Introduction
Lung cancer is the most frequent cause of
cancer-associated mortalities worldwide, and non-small-cell lung
cancer (NSCLC) is the most commonly occurring type of lung cancer
(1,2). NSCLC patients are often diagnosed at
a late stage of the disease, and exhibit a poor prognosis and
resistance to chemotherapy (3).
Despite advances associated with the use of targeted therapy,
chemotherapy and immunotherapy, the treatment of advanced NSCLC
remains challenging (2,4,5).
Therefore, it is urgent to elucidate the molecular mechanisms that
underlie the malignant behavior of NSCLC and identify novel
approaches to inhibit aggressiveness and increase chemosensitivity
of NSCLC to anticancer agents.
E3 ubiquitin ligases are engaged in the regulation
of the turnover of numerous target proteins and serve an important
role in controlling various biological processes (6-9).
Dysregulated expression of E3 ligases, such as Mdm2 RING finger E3
ligase (10) and Skp2 SCF E3
ligase (11,12), has been convincingly shown to
contribute to cancer development. Thus, targeting E3 ubiquitin
ligases for cancer therapy has gained increasing attention. Hakai,
a RING finger E3 ubiquitin ligase, was originally identified as an
E3 ligase for the E-cadherin complex in 2002 (13). Since its identification, an
increasing number of studies have focused on the role of Hakai in
cell-cell contacts and cell invasion (14). Recently, emerging evidence
suggested that Hakai promotes tumorigenesis. It was reported that
Hakai was highly expressed in human colon and gastric
adenocarcinomas, and regulated cell proliferation by enhancing the
RNA-binding function of polypyrimidine tract binding
protein-associated splicing factor (PSF) in an
E-cadherin-independent manner (15,16). Furthermore, several drugs, such as
vinflunine or silibinin, have been reported to inhibit cell
invasion by upregulation of E-cadherin and downregulation of Hakai
(17,18). These results demonstrated the
increasingly important role of Hakai in cancer development and
progression, and that Hakai may be a potential molecular target for
cancer treatment. However, little is known regarding the role of
Hakai and its direct mechanisms in human lung adenocarcinoma.
In the current study, the expression and potential
function of Hakai in NSCLC cell lines were investigated. The
results demonstrated that Hakai was significantly upregulated in
human NSCLC cell lines, while knockdown of Hakai inhibited the
proliferation, migration and invasion of NSCLC cells. Furthermore,
it was demonstrated that Hakai knockdown decreased the levels of
phosphorylated protein kinase B (pAKT) and enhanced the
cytotoxicity of cisplatin on NSCLC cells. The results suggested
that the development of small molecules or RNA interference
(RNAi)-based therapies targeting Hakai is promising for future
treatment of NSCLC.
Materials and methods
Reagents
Anti-Hakai antibody (21179-1-AP) was purchased from
ProteinTech Group, Inc. (Chicago, IL, USA). Anti-pAKT
(Ser473) antibody (sc-7985-R), anti-AKT antibody
(sc-8312), and the goat anti-rabbit (sc-2004) and anti-mouse
(sc-2005) horseradish peroxidase-conjugated IgG secondary
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Anti-N-cadherin (AF0243) and anti-E-cadherin
(AF0138) antibodies were obtained from Beyotime Institute of
Biotechnology (Shanghai, China). Anti-actin (A5316), cisplatin
(P4394) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT; Vetec V900888) were obtained from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany). Lipofectamine® 2000
(Invitrogen 11668-019) was purchased from Thermo Fisher Scientific,
Inc. (Waltham, MA, USA).
Cell lines and cell culture
The NSCLC cell lines A549 and NCI-H460 were obtained
from the American Type Culture Collection (Manassas, VA, USA). The
human normal bronchial epithelial cell line 16HBE was purchased
from the Cell Resource Center of the Chinese Academy of Medical
Sciences (Beijing, China) and cultured according to standard
protocols. The human normal bronchial epithelial cells Beas-2B were
provided by Professor Guangbiao Zhou at the Institute of Zoology,
Chinese Academy of Sciences. Cells were cultured in Dulbecco
modified Eagle medium (DMEM; HyClone; GE Healthcare Life Sciences,
Logan, UT, USA), supplemented with 10% fetal bovine serum (FBS;
HyClone; GE Healthcare Life Sciences), 100 U/ml penicillin and 0.1
mg/ml streptomycin (Beyotime Institute of Biotechnology) under 5%
CO2 and 37°C in an incubator.
Small interfering RNA (siRNA) assay
NSCLC cells were plated in 6-well plates with growth
medium without antibiotics at densities ranging between
1×105 and 2×105 cells/well. Following
incubation for 24 h, cells were transfected with synthesized Hakai
siRNA or negative control (NC) siRNA (GenePharma Co., Ltd.,
Shanghai, China) at a concentration of 100 nM using
Lipofectamine® 2000 for siRNA delivery, according to the
manufacturer's protocol. The specific transfection procedure was as
follows: First, siRNA oligomer was diluted in 250 µl
Opti-MEM I Reduced Serum Medium (Gibco; Thermo Fisher Scientific,
Inc.) without serum. Next, 4 µl Lipofectamine®
2000 was diluted in 250 µl Opti-MEM I Reduced Serum Medium,
mixed gently and incubated for 5 min at room temperature. After 5
min of incubation, the diluted oligomer and
Lipofectamine® 2000 solutions were combined, mixed
gently and incubated for 20 min at room temperature.
Oligomer-Lipofectamine® 2000 complexes were added to
each well containing the cells, along with 500 µl growth
medium without antibiotics, and cells were incubated at 37°C in a
CO2 incubator. After 6 h of transfection, the medium was
replaced with fresh growth medium, and the cells were harvest for
further analysis after 48-72 h of cultivation. The sequences for
Hakai RNAi were as follows: Hakai RNAi-1,
5′-CUCGAUCGGUCAGUCAGGAAA-3′; and Hakai RNAi-2,
5′-CACCGCGAACUCAAAGAACUA-3′. The efficiency of siRNA transfection
in downregulating the targeted gene was detected by a western blot
assay, with an >50% reduction observed in the targeted protein
expression.
