Introduction
Asthma is a common disease that is characterized by
reversible airflow obstruction, airway hyperresponsiveness (AHR),
airway inflammation, airway remodeling, mucus hypersecretion and
subepithelial fibrosis (1,2).
It is a heterogeneous syndrome affected by several factors,
including the environment, genetic background, and infection
(3). Multiple cell types and
cellular components are known to be involved in the
pathophysiological processes of asthma. One important link in the
pathogenesis of asthma is airway remodeling caused by recurrent
injury and repair processes initiated by chronic inflammation
(4). Changes in the morphology
and function of airway epithelial cells are key to airway
remodeling. This remodeling is considered to be the pathological
basis for irreversible AHR and airway obstruction (5). During airway remodeling the number
of goblet cells increases, resulting in increased mucus secretion,
which can cause airway obstruction and lead to asthma-related
mortality.
Repeated chronic inflammation in the asthmatic
airway induces epithelial cells to transdifferentiate into
myofibroblasts. This is an example of epithelial-mesenchymal
transition (EMT). EMT may be involved in the process of airway
remodeling and subepithelial fibrosis in asthma (6) and may be a possible mechanism of
airway inflammation (7).
Persistent EMT causes detrimental changes in pulmonary function
(8). Furthermore, EMT decreases
the sensitivity of airway epithelial cells to drug treatments and
thus decreases the therapeutic efficacy of glucocorticoids in
patients with severe asthma (9).
Abnormal EMT is now considered to be the central event in asthma
pathophysiology (10). EMT is a
current focus of investigations of the mechanisms underlying airway
remodeling in asthma. A variety of proteins are important in EMT,
including E-cadherin and vimentin, which represent epithelial and
mesothelial features, respectively. The decreased expression of
E-cadherin, the increased expression of vimentin, and the
transition from epithelial to mesenchymal cell morphology are
important manifestations of the EMT process. However, the molecular
mechanisms underlying these effects remain to be fully elucidated.
No effective treatments for asthma-related airway remodeling are
available (11).
Transforming growth factor β1 (TGFβ1) is an
important cytokine involved in airway remodeling that mediates
tissue inflammation in asthma. The mechanisms underlying the
effects of TGFβ1 on asthma remain to be fully elucidated.
Therefore, it is hypothesized that TGFβ1-induced EMT may be
involved in the processes of airway inflammation and airway
remodeling. Nuclear factor-κB (NF-κB) is an important
transcriptional factor in EMT and in the epithelial cell
inflammation of asthma (9).
TGFβ-induced EMT is driven by NF-κB-dependent cell signaling
(12). The persistent activation
of NF-κB has been observed in allergic airway inflammation
(13). Therefore, NF-κB has
emerged as a vital therapeutic molecular target, and the inhibition
of NF-κB activity may offer potential as a method to manage
asthma.
Several factors are involved in regulating the
expression and activity of NF-κB, providing several targets for the
pharmacological inhibition of NF-κB. One inhibitor target is
inhibitor of NF-κB (IκB) kinase (IKK), a kinase complex that
phosphorylates IκB, the inhibitory subunit of the NF-κB complex,
thereby releasing IκB and activating NF-κB. The IKK/NF-κB pathway
is important in regulating inflammation. IKKβ is particularly
important as a major upstream regulator of NF-κB activity, with an
important role in the immune response and inflammatory reactions
(14). Inhibitors of IKKβ,
including BMS-345541 [(2′-aminoethyl)amino-1,8-dimethy
limidazo(1,2-a) quinoxaline], have certain pharmacokinetic
characteristics, including 100% oral bioavailability and an
intravenous half-life of 2.2 h, which makes them particularly well
suited for use in investigating the utility of IKK inhibitors in
disease models (15). In various
cell and animal models of several diseases, BMS-345541 is key in
EMT and suppresses inflammatory responses by inhibiting the
activation of NF-κB (16-18). These results indicate that
BMS-345541 may have potent anti-inflammatory activity in the
context of asthma. However, whether BMS-345541 can be used to
inhibit asthma-induced airway inflammation, airway remodeling and
the EMT observed in asthma, and the exact therapeutic mechanisms of
BMS-345541 in asthma treatment remain to be fully elucidated.
