Introduction
Renal interstitial fibrosis (RIF) is the main
pathway leading to an irreversible loss of renal function, and has
a poor prognosis requiring dialysis or transplantation. In addition
to excessive deposition of extracellular matrix (ECM), RIF is also
associated with leukocyte infiltration, myofibroblast accumulation
and tubular atrophy (1).
Fibrogenesis refers to the deposition of pathological matrix by
cells, and is widely considered as a wound-healing response to
tissue injury (2). The process is
associated with inflammatory cell recruitment and the appearance of
myofibroblasts, which serve a key role in ECM remodeling.
Myofibroblasts may serve a beneficial role in wound
healing; however, myofibroblasts and inflammatory cells persist in
the case that the injury does not abate, resulting in the loss of
the reparative and healing processes (3). Previous studies (4,5)
investigating forkhead box D1 lineage cells have reported that
resident fibroblasts are one of the major precursors of
myofibroblasts, whereas other studies have concluded that resident
fibroblasts are equivalent to myofibroblasts cells in the kidney.
It is known that renal fibroblasts migrate, proliferate and then
proceed to differentiate into myofibroblasts, expressing α-smooth
muscle actin (α-SMA) (6,7).
The recruitment of inflammatory cells to the injury
tissue also serves a pivotal role in wound-healing responses.
Occurrence of RIF is reported when kidney repair is insufficient or
consistently suppressed by ongoing tissue injury and inflammation
(7). As a member of the
mononuclear phagocyte family, macrophages function as a key player
in renal injury, inflammation and fibrosis. In addition,
macrophages are pleiotropic inflammatory cells that participate in
inflammatory reactions (8). With
recruitment to the inflammatory milieu, macrophages interact with
other cell types, such as fibroblasts, which transdifferentiate
into matrix-secreting myofibroblasts, resulting in scar formation
and structure destruction (9).
The importance of macrophages in renal inflammation and fibrosis
responses has been reported in clinical and experimental studies.
Tubulointerstitial macrophage infiltration in renal biopsies from
patients is correlated with the severity of interstitial fibrosis
and progression of chronic renal failure to end-stage renal failure
(10). Experimental
hydronephrosis induced by unilateral ureteric obstruction (UUO) is
frequently used as a reliable model to investigate RIF in mice
(11,12). This model is characterized by
infiltrating tubulointerstitial macrophages, and RIF occurs rapidly
over the course of 5 days (13).
Furthermore, the inhibition of macrophage recruitment and
interstitial infiltration reduces the severity of renal fibrosis,
demonstrating that macrophages serve a pivotal role in promoting
RIF subsequent to UUO (14-16). Macrophages generate several
profibrotic factors, including galectin-3 (17,18), transforming growth factor (TGF)-β
(19), insulin-like growth
factor-1 (20), platelet-derived
growth factor (21,22) and basic fibroblast growth factor
(23), which support the
generation, survival and proliferation of myofibroblasts, and drive
tissue fibrosis. Therefore, increasing our understanding on the
mechanisms of macrophages in progressive renal fibrosis is a
critical step toward the design of novel diagnostic and therapeutic
targets.
Allograft inflammatory factor-1 (AIF-1) is a 17-kDa
cytoplasmic, calcium-binding, inflammation-responsive scaffold
protein that is mainly expressed in immunocytes (24). AIF-1 mRNA was cloned from
activated macrophages in rat and human cardiac allografts with
chronic rejection (25,26). In addition, three other proteins
appear to be identical to AIF-1, including ionized
Ca2+-binding adapter molecule-1 (27), microglia response factor-1
(28) and daintain (29). AIF-1 is expressed in macrophages
and upregulated in active macrophages, and its expression is
associated with inflammatory actions (30). Furthermore, AIF-1 promotes
macrophage proliferation, migration and the expression of
inflammatory mediators, such as cytokines and chemokines, which in
turn inhibits macrophage apoptosis (30-33). Given that AIF-1 is crucial for the
survival and proinflammatory activity of macrophages, subsequent
studies have indicated that numerous pathological processes are
regulated by AIF-1 in macrophages, including allograft rejection,
autoimmune diseases and vasculopathy (24,34). The correlation between AIF-1, as a
marker of active macrophages, and kidney disease has also been
receiving increasing attention. Subsequent to investigating the
correlation between serum AIF-1 concentrations and diabetic
nephropathy, a previous study has identified that serum AIF-1
concentration was correlated with albuminuria and the estimated
glomerular filtration rate in patients with type 2 diabetes, and
suggested that it may serve as a marker of diabetic nephropathy as
well as activated macrophages (35). Meanwhile, AIF-1 can be detected in
the liver tissue of mice infected with Schistosoma japonicum
and may alleviate hepatic fibrosis at the middle to advanced stages
of infection (36). Furthermore,
AIF-1 participates in the early pathogenesis of systemic sclerosis
by the production of cytokines capable of promoting fibroblasts to
a fibrotic phenotype (37), which
demonstrates that AIF-1 may be involved in the promotion of
fibrosis.
Aldosterone, as a steroid hormone regulating sodium
and potassium homeostasis, causes inflammation, tubulointerstitial
fibrosis and glomerular injury in the kidney. Based on these
findings, aldosterone increases the expression of a number of
profibrotic molecules that participated in aldosterone-induced
fibrosis (38). In the present
study, it was hypothesized that AIF-1 expressed by macrophages
drives the progression of RIF in a UUO animal model. Furthermore,
using a co-culture of macrophages induced by aldosterone and
fibroblasts, it was examined in vitro whether AIF-1
upregulated in macrophages promotes fibroblasts to a profibrotic
phenotype and induces RIF.
Materials and methods
Animals
A total of 45 male C57BL/6 mice (6-week-old) were
purchased from Charles River Laboratories, Inc. (Beijing, China).
The mice were kept at a conventional temperature (23±2°C) and a
humidity of 50% with a 12 h light/dark cycle. Mice had ad
libitum access to food and water. The mice were divided into
three groups (15 in each group) after 7 days of acclimatization,
including the experimental, spironolactone (SPI) and control
groups. In the experimental group, UUO was performed by complete
ligation of the left ureter as described previously (39) and saline was administered by
gavage. In the SPI group, UUO mice were administered SPI
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) by gavage (20
mg/kg/day) 24 h after UUO. Mice in the control group underwent sham
surgery in the ureter, which was dissociated with no ligation, and
saline by gavage. Mice were anesthetized and kidneys were harvested
at 14 days after UUO. Next, the kidney tissue was divided into
three sections. One section was fixed in Carnoy solution
(containing 60% methanol, 30% chloroform and 10% glacial acetic
acid) and embedded in paraffin for immunohistochemical analysis.
Another tissue section was perfused with optimum cutting
temperature reagent (Sakura Finetek, Inc., Torrance, CA, USA) and
immediately frozen in liquid nitrogen for use in immunofluorescence
assay. The remainder of the sample was snap-frozen in liquid
nitrogen for reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) analysis. All experimental procedures adhered to
the principles stated in the Guide for the Care and Use of
Laboratory Animals (updated 2011; National Institutes of Health,
Bethesda, MD, USA) and were approved by the Experimental Animal
Usage and Welfare Ethic Committee of Harbin Medical University
(Harbin, China).
Immunohistochemical and
immunofluorescence assays
Sections (4 µm) of the paraffin-embedded
kidney tissues were processed for immunohistochemistry. Serial
sections were deparaffinized in xylene, rehydrated in an alcohol
series and washed in phosphate-buffered saline (PBS)/Tween-20. All
sections underwent antigen retrieval with hydrated autoclaving for
20 min in citrate solution, and then blocked with 3%
H2O2 in PBS for 10 min at room temperature.
For blocking the antigen, 5% bovine serum albumin was used for 20
min at room temperature. Next, the slides were incubated overnight
at 4°C with the following primary antibodies: Rabbit anti-mouse
polyclonal AIF-1 (cat no. 10904-1-AP; 1:200 dilution; ProteinTech
Group, Inc., Chicago, IL, USA), rabbit anti-mouse polyclonal CD68
(cat no. ab125212; 1:400 dilution; Abcam, Cambridge, UK) and goat
anti-mouse polyclonal α-SMA (cat no. orb18863; 1:500 dilution;
Biorbyt Ltd., Cambridge, UK). All sections were then incubated with
peroxidase-conjugated goat (cat no. SV0003) or rabbit (cat no.
SV0002) IgG (ready-to-use; Boster Biological Technology, Ltd.,
Wuhan, China) as the secondary antibody for 30 min at room
temperature. The immune complex was visualized with DAB, serving as
the chromogenic substrate, and subsequently counterstained with
hematoxylin.
For immunofluorescence analysis, sections were
incubated with primary antibodies against AIF-1 (cat no. ab5076;
0.5 µg/ml) and CD68 (cat no. ab53444; 1.0 µg/ml) (all
from Abcam) for 1 h at room temperature, followed by a 30-min
incubation with secondary antibody conjugated to FITC (cat no.
BA1110; 1:32 dilution) and biotinylated secondary antibody followed
by SABC-Cy3 (cat no. SA1079; 1:100 dilution) in an
immunohistochemistry Elite kit (Boster Biological Technology, Ltd.)
at 37°C in the dark. Subsequent to washing with PBS, the samples
were observed using a NIKON ECLIPSE 80i confocal fluorescence
microscopy (Nikon Corporation, Tokyo, Japan).
Masson's trichrome stain
Serial sections were deparaffinized in xylene (10
min each, twice) and rehydrated in an alcohol series (100, 95 and
70%) for 5 min each, twice at room temperature. All sections were
washed in running water for 5 min and stained using a Masson's
trichrome-staining kit (cat no. BA-4079B; Baso Diagnostics, Inc.,
Hubei, China) according to the manufacturer's protocol. The
sections were treated as follows: All sections were stained with
Weigert-iron-hematoxylin (1:1) for 5 min at room temperature.
Following washing in running water for 10 min, the sections were
treated with hydrochloride-ethanol solution (1%) for 5 sec at room
temperature and rinsed under running tap water for 20 min. The
sections were stained in ponceau (1%) staining solution for 5-10
min at room temperature (observed under an electron microscope with
a magnification of ×200) and washed using phosphomolybdic acid
solution (Baso Diagnostics, Inc.) for 5 min at room temperature.