RNA isolation and reverse
transcription-polymerase chain reaction (RT-PCR)
Isolation of total RNA from cell lines was conducted
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
and the phenol-chloroform extraction method, according to the
manufacturer's protocols. The 28S and 18S bands were detected by
agarose gel electrophoresis for measuring the quality of the
extracted RNA. cDNA was subsequently generated from total RNA using
a PrimeScript II 1st Strand cDNA Synthesis kit (Takara
Biotechnology Co., Ltd., Dalian, China). The RT conditions were as
follows: 42°C for 1 h and 95°C for 5 min, followed by storage at
−20°C. Next, the resultant cDNAs were used as templates for PCR
amplification in 25 µl reaction mixture, including 10X
ExTaq PCR buffer, deoxynucleoside triphosphate mixture (2.5
mM each), 0.2 µM of each primer and 1 U ExTaq
polymerase (RR001A; Takara Biotechnology Co., Ltd.). The primers
for Hakai were synthesized by Jinsirui Biotechnology Co., Ltd.
(Nanjing, China), and the sequences were as follows: Hakai:
5′-GGACACCTTTTTTGGGACTT-3′ (forward) and
5′-CACCCTTGAACAATGCTACAC-3′ (reverse); Actin:
5′-ATCGTCCACCGCAAATGCTTCTA-3′ (forward) and
5′-AGCCATGCCAATCTCATCTTGTT-3′ (reverse). The PCR amplification
conditions for Hakai and Actin were 94°C for 10 sec, 55°C for 15
sec and 72°C for 30 sec, for a total of 35 and 22 cycles,
respectively. Actin was used as an internal control. PCR products
were separated by electrophoresis on 1.5% (w/v) agarose gels and
visualized under UV light via Goldview staining (Beijing Solarbio
Science & Technology Co. Ltd., Beijing, China).
Trypan blue exclusion assay
Cell viability was evaluated using the trypan blue
dye exclusion assay (cat. no. C0011; Beyotime Institute of
Biotechnology). Cells after 48 and 72 h of transfection were
respectively detached with a trypsin-EDTA solution (HyClone; GE
Healthcare Life Sciences). The mixture of detached cells was
centrifuged at 1,000 × g for 1 min at room temperature and
resuspended with fresh DMEM supplement with 10% FBS and
penicillin/streptomycin. Subsequently, 100 µl of cell
suspension was combined with 100 µl trypan blue solution
(2X) and mixed gently. After 3 min staining at room temperature,
viable cells that were not stained were counted manually using an
inverted light microscope (Leica Microsystems GmbH, Wetzlar,
Germany) using a x10 objective in 6-8 random fields of view in each
group.
Colony formation assay
A549 or NCI-H460 cells were transfected with NC or
Hakai-specific siRNA using the aforementioned method. After 48 h,
the transfected cells were digested with trypsin and seeded onto 35
mm plates in triplicate (1,000 cells per plate). The medium were
refreshed every 5 days. After 10 days of incubation, the cells were
fixed with methanol for 10 min and stained with 0.005% crystal
violet solution for ~20 min. Stained clones containing >50 cells
were counted under a microscope.
Wound healing assay
A549 (2×105/well) and NCI-H460
(4×105/well) cells were seeded into 6-well plates and
transfected the next day with Hakai RNAi or NC RNAi, while there
was also a control group without siRNA transfection. After 48 h,
when the cells approximated full confluence, a sterile
200-µl micropipette tip was used to create wounds in the
cell monolayer of the three groups, and the culture medium was
replaced with fresh serum-free medium. At 0 and 24 h, images of the
scratched areas were captured with an inverted microscope (Leica
Microsystems GmbH, Wetzlar, Germany) using a x4 objective. The
wound widths were measured, and the relative wound widths were
calculated.
In vitro cell invasion assay
A 24-well transwell unit (pore size, 8 µm;
Costar; Corning Life Science, Woburn, MA, USA) was used to evaluate
the ability of cell invasion. A total of 2×105
transfected cells were seeded into the upper chamber coated with
Matrigel (cat. no. 354248; BD Biosciences, Franklin Lakes, NJ,
USA), and the bottom chamber was filled with DMEM with 20% FBS.
After 24 h of incubation at 37°C in a 5% CO2 atmosphere,
invasive cells on the bottom sides of the membrane were fixed with
ethanol for 10 min and stained with 0.2% crystal violet for 20 min.
Invasive cells were manually counted under a microscope (Leica
Microsystems GmbH) at a ×20 objective in 4-5 random fields-of-view
in each group.
MTT assay
At 48 h post-transfection, A549 and NCI-H460 cells
from the two different groups (Hakai RNAi and NC RNAi) were seeded
into 96-well plates at a density of 5,000-10,000 cells/well, with
three replicate wells used per group. When the cells in each well
reached 70-80% confluence, different concentrations of cisplatin
(0, 5, 10 and 15 µM) were added followed by 48 h incubation.
Next, 10 µl MTT (5 mg/ml) was added to each well for an
additional 2-4 h. The medium was discarded and 150 µl DMSO
was added to dissolve the formazan crystals for measurement.
Subsequently, the optical density was measured at an absorbance
wavelength of 490 nm using a microplate reader (Multiskan FC;
Thermo Fisher Scientific, Inc.). The cell survival rate was
calculated as follows: Cell viability (%) = (average A490 of the
experimental group − average A490 of the blank group)/(average A490
of the control group − average A490 of the blank group) × 100.
Western blot analysis
Proteins from different cell lines in the different
transfection groups were extracted using radioimmunoprecipitation
assay buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1%
Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate
(SDS), 1 mM Na3VO4, 1 mM NaF and 1 mM PMSF
(19). The protein levels were
determined using Bradford Protein Assay kit (Beyotime Institute of
Biotechnology). Proteins (25-50 µg) were then subjected to
SDS-polyacrylamide gel electrophoresis (8 and 10%), and transferred
to a nitrocellulose membrane (Pall Corporation, East Hills, NY,
USA). Subsequent to blocking with 5% non-fat milk for 1 h at room
temperature, the membranes were incubated overnight at 4°C with the
indicated primary antibodies, then washed with Tris-buffered salin
containing 1% Tween-20 for 20 min, followed by incubation with the
corresponding secondary antibodies for 2 h at room temperature.
