Introduction
Diabetes mellitus (DM) is a common metabolic
disorder characterized by chronic hyperglycemia. The prevalence of
DM is increasing worldwide, and the International Diabetes
Federation has estimated that 592 million people will suffer from
diabetes by 2035 (1).
DM-associated chronic complications cause considerable morbidity
and mortality (2), and the
treatment of these complications imposes an enormous economic
burden on healthcare systems. The most common form of DM is type 2
DM (T2D), accounting for 90% of cases. It has been demonstrated
that genes serve an important role in the disease development by
interacting with environmental factors, such as a sedentary
life-style, diet and obesity (3).
Although T2D typically begins in middle-aged or older individuals,
early-onset T2D at <45 years of age has been reported in several
ethnic groups (4). Obesity in
young adults may be one of the factors driving the earlier onset of
T2D (5). The inheritance of a
stronger genetic influence within a family is another determinant,
with early-onset T2D individuals likely to have a multigenerational
family history of diabetes. The most definite form of familial,
autosomal dominant inheritance of diabetes is maturity-onset
diabetes of the young (MODY). Typically, MODY is characterized by a
young age at onset (usually prior to 25 years of age) and an
autosomal mode of inheritance in multigenerational pedigrees
(6).
The identification of T2D-causing genes will enable
precise disease classification, leading to better treatment and
prevention of the disease and its chronic complications. In 2007,
there was a breakthrough in the identification of T2D genetic risk
loci through a genome-wide association study (GWAS), and >100
such loci have been identified to date (7). However, several limitations of GWAS
have been proposed; for instance, the associated single nucleotide
polymorphisms fall outside coding regions. Numerous low-frequency
variants have never been directly tested for an association with
the trait and they may explain only ~11% of T2D heritability
(8,9). To address this issue, current
research is focusing on finding low-frequency to rare variants with
intermediate to large effects that are associated with diabetes
(10). Recent advances in nucleic
acid sequencing by next-generation sequencing technology have
facilitated the identification of genes causing diseases (10). Furthermore, whole-exome sequencing
(WES) has led to the successful discovery of mutations between rare
coding variants and T2D (11,12).
Thus, the aim of the present study was to identify
rare variant causing diabetes in Thai T2D families. Through the
application of WES, the study identified the causative variant in a
large family with autosomal dominant T2D in which a proband
developed T2D at an early age.
Materials and methods
Diabetes families
Diabetes families were recruited at the Diabetic
Clinic, Siriraj Hospital, Mahidol University (Bangkok, Thailand)
using the following criteria: i) The proband and at least one
first-degree relative were diagnosed with diabetes before the age
of 35 years; ii) two or more generations were affected by diabetes;
iii) glycemic control could be accomplished with diet and/or oral
agents; iv) there was no history of diabetic ketoacidosis; and v)
the proband was negative for anti-glutamic acid decarboxylase
antibody. A total of 91 autosomal dominant diabetes families were
recruited between January 2009 and 2017, 42 of whom fitted into the
classic MODY criteria, characterized by early age of onset (usually
before 25 years) and autosomal mode of inheritance in
multi-generational pedigree (13). The selected family was a large
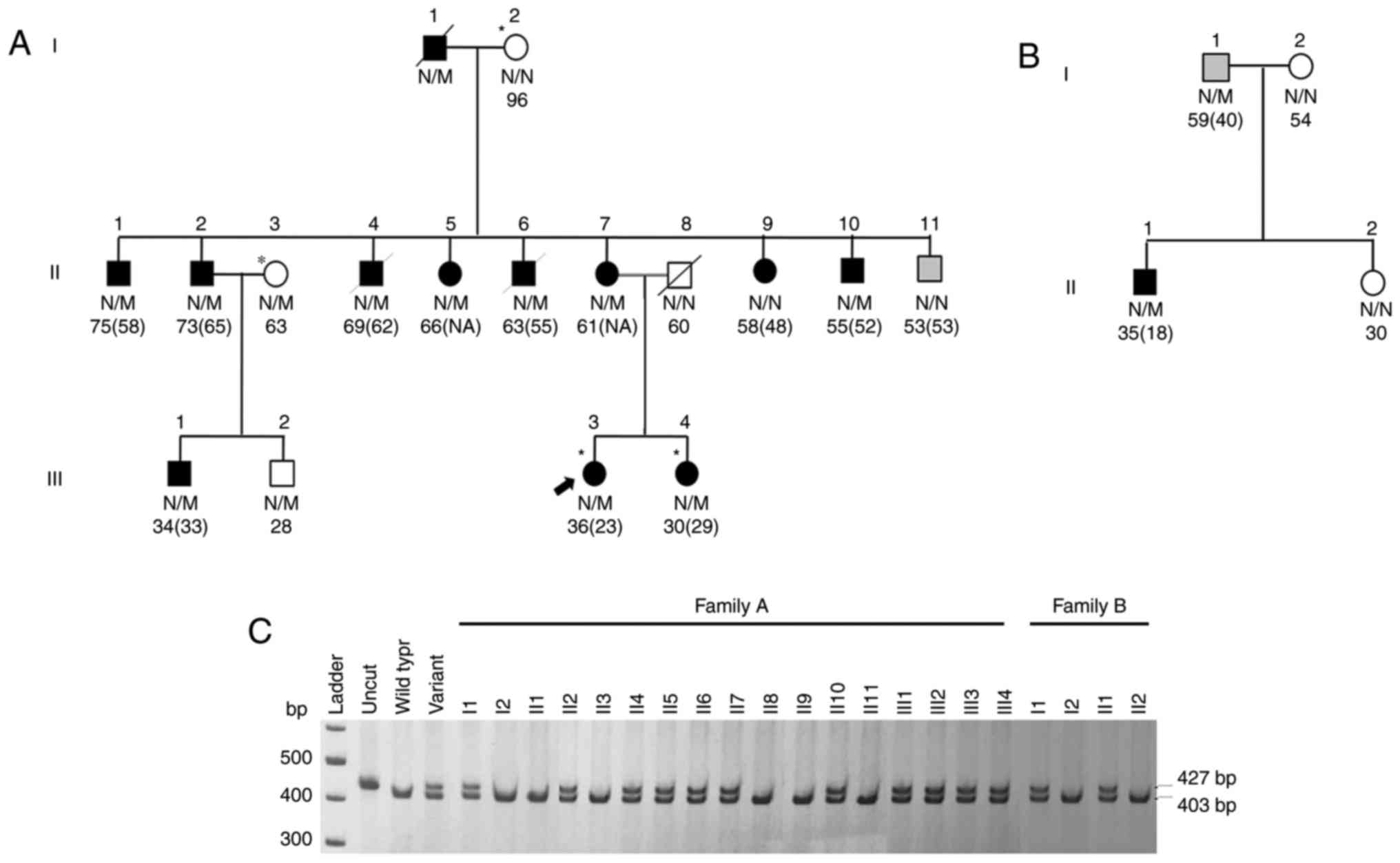
family composed of 17 members (Fig.