Therefore, to provide novel ideas and methods for
the clinical treatment of asthma and to investigate the mechanisms
of asthma, it was hypothesized that the secretion of TGFβ1 may
result in EMT, and the role of BMS-345541 on airway inflammation,
airway remodeling and potentially in the regulation of EMT marker
protein expression were investigated in an ovalbumin (OVA)-induced
asthma model in mice. If BMS-345541 inhibits asthma phenotypes in
the OVA mouse model, as expected, it is likely to be an important
tool in the elucidation of the roles of TGFβ1, EMT and NF-κB in
asthma pathophysiology and in the identification of targets for
novel drug therapies.
Materials and methods
Animals
A total of 32 female BALB/c mice aged 6-8 weeks and
weighing 25±3 g were maintained in the Laboratory Animal Science
Centre of Nanchang University (Nanchang, China). All the mice were
housed under specific pathogen-free laboratory conditions in a 12 h
light/dark cycle at a temperature of 20±5°C with 50±10% humidity.
Mice had ad libitum access to food and water. Care was taken
to alleviate any pain and suffering of the mice. All experiments
were performed according to institutional regulations, and all
procedures performed in the present study were approved by the
Ethics Commission of Jiangxi People's Hospital (Nanchang,
China).
Reagents
The reagents used were as follows: OVA
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), acetylcholine
chloride (Ach; Sigma-Aldrich; Merck KGaA), phosphate-buffered
saline (PBS; Sigma-Aldrich; Merck KGaA), sodium pentobarbital
(Sinopharm Chemical Reagent Co., Ltd., Shanghai, China), BMS-345541
(Abcam, Cambridge, MA, USA), E-cadherin antibody (cat. no. ab76055;
Abcam), vimentin antibody (cat. no. ab92547, Abcam), β-actin
antibody (cat. no. ab179467; Abcam), glyceraldehyde 3-phosphate
dehydrogenase (GADPH; Sigma-Aldrich; Merck KGaA), dimethyl
sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA), diaminobenzidine (DAB;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA), an enzyme-linked