Detection was performed using an enhanced chemiluminescence
substrate (CW0049M; Cwbiotech, Beijing, China). Bands intensities
were quantified using ImageJ software (version 1.48; National
Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data were collected and analyzed using GraphPad
Prism 6.0 software (GraphPad Software, Inc., La Jolla, CA, USA),
and are expressed as the mean ± standard deviation. Student's
t-test was used to compare data between two groups. One-way or
two-way analysis of variance, followed by Sidak post hoc test, was
used to analyze data of multiple groups. P<0.05 was considered
to be an indicator of a statistically significant difference.
Results
Elevated expression of Hakai in NSCLC
cell lines
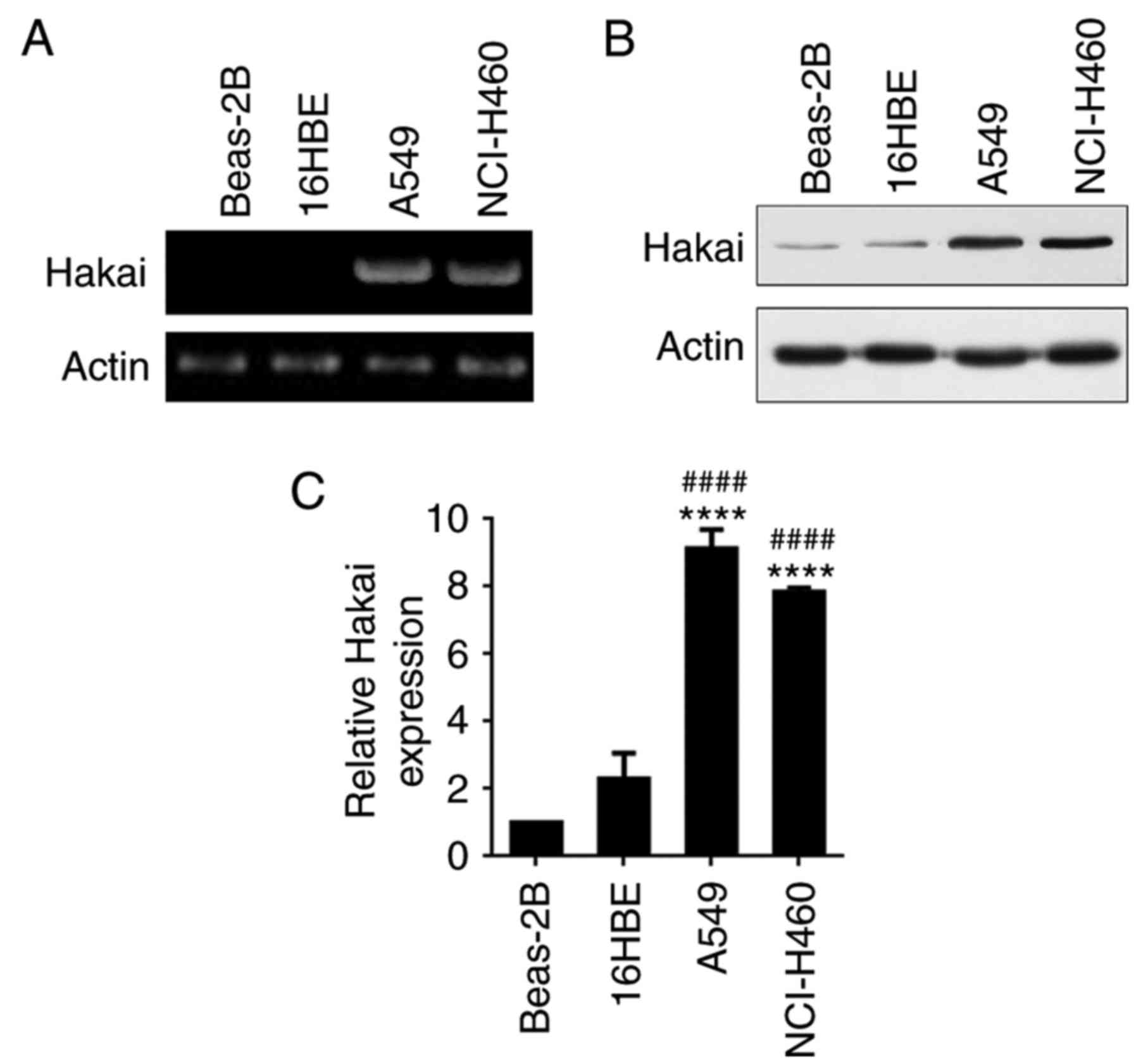
Hakai expression was assessed by RT-PCR and western
blot analysis in the NSCLC cell lines A549 and NCI-H460, as well as
in human normal bronchial epithelial cells 16HBE and Beas-2B. The
results demonstrated that Hakai mRNA was highly expressed in A549
and NCI-H460 cell lines compared with its expression in 16HBE and
Beas-2B cells (Fig. 1A).
Consistent with mRNA expression, western blot analysis revealed
significantly higher protein expression levels of Hakai in A549 and
NCI-H460 cell lines, with weaker expression observed in 16HBE and
Beas-2B cells (P<0.0001; Fig.
1B and C). These results indicated that Hakai is overexpressed
in NSCLC cells.
Downregulation of Hakai inhibits
proliferation in A549 and NCI-H460 cells
In order to investigate the potential function of
Hakai in NSCLC cell lines, A549 and NCI-H460 cells were transfected
with siRNA against Hakai, and the results revealed that the two
siRNAs were able to decrease the expression of Hakai (Fig. 2A). Thereafter, Hakai RNAi-1 was
used for further experiments, owing to its higher effect. Hakai
silencing was found to markedly inhibit the proliferation of A549
and NCI-H460 cells, and an enhanced inhibitory effect was observed
when the incubation time increased, as demonstrated by trypan blue
dye exclusion assay (P<0.05; Fig.
