. UVB irradiation of B16F10 cells
downregulated the protein expression levels of p-p38 and p-JNK.
Administration of UVB-irradiated B16F10 cells with juglanin induced
further inactivation of p-p38 and p-JNK (Fig. 8A). Furthermore, p-NF-κB was
downregulated in cells expose to UV and treated with juglanin
(Fig. 8A). Finally, p53, p27 and
p21 were assessed to explore the role of juglanin in human skin
cancer cells in vitro. The results indicated that p53, p27
and p21 were lowly expressed in the control group, and were
stimulated by UV exposure when combined with juglanin, in a
dose-dependent manner (Fig. 8B).
These data further indicated that juglanin may perform its role in
skin cancer suppression via cell proliferation inhibition in
vitro.
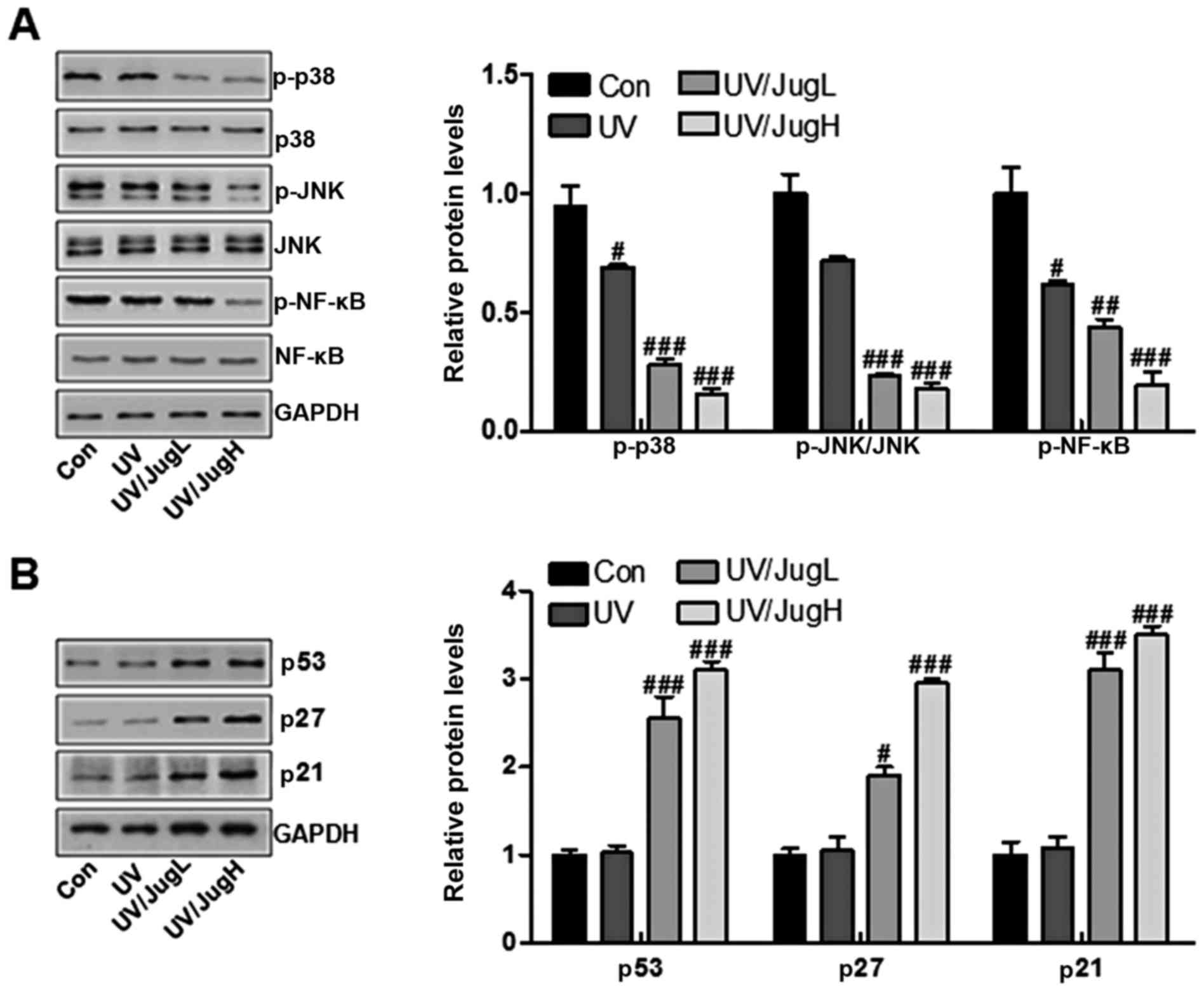 | Figure 8Effects of juglanin on major protein
regulators of p38/JNK and cell proliferation-associated signals in
UVB irradiated B16F10 cells. (A) Western blotting of p-p38, p-JNK
and p-NF-κB in B16F10 cells 24 h post-UVB/juglanin treatment. (B)
Western blotting of p53, p27 and p21 activation in B16F10 cells 24
h post-UVB/juglanin treatment. Data are presented as the means ±
standard deviation of three independent experiments.