All sections were stained with aniline blue solution for ~5 min at
room temperature (observed under an electron microscope with a
magnification of ×200) and followed by washing in glacial acetic
acid for 1 min. Subsequent to dehydration through an ethanol series
(95 and 100%, 10 min each, twice) and xylene (5 min each, twice),
followed by all sections being cover slipped.
Cell culture and stable
transfections
The mouse macrophage cell line RAW264.7 and renal
fibroblast cell line BHK-21 were purchased from the American Type
Culture Collection (Manassas, VA, USA). Cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine
serum at 37°C in 5% CO2 incubator. Equal numbers of
RAW264.7 cells were seeded into 100-mm culture dishes
(1×105 per dish) for western blot analysis and 6-well
plates (1×106 per well) for RT-qPCR. Confluent RAW264.7
cells were starved in serum-free DMEM for 24 h and then exposed to
different concentrations of aldosterone
(10−8-10−5 M; Sigma-Aldrich; Merck KGaA) for
72 h. Another part of cells was stimulated with 10−6 M
aldosterone for different incubation times (6, 24, 72 and 120 h).
Samples were subsequently processed for protein and RNA isolation
and further detection.
For AIF-1 gene knockdown, a small interfering RNA
(siRNA) construct was synthesized by GenePharma Co., Ltd.
(Shanghai, China), and its sequence was as follows: 5′-GCA AUG GAG
AUA UCG AUA UTT AUA UCG AUA UCC AUU GCT T-3′. The siRNA was
inserted into the expression vector pRNA-U6.1/shuttle (pShuttle).
RAW264.7 cells were transfected with the vector alone or with
pRNAU6.1/shuttle-siRNA/AIF-1 with Lipofectamine® 2000
reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) as described previously (40). Following antibiotic selection (400
g/ml G418; Thermo Fisher Scientific, Inc.), stably transduced
macrophages were pooled to avoid the effects of clonal selection.
The expression levels of AIF-1 in macrophages subsequent to
transfection were detected by western blot analysis and
RT-qPCR.
Co-culture of macrophages with renal
fibroblasts
Normal macrophages and macrophages transfected with
siRNA/AIF-1 stimulated with aldosterone were co-cultured with renal
fibroblasts. Prior to co-culture, RAW264.7 cells (1×106)
were cultured in 6-well plates, while 1×106 BHK-21 were
cultured in 100-mm culture dishes, and cells were incubated in
serum-free media for 24 h. Following digestion by trypsin/EDTA and
separation from the supernatant, the pellet of RAW264.7 cells
(1×107) was resuspended and added to the BHK-21 culture.
After aldosterone (10−6 M) was added, cells were
incubated for a further 72 h. Prior to the extraction of total RNA
and protein from the fibroblasts for expression detection,
macrophages were removed by rinsing extensively with PBS until a
few macrophages could be detected microscopically. The expression
levels of FN, α-SMA, p38 and phosphorylated-p38 (p-p38) from BHK-21
cells and AIF-1 from RAW264.7 cells were examined.
RT-qPCR
Total RNA was extracted from the samples using
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) according to
standard procedures. Next, the concentration of RNA
(CRNA=950-1,500 ng/µl) samples were tested using
a spectrophotometer (NanoDrop 2000; Thermo Fisher Scientific,
Inc.). When the A260/A280 ratio was 1.8 to 2.0, it was deemed to
have met the experimental requirements. The volumes of RNA
(VRNA=1 µg/CRNA) for RT were
calculated according to the concentrations of RNA. 1 µg RNA
was reverse-transcribed into cDNA (20-µl reactions), and
qPCR was performed using SYBR Premix Ex Taq™ II (Takara Bio, Inc.,
Otsu, Japan) with LightCycler 2.0 instrument (Roche Diagnostics
GmbH, Mannheim, Germany). A total of 0.4 µM of the following
primers was used for qPCR: FN forward, 5′-CAA AGA TGA CAA GGA AAG
TGC C-3′, and reverse, 5′-CCC GAT AAT GGT GGA AGA GT-3′; α-SMA
forward, 5′-GCA TCC GAC CTT GCT AAC G-3′, and reverse, 5′-CAT CTC
CAG AGT CCA GCA CAA T-3′; p38 forward, 5′-ACC TAA AGC CCA GCA ACC
T-3′, and reverse, 5′-CAG CCC ACG GAC CAA ATA-3′; AIF-1 forward,
5′-GTT CCC AAG ACC CAT CTA GAG CTG-3′, and reverse, 5′-AGT TGG CTT
CTG GTG TTC TTT GTT T-3′; GAPDH forward, 5′-ACC ACA GTC CAT GCC ATC
AC-3′, and reverse, 5′-TCC ACC ACC CTG TTG CTG TA-3′. GAPDH served
as the endogenous reference gene. According to the manufacturer's
protocol, the relative gene expression levels were presented with
the 2−ΔΔCt method, defined as the comparative
quantification cycle (41).
Western blotting
Cultured cells were lysed by adding lysis buffer
(radioimmunoprecipitation assay to phenylmethane sulfonyl fluoride
ratio, 99:1), and protein was extracted in 1X SDS sample buffer
following centrifugation at 12,000 × g for 15 min at 4°C. The
protein concentration of each sample was measured using a BCA
protein assay kit (cat no. ab207002; Abcam). Equal amounts of
protein were separated using SDS-PAGE (12%), transferred onto
polyvinylidene difluoride membranes, and then blocked in
Tris-buffered saline/Tween-20 containing 5% non-fat milk for 1 h at
room temperature. The membranes were incubated overnight at 4°C
with the following primary antibodies: Rabbit anti-mouse polyclonal
p38 (cat no. 8690S; 1:1,000 dilution), p-p38 (cat no. 4511S;
1:1,000 dilution) (both from Cell Signaling Technology, Inc.), FN
(cat no. 15613-1-AP; 1:1,000 dilution; ProteinTech Group, Inc.) and
GAPDH (cat no. 10494-1-AP; 1:5,000 dilution; ProteinTech Group,
Inc.) antibodies; AIF-1 and α-SMA antibodies were used as described
earlier. Following incubation with horseradish peroxidase
conjugated-secondary antibodies (cat no. ZB2301; 1:2,000 dilution;
OriGene Technologies, Inc., Rockville, MD, USA) for 1 h at 37°C,
the membranes were treated with an enhanced chemiluminescence
substrate kit to obtain visible bands, which were observed with a
ImageQuant LAS-4000 device (GE Healthcare, Chicago, IL, USA).
Enzyme-linked immunosorbent assay
(ELISA)
The concentration of AIF-1 and TGF-β in culture
supernatants were determined using a mouse AIF-1 ELISA kit (cat no.
0000768; Tsz Biosciences, San Francisco, CA, USA) and TGF-β ELISA
kit (cat no. E-EL-M1192c; Elabscience Biotechnology Co., Ltd.,
Wuhan, China) according to the manufacturer's protocol. Briefly,
the supernatants were obtained following centrifugation at 1,000 ×
g for 20 min at 4°C. Standard solution (4-500 pg/ml) and
supernatant samples (1×104) were added to the 96 well
plates, and the plate was incubated at 37°C for 45 min. Subsequent
to washing with PBS with 10% Tween-20 (PBST), 50 µl
anti-biotin IgG antibody (part of the kit) was added to each well
and incubated at 37°C for 60 min. Following further washing with
PBST, horseradish peroxidase-conjugated pretitrated avidin was
added to each well and incubated at 37°C for 30 min. After final
washing with PBST, the substrate solution was added to each well
and allowed to react at 37°C for 15 min. The reaction was stopped
by the addition of the termination solution, after which the
optical density values at 450 nm were read with an ELISA plate
reader. The detection limit of the assay was 4 pg/ml.
Statistical analysis
Experiments were repeated three times. Results are
presented as the mean ± standard deviation. SPSS 17 software (SPSS
Inc., Chicago, IL, USA) was used to perform statistical analysis.
Statistically significant differences between the means were
assessed using one-way analysis of variance. The differences
between multiple groups were determined by Tukey's post hoc test
and an unpaired Student's t-test was used for the comparison of two
groups. P≤0.01 was considered to indicate a statistically
significant difference.