2B and C). Similarly, a flat colony formation assay indicated
that Hakai knockdown in A549 and NCI-H460 cells led to a
significant decrease in the colony numbers (P<0.01 and
P<0.0001, respectively; Fig.
2D). These data demonstrated that Hakai serves a critical role
in NSCLC cell proliferation.
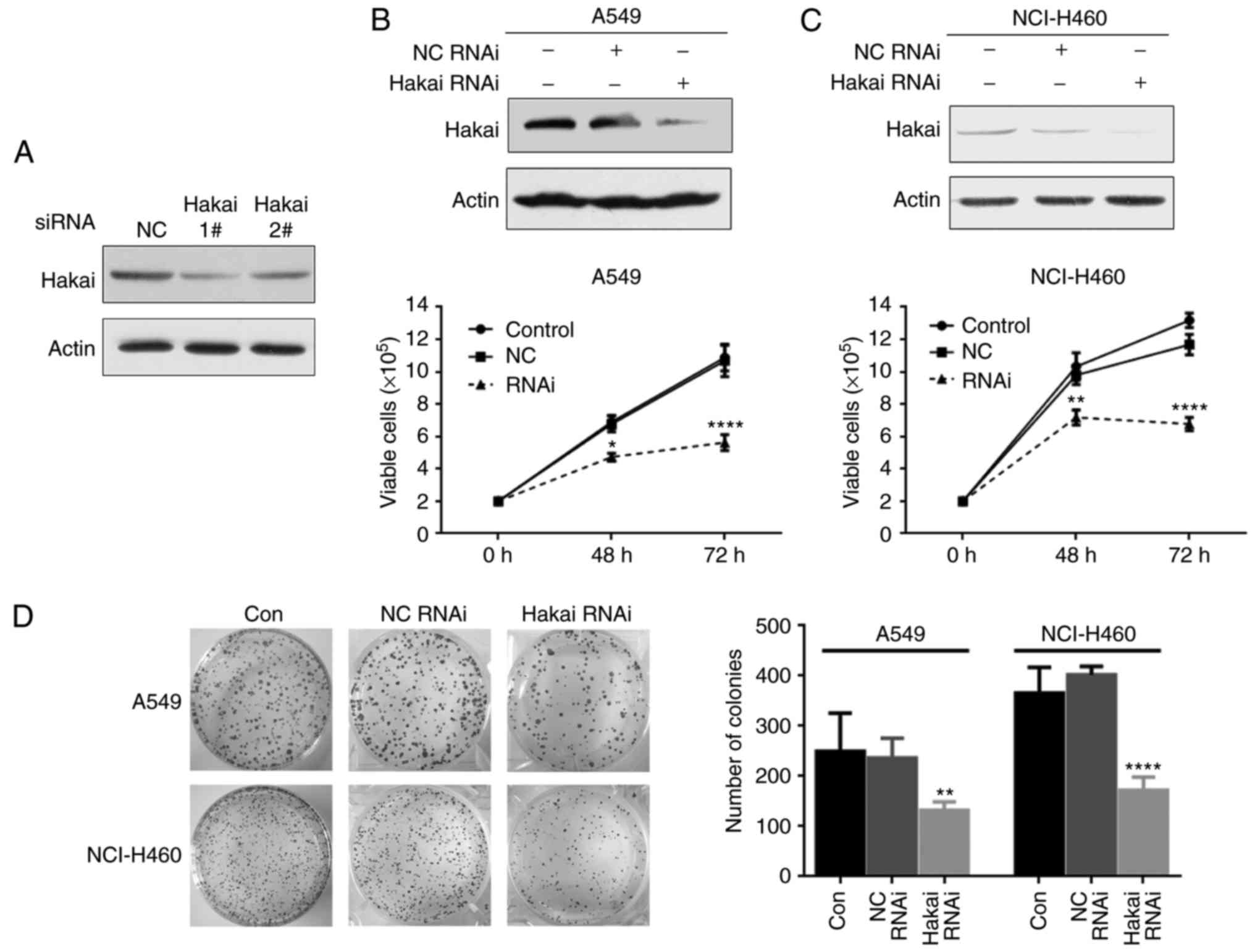 | Figure 2Knockdown of Hakai inhibits
proliferation in A549 and NCI-H460 cells. (A) A549 cells were
transfected with the indicated siRNAs (100 nM) for 72 h, and the
protein levels of Hakai were detected by western blotting. (B) A549
and (C) NCI-H460 cells in the three groups (non-transfected
control, NC RNAi and Hakai RNAi) were examined by a trypan blue
exclusion assay to determine the proliferation abilities. Upper
panels indicate the expression levels of Hakai protein in the three
groups, while lower panels indicate the results of trypan blue
exclusion assay (two-way ANOVA). (D) Flat plate clone formation
assay was used to detect the clonogenic activity of A549 and
NCI-H460 cells at 48 h after transfection with NC or Hakai-specific
siRNA. The left panel shows representative images, and the right
panel shows the quantification of the number of colonies with
>50 cells from three separate experiments (one-way ANOVA).