1A; Table I), in which the
proband and third generation family members developed diabetes at a
young age of <35 years, and diabetes had been transmitted
through three generations. This selected family did not fit classic
MODY criteria and was one of the 49 early-onset T2D families. The
proband and her first degree relative were diagnosed with diabetes
before 35 years of age and the diabetes was transmitted in an
autosomal dominant fashion. Mutations of 14 known MODY genes
[HNF4A (MIM: 600281), GCK (MIM: 138079), HNF1A
(MIM: 142410), PDX1(MIM: 600733), HNF1B (MIM:
189907), NEUROD1 (MIM: 601724), KLF11 (MIM: 603301),
CEL (MIM: 114840), PAX4 (MIM: 167413), INS
(MIM: 176730), BLK (MIM: 191305), ABCC8 (MIM:
600509), KCNJ11 (MIM: 600937) and APPL1 (MIM:
604299)] were excluded as a cause of diabetes in the family.
 | Table IClinical characteristics and
genotypes of each member of the type 2 diabetes families. |
Table I
Clinical characteristics and
genotypes of each member of the type 2 diabetes families.
A, Family A
|
|---|
| Member | Genotype | Sex | Age at diagnosis
(years) | BMI
(km/m2) | Glycemic
status | FPG (mmol/l) | HbA1c
(mmol/mol) | Current
treatment |
|---|
| I1 | CA | M | 49 | NA | DM | NA | NA | OHA |
| I2 | CC | F | – | 15.3 | NG | 4.848 | 37 | – |
| II1 | CC | M | 58 | 29.4 | DM | 7.052 | 56 | OHA |
| II2 | CA | M | 65 | 21.3 | DM | 12.507 | 98 | – |
| II3 | CC | F | – | 20.8 | NG | 4.628 | NA | – |
| II4 | CA | M | 62 | 24.2 | DM | 10.744 | 63 | – |
| II5 | CA | F | NA | 31.2 | DM | 5.069 | 42 | – |
| II6 | CA | M | 55 | 32.8 | DM | 15.702 | 142 | – |
| II7 | CA | F | NA | 28.4 D | M | 8.65 | 69 | OHA |
| II8 | CC | M | – | 31.3 | NG | 5.124 | NA | – |
| II9 | CC | F | 48 | 35.9 | DM | 6.942 | 81 | OHA |
| II10 | CA | M | 52 | NA | DM | NA | NA | OHA, ins |
| II11 | CC | M | 53 | 19.1 | PD | 5.014 | 39 | – |
| III1 | CA | M | 33 | NA | DM | 6.612 | NA | – |
| III2 | CA | M | – | 20.3 | NG | 4.408 | NA | – |
| III3 | CA | F | 23 | 36.7 | DM | 13.113 | 98 | OHA |
| III4 | CA | F | 29 | 36.3 | DM | 9.091 | 53 | – |
B, Family B
|
|---|
| Member | Genotype | Sex | Age at diagnosis
(years) | BMI
(km/m2) | Glycemic
status | FPG (mmol/l) | HbA1c
(mmol/mol) | Current
treatment |
|---|
| I1 | CA | M | 40 | NA | PD | 5.675 | NA | – |
| I2 | CC | F | – | NA | NG | 4.904 | NA | – |
| II1 | CA | M | 18 | 27.3 | DM | 11.295 | NA | OHA |
| II2 | CC | F | – | NA | NG | 4.628 | NA | – |
Unrelated T2D patients
A total of 1,000 unrelated T2D patients, diagnosed
according to the American Diabetes Association (ADA) 2017 criteria
(14), were enrolled at the
Diabetic Clinic of Siriraj Hospital, Mahidol University. The age at
onset was >45 years (mean age ± standard deviation=54.6±11.0
years).
Non-diabetic subjects
In total, 500 non-diabetic subjects with an age of
>40 years (mean age ± standard deviation=50.69±8.5), were
recruited from the health-checkup facility at the Department of
Preventive and Social Medicine, Siriraj Hospital, Mahidol
University. All participants had a fasting plasma glucose of
<5.55 mmol/l (100 mg/dl), a glycated hemoglobin (HbA1c) level of
≤38 mmol/mol (5.6%), and normal blood pressure. These subjects had
no family history of diabetes among their first-degree
relatives.
Ethical approval and consent to
participate
All subjects were informed of the purpose of the
study prior to signing an informed consent form. The entire study
was approved by the Ethics Committee, Faculty of Medicine, Siriraj
Hospital, Mahidol University (COA no. Si491/2014), and was
conducted according to the Declaration of Helsinki, the Belmont
Report, Council for International Organizations of Medical Sciences
Guidelines and the International Conference on Harmonization in
Good Clinical Practice.
Exome sequencing
DNA samples of 2 diabetic and 2 non-diabetic family
members were subjected to exome sequencing. Exome capture was
performed using the Agilent SureSelect Human All Exon 50 Mb kit
(Agilent Technologies, Inc., Santa Clara, CA, USA) according to the
manufacturer's protocol. The captured library was then loaded onto
the Illumina HiSeq 2000 platform (Illumina, Inc., San Diego, CA,
USA) for amplifying and sequencing. The sequence reads were mapped
to the reference human genome (UCSC NCBI37/hg19) using the
Burrows-Wheeler Aligner (15).
Variant detections and annotations were performed by SAMtools
(16) and the Genome Analysis
Toolkit (17), respectively.
Variants with low quality scores and those covered by <5 reads
were excluded.
Candidate variant selection
Candidate variants were selected using an autosomal
dominant mode of inheritance according to the following criteria:
i) Variants identified only in diabetic subjects; ii) heterozygous,
non-synonymous variants with a minor allele frequency (MAF) of
<0.01 were selected from the 1000 Genome Project (http://www.1000genomes.org/), the National Institute
of Environmental Health Sciences Exome Project (http://evs.gs.washington.edu/niehsExome/) and the
dbSNP147 database from UCSC Genome Browser Gateway (http://genome.ucsc.edu/cgi-bin/hgGateway?hgsid=662106975_rQ8ylYRLVt-bw0NVxwJnh1AhFhb5S&redirect=manual&source=genome.ucsc.edu);
and iii) variants predicted by at least one out of four in
silico programs (including MutationTaster, VarioWatch,
PolyPhen2 and SIFT) (18) as
possible pathogenic mutations, and residing in genes involved in
diabetes and metabolism as determined using the GeneDistiller 2014
(http://www.genedistiller.org/) (19). Candidate variants passing the
criteria were validated by Sanger sequencing (Macrogen, Inc.,
Seoul, Republic of Korea). Furthermore, protein stability of
candidate variants was predicted by DUET algorithms (http://structure.bioc.cam.ac.uk/duet)
(20).