Results
AIF-1 expression is upregulated in the
mouse model of RIF
UUO, a classic experimental model of progressive RIF
(11), was conducted in the
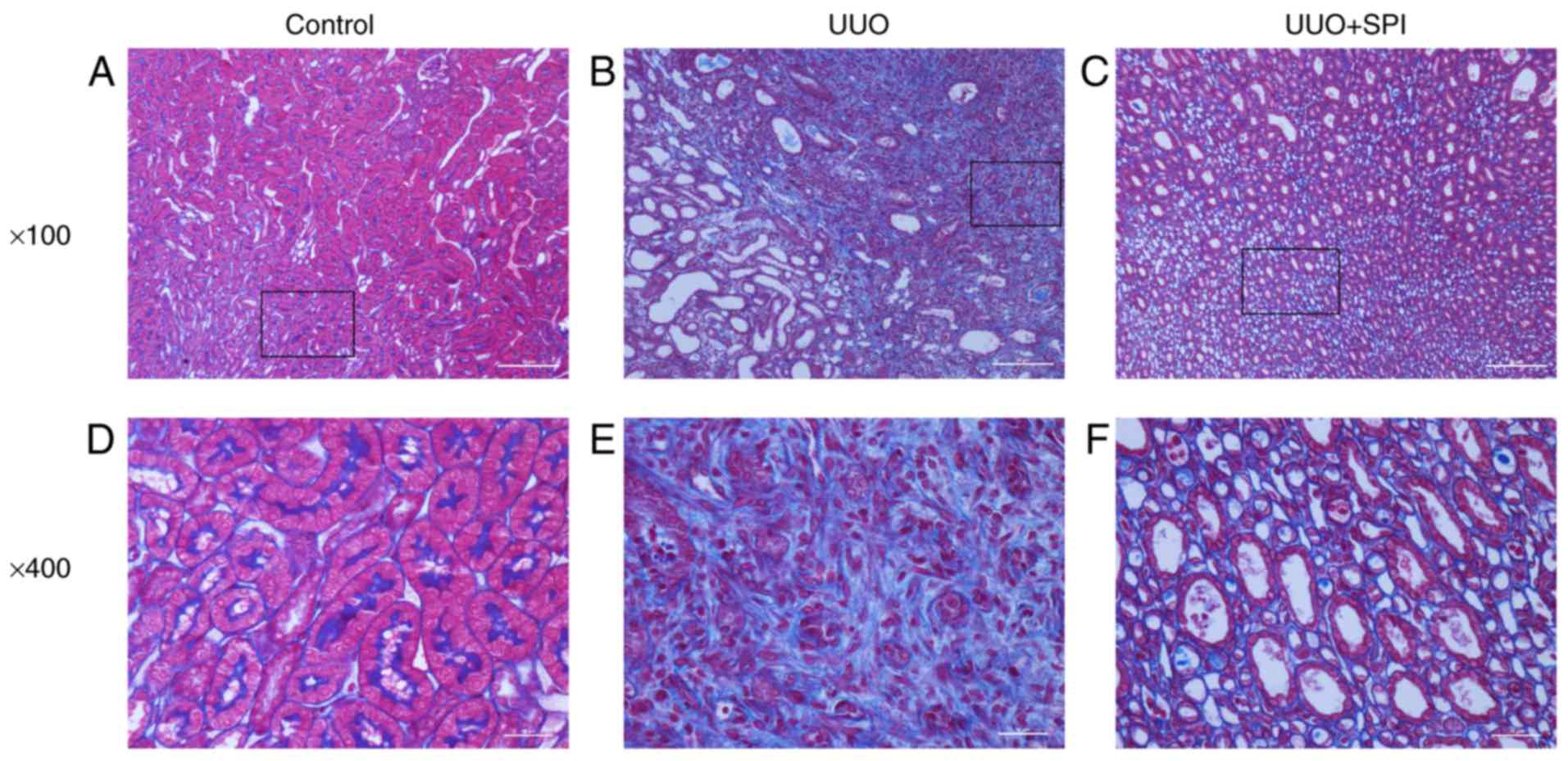
present study. Histopathological changes of the kidney sections
were evaluated by Masson's trichrome. The results revealed
interstitial fibrosis characterized by interstitial collagen
deposition and tubular cell atrophy at 14 days after UUO. Compared
with the experimental UUO group, SPI treatment reduced the renal
tubulointerstitial fibrosis. Interstitial edema and inflammation
were also observed in the UUO and SPI group (Fig. 1). Furthermore, α-SMA positivity
was examined as a marker of activated myofibroblasts involved in
ECM production. Immunohistochemical analysis revealed an evidently
increased expression of α-SMA in the renal interstitium of UUO
mice. Compared with the experimental group, the α-SMA expression
decreased with SPI treatment (Fig.
2A). As assessed by RT-qPCR, α-SMA mRNA expression levels in
UUO mice were also significantly increased compared with those in
sham surgery kidneys. The α-SMA expression of the SPI group was
significantly decreased compared with the experimental group
(Fig. 2B).
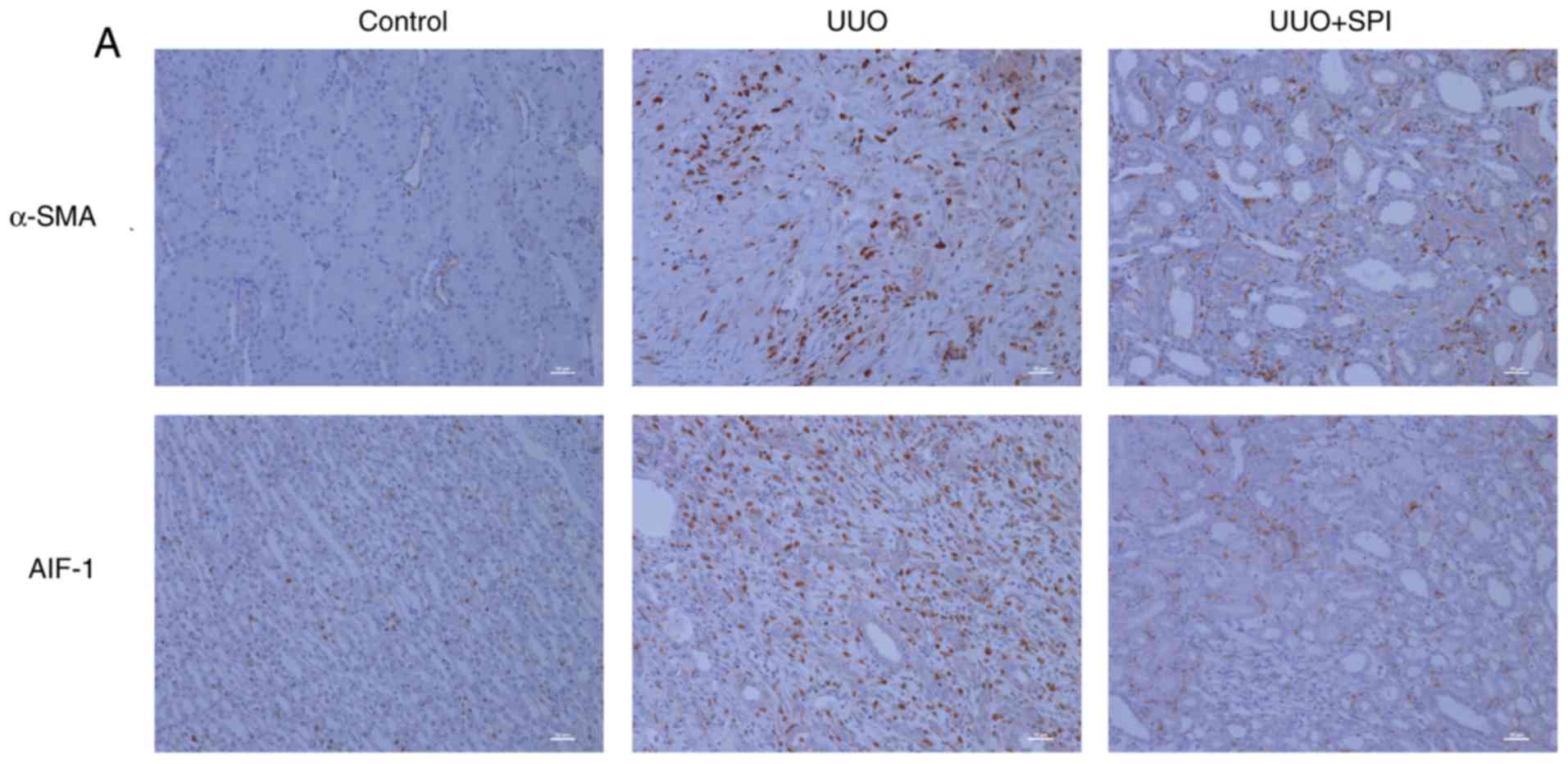
To evaluate the role of AIF-1 in promoting RIF,
AIF-1 expression in kidney tissues was analyzed.
Immunohistochemical results demonstrated that AIF-1 expression was
minimal in the normal renal interstitium and notably increased
subsequent to UUO. AIF-1 expression of SPI group decreased compared
with the experimental group (Fig.
2A). This increase in AIF-1 expression was further confirmed by
RT-qPCR, while the expression of AIF-1 was reduced following SPI
treatment (Fig. 2B). These
results demonstrated that α-SMA and AIF-1 expression in kidney
tissue were upregulated in the mouse model of progressive RIF.
α-SMA reflects the degree of renal interstitial fibrosis. AIF-1 may
cause the change of α-SMA expression and participate in the
development of RIF.
AIF-1 is localized in the macrophages
infiltrating in the renal interstitium of UUO mice
A previous study has suggested that macrophages that
underwent recruitment and infiltration subsequent to injury served
a vital role in the development of renal fibrosis (14). The expression of CD68, a
macrophage surface marker, was examined in the present study.
Compared with the control group, an increased number of
CD68-positive cells were observed in the renal interstitium of the
UUO group (Fig. 3A). The results
revealed that interstitial macrophage infiltration was increased in
the UUO kidney, and this was inhibited by SPI treatment. Although
AIF-1 expression has been reported to be immunolocalized in
podocytes of the glomerular capillary wall in an anti-glomerular
basement membrane nephritis model (42), its localization in renal
tubulointerstitial injury has not been reported. To detect whether
AIF-1 was localized in the interstitial infiltrating macrophages,
co-immunofluorescence staining with anti-AIF-1 and anti-CD68
antibodies was performed (Fig.
3B). In kidney sections of UUO mice, the immunoreactivity for
AIF-1 was detected on the same cells that were positive for CD68,
indicating that AIF-1 protein was localized in macrophages
infiltrating the renal interstitium.