#P<0.05, ##P<0.01 and
###P<0.001 compared with the Con group. Con, control;
JNK, c-Jun N-terminal kinase; JugH, high juglanin; JugL, low
juglanin; NF-κB, nuclear factor-κB; p-, Figure 1. Juglanin ameliorates
UVB-induced skin carcinogenesis. (A) Photographs of Con, Mod and
juglanin-treated hairless mice exhibiting erythema following
irradiation. (B) Representative images of dorsal skin vasculature
of 6 individual animals from each group. (C) Hematoxylin and eosin
staining. The representative photomicrographs from each group were
shown (scale bar, 100 μm). (D) Immunohistochemical analysis
of KI-67 was conducted on 5-μm sections. Representative
photomicrographs from each group were shown (scale bar, 100
μm). Data are presented as the means ± standard deviation of
three independent experiments. ###P<0.001 compared
with the Con group; **P<0.01 and
***P<0.001 compared with the Mod group. Con, control;
JugH, high juglanin; JugL, low juglanin; Mod, model; UVB,
ultraviolet B. |
Discussion
Skin cancer is one of the most common cancers
worldwide, particularly in USA, which accounts for ~50% of all
human cancers (30). Solar UVB
radiation is an ubiquitous environmental carcinogen, which leads to
various cutaneous disorders, including melanoma and NMSCs (31). Previous studies have indicated
that plant-derived compounds possess potential anti-mutagenic,
anti-inflammatory and anticarcinogenic properties, and are gaining
considerable attention regarding the prevention and inhibition of
UVB-induced skin damage (32).
Juglanin is a natural compound extracted from crude Polygonum
aviculare, which exhibits inhibitory activity against
inflammation and cancer growth (7). In the present study, juglanin was
topically applied to UVB-exposed SKH-1 hairless mice by gavage in
order to explore its effects on inflammatory markers, and the
p38/JNK and PI3K/AKT-associated apoptosis signaling pathways
(33). In response to UVB
irradiation, activation of p38 MAPK and JNK is considered to induce
apoptosis (34). Therefore, the
inactivation of p38 and JNK is likely to enhance the therapeutic
efficacy of juglanin in UVB-irradiated mice and cells. In our
study, we found that in vivo p38 MAPK and JNK MAPK pathway
was inactivated by juglanin in UVB-treated animals, while apoptosis
was induced by juglanin. Herein, we supposed that there might be
other signals involved in juglanin-regulated apoptosis in the skin
of mice with UVB irradiation. Furthermore, in vivo, UV
reduced caspase activation and PARP cleavage, whereas in
vitro, it increased them. As for this, we hypothesized that
there were different types of cells in tissue composition, while
in vitro, only skin cancer cells were included (35). Consequently, different results
were observed. Thus, further study is still required in future to
comprehensively reveal the underlying molecular mechanism by which
juglanin modulates apoptotic response in multiple types of
cells.
Increasing evidence has indicated that exposure of
the skin to UVB leads to the induction of inflammatory cytokine
expression and production (36).