immunosorbent assay (ELISA) kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), Pierce Enhanced Chemiluminescence (ECL) western
blot substrate (Thermo Fisher Scientific, Inc., Waltham, MA, USA),
a reverse transcription kit (Promega Corporation, Madison, WI,
USA), a quantitative polymerase chain reaction (qPCR) kit
(TransStart Green; Beijing Transgen Biotech Co., Ltd., Beijing,
China), a bicinchoninic acid (BCA) protein assay kit and
radioimmunoprecipitation assay (RIPA) lysis buffer (Beijing ComWin
Biotech Co., Ltd., Beijing, China). The qPCR primers were designed
and synthesized by Nanjing Kingsy Biotechnology (Nanjing,
China).
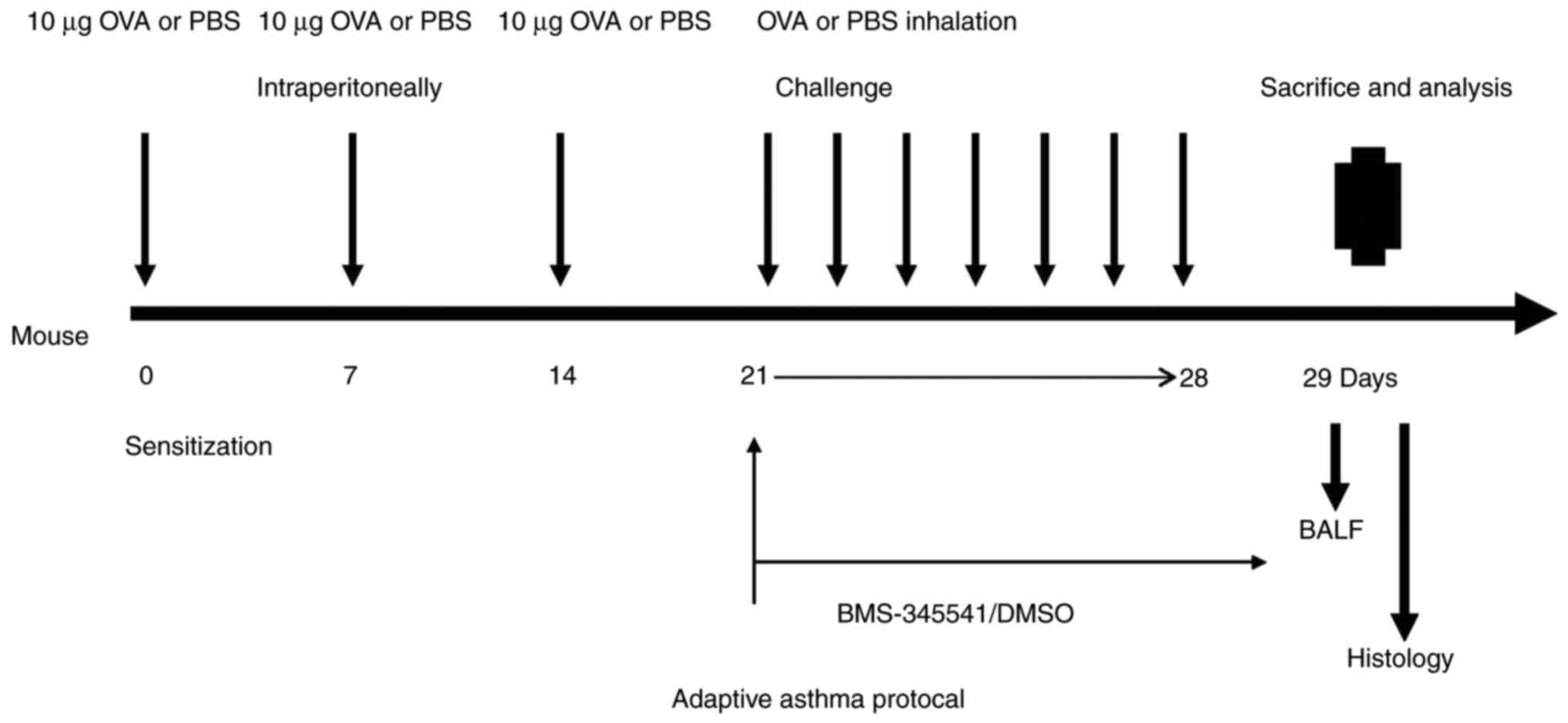
Asthma sensitization, challenge and
treatment protocol
The 32 mice were divided into four groups of eight
mice: Control, OVA, OVA + DMSO, and OVA + BMS-345541 groups. First,
eight mice were randomly selected from the 32 mice as the normal
control group. The remaining 24 mice were then selected to be
sensitized and challenged with OVA. The mice were sensitized with
intraperitoneal (i.p.) injections of 10 µg of OVA emulsified in
aluminium hydroxide in a total volume of 0.2 ml on days 0, 7 and
14. Between days 21 and 28, they were challenged by aerosol
exposure to 5% OVA for 30 min daily for 7 days. The control group
mice were treated with PBS for sensitization and stimulation using
the same protocol.
Prior to OVA challenge, the 24 sensitized mice were
equally divided into three groups of eight mice: OVA group, OVA +
DMSO group and OVA + BMS-345541 group. The OVA + BMS-345541 group
comprised mice treated with BMS-345541. The BMS-345541 was
dissolved in 1% DMSO, and the resulting solution was diluted with
sterile water to a final concentration of 10 µg/µl. The OVA + DMSO
group was the DMSO vehicle control group. BMS-345541 (50 mg/kg) or
vehicle (20 µl DMSO in a total of 200 µl saline,
without BMS-345541) was administered to the mice by oral gavage
using a feeding needle (18 gauge, 5 cm) for 7 days (19). No such intervention was performed
in the OVA group mice. The detailed experimental protocol is shown
in Fig. 1.