Segregation analysis
The validated candidate variants were investigated
for segregation with diabetes in the family by means of polymerase
chain reaction (PCR)-restriction fragment length polymorphism.
Restriction enzymes for the genotyping of each candidate variant
were determined using NEBcutter (http://nc2.neb.com/NEBcutter2/). Candidate variants
segregated with diabetes were genotyped in other 90 early-onset
T2D/MODY-X patients, 1,000 T2D patients and in 500 non-diabetic
subjects.
Three-dimensional (3D) structure of
protein and in silico mutagenesis
The 3D structure of the wild-type human DnaJ homolog
subfamily C member 3 (DNAJC3) protein (2Y4T) (21) was obtained from The Protein Data
Bank (http://www.rcsb.org/) (22). In silico mutagenesis and
polar contact displays were performed using the PyMOL software
version 1.8.4.0-r2 (Schrödinger, LLC, NY, USA).
Cell culture
The human pancreatic β-cell line 1.1b4 was purchased
from the American Type Culture Collection (Manassas, VA, USA).
Cells were grown in RPMI-1640 medium (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) and 1% antibiotics
(100 IU/ml penicillin and 100 mg/ml streptomycin) at 37°C in a
humidified atmosphere containing 5% CO2.
Plasmid construct and transfection
In order to generate DNAJC3 plasmids, a DNAJC3
coding sequence (NM_006260) and a FLAG-tag sequence (DYKDDDDK) were
amplified and cloned into the pcDNA3.1 plasmid (Invitrogen; Thermo
Fisher Scientific, Inc.). All plasmids were amplified in E.
coli (D5α) (Invitrogen; Thermo Fisher Scientific, Inc.) and
purified using a mini kit (Qiagen GmbH, Hilden, Germany). For
generating the DNAJC3 p.H238N clone, wild-type DNAJC3 plasmids were
used as the template the wild-type was subjected to mutagenesis to
obtain the mutant plasmid. On the day prior to transfection,
2×105 cells/well were seeded into 6-well plates. After
24 h, cells were transfected by 1 µg DNAJC3 wild-type or
p.H238N plasmid using Lipofectamine® 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to
manufacturer's protocol. After 48 h of transfection, cells were
harvested and the stability of protein was measured by western blot
analysis. The efficiency of plasmid transfection was determined
using quantitative PCR by detection Neomycin DNA region of pcDNA3.1
plasmid. The forward primer (TGA ATG AAC TGC AGG ACG AG) and
reverse primer (ATA CTT TCT CGG CAG GAG CA) were used to amplified
the PCR product. qPCR was performed in a LightCycler 480 Instrument
(Roche Diagnostics, Mannheim, Germany) using
LightCycler® 480 SYBR Green I Master (Roche Diagnostics,
Mannheim, Germany). Initial enzyme activation proceeded at 95°C for
10 min, followed by 45 cycles at 95°C for 30 sec, 60°C for 20 sec,
and 72°C for 20 sec. β-actin was used as an internal control to
normalize input cDNA. Relative mRNA expression levels were
calculated using the 2−ΔΔCq method (23). The assay was performed in
triplicate.
RNA isolation and RNA transcripts level
measurement
Total RNA from 1.1b4 cells transfected with DNAJC3
wild-type, p.H238N, and empty pcDNA3.1 plasmid constructs were
isolated using TRizol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). RNA was reverse-transcribed into cDNA
using a Thermo Scientific Revert Aid First Strand cDNA Synthesis
kit (Thermo Fisher Scientific, Inc.) and quantitated by means of
qPCR (forward primer: 5′-GTC CTC TCT GAT CCA GAA ATG A-3′ and
reverse primer 5′-TCA TCG TCT TTG TAG TCA TTG AAG-3′). The
materials, methods, instrument, and calculation used for
quantitative PCR were the same as those previously described for
detection of the Neomycin DNA region.
Western blot analysis
Cells lysis was performed with a
radioimmunoprecipitation assay reagent (Thermo Fisher Scientific,
Inc.). Protein concentrations were then quantified using a NanoDrop
2000 spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington,
DE, USA) by Bradford protein assay (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Proteins were separated on a 10%
SDS-polyacrylamide gel by electrophoresis and then electroblotted
to a nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) by a Semi-Dry Electrophoretic Transfer Cell system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membrane was
blocked with 5% BSA (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
for 1 h at room temperature. Subsequently, the samples were
incubated with primary mouse monoclonal antibody against FLAG (cat.
no. F3165; 1:2,000; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
for 2 h and mouse monoclonal antibody against β-actin (cat. no.
s-47778; 1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
for 1 h at room temperature. Membranes were incubated with
horseradish peroxidase-linked goat anti-mouse secondary antibody
(cat. no. P044701-2; 1:1,000; Dako; Agilent Technologies, Inc.,
Santa Clara, CA, USA). The binding antibodies were visualized using
an enhanced chemiluminescence detection kit (SuperSignal™ West Pico
PLUS Chemiluminescent Substrate; Thermo Fisher Scientific, Inc.)
and bands were detected by Biomolecular Imager (ImageQuant LAS
4010; GE Healthcare Life Sciences, Little Chalfont, UK). β-actin
staining was utilized as an internal control in all experiments.
The expressed bands were quantified by computer-assisted scanning
densitometry using ImageJ software version 1.4.3.x (National
Institutes of Health, Bethesda, MD, USA). The experiments were
conducted three times independently.
Statistical analysis
SPSS software version 17.0 (SPSS, Inc., Chicago, IL,
USA) was used for data analysis. A χ2 test was used to
examine the significance of the association between each variation
and T2D. In case of a sample size of <5, Fisher's exact test was
used to validate the association. A Student's t-test was conducted
to evaluate mean differences in protein expression between the
wild-type and mutant groups following transfection of the plasmids
into 1.1b4 β-cells. A P-value of ≤0.05 was considered to indicate a
statistically significant difference.