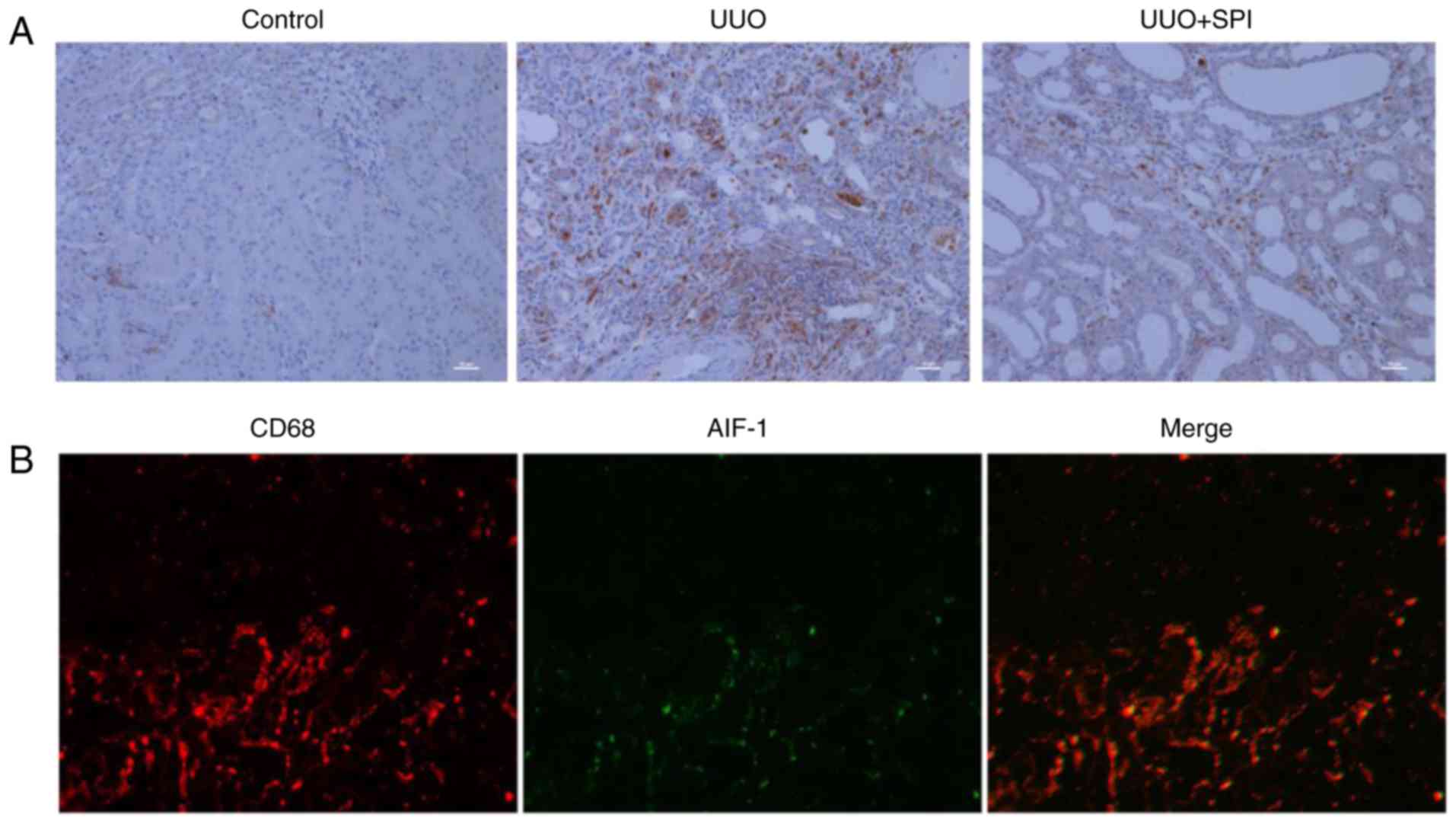 | Figure 3AIF-1 is localized in the macrophages
infiltrating in the renal interstitium of UUO mice. (A) The
expression of CD68, a macrophage surface marker, was detected by
immunohistochemical stain in kidney tissue. Compared with the
control group, more CD68-positive cells infiltrating the renal
interstitium of the UUO group were detected, which decreased by SPI
treatment (original magnification, ×200; scale bar, 10 µm).
(B) Co-localization of AIF-1 and CD68 in kidney tissue of UUO mice
was detected by immunofluorescence staining with anti-AIF-1
antibody (green) and anti-CD68 antibody for macrophages (red), and
merging of AIF-1 and CD68 images. UUO, unilateral ureteric
obstruction; SPI, spironolactone; AIF-1, allograft inflammatory
factor-1. |
AIF-1 expression and secretion by
macrophages are upregulated with aldosterone stimulation
AIF-1 expression in macrophages can be induced by
inflammatory cytokines, as has been described previously (43); however, induction by aldosterone
has not been reported. Physiological concentration of aldosterone
increases the expression of proinflammatory genes in cultured
macrophages (44). The present
study investigated whether aldosterone is capable of inducing AIF-1
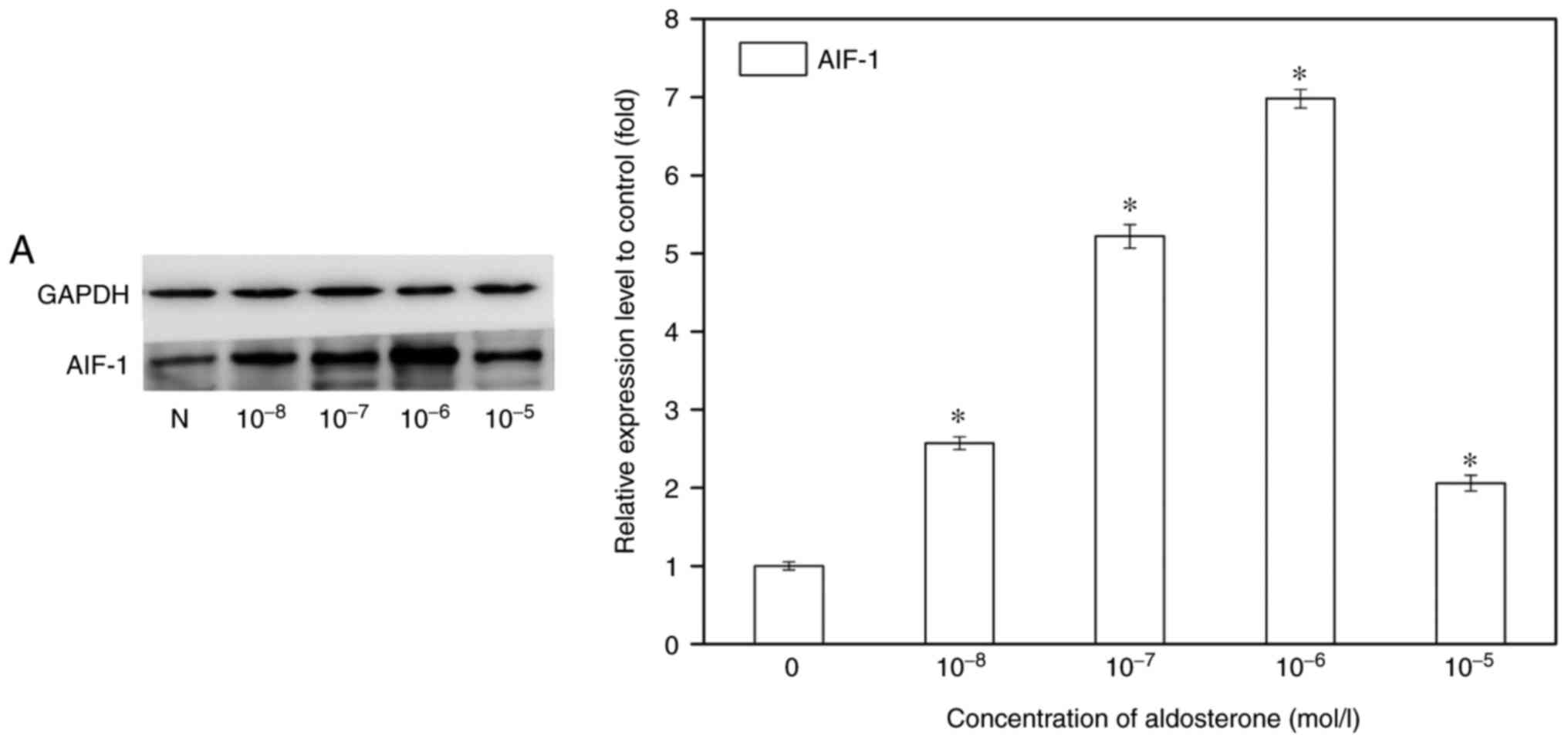
expression in macrophages. RAW264.7 cells, a commonly used
macrophage cell line, were stimulated with different concentrations
of aldosterone (10−8-10−5 M) for 72 h.
Following incubation with aldosterone, the protein and mRNA
transcription levels of AIF-1 in macrophages were upregulated, and
the maximum effect occurred at the concentration of 10−6
M (Fig. 4A). AIF-1 induction at
different time-points was also detected through the treatment of
macrophages with aldosterone (10−6 M) for 6, 24, 72 and
120 h. In response to aldosterone stimulation, the mRNA
transcription level of AIF-1 was significantly increased and
reached a peak value at 72 h, with a 5-fold increase above the
basal level, which was also confirmed in the protein level of AIF-1
detected by western blot analysis (Fig. 4B). ELISA was conducted to examine
AIF-1 excretion in the supernatant of RAW264.7 cells, and it
demonstrated similar results (Fig. 4C
and D). Meanwhile, AIF-1 in renal fibroblast cells was also
detected. AIF-1 exhibited low expression or secretion by BHK-21
cells with or without aldosterone stimulation.