Evaluation of asthma symptom severity,
and measurement of airway responsiveness
A symptom assessment of asthma in mice was performed
with regard to specific criteria, as previously described (20). The asthma symptoms were observed
in 15 min during inhaled OVA. Airway responsiveness to Ach was
measured with a whole-body and invasive plethysmography (Buxco
Electronics, Inc., Troy, NY, USA) and analyzed as previously
described (21). Lung resistance
(RL) was measured to assess the change in AHR. At 24 h following
the final challenge, all mice were weighed and anesthetized with 2%
sodium pentobarbital (30 mg/kg) via i.p. injection. The cervical
trachea was completely exposed by blunt dissection, a tracheal tube
(2-mm internal diameter) was inserted into the trachea via a
tracheotomy. The mice were then placed into the whole-body
plethysmography chamber, and the tracheal tube was connected to the
ventilator for mechanical ventilation with a tidal volume of 0.2 ml
and a frequency of 140 breaths/min. After 5 min of equilibration on
the ventilator, PBS and a series of increasing doses of Ach (3.125,
6.25, 12.5 and 25 mg/ml) were administered through the ventilator
with an ultrasonic nebulizer. The data were collected through the
sensor.
Serum, bronchoalveolar lavage fluid
(BALF) and lung tissue specimen preparation
The mice were sacrificed, following which one side
of the bronchus was ligated and the airway on other side was
lavaged three times with 0.5 ml of normal saline; 80% of the input
volume was recovered. BALF was centrifuged at 500 × g for 10 min at
4°C. The total number of cells in the BALF was counted with a
hemocytometer, and the percentages of the inflammatory cells were
determined by counting 400 cells in randomly selected areas of the
slide under a light microscope. All counts were performed in a
blinded manner and in a randomized order by the same observer at
the end of the experiment. The BALF supernatants were stored at
−80°C for ELISA assessment.
Following the collection of BALF, all mice underwent
an abdominal surgical procedure in which the abdominal anatomy was
carefully examined to identify and expose the inferior vena cava,
from which 0.5–1 ml of venous blood was withdrawn into a sterile
Eppendorf tube. Following standing for 1 h, the sample was
centrifuged at 1,000 × g at 4°C for 10 min, and the supernatant was
stored at −80°C for further analysis.
The non-lavaged side of the lungs was harvested, the
lower lobe was isolated, and fixed in 0.1 mmol/l paraformaldehyde,
and was made into 4-µm thick paraffin-embedded tissue
sections for histopathological analysis via immunohistochemistry.
The upper and middle lobes were stored at −80°C in a refrigerator
for reverse transcription-qPCR (RT-qPCR) and western blot
analyses.
ELISA for TGFβ1 in serum and BALF
The concentrations of TGFβ1 in the serum and BALF
were measured with an ELISA kit according to the manufacturer's
protocol. A microplate reader was used to detect the optical
density (OD) for the ELISA. TGFβ1 levels were determined by the OD
values.
Lung tissue histopathology
The paraffin-embedded sections were stained with
hematoxylin and eosin (H&E) to observe changes in airway
inflammation and airway remodeling. Periodic acid-Schiff (PAS)
staining was performed to evaluate airway goblet cell hyperplasia
and mucus production. The stained sections were mounted on slides
and examined under a light microscope (Olympus BX50, Olympus,
Tokyo, Japan). Images were captured with a Nikon DS-Ri2 digital
camera.
Epithelial thickness, the area between the luminal
cell membrane and the basement membrane, was measured at four sites
in five different medium-sized bronchi per slide using Image Pro
Plus 6.0 image analysis software (Media Cybernetics, Inc.,
Rockville, MD, USA). All the measurements are provided as the
average epithelial thickness per group.
Immunohistochemical analysis of the
expression of E-cadherin and vimentin in lung tissues
The sections of lung tissues were deparaffinized in
xylene and rehydrated in graduated ethanol solutions. Following
microwave-based antigen retrieval with citric acid pretreatment,
the sections were incubated in 1% hydrogen peroxide for 15 min to
block endogenous peroxidase. Subsequently, the specimens were
incubated with a mouse polyclonal antibody to E-cadherin (1:200) or
a rabbit polyclonal antibody to vimentin (1:200) at 4°C overnight,
respectively. The sections were then incubated with anti-mouse
(cat. no. CW01025) or anti-rabbit (cat. no. CW01035) horseradish
peroxidase (HRP)-conjugated secondary antibody (1:100; Beijing Com
Win Biotech Co., Ltd.) for 30 min at room temperature, followed by
staining with DAB (22). For the
negative control, the primary antibody was replaced with PBS. The
sections were observed under a light microscope (magnification,
×400). E-cadherin was mainly expressed in the cell membrane and
cytoplasm, whereas vimentin was mainly expressed in the cytoplasm.