*P<0.05, **P<0.01 and
****P<0.0001, vs. NC RNAi group. Con, non-transfected
control; RNAi, RNA interference; NC, negative control; siRNA, small
interfering RNA. |
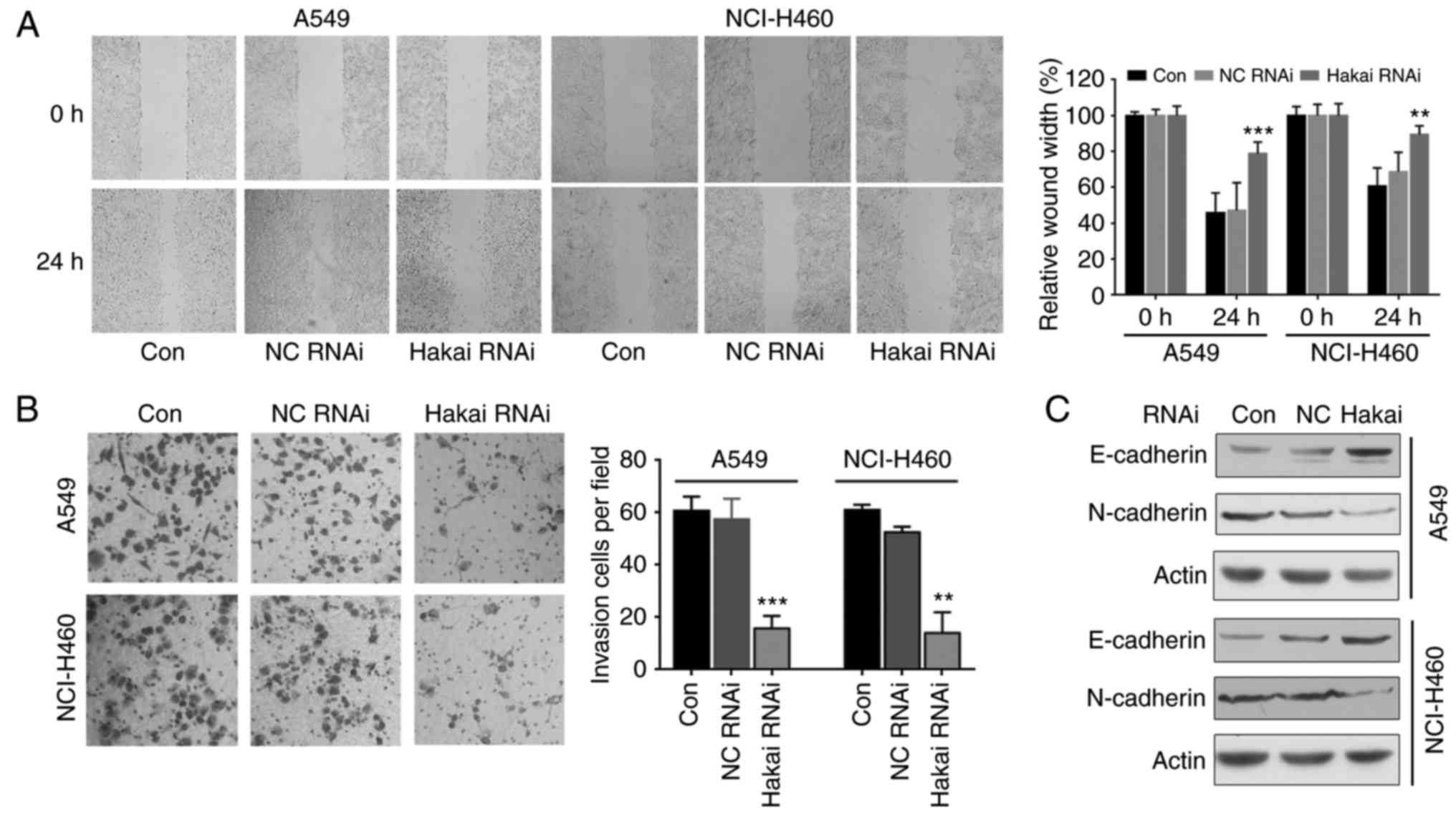
Hakai depletion inhibits migration and
invasion in NSCLC cells
Hakai is implicated in the regulation of cell
substratum adhesions and in epithelial cell invasion (14). Thus, the present study next
investigated whether Hakai was involved in the cell migration
progress in NSCLC cells. When comparing with the cells of the NC
and non-transfected control groups, the results of wound healing
assay demonstrated that Hakai depletion led to a significant
decrease of the migration ability of A549 and NCI-H460 cells
transfected with Hakai RNAi (P<0.001 and P<0.01,
respectively; Fig. 3A). A
transwell invasion assay revealed a low level of invasion in Hakai
RNAi cells compared with the NC and non-transfected control cells
(P<0.01 and P<0.001; Fig.
3B).
Hakai was firstly described as the E3 ubiquitin
ligase for the E-cadherin and mediated its degradation (13). Thus, the current study explored
whether downregulation of Hakai was able to regulate the expression
levels of E-cadherin and N-cadherin in NSCLC cells. The results of
western blot analysis demonstrated that Hakai depletion upregulated
E-cadherin and downregulated N-cadherin in both A549 and NCI-H460
cells (Fig. 3C), which may
contribute to the inhibitory effect of Hakai decrease on cell
migration and invasion. These results indicate that Hakai
influences cell migration and invasion in NSCLC cells.