AIF-1-positive macrophages promote renal
fibroblast activation to a profibrotic phenotype
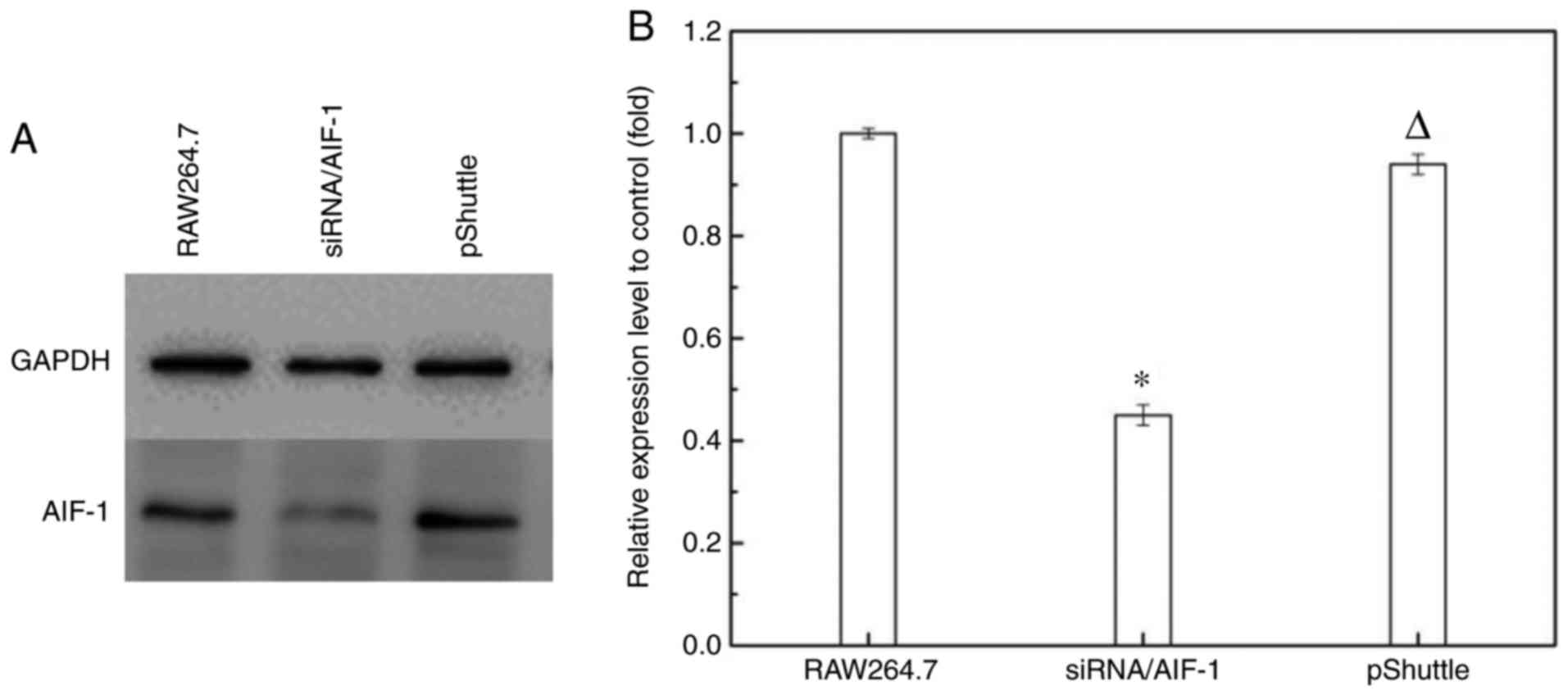
To test the role of AIF-1 in promoting RIF, AIF-1
expression in macrophages was inhibited by stable transfection with
siRNA/AIF-1 vector. The macrophage cell line RAW264.7 was selected
since it constitutively expresses low levels of AIF-1 mRNA and
facilitates positive selection of stable transfectants for AIF-1
gene knockdown (45). The AIF-1
siRNA construct was cloned into the pShuttle vector and stable
transfectants were isolated by antibiotic selection. AIF-1
expression was determined using western blot analysis and RT-qPCR.
It was demonstrated that stable transfection with the siRNA/AIF-1
construct reduced AIF-1 expression, whereas transfection with
vector alone (pShuttle) did not have an evident effect on AIF-1
expression (Fig. 5).
Recent findings have identified that activated renal
fibroblasts were the main origin of myofibroblasts, a primary
source of ECM in scar tissue formation causing RIF. It has been
demonstrated that co-culture of activated immune cells with
fibroblasts in vitro induced the production of ECM and
expression of α-SMA (5,46). To verify the hypothesis that
AIF-1-positive macrophages may promote renal fibroblast activation
to a profibrotic phenotype, a mouse renal fibroblast BHK-21 cell
line was incubated with RAW264.7 cells with aldosterone
stimulation. Expression of α-SMA, p-p38 and FN in fibroblasts was
examined following co-culture with normal macrophages or
macrophages transfected with siRNA/AIF-1. The results revealed
that, after 72 h of co-culture of fibroblasts with macrophages
activated by aldosterone, AIF-1 expression in macrophages and
supernatants was upregulated, compared with that in the co-culture
of two cells without aldosterone (Fig. 6A–C). The protein concentration of
TGF-β in the supernatants of macrophages was also increased
following aldosterone stimulation, but was not significantly
different compared with the siRNA/AIF-1 macrophages (Fig. 6C). In addition, the α-SMA
expression was induced in fibroblasts, with significantly increased
expression levels of FN and p-p38 also observed. These levels were
reduced significantly in fibroblasts co-cultured with siRNA/AIF-1
macrophages, compared with those in AIF-1-positive macrophages
(Fig. 6D and E). These data
demonstrate that AIF-1 expression or excretion by macrophages is an
important mechanism in the promotion of the profibrotic phenotype
in renal fibroblasts, which may occur via the p38 signaling
pathway.
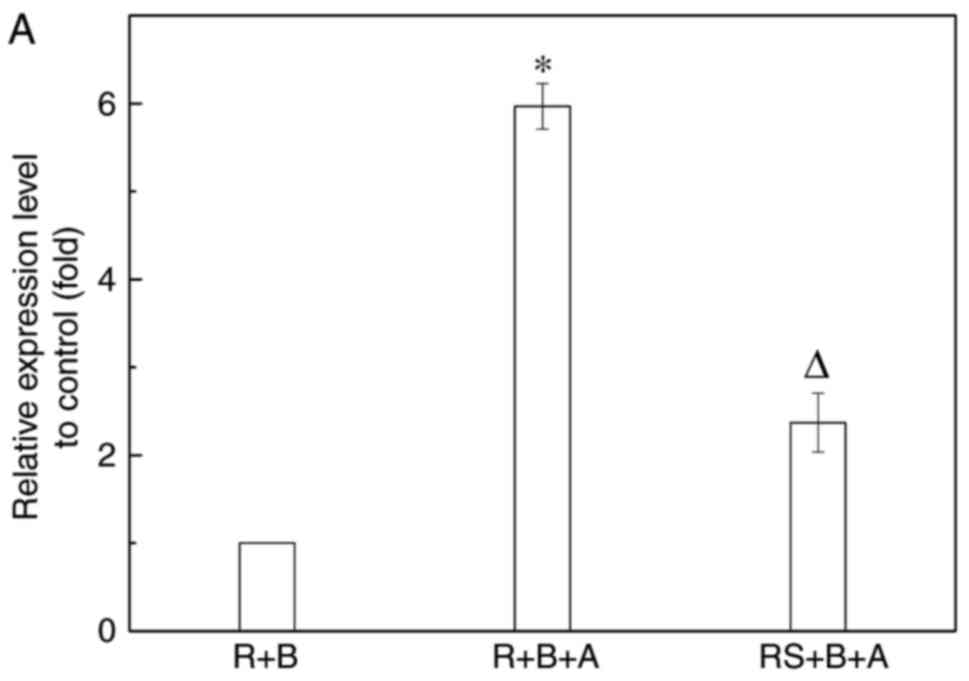 | Figure 6Fibroblasts were activated to a
profibrotic phenotype following co-culture with macrophages induced
by aldosterone. This phenotypic transition was inhibited by AIF-1
knockdown through transfection with siRNA/AIF-1. Macrophages and
renal fibroblasts were separated after incubation for 72 h, and the
(A) mRNA and (B) protein expression levels of AIF-1 in macrophages
were examined. (C) Secretions of AIF-1 and TGF-β in the
supernatants of macrophages were also detected. The expression
levels of protein kinase (p38 and p-p38), FN and α-SMA in
fibroblasts were evaluated by (D) western blot analysis and (E)
reverse transcription-quantitative polymerase chain reaction.
Following co-culture, BHK-21 and RAW264.7 cells were separated and
used for further detection. *P≤0.01 vs. R+B group;
∆P≤0.01 vs. R+B+A group. R, RAW264.7 cells
(macrophages); B, BHK-21 cells (renal fibroblasts); A, aldosterone
stimulation; RS, RAW264.7 cells transfected with siRNA/AIF-1;
AIF-1, allograft inflammatory factor-1; siRNA, small interfering
RNA; p-p38, phosphorylated p38; FN, fibronectin; α-SMA, smooth
muscle actin. |
Discussion
In the present study, the potential link of AIF-1
expression in macrophages with the promotion of RIF was examined.
The results demonstrated that AIF-1 expression was upregulated in
the animal model of RIF (UUO), and that it was co-localized with
CD68-positive macrophages infiltrating in the renal interstitium of
UUO mice. In addition, AIF-1 was detected at low levels in
unstimulated macrophages, and upregulated in response to
aldosterone treatment. The peak expression of AIF-1 was observed at
72 h after aldosterone (10−6 M) stimulation.
Furthermore, α-SMA, p-p38, and FN expression levels in renal
fibroblasts were markedly increased following co-culture with
macrophages activated by aldosterone. Finally, inhibition of AIF-1
in macrophages transfected with siRNA/AIF-1 reduced the expression
levels of profibrotic molecules and p-p38 in fibroblasts. These
novel findings demonstrate that AIF-1 expression in macrophages
promotes renal fibroblast activation to a profibrotic phenotype and
participates in the progression of RIF induced by aldosterone.
As an inflammation-associated protein, AIF-1 has
been identified as an important regulator in various inflammatory
pathological processes in multiple organs (24). However, there are few studies
investigating the function of AIF-1 in kidney diseases,
particularly in RIF. A previous study reported that AIF-1-activated
macrophages were increased in the infiltrate of clinical rejection
biopsies and were associated with clinical rejection episodes,
which was the first report on AIF-1 expression in kidney tissue
(47). Subsequent studies have
demonstrated that AIF-1 involved in the inflammatory signaling
network regulated the innate immune responses (48), while the genetic variant TT/CT of
the AIF-1 gene was associated with a lower risk of renal rejection
(49). Previous studies also
indicated that AIF-1 expressed in podocytes and infiltrating
inflammatory cells of kidney tissues (42) was associated with inflammatory
reaction, immune regulation and allograft rejection (48,50). The results of the present study
revealed that AIF-1 expression and infiltration of macrophages
occurred in a progressive renal fibrosis model. In accordance with
a previous study (42), AIF-1
expression was localized in infiltrating interstitial macrophages
of the kidney. Furthermore, as renal fibrosis progresses (collagen
accumulation and α-SMA expression upregulation), a continued
increase in AIF-1 expression was reported in the current study,
which demonstrates that AIF-1 participates in the progression of
RIF. To the best of our knowledge, this is the first link reported
between AIF-1 expression and RIF.
Macrophages are found in normal kidneys and
increased numbers are observed in diseased kidney, where they are
key players in renal injury, inflammation and fibrosis.