The brown staining of a cell membrane or cytoplasm is a positive
signal in protein immunohistochemistry. The mean integrated OD
(IOD) was then detected using the Image Pro Plus 6.0 image analysis
system for all the sections. The IOD value represented the
expression of each protein. The mean IOD values of each group were
compared.
RT-qPCR analysis of E-cadherin and
vimentin expression
Total RNA was extracted from the lung tissues using
TRIzol. The mRNA was then reverse transcribed using a reverse
transcription kit, according to the manufacturer's protocol. The
reaction system was made up to 20 µl with RNAse free water
and contained 2.5 mM MgCl2 (4 µl), reverse
transcription 10X buffer (2 µl), 0.5 mg/ml primers (1
µl), dNTP mix (2 µl) and AMV reverse transcriptase
(0.7 µl). The RT reaction occurred at 42°C for 1 h and was
subsequently inactivated at 95°C for 5 min. A qPCR kit was used to
amplify the resulting cDNAs, and fluorescence was detected with an
7500 PCR detection system (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The PCR reaction system was made up to 20
µl with RNAse free water and contained 2X TransStart Green
qPCR Super MIX UDG (10 µl), 10 µM forward primer (0.4
µl), 10 µM reverse primer (0.4 µl), 50X
passive reference dye (0.4 µl) and cDNA (1 µl). The
thermocycling conditions were as follows: 50°C for 2 min, 94°C for
10 min, followed by 40–45 cycles of 94°C for 5 sec and 60°C for 30
sec. GADPH was used as an internal control. The PCR products were
subjected to melting curve analysis to ensure that a single
amplification product was produced. The relative expression levels
of E-cadherin and vimentin were calculated using the quantification
cycle (2−ΔΔCq) method (23). The sequences of primers used for
RT-qPCR analysis are listed in Table
I.
 | Table IPrimers and sequences. |
Table I
Primers and sequences.
| Name | Sequence
(5′-3′) | Fragment length
(bp) |
|---|
| E-cadherin | F:
AAAAGAAGGCTGTCCTTGGC | 106 |
| R:
GAGGTCTACACCTTCCCGGT | |
| Vimentin | F:
TCCACTTTCCGTTCAAGGTC | 91 |
| R:
AGAGAGAGGAAGCCGAAAGC | |
| GAPDH | F:
ATGGAGGGGAATACAGCCC | |
| R:
TTCTTTGCAGCTCCTTCGTT | 149 |
Western blot analysis of E-cadherin and
vimentin expression
Total protein was extracted from lung tissues using
RIPA lysis buffer. Protein concentration was determined with a BCA
protein assay, following which the proteins (50 µg/lane)
were separated on 15% SDS-PAGE gels and transferred onto PVDF
membranes. The membranes were blocked with 5% non-fat milk for 1 h
at room temperature and then incubated with anti-E-cadherin
(1:1,000) and anti-vimentin (1:1,000) antibodies overnight at 4°C.
The β-actin antibody (1:2,000) was used as an internal control.
Following three washes, the membranes were probed with
HRP-conjugated secondary antibodies (1:3,000 in blocking buffer) at
room temperature for 2 h and visualized with ECL reagent. The
relative expression levels of E-cadherin and vimentin were
quantified using Image lab Version 6.0 software (Bio-Rad
Laboratories, Inc.) and were normalized to levels of β-actin. The
normalized level of expression in the control group was set to
1.00, and the normalized expression in the OVA and OVA + BMS-345541
groups is expressed relative to the control.