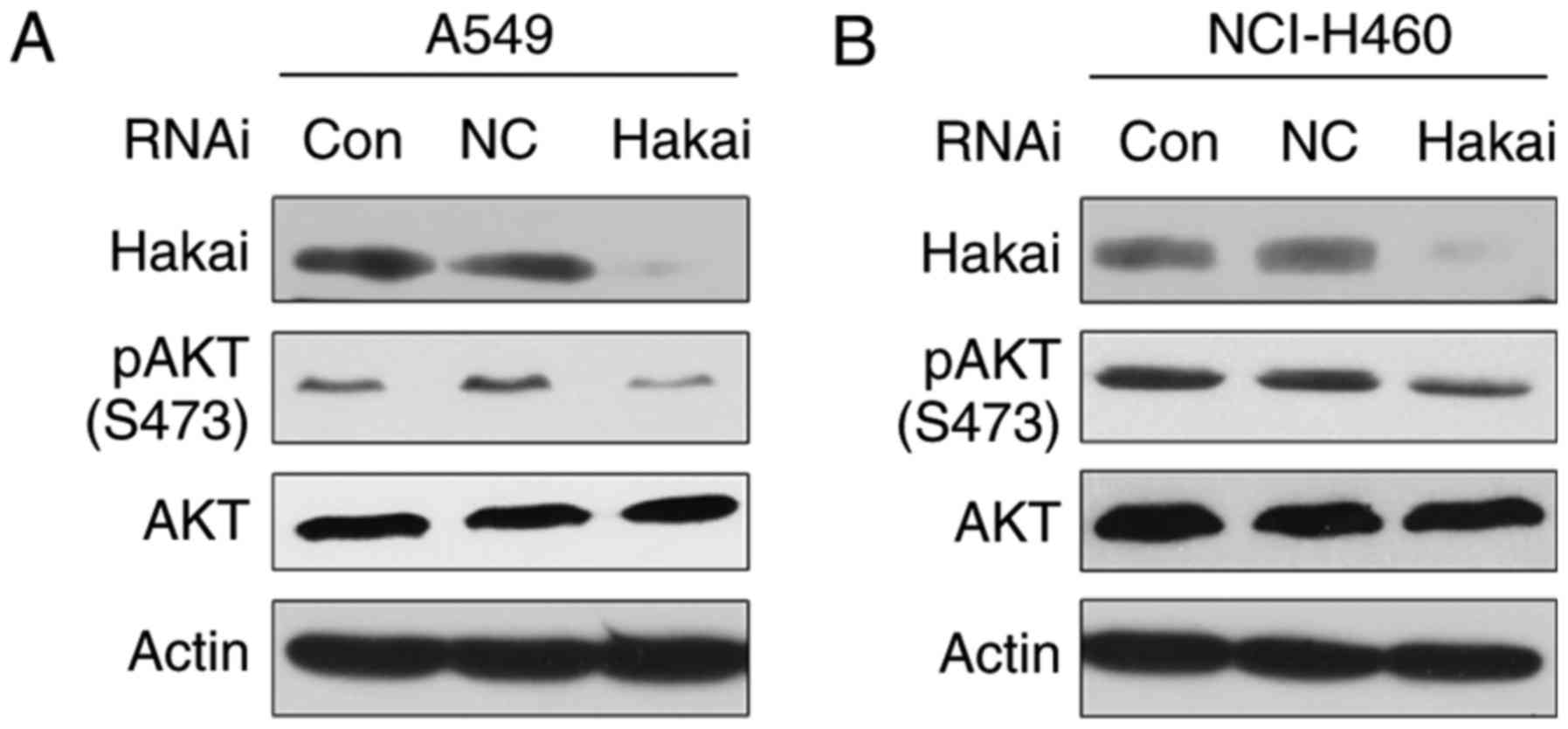
Silencing of Hakai expression suppresses
AKT activity in NSCLC cells
A growing body of evidence suggests that AKT
perturbations serve an important role in human malignancy (20). Consistently, high activation of
AKT has been reported in various types of human cancer, including
NSCLC (21,22). Thus, whether Hakai inhibition
regulated the activity of the AKT signaling pathway was examined in
NSCLC cells. As shown in Fig. 4A
and B, the western blot results revealed that downregulation of
Hakai decreased AKT phosphorylation at the Ser-473 site in both
A549 and NCI-H460 cells, with no apparent change in total AKT
level.
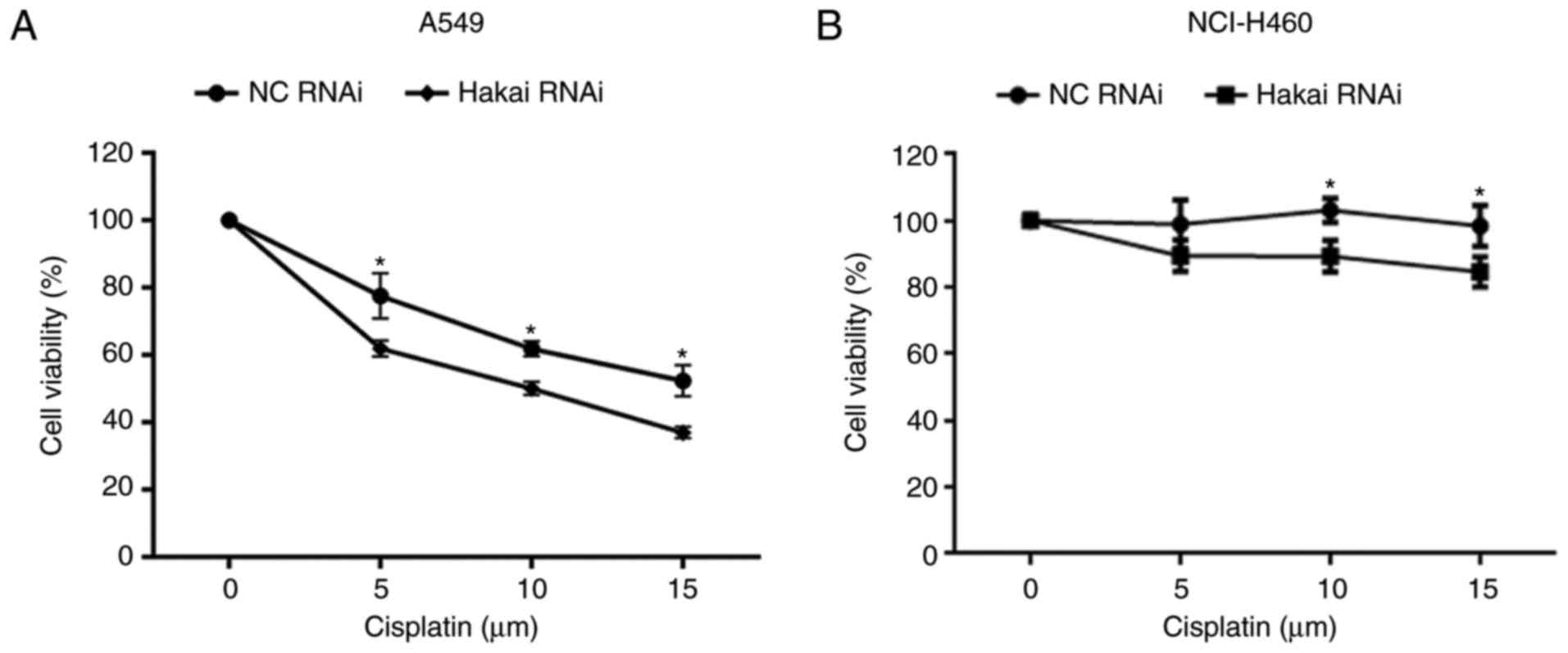
Downregulation of Hakai sensitizes NSCLC
cells to cisplatin treatment
Cisplatin resistance was associated with AKT
overexpression and gene amplification in human lung cancer cells
that acquired drug resistance (23). Next, the current study tested
whether Hakai was involved in the sensitivity of NSCLC cells to
cisplatin. The results demonstrated that silencing of Hakai
potentiated the susceptibility to cisplatin in A549 and NCI-H460
cells (P<0.05; Fig. 5A and B).