Investigation of of experimental and human renal disease has
demonstrated that the involvement of macrophages in renal fibrosis
results from diverse disease processes. A recent study has explored
the nature of both circulating monocytes and tissue macrophages,
exhibiting distinct phenotypic and functional characteristics in
response to various stimuli in renal fibrosis (51). There are also studies suggesting
that there is a subpopulation of macrophages with an antifibrotic
role in the UUO model (52,53), although infiltrating macrophages
promoting renal fibrosis have been confirmed by one study (54). The macrophage phenotype is altered
in response to signals from the local kidney milieu; however, the
mechanisms by which macrophages are polarized are not well
understood. Macrophage migration and phenotype transitions can be
mediated by kidney injury molecule-1 (55). In addition, macrophages
overexpressing neutrophil gelatinase-associated lipocalin-2
ameliorated renal inflammation and fibrosis in a UUO mouse model
(56). These previous
observations indicated that certain factors affect the change of
macrophages to different phenotypes. The role and regulated
mechanism of macrophages in renal fibrosis are complex and need to
be extensively explored. The results of the present study
demonstrated that the major tissue source of AIF-1 driving RIF is
derived from infiltrating macrophages. Macrophages with distinct
phenotypes have a different influence on kidney injury or repair
process through the secretion of various cytokines. M1 macrophages
produce proinflammatory cytokines, including tumor necrosis factor
α, interleukin (IL)-6 and inducible isoform nitric oxide synthase,
whereas M2 macrophages can synthesize profibrotic factors, such as
TGF-β, platelet-derived growth factor and galectin-3 (9,51,57). TGF-β is a pleiotropic cytokine
with multiple effects on cellular behavior, including
proliferation, migration and immune response. Aberrant TGF-β
signaling is considered as a hallmark of certain diseases. Three
isoforms of TGF-β exist, including TGF-β1, TGF-β2 and TGF-β3, and
targeting TGF-β3 represents a promising strategy interfering with
aberrant TGF-β signaling in glioblastoma (58). TGF-β has also been implicated as
an important mediator of renal fibrosis (59), and thus, the concentration of
TGF-β was detected in the current study. Following inhibition of
the expression of AIF-1 by gene knockdown, the secretion of TGF-β
in the supernatant of macrophages was not altered as compared with
that in the control group. This indicates that AIF-1 promotes renal
fibrosis via a TGF-β-independent signaling pathway, which may
provide a new mechanism of macrophages linked with fibrosis and a
theoretical foundation to clarify the role of AIF-1 in kidney
disease.
Through macrophage co-culture with renal fibroblasts
in vitro, the mechanism of the promotion of AIF-1 expression
to RIF was further investigated in the current study. Fibroblasts
are quiescent cells in the interstitial space of the kidney, which
can be activated by inflammatory cells and cytokines. It is
considered that α-SMA expression of fibroblasts is indicative of
myofibroblasts contributing to the pathogenesis of RIF (5). However, further investigation is
required to elucidate the cellular mechanisms underlying the renal
fibroblast transition to myofibroblasts. It has been reported that
AIF-1, which can induce the migration of fibroblasts, and the
production of IL-6, is an important molecule promoting fibrosis in
chronic graft versus host disease (60). Therefore, the present study
hypothesize that AIF-1 participates in the mechanism of renal
fibroblast transition to myofibroblasts. The results demonstrated
that upregulation of AIF-1 in active macrophages served a key role
in promoting FN and α-SMA expression levels in fibroblasts.
Furthermore, AIF-1 has been reported to be associated with
fibrosis-associated diseases. A previous study indicated that AIF-1
played an important role in the pathogenesis of systemic sclerosis
(61). AIF-1 also promoted tissue
T cell production of cytokines capable of inducing the expression
of IL-6, TGF-β and α-SMA in normal dermal fibroblasts, and
increasing their collagen production (37). These findings implied that AIF-1
promotes normal fibroblasts to the fibrotic phenotype. Our previous
study indicated that the interaction between renal fibroblasts and
peripheral blood mononuclear cells was mediated through direct
cell-to-cell contact involved in renal interstitial inflammation
(62). The current study further
demonstrated that AIF-1 expression by macrophages induced the
activation of normal renal fibroblasts, increasing the expression
of fibrosis markers, including FN and α-SMA, in these cells, and
this effect may direct cell-to-cell contact between macrophages and
renal fibroblasts.
AIF-1 participates in signaling cascades in
macrophages, vascular smooth muscle cells and endothelial cells
(ECs). The absence of AIF-1 leads to the activation of suppressed
kinases, which interrupts the signaling cascades. However, the
AIF-1 mediated signal transduction pathway involves different types
of cells. In macrophages, AIF-1 attenuation reduced the activation
of p38/MAPK, AKT and p90RSK kinases (30). In murine vascular smooth muscle
cells, overexpression of AIF-1 increased p38 activation (63). However, a study on ECs reported
that abrogation or overexpression of AIF-1 does not alter p38/MAPK
activation, while the reduction of AIF-1 expression significantly
inhibited the activation of p44/42 in ECs (64). As a classic signaling pathway
participating in a variety of reactions, p38/MAPK is used as a
target to downstream alterations in the pathophysiological process.
A study revealed that microRNA mimics targeting p38/MAPK signaling
pathway inhibited broad-spectrum respiratory virus infection
(65). Inhibition of p38/MAPK
also attenuated Hg-induced FN of renal interstitial fibroblasts and
renal fibrosis in mice (66,67). Due to the upregulation of
fibroblast activation and renal interstitial fibrosis induced by
AIF-1, the present study focused on p38 kinase and observed that
reduction of AIF-1 expression significantly inhibited the
activation of p38/MAPK. This suggests that decreased profibrotic
phenotype transformation in fibroblasts with AIF-1 abrogation may
be attributed to the reduction of p38/MAPK activation.
Aldosterone and activation of the mineralocorticoid
receptor (MR) serve a key role in macrophages, and cause
tubulointerstitial fibrosis and glomerular injury in the kidney
(68). Aldosterone at a
physiological concentration increases the expression of
pro-inflammatory genes in cultured macrophages. In addition,
aldosterone receptor blocker suppresses the release of interstitial
inflammatory cytokines from macrophages, which may serve to delay
the progression of renal fibrosis. Aldosterone also induces the
activation of macrophage, while MR controls macrophage polarization
(44,69). As an inflammation-associated
protein in macrophages, AIF-1 is upregulated and promotes RIF with
aldosterone stimulation, which was verified in the present study.
Aldosterone is known to bind to the cytoplasmic MR, which functions
as a transcription factor to regulate gene transcription (70). In addition to this traditional
genomic pathway, aldosterone can also exert rapid nongenomic
effects that are not blocked by inhibitors of transcription. These
rapid actions (requiring seconds to minutes) are coupled to MR or
to a specific membrane aldosterone receptor. Systemic aldosterone
administration induces phosphorylation of epidermal growth factor
receptor and extracellular signal-regulated kinase (ERK) in the
kidney within 30 min. Aldosterone rapidly activates ERK1/2 in
vascular smooth muscle cells in order to promote a mitogenic and
profibrotic phenotype. This mechanism is MR-independent, and
certain rapid effects of aldosterone cannot be blocked by SPI, an
MR antagonist (38).
In subsequent studies, our study will focus on the
definite mechanism of AIF-1 expression regulated by aldosterone and
will determine whether it is MR-dependent. AIF-1 expression in
macrophages and its regulation effect on proliferation, migration
and inflammation has previously been explored (30). However, the function and mechanism
of AIF-1 on the development of fibrosis is undefined in previous
studies. To the best of our knowledge, the present study is the
first to prove that AIF-1 expression in macrophages promotes RIF.
Through AIF-1 abrogation by stable transfection with siRNA and
co-culture of macrophages with renal fibroblasts, the current study
demonstrated the key role of AIF-1 in the renal fibroblast
activation to a profibrotic phenotype and in the promotion of RIF.
The profibrotic signaling axis between macrophages and renal
fibroblasts was mediated by AIF-1. Therefore, targeted inhibition
of AIF-1 expression in macrophages may result in the development of
novel antifibrotic therapies. Renal fibroblasts are the main
inherent cells with an important role in promoting RIF (5). Given that the functional
consequences of AIF-1 expression in fibroblasts have been reported
(71), there is the implication
that the association between AIF-1 and renal fibrosis is complex
and the exact mechanism requires further investigation.
In conclusion, the present study illustrated that
upregulation of AIF-1 expression and secretion by macrophage
stimulated with aldosterone promoted renal fibroblast to a
profibrotic phenotype. This is a novel mechanism linking
macrophages to the promotion of RIF, which may occur via the p38
signaling pathway.
Acknowledgments
The authors would like to thank Mrs Cailing Guo
(Microbiology Department of Harbin Medical University, Harbin,
China) and Miss Yiqun Ren (Pathology Laboratory of the Nephrology
Department, The First Affiliated Hospital of Harbin Medical
University, Harbin, China) for their technical assistance.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81570638) and the
Scientific Funding Project for the Returned Overseas of Education
Department of Heilong Jiang Province (grant no. 1253HQ006).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XW was a major contributor in experimental design.
LZ established the animal models. XY performed the protein test. JH
participated in cell culture. JN guided the writing of the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental procedures adhered to the
principles stated in the Guide for the Care and Use of Laboratory
Animals (updated 2011; National Institutes of Health, Bethesda, MD,
USA) and were approved by the Experimental Animal Usage and Welfare
Ethics Committee of Harbin Medical University (Harbin, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Farris AB and Colvin RB: Renal
interstitial fibrosis: Mechanisms and evaluation. Curr Opin Nephrol
Hypertens. 21:289–300. 2012. View Article : Google Scholar
|
|
2
|
Zeisberg M and Neilson EG: Mechanisms of
tubulointerstitial fibrosis. J Am Soc Nephrol. 21:1819–1834. 2010.
View Article : Google Scholar
|
|
3
|
Duffield JS: Cellular and molecular
mechanisms in kidney fibrosis. J Clin Invest. 124:2299–2306. 2014.
View Article : Google Scholar
|
|
4
|
Strutz F and Zeisberg M: Renal fibroblasts
and myofibroblasts in chronic kidney disease. J Am Soc Nephrol.