The inflammatory response promotes skin cancer progression. In
addition, TNF-α, IL-1β and IL-18 serve a critical role in induction
of UVB-induced skin hyperplasia and inflammation (37). In the present study, the
expression levels of TNF-α, IL-1β and IL-18 were increased
following UVB exposure, whereas juglanin treatment significantly
inhibited TNF-α, IL-1β and IL-18 expression in the skin of
UVB-exposed mice. Increased expression of proinflammatory cytokines
further promotes infiltration of inflammatory cells (38). In addition, the NF-κB signaling
pathway is associated with the transcription of numerous
proinflammatory genes, including IL-1β, IL-18 and TNF-α (39). It has previously been suggested
that UVB radiation enhances NF-κB signaling in mouse skin (40). PI3K/AKT has been suggested to have
an important role in modulating the inflammatory response via
regulation of NF-κB (41). A
previous study reported that the PI3K/AKT-regulated NF-κB pathway
was involved in myocardial injury following chronic stress
(42). In addition, UVB radiation
has been reported to stimulate numerous signal transduction
pathways, including the PI3K/AKT pathway in vivo and in
vitro (43). Given the
possible causal relationship of inflammation with cancer, the
present study hypothesized that the PI3K/AKT-regulated NF-κB
pathway may be involved in UVB-induced skin cancer. The in
vivo results of the present study indicated that the NF-κB
signaling pathway was activated, accompanied with increased
PI3K/AKT phosphorylation, in UVB-exposed mice. Notably, juglanin
was able to suppress inflammation via NF-κB inactivation and AKT
inhibition.
DNA injury caused by UVB radiation results in
stabilization and accumulation of p53 protein via transcriptional
activation, which serves an essential role in cell cycle arrest.
Once activated, the p53 protein translocates to the nucleus and
activates numerous DNA-binding proteins and downstream effectors
associated with cell cycle arrest and apoptotic induction (44). In UVB-exposed skin, p53 activation
leads to p27 and p21 activation, which in turn suppresses cyclin
D/CDK1 kinases in skin cancer. Blockage of the cell cycle helps to
repair damaged DNA via nucleotide excision repair mechanisms
(45). In the present study,
western blotting clearly demonstrated that p53, p27 and p21
expression was decreased in the skin of UVB-exposed mice and cells.
Juglanin treatment further augmented protein expression of p53, p27
and p21 in UVB-exposed mice and cells in vivo and in
vitro. PCNA enhances degradation of the replication initiation
factor complex induced by DNA damage; upregulated PCNA expression
in UVB-induced DNA-injured cells is a result of the combination of
p53 and the PCNA promoter (46).
In the present study, treatment with juglanin significantly
suppressed the protein expression levels of PCNA in the skin of
UVB-exposed mice. In addition, cyclin D1 is an important
proliferative marker, which regulates cell proliferation (47). Overexpression of cyclin D1 is
associated with the development and progression of numerous types
of cancer, including UVB-induced skin carcinogenesis (48). In the present study, UVB exposure
significantly upregulated cyclin D1 expression. However, juglanin
treatment significantly inhibited the expression of cyclin D1 in
the skin of UVB-exposed mice.
In conclusion, the present study indicated that
juglanin treatment of SKH-1 mice following UVB exposure may lead to
a significant decrease in the expression of inflammatory mediators,
inflammatory cytokines, including TNF-α, IL-1β and IL-18, and cell
proliferative markers. In addition, juglanin suppressed the p38/JNK
and PI3K/AKT/NF-κB signaling pathways, which are involved in
UVB-induced inflammation, cell survival, apoptosis and
proliferation. Juglanin treatment also enhanced UVB-mediated p53,
p27 and p21 protein expression. Notably, juglanin treatment is not
toxic to mouse skin. Overall, these results indicated that juglanin
may be developed as a natural and novel chemopreventive agent for
the therapeutic treatment of UVB-induced skin cancer.
References
|
1
|
D'Orazio J, Jarrett S, Amaro-Ortiz A and
Scott T: UV radiation and the skin. Int J Mol Sci. 14:12222–12248.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen ST, Geller AC and Tsao H: Update on
the epidemiology of melanoma. Curr Dermatol Rep. 2:24–34. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saladi RN and Persaud AN: The causes of
skin cancer: a comprehensive review. Drugs Today (Barc). 41:37–53.