This finding indicates that the level of Hakai expression
influences the sensitivity to cisplatin in NSCLC cells.
Discussion
Hakai was initially identified as a RING finger type
E3 ubiq-uitin ligase for E-cadherin complex in 2002 (13). Since then, researchers have
highlighted its crucial role in cell adhesion and invasion during
carcinogenesis. Emerging evidence has also supported the critical
role of Hakai in cell proliferation. Figueroa et al
(15,16) reported that overexpression of
Hakai increased the proliferation of MDCK stable cell lines, while
transient knockdown of Hakai expression in MCF-7 and HEK293 cells
significantly decreased cell proliferation. The authors also
demonstrated that Hakai was upregulated in colon and gastric cancer
(16). In accordance with these
observations, the present study found a significantly increased
expression of Hakai in NSCLC cell lines, compared with that in
normal bronchial epithelial cells 16HBE and Beas-2B. To further
investigate the oncogenic potential of Hakai, the expression of
Hakai was downregulated in vitro. The results revealed that
the proliferation of NSCLC cells was inhibited by silencing the
expression of Hakai. These findings suggest that Hakai may function
as an oncoprotein in the development and progression of NSCLC.
The poor outcome of NSCLC is crucially correlated to
the onset of tumor metastasis (24). Therefore, reducing cell
invasiveness is a potential therapeutic strategy for attenuating
the progression of NSCLC. Epithelial-mesenchymal transition (EMT)
is a crucial step for cell metastasis, and has been reported to be
associated with a poor clinical outcome in NSCLC (25,26). A characteristic of cells that
undergo EMT is an increase of E-cadherin expression and a loss of
N-cadherin expression (27,28). It was reported that Hakai
overexpression enhances invasiveness and is implicated in several
processes that often occur during EMT (14). In the present study, knockdown of
Hakai was found to suppress NSCLC cell migration and invasion,
accompanied by an E-cadherin increase and an N-cadherin decrease,
indicating that Hakai regulates the invasive and metastatic ability
of NSCLC cells, partially through regulation of EMT.
The molecular mechanisms involved in the oncogenic
role of Hakai are largely unknown. Studies reported that there were
two proteins influencing proliferation through Hakai, namely PSF
and cyclin D1, which may function independently (15,16). Hakai can affect the oncogenic
phenotype by increasing the ability of PSF to bind to RNAs that
promote cancer-associated gene expression. Knockdown of Hakai
specifically inhibited the expression of cyclin D1 in MCF-7, HEK293
and MDA-MB231 cells (16). In the
current study, it was reported that Hakai knockdown decreased the
pAKT (Ser473) levels in A549 and NCI-H460 cells.
Hyperactivation of AKT is detected in the majority of NSCLC cell
lines, and certain studies have reported growth inhibition due to
blockade of constitutive AKT activity in NSCLC cells (21,29,30). The current study has established a
functional link between the pathways of Hakai and AKT, and provides
a possible explanation for the oncogenic activity of Hakai in
NSCLC. Further investigation is warranted to elucidate the
molecular mechanism on how Hakai regulates the AKT signaling
pathway.
Cisplatin is the cornerstone of lung cancer therapy;
however, its efficacy is limited due to the development of drug
resistance in cancer cells. Increasing studies have suggested AKT
phosphorylation as a novel mechanism in promoting the resistance of
tumor cells against cisplatin (21,23,31,32). As the current study results
demonstrated that the protein levels of pAKT (Ser473)
were reduced by Hakai depletion, the therapeutic role of Hakai
silencing in combination with cisplatin was then explored. To the
best of our knowledge, it was revealed for the first time that
Hakai downregulation sensitized NSCLC cells to cisplatin. The
present data suggest that cisplatin chemotherapy may be more
effective in combination with knockdown of Hakai for NSCLC
therapy.
In conclusion, the present study reported that Hakai
serves an important role in NSCLC progression by regulating the
growth, migration and invasion of NSCLC cells. Mechanistically, the
results revealed that the oncogenic profile of Hakai may be
partially mediated through AKT-associated pathways.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81402511 and
81201577) and the Student Research Training Program of Anhui
University of Technology (grant nos. 201510360171 and
2015024Z).
Availability of data and materials
The datasets generated and analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
LM designed the study. ZL and YW performed the
experiments. LM and ZT analyzed the data. LM and ZL wrote the
paper. All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirsch FR, Scagliotti GV, Mulshine JL,
Kwon R, Curran WJ Jr, Wu YL and Paz-Ares L: Lung cancer: Current
therapies and new targeted treatments. Lancet. 389:299–311. 2017.
View Article : Google Scholar
|
|
3
|
Saintigny P and Burger JA: Recent advances
in non-small cell lung cancer biology and clinical management.
Discov Med. 13:287–297. 2012.PubMed/NCBI
|
|
4
|
Dy GK and Adjei AA: Emerging therapeutic
targets in non-small cell lung cancer. Proc Am Thorac Soc.
6:218–223. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reck M, Heigener DF, Mok T, Soria JC and
Rabe KF: Management of non-small-cell lung cancer: Recent
developments. Lancet. 382:709–719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ciechanover A and Iwai K: The ubiquitin
system: From basic mechanisms to the patient bed. IUBMB life.