Statistical analysis
All data were processed using SPSS 19.0 (IBM SPSS,
Armonk, NY, USA), and all quantitative data are presented as the
mean ± standard deviation (n=8 mice/sample per group). The data
were compared using a single-factor analysis of variance following
variance determination. Comparisons among multiple groups were
assessed using the Least Significant Difference method. If the
variance was not homogeneous, Tamhane's multiple comparison
procedure was used to assess comparisons among multiple groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effect of BMS-345541 on OVA-induced
symptoms
During OVA challenge, the OVA-group mice showed a
variety of asthma-related symptoms, including dysphoria, shortness
of breath and irregular breath rhythm, cyanosis, coughing, nose
scratching and ear grasping, shrinking forelimb lift, and decreased
activity. However, following treatment with BMS-345541, the
asthmatic symptoms in the OVA + BMS-345541 group were significantly
reduced, compared with those in the OVA and OVA + DMSO groups. The
control group mice exhibited no asthma attack or allergic
symptoms.
Effect of BMS-345541 on airway
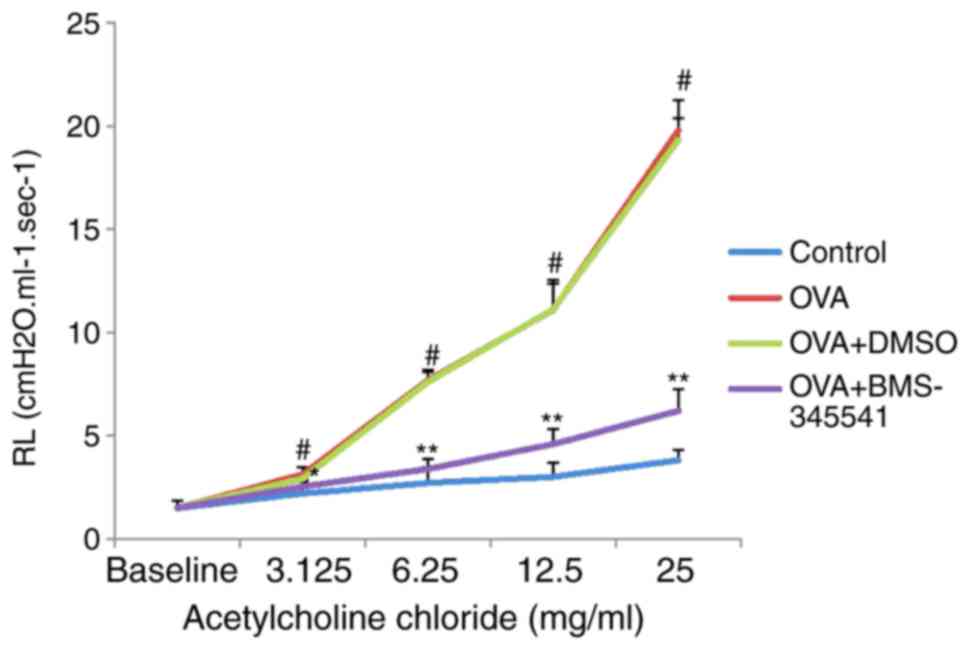
responsiveness
To evaluate the effect of BMS-345541 on AHR in
response to Ach, the RL was measured in an invasive whole-body
plethysmography chamber in anesthetized mice. No significant
differences in baseline airway resistance were observed among the
four groups. The administration of Ach at doses increasing
progressively between 3.125 and 25 mg/ml led to markedly increased
RL values in the OVA group compared with those in the control group
(P<0.001). However, compared with the OVA group, treatment with
BMS-345541 resulted in a significant decrease in RL (P<0.01).
The change in RL in the OVA + DMSO group was close to that in the
OVA group (Fig. 2, Table II).
| Table IILung resistance (cm
H2O.ml−1.sec−1). |
Table II
Lung resistance (cm
H2O.ml−1.sec−1).