17:2992–2998. 2006. View Article : Google Scholar
|
|
5
|
Sun YB, Qu X, Caruana G and Li J: The
origin of renal fibroblasts/myofibroblasts and the signals that
trigger fibrosis. Differentiation. 92:102–107. 2016. View Article : Google Scholar
|
|
6
|
Asada N, Takase M, Nakamura J, Oguchi A,
Asada M, Suzuki N, Yamamura K, Nagoshi N, Shibata S, Rao TN, et al:
Dysfunction of fibroblasts of extrarenal origin underlies renal
fibrosis and renal anemia in mice. J Clin Invest. 121:3981–3990.
2011. View
Article : Google Scholar
|
|
7
|
Li X and Zhuang S: Recent advances in
renal interstitial fibrosis and tubular atrophy after kidney
transplantation. Fibrogenesis Tissue Repair. 7:152014. View Article : Google Scholar
|
|
8
|
Han HI, Skvarca LB, Espiritu EB, Davidson
AJ and Hukriede NA: The role of macrophages during acute kidney
injury: Destruction and repair. Pediatr Nephrol. 2018. View Article : Google Scholar
|
|
9
|
Cao Q, Harris DC and Wang Y: Macrophages
in kidney injury, inflammation, and fibrosis. Physiology
(Bethesda). 30:183–194. 2015.
|
|
10
|
Yonemoto S, Machiguchi T, Nomura K,
Minakata T, Nanno M and Yoshida H: Correlations of tissue
macrophages and cytoskeletal protein expression with renal fibrosis
in patients with diabetes mellitus. Clin Exp Nephrol. 10:186–192.
2006. View Article : Google Scholar
|
|
11
|
Chevalier RL, Forbes MS and Thornhill BA:
Ureteral obstruction as a model of renal interstitial fibrosis and
obstructive nephropathy. Kidney Int. 75:1145–1152. 2009. View Article : Google Scholar
|
|
12
|
Wang M, Liu R, Jia X, Mu S and Xie R:
N-acetyl-seryl-aspartyllysyl-proline attenuates renal inflammation
and tubulointerstitial fibrosis in rats. Int J Mol Med. 26:795–801.
2010.
|
|
13
|
Diamond JR: Macrophages and progressive
renal disease in experimental hydronephrosis. Am J Kidney Dis.
26:133–140. 1995. View Article : Google Scholar
|
|
14
|
Kitamoto K, Machida Y, Uchida J, Izumi Y,
Shiota M, Nakao T, Iwao H, Yukimura T, Nakatani T and Miura K:
Effects of liposome clodronate on renal leukocyte populations and
renal fibrosis in murine obstructive nephropathy. J Pharmacol Sci.
111:285–292. 2009. View Article : Google Scholar
|
|
15
|
Eis V, Luckow B, Vielhauer V, Siveke JT,
Linde Y, Segerer S, Perez De Lema G, Cohen CD, Kretzler M, Mack M,
et al: Chemokine receptor CCR1 but not CCR5 mediates leukocyte
recruitment and subsequent renal fibrosis after unilateral ureteral
obstruction. J Am Soc Nephrol. 15:337–347. 2004. View Article : Google Scholar
|
|
16
|
Kitagawa K, Wada T, Furuichi K, Hashimoto
H, Ishiwata Y, Asano M, Takeya M, Kuziel WA, Matsushima K, Mukaida
N and Yokoyama H: Blockade of CCR2 ameliorates progressive fibrosis
in kidney. Am J Pathol. 165:237–246. 2004. View Article : Google Scholar
|
|
17
|
MacKinnon AC, Farnworth SL, Hodkinson PS,
Henderson NC, Atkinson KM, Leffler H, Nilsson UJ, Haslett C, Forbes
SJ and Sethi T: Regulation of alternative macrophage activation by
galectin-3. J Immunol. 180:2650–2658. 2008. View Article : Google Scholar
|
|
18
|
Henderson NC, Mackinnon AC, Farnworth SL,
Kipari T, Haslett C, Iredale JP, Liu FT, Hughes J and Sethi T:
Galectin-3 expression and secretion links macrophages to the
promotion of renal fibrosis. Am J Pathol. 172:288–298. 2008.
View Article : Google Scholar
|
|
19
|
Hugo C and Daniel C: Thrombospondin in
renal disease. Nephron Exp Nephrol. 111:e61-e662009. View Article : Google Scholar
|
|
20
|
Wynes MW, Frankel SK and Riches DW:
IL-4-induced macrophage-derived IGF-I protects myofibroblasts from
apoptosis following growth factor withdrawal. J Leukoc Biol.
76:1019–1027. 2004. View Article : Google Scholar
|
|
21
|
Lin SL and Kisseleva T: Pericytes and
perivascular fibroblasts are the primary source of
collagen-producing cells in obstructive fibrosis of the kidney. Am
J Pathol. 173:1617–1627. 2008. View Article : Google Scholar
|
|
22
|
Mori R, Shaw TJ and Martin P: Molecular
mechanisms linking wound inflammation and fibrosis: Knockdown of
osteopontin leads to rapid repair and reduced scarring. J Exp Med.
205:43–51. 2008. View Article : Google Scholar
|
|
23
|
Chujo S, Shirasaki F, Kondo-Miyazaki M,
Ikawa Y and Takehara K: Role of connective tissue growth factor and
its interaction with basic fibroblast growth factor and macrophage
chemoattractant protein-1 in skin fibrosis. J Cell Physiol.
220:189–195. 2009. View Article : Google Scholar
|
|
24
|
Zhao YY, Yan DJ and Chen ZW: Role of AIF-1
in the regulation of inflammatory activation and diverse disease
processes. Cell Immunol. 284:75–83. 2013. View Article : Google Scholar
|
|
25
|
Utans U, Arceci RJ, Yamashita Y and
Russell ME: Cloning and characterization of allograft inflammatory
factor-1: A novel macrophage factor identified in rat cardiac
allografts with chronic rejection. J Clin Invest. 95:2954–2962.
1995. View Article : Google Scholar
|
|
26
|
Utans U, Quist WC, McManus BM, Wilson JE,
Arceci RJ, Wallace AF and Russell ME: Allograft inflammatory
factory-1. A cytokine-responsive macrophage molecule expressed in
transplanted human hearts. Transplantation. 61:1387–1392. 1996.
View Article : Google Scholar
|
|
27
|
Imai Y, Ibata I, Ito D, Ohsawa K and
Kohsaka S: A novel gene iba1 in the major histocompatibility
complex class III region encoding an EF hand protein expressed in a
monocytic lineage. Biochem Biophys Res Commun. 224:855–862. 1996.
View Article : Google Scholar
|
|
28
|
Tanaka S, Suzuki K, Watanabe M, Matsuda A,
Tone S and Koike T: Upregulation of a new microglial gene, mrf-1,
in response to programmed neuronal cell death and degeneration. J
Neurosci. 18:6358–6369. 1998. View Article : Google Scholar
|
|
29
|
Chen ZW, Ahren B, Ostenson CG, Cintra A,
Bergman T, Möller C, Fuxe K, Mutt V, Jörnvall H and Efendic S:
Identification, isolation, and characterization of daintain
(allograft inflammatory factor 1), a macrophage polypeptide with
effects on insulin secretion and abundantly present in the pancreas
of prediabetic BB rats. Proc Natl Acad Sci USA. 94:13879–13884.
1997. View Article : Google Scholar
|
|
30
|
Tian Y, Kelemen SE and Autieri MV:
Inhibition of AIF-1 expression by constitutive siRNA expression
reduces macrophage migration, proliferation, and signal
transduction initiated by atherogenic stimuli. Am J Physiol Cell
Physiol. 290:C1083–C1091. 2006. View Article : Google Scholar
|
|
31
|
Watano K, Iwabuchi K, Fujii S, Ishimori N,
Mitsuhashi S, Ato M, Kitabatake A and Onoé K: Allograft
inflammatory factor-1 augments production of interleukin-6, -10 and
-12 by a mouse macrophage line. Immunology. 104:307–316. 2001.
View Article : Google Scholar
|
|
32
|
Yang ZF, Ho DW, Lau CK, Lam CT, Lum CT,
Poon RT and Fan ST: Allograft inflammatory factor-1 (AIF-1) is
crucial for the survival and pro-inflammatory activity of
macrophages. Int Immunol. 17:1391–1397. 2005. View Article : Google Scholar
|
|
33
|
Kadoya M, Yamamoto A, Hamaguchi M,
Obayashi H, Mizushima K, Ohta M, Seno T, Oda R, Fujiwara H, Kohno M
and Kawahito Y: Allograft inflammatory factor-1 stimulates
chemokine production and induces chemotaxis in human peripheral
blood mononuclear cells. Biochem Biophys Res Commun. 448:287–291.
2014. View Article : Google Scholar
|
|
34
|
Mishima T, Iwabuchi K, Fujii S, Tanaka SY,
Ogura H, Watano-Miyata K, Ishimori N, Andoh Y, Nakai Y, Iwabuchi C,
et al: Allograft inflammatory factor-1 augments macrophage
phagocytotic activity and accelerates the progression of
atherosclerosis in ApoE−/− mice. Int J Mol Med.
21:181–187. 2008.
|
|
35
|
Fukui M, Tanaka M, Asano M, Yamazaki M,
Hasegawa G, Imai S, Fujinami A, Ohta M, Obayashi H and Nakamura N:
Serum allograft inflammatory factor-1 is a novel marker for
diabetic nephropathy. Diabetes Res Clin Pract. 97:146–150. 2012.
View Article : Google Scholar
|
|
36
|
Chen QR, Guan F, Song SM, Jin JK, Lei DS,
Chen CM, Lei JH, Chen ZW and Niu AO: Allograft inflammatory
factor-1 alleviates liver disease of BALB/c mice infected with
Schistosoma japonicum. Parasitol Res. 113:2629–2639. 2014.