Results
Exome sequencing performance
An average of 2.8 Gb on-target yields were generated
from each sample of four family members (data not shown), including
two subjects with diabetes (III3 and III4) and two with normal
glucose tolerance (I2 and II3). These four family members were
selected for exome sequencing as representatives of this large
early-onset T2D family because they were able to provide valuable
information regarding diabetes-predisposing variants. The mean read
depth of the target region was 45x, while 93 and 83% of the target
region were covered more than 1 and 10x which mean that 93 and 83%
of each base in the target region were at least 1 and 10x,
respectively. A total of 72,723, 61,409, 64,723 and 76,413 sequence
variants were detected in the samples from the I2, II3, III3 and
III4 subjects, respectively. Among those, 3,451 variants were
identified only in diabetic (III3 and III4) family members.
Candidate variant selection and
validation
The number of candidate variants falling under each
selection criteria is shown in Table
II. In total, 152 of those variants were novel or rare
heterozygous non-synonymous variants (MAF<0.01). Among these,
122 variants from 117 genes were predicted as having deleterious
effects on protein functions by at least one of the four in
silico mutation prediction programs. Furthermore, of these 122
variants, the GeneDistiller software revealed that 34 variants
residing in 32 genes were implicated in diabetes and
metabolism.
| Table IINumber of candidate variants under
different selection criteria. |
Table II
Number of candidate variants under
different selection criteria.
| Selection
criteria | Number of
variants |
|---|
| Variants from exome
sequencing | 68,817 |
| Variants identified
only in two diabetic samples | 3,451 |
| Coding
variants | 2,551 |
| Non-synonymous
variants | 393 |
| Heterozygous
non-synonymous variants | 245 |
| Heterozygous
non-synonymous variants with MAF <0.01 | 152 |
| Variants predicted
as having deleterious effect on protein function by in
silico programs | 122 |
| Segregation
analysis | DNAJC3
p.H238N |
Segregation analysis
The 122 variants which were considered as possible
pathogenic variants were subjected to segregation analysis with
diabetes in family A. This analysis indicated that a missense
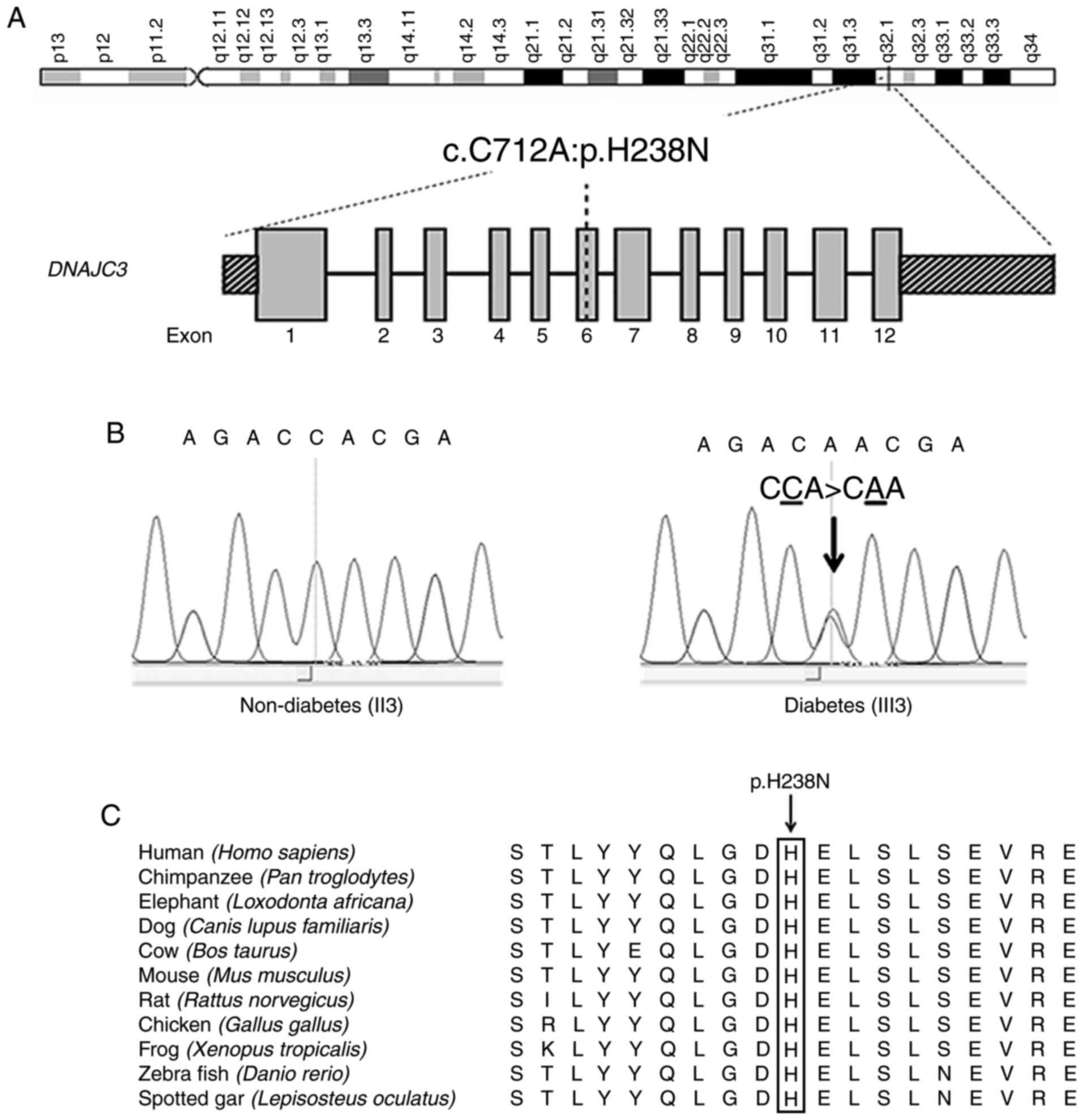
variant in DNAJC3 (NM_006260; c.C712A p.H238N) exhibited
partial segregation with diabetes in this family (Fig. 1A and C). DNAJC3 encodes a
co-chaperone of BiP and is involved in diabetes and metabolism, as
demonstrated using the GeneDistiller software. Mutations of
DNAJC3 have been attributed to DM, as well as multisystem
neurodegeneration (24).
DNAJC3 H238N was reported as rs527330902 in the Exome
Aggregation Consortium (http://exac.broadinstitute.org/) database, with a
frequency of 0.003% (3 out of a total of 119,086 alleles).
Subsequent genotyping this variant in other 90 early-onset T2D/MODY
X probands, demonstrated that DNAJC3 p.H238N segregated with
diabetes in another family (Fig. 1B
and C and Table I) but not in
the other 89 early-onset T2D/MODYX probands.