56:193–201. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nakayama KI and Nakayama K: Ubiquitin
ligases: Cell-cycle control and cancer. Nat Rev Cancer. 6:369–381.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pal A, Young MA and Donato NJ: Emerging
potential of therapeutic targeting of ubiquitin-specific proteases
in the treatment of cancer. Cancer Res. 74:4955–4966. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen M, Schmitt S, Buac D and Dou QP:
Targeting the ubiq-uitin-proteasome system for cancer therapy.
Expert Opin Ther Targets. 17:1091–1108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Duncan SJ, Cooper MA and Williams DH:
Binding of an inhibitor of the p53/MDM2 interaction to MDM2. Chem
Commun. 7:316–317. 2003. View
Article : Google Scholar
|
|
11
|
Yokoi S, Yasui K, Mori M, Iizasa T,
Fujisawa T and Inazawa J: Amplification and overexpression of SKP2
are associated with metastasis of non-small-cell lung cancers to
lymph nodes. Am J Pathol. 165:175–180. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang F, Caraway NP, Li R and Katz RL: RNA
silencing of S-phase kinase-interacting protein 2 inhibits
proliferation and centrosome amplification in lung cancer cells.
Oncogene. 24:3409–3418. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fujita Y, Krause G, Scheffner M, Zechner
D, Leddy HE, Behrens J, Sommer T and Birchmeier W: Hakai, a
c-Cbl-like protein, ubiquitinates and induces endocytosis of the
E-cadherin complex. Nat Cell Biol. 4:222–231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rodríguez-Rigueiro T, Valladares-Ayerbes
M, Haz-Conde M, Aparicio LA and Figueroa A: Hakai reduces
cell-substratum adhesion and increases epithelial cell invasion.
BMC Cancer. 11:4742011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Figueroa A, Fujita Y and Gorospe M:
Hacking RNA: Hakai promotes tumorigenesis by enhancing the
RNA-binding function of PSF. Cell Cycle. 8:3648–3651. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Figueroa A, Kotani H, Toda Y,
Mazan-Mamczarz K, Mueller EC, Otto A, Disch L, Norman M, Ramdasi
RM, Keshtgar M, et al: Novel roles of hakai in cell proliferation
and oncogenesis. Mol Biol Cell. 20:3533–3542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deep G, Gangar SC, Agarwal C and Agarwal
R: Role of E-cadherin in antimigratory and antiinvasive efficacy of
silibinin in prostate cancer cells. Cancer Prev Res. 4:1222–1232.
2011. View Article : Google Scholar
|
|
18
|
Aparicio LA, Castosa R, Haz-Conde M,
Rodríguez M, Blanco M, Valladares M and Figueroa A: Role of the
microtubule-targeting drug vinflunine on cell-cell adhesions in
bladder epithelial tumour cells. BMC cancer. 14:5072014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma L, Wen ZS, Liu Z, Hu Z, Ma J, Chen XQ,
Liu YQ, Pu JX, Xiao WL, Sun HD and Zhou GB: Overexpression and
small molecule-triggered downregulation of CIP2A in lung cancer.
PLoS One. 6:e201592011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Testa JR and Bellacosa A: AKT plays a
central role in tumorigenesis. Proc Natl Acad Sci USA.
98:10983–10985. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brognard J, Clark AS, Ni Y and Dennis PA:
Akt/protein kinase B is constitutively active in non-small cell
lung cancer cells and promotes cellular survival and resistance to
chemotherapy and radiation. Cancer Res. 61:3986–3997.
2001.PubMed/NCBI
|
|
22
|
Tang JM, He QY, Guo RX and Chang XJ:
Phosphorylated Akt overexpression and loss of PTEN expression in
non-small cell lung cancer confers poor prognosis. Lung Cancer.
51:181–191. 2006. View Article : Google Scholar
|
|
23
|
Liu LZ, Zhou XD, Qian G, Shi X, Fang J and
Jiang BH: AKT1 amplification regulates cisplatin resistance in
human lung cancer cells through the mammalian target of
rapamycin/p70S6K1 pathway. Cancer Res. 67:6325–6332. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schmidt LH, Spieker T, Koschmieder S,
Schäffers S, Humberg J, Jungen D, Bulk E, Hascher A, Wittmer D,
Marra A, et al: The long noncoding MALAT-1 RNA indicates a poor
prognosis in non-small cell lung cancer and induces migration and
tumor growth. J Thorac Oncol. 6:1984–1992. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu D, Huang C, Kameyama K, Hayashi E,
Yamauchi A, Kobayashi S and Yokomise H: E-cadherin expression
associated with differentiation and prognosis in patients with
non-small cell lung cancer. Ann Thorac Surg. 71:949–954. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bremnes RM, Veve R, Gabrielson E, Hirsch
FR, Baron A, Bemis L, Gemmill RM, Drabkin HA and Franklin WA:
High-throughput tissue microarray analysis used to evaluate biology
and prognostic significance of the E-cadherin pathway in
non-small-cell lung cancer. J Clin Oncol. 20:2417–2428. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: Twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Soltermann A: Epithelial-mesenchymal
transition in non-small cell lung cancer. Der Pathologe. 33(Suppl
2): S311–S317. 2012.In German. View Article : Google Scholar
|
|
29
|
Kurie JM: Role of protein kinase
B-dependent signaling in lung tumorigenesis. Chest. 125(Suppl):
S141–S144. 2004. View Article : Google Scholar
|
|
30
|
Scrima M, De Marco C, Fabiani F, Franco R,
Pirozzi G, Rocco G, Ravo M, Weisz A, Zoppoli P, Ceccarelli M, et
al: Signaling networks associated with AKT activation in non-small
cell lung cancer (NSCLC): New insights on the role of
phosphatydil-inositol-3 kinase. PLoS One. 7:e304272012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang X, Fraser M, Abedini MR, Bai T and
Tsang BK: Regulation of apoptosis-inducing factor-mediated,
cisplatin-induced apoptosis by Akt. Br J Cancer. 98:803–808. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin Y, Wang Z, Liu L and Chen L: Akt is
the downstream target of GRP78 in mediating cisplatin resistance in
ER stress-tolerant human lung cancer cells. Lung Cancer.
71:291–297. 2011. View Article : Google Scholar
|