| Group | N | Baseline | 3.125 | 6.25 | 12.5 | 25 |
|---|
| Control | 8 | 1.49±0.36 | 2.20±0.55 | 2.71±0.74 | 3.00±0.71 | 3.81±0.48 |
| OVA | 8 | 1.49±0.36 | 3.11±0.34a | 7.66±0.53a | 11.09±1.28a | 19.79±1.48a |
| OVA + DMSO | 8 | 1.49±0.36 | 2.91±0.27 | 7.59±0.50 | 11.11±1.41 | 19.31±1.04 |
| OVA +
BMS-345541 | 8 | 1.49±0.36 | 2.52±0.31b | 3.38±0.49c | 4.59±0.72c | 6.20±1.04c |
| F-value | | <0.001 | 9.037 | 172.304 | 125.589 | 499.166 |
| P-value | | 1.00 | <0.001 | <0.001 | <0.001 | <0.001 |
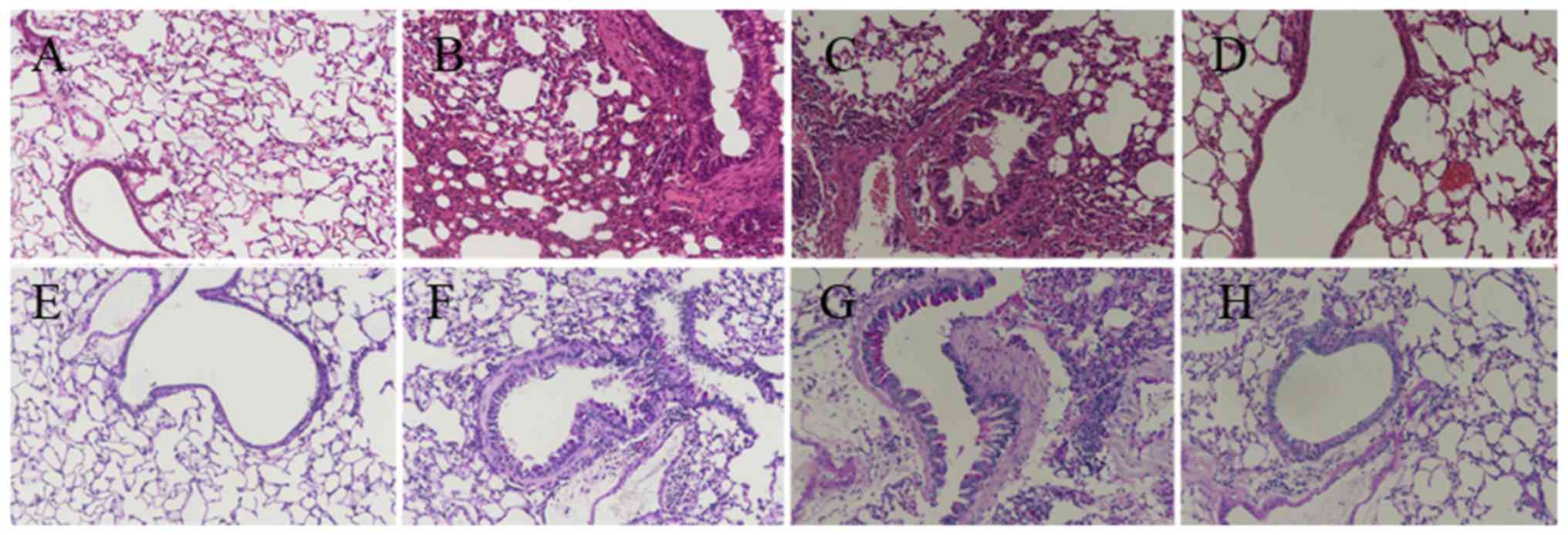
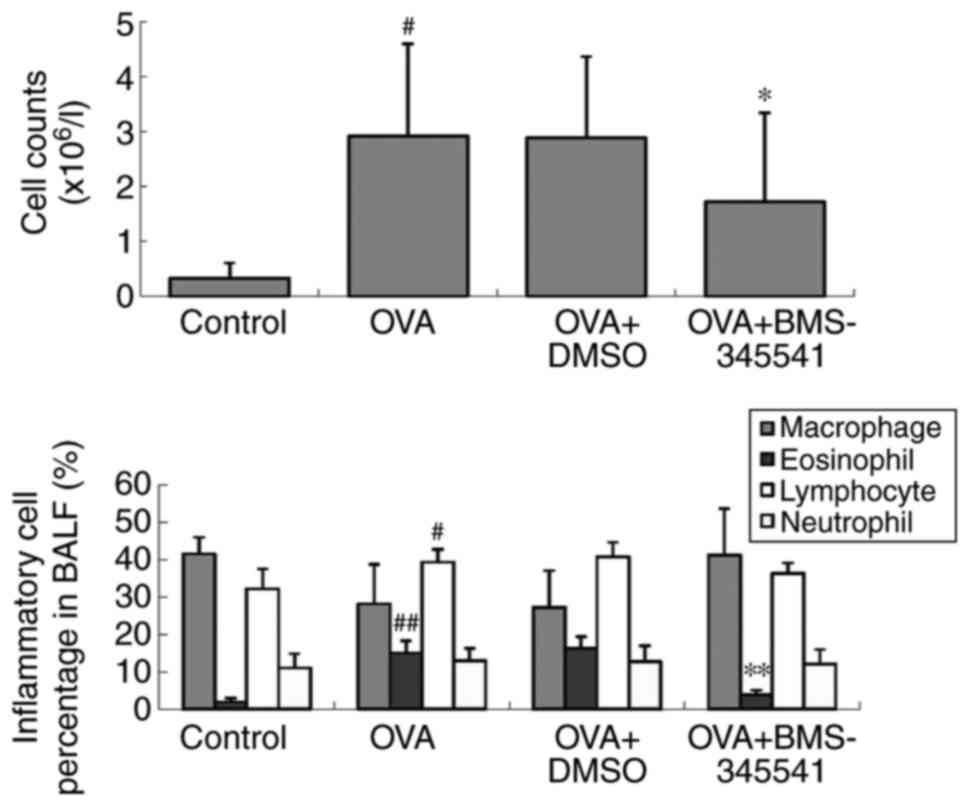
Effect of BMS-345541 on airway
inflammation and remodeling
Airway inflammation is characterized by the
infiltration of inflammatory cells. The degree of airway
inflammation was evaluated via histopathological analysis of lung
lesions (H&E staining; Fig.
3A–D), and by total cell counts and inflammatory cell
percentage, including eosinophils, lymphocytes, neutrophils and
macrophages, in the BALF. The mice in the OVA and OVA + DMSO groups
exhibited an influx of inflammatory cells around the bronchi and
bronchioles along with submucosal edema, as shown in Fig. 3B and C. The total cell numbers and
the percentages of eosinophil and lymphocytes were higher in the
OVA-induced mice than those in the control group. By contrast, the
BMS-345541 treatment group exhibited less inflammatory cell
infiltration and edema.
Goblet cell hyperplasia is one of the critical
pathophysiologic changes in the airway remodeling of asthma
(24). PAS staining was used to