Prevalence of DNAJC3 p.H238N in unrelated
T2D patients and non-diabetic subjects
DNAJC3 p.H238N was also genotyped in
unrelated T2D patients and individuals with normal glucose
tolerance. This variant was identified in 14 out of 1,000 T2D
patients (MAF=0.007), in 2 out of 500 non-diabetic subjects
(MAF=0.002), and in 3 prediabetic individuals who had previously
been classified as non-diabetic subjects. In addition,
DNAJC3 p.H238N exhibited a trend towards increasing the risk
of developing T2D (odds ratio, 3.519; 95% confidence interval,
0.80-15.62; Table III),
although a statistically significant difference was not observed
(P=0.107).
| Table IIIGenotype and allele frequencies of
DNAJC3 p.H238N identified in Thai subjects, including 1,000
type 2 diabetic patients and 500 non-diabetic subjects. |
Table III
Genotype and allele frequencies of
DNAJC3 p.H238N identified in Thai subjects, including 1,000
type 2 diabetic patients and 500 non-diabetic subjects.
| Genotypes | Genotype
frequencies, n (%)
| P-value | Allele frequencies
(%)
| Non-diabetic
subjects | P-value |
|---|
| T2D | Non-diabetic
subjects | OR (95% CI) | T2D |
|---|
| CC | 986 (98.6) | 498 (99.6) | 0.107 | 3.519
(0.80-15.62) | C: 99.3 | C: 99.8 | 0.108 |
| CA | 14 (1.4) | 2 (0.4) | | | A: 0.7 | A: 0.2 | |
| AA | 0 (0) | 0 (0) | | | | | |
| Total | 1,000 (100) | 500 (100) | | | | | |
Functional impact of the mutation
Of the 4 in silico programs, used for
prediction of functional impact of the mutation, 3 suggested
DNAJC3 p.H238N as a deleterious mutation. These included the
results from Mutation taster (score=0.99; cut-off >0.5),
Variowatch (protein domain abolish), Polyphen2 (score=0.988;
cut-off >0.5). In addition, DUET algorithms predicted
instability of the mutant protein (ΔΔG= −2.009 kcal/mol; Table IV).
| Table IVBioinformatics analysis of DNAJC3
p.H238N. |
Table IV
Bioinformatics analysis of DNAJC3
p.H238N.
| Program | Prediction | Score | Cut-off value |
|---|
| MutationTaster | Disease
causing | 0.99 | >0.5 |
| VarioWatch | High, missense
(protein domain abolished) | – | – |
| PolyPhen2 | Deleterious | 0.988 | >0.5 |
| SIFT | Tolerated | 0.31 | <0.05 |
| DUET (predicted
stability changes) | Destabilizing | ΔΔG=−2.009
kcal/mol | Negative values
denote destabilizing mutations |
3D structure of mutant protein
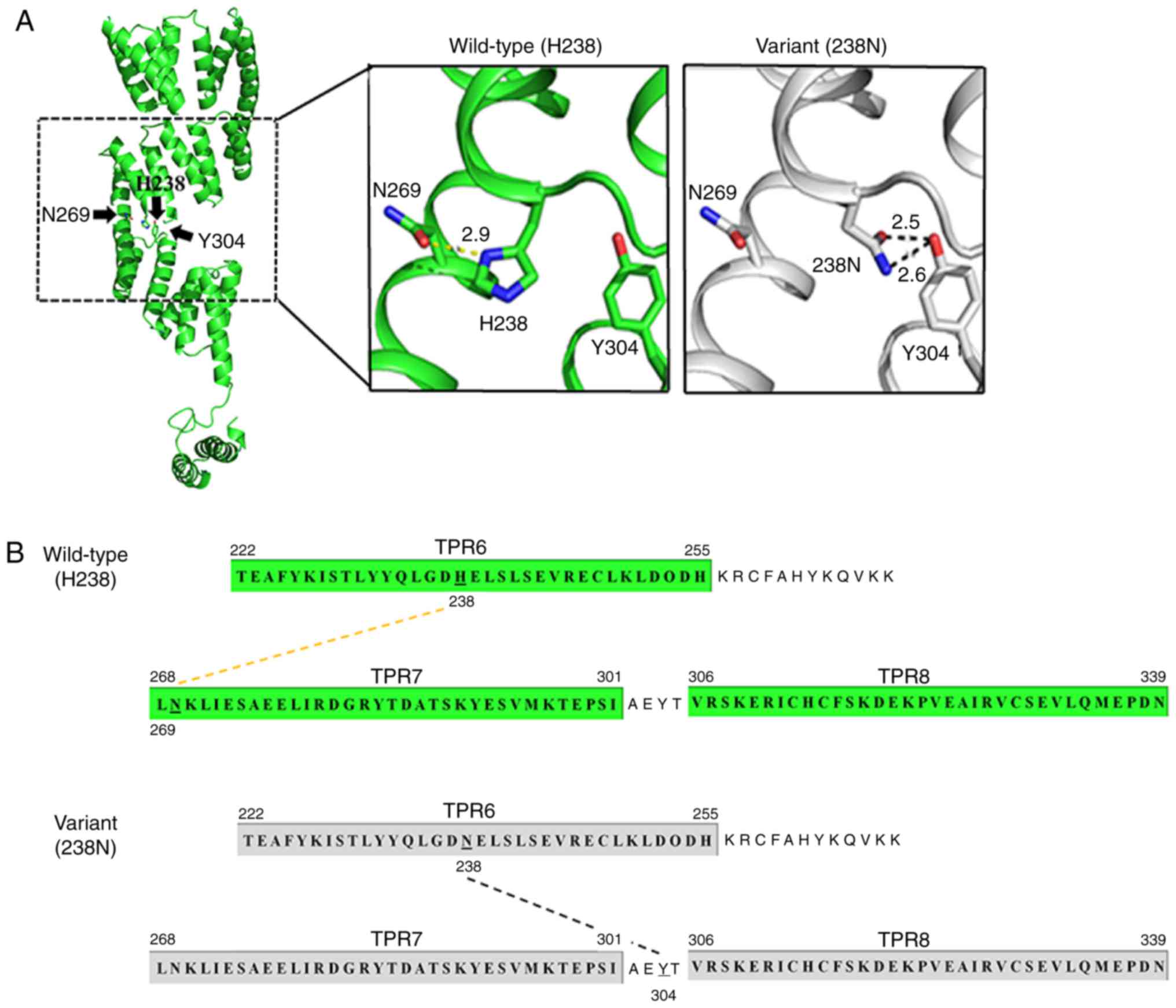
DNAJC3 comprises 504 amino acids which are divided
into an N-terminal ER-targeting tetratricopeptide repeat (TPR)
domain, a peptide sequence that has 9 TPR motifs and C-terminal J
domains (21). The crystal
structure revealed the presence of 9 TPR motifs forming 3 TPR
subdomains (21). Histidine
residue at position 238 (H238) residing in TPR6 of subdomain II is
conserved among humans, chimpanzees, elephants, cows, dogs, mice,
rats, chicken, frogs and zebrafish (Fig. 2). Using the PyMol software, a
polar contact between H238 and N269 of TPR7 of the protein
subdomain III was detected. However, substitution of H238 with 238N
disturbed the polar contact by creating a new, stronger hydrogen
bond with Y304, residing in the region between TPR7 and TPR8 of
subdomain III (Fig. 3).