2005. View Article : Google Scholar
|
|
4
|
Balk SJ; Section on Dermatology; Council
on Environmental Health: Ultraviolet radiation: a hazard to
children and adolescents. Pediatrics. 127:588–597. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou GY, Yi YX, Jin LX, Lin W, Fang PP,
Lin XZ, Zheng Y and Pan CW: The protective effect of juglanin on
fructose-induced hepatitis by inhibiting inflammation and apoptosis
through TLR4 and JAK2/STAT3 signaling pathways in fructose-fed
rats. Biomed Pharmacother. 81:318–328. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang HH, Hwangbo K, Zheng MS, Son JK, Kim
HY, Baek SH, Choi HC, Park SY and Kim JR: Inhibitory effects of
juglanin on cellular senescence in human dermal fibroblasts. J Nat
Med. 68:473–480. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Phan VK, Nguyen TM, Minh CV, Nguyen HK,
Nguyen HD, Nguyen PT, Nguyen XC, Nguyen HN, Nguyen XN, Heyden YV,
et al: Two new C-glucosyl benzoic acids and flavonoids from
Mallotus nanus and their antioxidant activity. Arch Pharm Res.
33:203–208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang H, Sung SH, Kim J and Kim YC:
Neuroprotective diarylheptanoids from the leaves and twigs of
juglans sinensis against glutamate-induced toxicity in HT22 cells.
Planta Med. 77:841–845. 2011. View Article : Google Scholar
|
|
9
|
Kim HH, Oh MH, Park KJ, Heo JH and Lee MW:
Anti-inflammatory activity of sulfate-containing phenolic compounds
isolated from the leaves of Myrica rubra. Fitoterapia. 92:188–193.
2014. View Article : Google Scholar
|
|
10
|
Nguelefack TB, Mbakam FH, Tapondjou LA,
Watcho P, Nguelefack-Mbuyo EP, Ponou BK, Kamanyi A and Park HJ: A
dimeric triterpenoid glycoside and flavonoid glycosides with free
radical-scavenging activity isolated from Rubus rigidus var.
camerunensis. Arch Pharm Res. 34:543–550. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schwarz S, Sauter D, Wang K, Zhang R, Sun
B, Karioti A, Bilia AR, Efferth T and Schwarz W: Kaempferol
derivatives as antiviral drugs against the 3a channel protein of
coronavirus. Planta Med. 80:177–182. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Asgari M, White E and Chren MM:
Nonsteroidal anti-inflammatory drug use in the prevention and
treatment of squamous cell carcinoma. Dermatol Surg. 30:1335–1342.
2004.PubMed/NCBI
|
|
13
|
Khan N, Syed DN, Pal HC, Mukhtar H and
Afaq F: Pomegranate fruit extract inhibits UVB-induced inflammation
and proliferation by modulating NF-κB and MAPK signaling pathways
in mouse skin. Photochem Photobiol. 88:1126–1134. 2012. View Article : Google Scholar :
|
|
14
|
Thompson EJ, MacGowan J, Young MR, Colburn
N and Bowden GT: A dominant negative c-jun specifically blocks
okadaic acid-induced skin tumor promotion. Cancer Res.
62:3044–3047. 2002.PubMed/NCBI
|
|
15
|
Pal HC, Sharma S, Elmets CA, Athar M and
Afaq F: Fisetin inhibits growth, induces G2/M arrest and
apoptosis of human epidermoid carcinoma A431 cells: role of
mitochondrial membrane potential disruption and consequent caspases
activation. Exp Dermatol. 22:470–4752. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meek DW: Tumour suppression by p53: a role
for the DNA damage response? Nat Rev Cancer. 9:714–723. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Q, Bian H, Li Y, Guo L, Tang Y and
Zhu H: Preconditioning with the traditional Chinese medicine
Huang-Lian-Jie-Du-Tang initiates HIF-1α-dependent neuroprotection
against cerebral ischemia in rats. J Ethnopharmacol. 154:443–452.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
19
|
Pal HC, Athar M, Elmets CA and Afaq F:
Fisetin inhibits UVB-induced cutaneous inflammation and activation
of PI3K/AKT/NFκB signaling pathways in SKH-1 hairless mice.