View Article : Google Scholar
|
|
37
|
Del Galdo F and Jimenez SA: T cells
expressing allograft inflammatory factor 1 display increased
chemotaxis and induce a profibrotic phenotype in normal fibroblasts
in vitro. Arthritis Rheum. 56:3478–3488. 2007. View Article : Google Scholar
|
|
38
|
Brown NJ: Contribution of aldosterone to
cardiovascular and renal inflammation and fibrosis. Nat Rev
Nephrol. 9:459–469. 2013. View Article : Google Scholar
|
|
39
|
Ranjit S, Dvornikov A, Levi M, Furgeson S
and Gratton E: Characterizing fibrosis in UUO mice model using
multiparametric analysis of phasor distribution from FLIM images.
Biomed Opt Express. 7:3519–3530. 2016. View Article : Google Scholar
|
|
40
|
Autieri MV, Carbone C and Mu A: Expression
of allograft inflammatory factor-1 is a marker of activated human
vascular smooth muscle cells and arterial injury. Arterioscler
Thromb Vasc Biol. 20:1737–1744. 2000. View Article : Google Scholar
|
|
41
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
42
|
Tsubata Y, Sakatsume M, Ogawa A, Alchi B,
Kaneko Y, Kuroda T, Kawachi H, Narita I, Yamamoto T and Gejyo F:
Expression of allograft inflammatory factor-1 in kidneys: A novel
molecular component of podocyte. Kidney Int. 70:1948–1954. 2006.
View Article : Google Scholar
|
|
43
|
Yamamoto A and Kawahito Y: The immunologic
function and role of allograft inflammatory factor-1. Nihon Rinsho
Meneki Gakkai Kaishi. 37:139–145. 2014.In Japanese. View Article : Google Scholar
|
|
44
|
Usher MG, Duan SZ, Ivaschenko CY, Frieler
RA, Berger S, Schutz G, Lumeng CN and Mortensen RM: Myeloid
mineralocorticoid receptor controls macrophage polarization and
cardiovascular hypertrophy and remodeling in mice. J Clin Invest.
120:3350–3364. 2010. View Article : Google Scholar
|
|
45
|
Sibinga NE, Feinberg MW, Yang H, Werner F
and Jain MK: Macrophage-restricted and interferon gamma-inducible
expression of the allograft inflammatory factor-1 gene requires
Pu.1. J Biol Chem. 277:16202–16210. 2002. View Article : Google Scholar
|
|
46
|
Cook HT: The origin of renal fibroblasts
and progression of kidney disease. Am J Pathol. 176:22–24. 2010.
View Article : Google Scholar
|
|
47
|
Grimm PC, McKenna R, Nickerson P, Russell
ME, Gough J, Gospodarek E, Liu B, Jeffery J and Rush DN: Clinical
rejection is distinguished from subclinical rejection by increased
infiltration by a population of activated macrophages. J Am Soc
Nephrol. 10:1582–1589. 1999.
|
|
48
|
McDaniel DO, Rigney DA, McDaniel KY,
Windham WJ, Redmond P, Williams B, Zhou X, Hawxby A and Butt F:
Early expression profile of inflammatory markers and kidney
allograft status. Transplant Proc. 45:1520–1523. 2013. View Article : Google Scholar
|
|
49
|
Vu D, Tellez-Corrales E, Shah T,
Hutchinson I and Min DI: Influence of Cyclooxygenase-2 (COX-2) gene
promoter-1195 and allograft inflammatory factor-1 (AIF-1)
polymorphisms on allograft outcome in Hispanic kidney transplant
recipients. Hum Immunol. 74:1386–1391. 2013. View Article : Google Scholar
|
|
50
|
Brauner A, Hertting O, Alkstrand E,
Sandberg E, Chromek M, Chen ZW and Ostenson CG: CAPD peritonitis
induces the production of a novel peptide, daintain/allograft
inflammatory factor-1. Perit Dial Int. 23:5–13. 2003.
|
|
51
|
Meng XM, Tang PM, Li J and Lan HY:
Macrophage phenotype in kidney injury and repair. Kidney Dis
(Basel). 1:138–146. 2015. View Article : Google Scholar
|
|
52
|
Nishida M, Okumura Y, Fujimoto S,
Shiraishi I, Itoi T and Hamaoka K: Adoptive transfer of macrophages
ameliorates renal fibrosis in mice. Biochem Biophys Res Commun.
332:11–16. 2005. View Article : Google Scholar
|
|
53
|
López-Guisa JM, Cai X, Collins SJ,
Yamaguchi I, Okamura DM, Bugge TH, Isacke CM, Emson CL, Turner SM,
Shankland SJ, et al: Mannose receptor 2 attenuates renal fibrosis.
J Am Soc Nephrol. 23:236–251. 2012. View Article : Google Scholar
|
|
54
|
Huen SC and Cantley LG: Macrophages in
Renal Injury and Repair. Annu Rev Physiol. 79:449–469. 2017.
View Article : Google Scholar
|
|
55
|
Tian L, Shao X, Xie Y, Wang Q, Che X,
Zhang M, Xu W, Xu Y, Mou S and Ni Z: Kidney injury molecule-1 is
elevated in nephropathy and mediates macrophage activation via the
Mapk signalling pathway. Cell Physiol Biochem. 41:769–783. 2017.
View Article : Google Scholar
|
|
56
|
Guiteras R, Sola A, Flaquer M, Hotter G,
Torras J, Grinyó JM and Cruzado JM: Macrophage overexpressing NGAL
ameliorated kidney fibrosis in the UUO mice model. Cell Physiol
Biochem. 42:1945–1960. 2017. View Article : Google Scholar
|
|
57
|
Xu Y, Yang S, Huang J, Ruan S, Zheng Z and
Lin J: Tgf-β1 induces autophagy and promotes apoptosis in renal
tubular epithelial cells. Int J Mol Med. 29:781–790. 2012.
|
|
58
|
Seystahl K, Papachristodoulou A, Burghardt
I, Schneider H, Hasenbach K, Janicot M, Roth P and Weller M:
Biological role and therapeutic targeting of TGF-β3 in
glioblastoma. Mol Cancer Ther. 16:1177–1186. 2017. View Article : Google Scholar
|
|
59
|
Vega G, Alarcon S and San Martín R: The
cellular and signalling alterations conducted by TGF-β contributing
to renal fibrosis. Cytokine. 88:115–125. 2016. View Article : Google Scholar
|
|
60
|
Yamamoto A, Ashihara E, Nakagawa Y,
Obayashi H, Ohta M, Hara H, Adachi T, Seno T, Kadoya M, Hamaguchi
M, et al: Allograft inflammatory factor-1 is overexpressed and
induces fibroblast chemotaxis in the skin of sclerodermatous GVHD
in a murine model. Immunol Lett. 135:144–150. 2011. View Article : Google Scholar
|
|
61
|
Del Galdo F, Maul GG, Jiménez SA and
Artlett CM: Expression of allograft inflammatory factor 1 in
tissues from patients with systemic sclerosis and in vitro
differential expression of its isoforms in response to transforming
growth factor beta. Arthritis Rheum. 54:2616–2625. 2006. View Article : Google Scholar
|
|
62
|
Hao L, Okada H, Kanno Y, Inoue T,
Kobayashi T, Watanabe Y, Strutz F, Müller GA and Suzuki H: Direct
contact between human peripheral blood mononuclear cells and renal
fibroblasts facilitates the expression of monocyte chemoattractant
protein-1. Am J Nephrol. 23:208–213. 2003. View Article : Google Scholar
|
|
63
|
Sommerville LJ, Kelemen SE and Autieri MV:
Increased smooth muscle cell activation and neointima formation in
response to injury in AIF-1 transgenic mice. Arterioscler Thromb
Vasc Biol. 28:47–53. 2008. View Article : Google Scholar
|
|
64
|
Tian Y, Jain S, Kelemen SE and Autieri MV:
AIF-1 expression regulates endothelial cell activation, signal
transduction, and vasculogenesis. Am J Physiol Cell Physiol.
296:C256–C266. 2009. View Article : Google Scholar
|
|
65
|
McCaskill JL, Ressel S, Alber A, Redford
J, Power UF, Schwarze J, Dutia BM and Buck AH: Broad-spectrum
inhibition of respiratory virus infection by MicroRNA mimics
targeting p38 MAPK signaling. Mol Ther Nucleic Acids. 7:256–266.
2017. View Article : Google Scholar
|
|
66
|
Wang D, Warner GM, Yin P, Knudsen BE,
Cheng J, Butters KA, Lien KR, Gray CE, Garovic VD, Lerman LO, et
al: Inhibition of p38 MAPK attenuates renal atrophy and fibrosis in
a murine renal artery stenosis model. Am J Physiol Renal Physiol.
304:F938–F947. 2013. View Article : Google Scholar
|
|
67
|
Huang JS, Chuang CT, Liu MH, Lin SH, Guh
JY and Chuang LY: Klotho attenuates high glucose-induced
fibronectin and cell hypertrophy via the ERK1/2-p38 kinase
signaling pathway in renal interstitial fibroblasts. Mol Cell
Endocrinol. 390:45–53. 2014. View Article : Google Scholar
|
|
68
|
Blasi ER, Rocha R, Rudolph AE, Blomme EA,
Polly ML and McMahon EG: Aldosterone/salt induces renal
inflammation and fibrosis in hypertensive rats. Kidney Int.
63:1791–1800. 2003. View Article : Google Scholar
|
|
69
|
Kadoya H, Satoh M, Sasaki T, Taniguchi S,
Takahashi M and Kashihara N: Excess aldosterone is a critical
danger signal for inflammasome activation in the development of
renal fibrosis in mice. FASEB J. 29:3899–3910. 2015. View Article : Google Scholar
|
|
70
|
Bengough AG, Hans J, Bransby MF and
Valentine TA: PIV as a method for quantifying root cell growth and
particle displacement in confocal images. Microsc Res Tech.
73:27–36. 2010.
|
|
71
|
Nagahara H, Yamamoto A, Seno T, Obayashi
H, Kida T, Nakabayashi A, Kukida Y, Fujioka K, Fujii W, Murakami K,
et al: Allograft inflammatory factor-1 in the pathogenesis of
bleomycin-induced acute lung injury. Biosci Trends. 10:47–53. 2016.
View Article : Google Scholar
|