Protein stability of DNAJC3 p.H238N
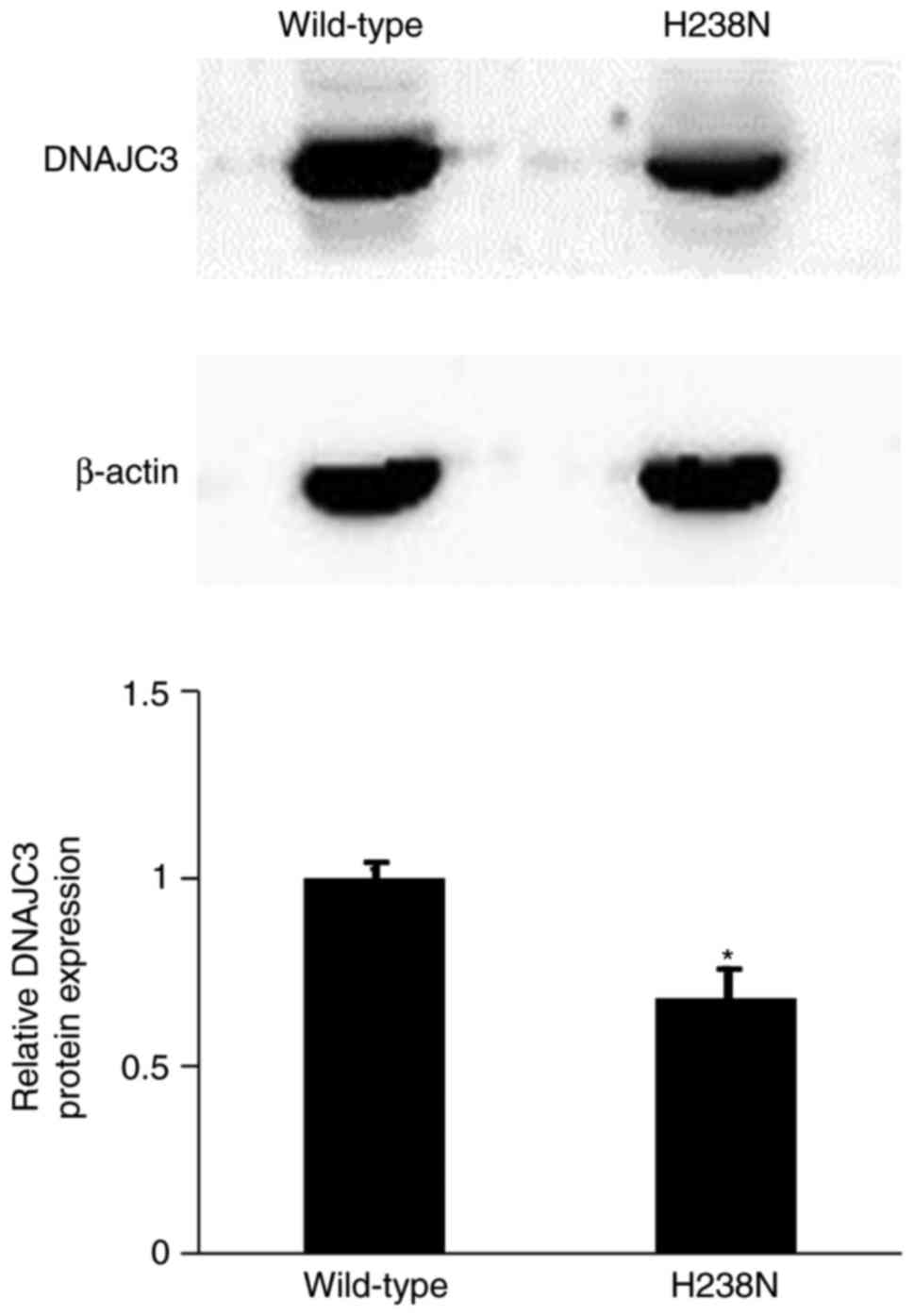
DNAJC3 wild-type and mutant plasmid tag with FLAG
sequence were constructed for detection of the expression of DNAJC3
p.H238N protein without the effect of endogenous protein in the
1.1b4 human pancreatic β-cell line. The results demonstrated that
the expression of DNAJC3 p.H238N protein was significantly lower
compared with that of the wild-type (0.68±0.08-fold change;
P<0.05; Fig. 4). Furthermore,
the efficiency of plasmid transfection was not significantly
different between DNAJC3 wild-type and mutant plasmid (data not
shown).
Discussion
The aim of the current study was to identify a
pathogenic mutation causing diabetes in a large Thai
multigenerational pedigree with diabetic family members in 3
generations, consistent with an autosomal dominant mode of
inheritance by exome sequencing. The proband (female), the
proband's sibling (female) and one of their relatives in generation
III were diagnosed with diabetes before 35 years of age. The
identified rare DNAJC3 p.H238N was partially segregated with
diabetes in the family. Although subject III2 carried DNAJC3
p.H238N, he exhibited a normal fasting plasma glucose level. The
patient was young (28-years-old) and had a normal BMI at the time
of diagnosis; therefore, this patient may develop diabetes later in
life, or due to being overweight or obese. The other two diabetic
family members (II1 and II9) who did not carry the DNAJC3
mutation were probably phenocopies (25,26). In subject II11 (male), although
DNAJC3 p.H238N was detected, the patient displayed a normal
fasting plasma glucose level (4.959 mmol/l); however, the HbA1c
level of this patient was in the prediabetes range (39 mmol/mol).
Therefore, follow-up of the glucose tolerance status is mandatory.
In addition, this variant was identified in another early-onset T2D
family without mutation of known MODY genes in which diabetes had
been transmitted from father to son, emphasizing the important role
of DNAJC3 p.H238N and glycemic traits. Furthermore,
DNAJC3 p.H238N was identified in unrelated T2D patients
(MAF=0.007) and in non-diabetic subjects (MAF=0.002), as well as 3
prediabetes cases. However, the allele frequencies of DNAJC3
p.H238N in T2D and non-diabetic subjects were not statistically
different, which is possibly due to the relatively small sample
size. It is possible that there are modifier genes or environmental
factors that can alter the effect or penetrance of this variant.