Photochem Photobiol. 91:225–234. 2015. View Article : Google Scholar
|
|
20
|
Afaq F, Adhami VM and Mukhtar H:
Photochemoprevention of ultraviolet B signaling and
photocarcinogenesis. Mutat Res. 571:153–173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bowden GT: Prevention of non-melanoma skin
cancer by targeting ultraviolet-B-light signalling. Nat Rev Cancer.
4:23–35. 2004. View
Article : Google Scholar
|
|
22
|
Segrelles C, Lu J, Hammann B, Santos M,
Moral M, Cascallana JL, Lara MF, Rho O, Carbajal S, Traag J, et al:
Deregulated activity of Akt in epithelial basal cells induces
spontaneous tumors and heightened sensitivity to skin
carcinogenesis. Cancer Res. 67:10879–10888. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neagu M, Constantin C, Dumitrascu GR, Lupu
AR, Caruntu C, Boda D and Zurac S: Inflammation markers in
cutaneous melanoma - edgy biomarkers for prognosis. Discoveries.
3:e382015. View Article : Google Scholar
|
|
24
|
Fujiki H, Sueoka E and Suganuma M: Tumor
promoters: from chemicals to inflammatory proteins. J Cancer Res
Clin Oncol. 139:1603–1614. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yan C, Grimm WA, Garner WL, Qin L, Travis
T, Tan N and Han YP: Epithelial to mesenchymal transition in human
skin wound healing is induced by tumor necrosis factor-alpha
through bone morphogenic protein-2. Am J Pathol. 176:2247–2258.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rogers HW, Weinstock MA, Harris AR,
Hinckley MR, Feldman SR, Fleischer AB and Coldiron BM: Incidence
estimate of nonmelanoma skin cancer in the United States, 2006.
Arch Dermatol. 146:283–287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tak PP and Firestein GS: NF-kappaB: a key
role in inflammatory diseases. J Clin Invest. 107:7–11. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li J, Cheng Y, Qu W, Sun Y, Wang Z, Wang H
and Tian B: Fisetin, a dietary flavonoid, induces cell cycle arrest
and apoptosis through activation of p53 and inhibition of NF-kappa
B pathways in bladder cancer cells. Basic Clin Pharmacol Toxicol.
108:84–93. 2011. View Article : Google Scholar
|
|
29
|
Kassi E, Sourlingas TG, Spiliotaki M,
Papoutsi Z, Pratsinis H, Aligiannis N and Moutsatsou P: Ursolic
acid triggers apoptosis and Bcl-2 downregulation in MCF-7 breast
cancer cells. Cancer Invest. 27:723–733. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kornek T and Augustin M: Skin cancer
prevention. J Dtsch Dermatol Ges. 11:283–296. 2013.PubMed/NCBI
|
|
31
|
Nijsten T, Colpaert CG, Vermeulen PB,
Harris AL, Van Marck E and Lambert J: Cyclooxygenase-2 expression
and angiogenesis in squamous cell carcinoma of the skin and its
precursors: a paired immunohistochemical study of 35 cases. Br J
Dermatol. 151:837–845. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim RH and Armstrong AW: Nonmelanoma skin
cancer. Dermatol Clin. 30:125–139. 2012. View Article : Google Scholar
|
|
33
|
Li B, Zhang G, Li R and Duan C:
Phosphoinositide 3-kinase/Akt Pathway Mediates
Fip1-like1-platelet-derived Growth Factor Receptor α-induced Cell
Infiltration and Activation: Possible Molecular Mechanism for the
Malignant Phenotype of Chronic Eosinophilic Leukemia. Cancer Transl
Med. 1:31–34. 2015. View Article : Google Scholar
|
|
34
|
George J, Singh M, Srivastava AK, Bhui K,
Roy P, Chaturvedi PK and Shukla Y: Resveratrol and black tea
polyphenol combination synergistically suppress mouse skin tumors
growth by inhibition of activated MAPKs and p53. PLoS One.