The results of the current study implied that this variant was
neither a rare allele causing a Mendelian disease, nor a common
variant implicated in the common T2D. Instead, it is a
low-frequency variant with an intermediate effect that confers a
higher risk of developing a chronic disease (such as T2D), as
described by Manolio et al (27).
DNAJC3 mRNA and protein are expressed in the
pancreatic β-cells. This protein is involved in an unfolded protein
response (UPR) during endoplasmic reticulum (ER) stress (28). The ER can transmit apoptotic
signals in the pancreas during ER stress, which mediates the
apoptosis of β-cells during the development of diabetes (29-32). Under the ER stress condition,
cells activate the adaptive UPR to resolve the protein-folding
defect (33). In addition, an
increased expression of UPR genes is observed in the islets of T2D
patients (34,35). Several studies have indicated that
DNAJC3 is regularly induced during ER stress, which is a key
component of a negative feedback used by the cell to inhibit
eukaryotic initiation factor-2 signaling and attenuate the UPR,
leading to a reduction of cell apoptosis (36,37). Ladiges et al (28) generated DnajC3 knockout
mice, demonstrating an ongoing onset of glycosuria and
hyperglycemia associated with an increasing apoptosis of the
pancreatic islet cells. A recent study by Han et al
(38) revealed that defective
protein folding generated oxidative stress, which serves as an
essential signal for apoptosis in response to ER stress in the
pancreatic β-cells. The authors demonstrated that homozygous
DNAJC3 knockout mice become diabetic due to decreased β-cell
mass and function as a result of the oxidative stress and apoptosis
signaling (38). Furthermore,
Synofzik et al (24)
demonstrated that a homozygous DNAJC3 p.R194* mutation
identified in three siblings from a Turkish family caused
juvenile-onset diabetes with central and peripheral
neurodegeneration. Also, they discovered that the homozygous
frameshift deletion DNAJC3 p.N34Mfs*20 caused juvenile-onset
diabetes with hearing impairment and ataxia in a German family with
a consanguineous marriage (24).
These two diabetes-associated mutations caused loss of the DNAJC3
protein function. Furthermore, 39 missense homozygous variants of
DNAJC3 were identified in 506 unrelated subjects with a
family history consistent with recessive disease, with one or more
features of the DNAJC3 phenotype in this cluster (diabetes,
neurodegeneration, hearing impairment or ataxia) (24).
Notably, the present study identified DNAJC3
p.H238N, which segregated with diabetes in autosomal dominant mode.
Since it was a heterozygous mutation, the disruption of the protein
function may be less harmful compared with its homozygous
counterpart. In silico mutagenesis of DNAJC3 H238N
revealed alteration of the polar contact across TPR motifs and
subdomains, which may affect the tertiary structure of the DNAJC3
protein. The DNAJC3 RNA levels of cells transfected with the
wild-type and mutant plasmid constructs were not statistically
different (data not shown). By contrast, the expression of
DNAJC3 H238N protein was lower compared with that in the
wild-type, which may be due to protein instability. This is
consistent with the 'Destabilizing' result predicted by DUET
predicted stability changes program. Since DNAJC3 R194* and
N34Mfs*20 caused loss of protein expression and function, the
clinical manifestations of patients harboring these mutations were
more severe in comparison with those harboring DNAJC3
p.H238N (24). Thus, the patients
in the present study became diabetic at a later age and did not
exhibit neurodegeneration, hearing impairment or ataxia. Further
studies considering the molecular and cell biology of this variant,
particularly in pancreatic β-cells, should promote a better
understanding of the mechanism underlying the pathogenesis of T2D,
and such research may lead to the development of novel therapies
for DM. Furthermore, screening for this variant in a larger sample
size of T2D patients and non-diabetic subjects should be conducted
to clarify the association between this variant and the risk of
developing diabetes.
In conclusion, in the present study, exome
sequencing successfully identified DNAJC3 p.H238N, which is
a low-frequency variant with an intermediate effect, causing
familial T2D in Thai individuals. The findings of the present study
were the first to demonstrate that DNAJC3 variants were
involved in pathogenesis of early-onset autosomal dominant T2D
while a previous report identified that different variants of this
gene were involved in syndromic autosomal recessive diseases with
diabetes (24).
Acknowledgments
Not applicable.
Funding
This research project was supported by the Mahidol
University Grant, Siriraj Research Grant for Research and
Development, Faculty of Medicine, Siriraj Hospital (awarded to NP,
WT, PJ and CC), the Thailand Research Fund (grant nos. BRG5280008,
TRG5780113 and IRG5980006; awarded to NP, WT and PTY), and a
Mahidol University Postdoctoral Fellowship Grant (awarded to SK).
PJ and PY were supported by a Chalermprakiat Grant from the Faculty
of Medicine, Siriraj Hospital.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SK, PTY and NP conceived and designed the study. WT
and PJ analyzed and interpreted the patient data and modeling. CC,
NB and JS performed the cell experiments and molecular biology
analysis. SK was a major contributor in writing the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee, Faculty of Medicine, Siriraj Hospital, Mahidol
University (COA no. Si491/2014). Written informed consent was
obtained.
Consent for publication
All subjects were informed of the purpose of the
study prior to signing an informed consent form.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Guariguata L, Whiting DR, Hambleton I,
Beagley J, Linnenkamp U and Shaw JE: Global estimates of diabetes
prevalence for 2013 and projections for 2035. Diabetes Res Clin
Pract. 103:137–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roglic G, Unwin N, Bennett PH, Mathers C,
Tuomilehto J, Nag S, Connolly V and King H: The burden of mortality
attributable to diabetes: Realistic estimates for the year 2000.
Diabetes Care. 28:2130–2135. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Murea M, Ma L and Freedman BI: Genetic and
environmental factors associated with type 2 diabetes and diabetic
vascular complications. Rev Diabet Stud. 9:6–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wilmot E and Idris I: Early onset type 2
diabetes: Risk factors, clinical impact and management. Ther Adv
Chronic Dis. 5:234–244. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bridger T: Childhood obesity and
cardiovascular disease. Paediatr Child Health. 14:177–182. 2009.
View Article : Google Scholar :
|
|
6
|
Fajans SS, Bell GI and Polonsky KS:
Molecular mechanisms and clinical pathophysiology of maturity-onset
diabetes of the young. N Engl J Med. 345:971–980. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sanghera DK and Blackett PR: Type 2
diabetes genetics: Beyond GWAS. J Diabetes Metab. 3:69482012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee S, Abecasis GR, Boehnke M and Lin X:
Rare-variant association analysis: Study designs and statistical
tests. Am J Hum Genet. 95:5–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Morris AP, Voight BF, Teslovich TM,
Ferreira T, Segrè AV, Steinthorsdottir V, Strawbridge RJ, Khan H,
Grallert H, Mahajan A, et al: Large-scale association analysis
provides insights into the genetic architecture and pathophysiology
of type 2 diabetes. Nat Genet. 44:981–990. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cirulli ET and Goldstein DB: Uncovering
the roles of rare variants in common disease through whole-genome
sequencing. Nat Rev Genet. 11:415–425. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lohmueller KE, Sparsø T, Li Q, Andersson
E, Korneliussen T, Albrechtsen A, Banasik K, Grarup N,
Hallgrimsdottir I, Kiil K, et al: Whole-exome sequencing of 2,000
Danish individuals and the role of rare coding variants in type 2
diabetes. Am J Hum Genet. 93:1072–1086. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kwak SH, Jung CH, Ahn CH, Park J, Chae J,
Jung HS, Cho YM, Lee DH, Kim JI and Park KS: Clinical whole exome
sequencing in early onset diabetes patients. Diabetes Res Clin
Pract. 122:71–77. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Plengvidhya N, Boonyasrisawat W,
Chongjaroen N, Jungtrakoon P, Sriussadaporn S, Vannaseang S,
Banchuin N and Yenchitsomanus PT: Mutations of maturity-onset
diabetes of the young (MODY) genes in Thais with early-onset type 2
diabetes mellitus. Clin Endocrinol (Oxf). 70:847–853. 2009.
View Article : Google Scholar
|
|
14
|
Standards of medical care in
diabetes-2017: Summary of revisions. Diabetes care. 40:S4–S5. 2017.
View Article : Google Scholar
|
|
15
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup: The sequence alignment/map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A mapreduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle
E, Wang K and Liu X: Comparison and integration of deleteriousness
prediction methods for nonsynonymous SNVs in whole exome sequencing
studies. Hum Mol Genet. 24:2125–2137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seelow D, Schwarz JM and Schuelke M:
GeneDistiller-distilling candidate genes from linkage intervals.
PloS One. 3:e38742008. View Article : Google Scholar
|
|
20
|
Pires DE, Ascher DB and Blundell TL: DUET:
A server for predicting effects of mutations on protein stability
using an integrated computational approach. Nucleic Acids Res.
42:W314–W319. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Svärd M, Biterova EI, Bourhis JM and Guy
JE: Crystal structure of the human co-chaperone P58(IPK). PLoS One.
6:e223372011. View Article : Google Scholar
|
|
22
|
Berman HM, Westbrook J, Feng Z, Gilliland
G, Bhat TN, Weissig H, Shindyalov IN and Bourne PE: The protein
data bank. Nucleic Acids Res. 28:235–242. 2000. View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Synofzik M, Haack TB, Kopajtich R, Gorza
M, Rapaport D, Greiner M, Schönfeld C, Freiberg C, Schorr S, Holl
RW, et al: Absence of BiP co-chaperone DNAJC3 causes diabetes
mellitus and multisystemic neurodegeneration. Am J Hum Genet.
95:689–697. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamagata K, Oda N, Kaisaki PJ, Menzel S,
Furuta H, Vaxillaire M, Southam L, Cox RD, Lathrop GM, Boriraj VV,
et al: Mutations in the hepatocyte nuclear factor-1alpha gene in
maturity-onset diabetes of the young (MODY3). Nature. 384:455–458.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stoffers DA, Ferrer J, Clarke WL and
Habener JF: Early-onset type-II diabetes mellitus (MODY4) linked to
IPF1. Nat Genet. 17:138–139. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Manolio TA, Collins FS, Cox NJ, Goldstein
DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR,
Chakravarti A, et al: Finding the missing heritability of complex
diseases. Nature. 461:747–753. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ladiges WC, Knoblaugh SE, Morton JF, Korth
MJ, Sopher BL, Baskin CR, MacAuley A, Goodman AG, LeBoeuf RC and
Katze MG: Pancreatic β-cell failure and diabetes in mice with a
deletion mutation of the endoplasmic reticulum molecular chaperone
gene P58IPK. Diabetes. 54:1074–1081. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mao C, Dong D, Little E, Luo S and Lee AS:
Transgenic mouse model for monitoring endoplasmic reticulum stress
in vivo. Nat Med. 10:1013–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Iwawaki T, Akai R, Kohno K and Miura M: A
transgenic mouse model for monitoring endoplasmic reticulum stress.
Nat Med. 10:98–102. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ron D: Translational control in the
endoplasmic reticulum stress response. J Clin Invest.
110:1383–1388. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang K and Kaufman RJ: Signaling the
unfolded protein response from the endoplasmic reticulum. J Biol
Chem. 279:25935–25938. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Manié SN, Lebeau J and Chevet E: Cellular
mechanisms of endoplasmic reticulum stress signaling in health and
disease. 3. Orchestrating the unfolded protein response in
oncogenesis: An update. Am J Physiol Cell Physiol. 307:C901–C907.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang CJ, Lin CY, Haataja L, Gurlo T,
Butler AE, Rizza RA and Butler PC: High expression rates of human
islet amyloid polypeptide induce endoplasmic reticulum stress
mediated β-cell apoptosis, a characteristic of humans with type 2
but not type 1 diabetes. Diabetes. 56:2016–2027. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Laybutt DR, Preston AM, Akerfeldt MC,
Kench JG, Busch AK, Biankin AV and Biden TJ: Endoplasmic reticulum
stress contributes to β-cell apoptosis in type 2 diabetes.
Diabetologia. 50:752–763. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
van Huizen R, Martindale JL, Gorospe M and
Holbrook NJ: P58IPK, a novel endoplasmic reticulum stress-inducible
protein and potential negative regulator of eIF2alpha signaling. J
Biol Chem. 278:15558–15564. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yan W, Frank CL, Korth MJ, Sopher BL,
Novoa I, Ron D and Katze MG: Control of PERK eIF2alpha kinase
activity by the endoplasmic reticulum stress-induced molecular
chaperone P58IPK. Proc Natl Acad Sci USA. 99:15920–15925. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han J, Song B, Kim J, Kodali VK, Pottekat
A, Wang M, Hassler J, Wang S, Pennathur S, Back SH, et al:
Antioxidants complement the requirement for protein chaperone
function to maintain β-cell function and glucose homeostasis.
Diabetes. 64:2892–2904. 2015. View Article : Google Scholar : PubMed/NCBI
|