6:e233952011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Larisch P, Verwanger T, Linecker M and
Krammer B: The interrelation between a pro-inflammatory milieu and
fluorescence diagnosis or photodynamic therapy of human skin cell
lines. Photodiagnosis Photodyn Ther. 11:91–103. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Joosse A, Koomen ER, Casparie MK, Herings
RM, Guchelaar HJ and Nijsten T: Non-steroidal anti-inflammatory
drugs and melanoma risk: large Dutch population-based case-control
study. J Invest Dermatol. 129:2620–2627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Johannesdottir SA, Chang ET, Mehnert F,
Schmidt M, Olesen AB and Sørensen HT: Nonsteroidal
anti-inflammatory drugs and the risk of skin cancer: a
population-based case-control study. Cancer. 118:4768–4776. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nedoszytko B, Sokołowska-Wojdyło M,
Ruckemann-Dziurdzińska K, Roszkiewicz J and Nowicki RJ: Chemokines
and cytokines network in the pathogenesis of the inflammatory skin
diseases: atopic dermatitis, psoriasis and skin mastocytosis.
Postepy Dermatol Alergol. 31:84–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shen J, Abel EL, Riggs PK, Repass J,
Hensley SC, Schroeder LJ, Temple A, Chau A, McClellan SA, Rho O, et
al: Proteomic and pathway analyses reveal a network of inflammatory
genes associated with differences in skin tumor promotion
susceptibility in DBA/2 and C57BL/6 mice. Carcinogenesis.
33:2208–2219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Suganuma M, Okabe S, Kurusu M, Iida N,
Ohshima S, Saeki Y, Kishimoto T and Fujiki H: Discrete roles of
cytokines, TNF-α IL-1, IL-6 in tumor promotion and cell
transformation. Int J Oncol. 20:131–136. 2002.
|
|
41
|
Yang C, Zhao T, Lin M, Zhao Z, Hu L, Jia
Y, Xue Y, Xu M, Tang Q, Yang B, et al: Helix B surface peptide
administered after insult of ischemia reperfusion improved renal
function, structure and apoptosis through beta common
receptor/erythropoietin receptor and PI3K/Akt pathway in a murine
model. Exp Biol Med (Maywood). 238:111–119. 2013. View Article : Google Scholar
|
|
42
|
Moore T, Beltran L, Carbajal S, Strom S,
Traag J, Hursting SD and DiGiovanni J: Dietary energy balance
modulates signaling through the Akt/mammalian target of rapamycin
pathways in multiple epithelial tissues. Cancer Prev Res (Phila).
1:65–76. 2008. View Article : Google Scholar
|
|
43
|
Wilker E, Lu J, Rho O, Carbajal S, Beltrá
L and DiGiovanni J: Role of PI3K/Akt signaling in insulin-like
growth factor-1 (IGF-1) skin tumor promotion. Mol Carcinog.
44:137–145. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kleiner HE, Vulimiri SV, Starost MF, Reed
MJ and DiGiovanni J: Oral administration of the citrus coumarin,
isopimpinellin, blocks DNA adduct formation and skin tumor
initiation by 7,12-dimethylbenz[a]anthracene in SENCAR mice.
Carcinogenesis. 23:1667–1675. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen T: The role of microRNA in chemical
carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol
Rev. 28:89–124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li J, Qu W, Cheng Y, Sun Y, Jiang Y, Zou
T, Wang Z, Xu Y and Zhao H: The inhibitory effect of intravesical
fisetin against bladder cancer by induction of p53 and
down-regulation of NF-kappa B pathways in a rat bladder
carcinogenesis model. Basic Clin Pharmacol Toxicol. 115:321–329.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wong YK, Lin SC, Chang CS, Tseng YH, Liu
CJ, Lin HC and Chang KW: Cyclin D1 genotype in areca-associated
oral squamous cell carcinoma. J Oral Pathol Med. 32:265–270. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yaylim-Eraltan I, Arikan S, Yildiz Y,
Cacina C, Ergen HA, Tuna G, Görmüs U, Zeybek U and Isbir T: The
influence of cyclin D1 A870G polymorphism on colorectal cancer risk
and prognosis in a Turkish population. Anticancer Res.
30:2875–2880. 2010.PubMed/NCBI
|