evaluate airway global cell hyperplasia and mucus production
(Fig. 3E–H). The lung tissue from
the OVA mice (Fig. 3F) exhibited
airway remodeling compared with that in the control group (Fig. 3E), including the enlargement of
airway smooth muscle, basement membrane thickening, goblet cell
hyperplasia, mucus hypersecretion, and epithelium damage. The OVA +
DMSO group (Fig. 3G) exhibited
similar changes. However, the development of airway remodeling was
significantly attenuated by the administration of BMS-345541.
Despite inflammatory changes, the mice in the OVA + BMS-345541
group showed only marginal bronchial wall thickening, airway
stenosis and epithelial damage (Fig.
3H).
Following BMS-345541 treatment, the increased cell
counts and eosinophil percentages were markedly decreased compared
with the corresponding values in the OVA mice (P<0.05). There
was no change between the OVA and OVA + DMSO groups (Fig. 4, Table III).
| Table IIICell counts and percentages of
inflammatory cells in the BALF. |
Table III
Cell counts and percentages of
inflammatory cells in the BALF.
| Group | N | Total cells
(106/l) | Macrophages
(%) | Eosinophil (%) | Lymphocytes
(%) | Neutrophils
(%) |
|---|
| Control | 8 | 0.33±0.28 | 41.56±4.44 | 1.94±0.90 | 32.19±5.80 | 11.06±3.73 |
| OVA | 8 | 2.92±1.68a | 28.19±10.50 | 15.08±3.26b | 39.31±3.39a | 12.94±3.39 |
| OVA + DMSO | 8 | 2.89±1.48 | 38.31±9.86 | 16.20±3.18 | 39.94±5.41 | 10.00±3.30 |
| OVA +
BMS-345541 | 8 | 1.72±1.63c | 41.25±12.37 | 3.81±1.25d | 36.81±2.80 | 12.00±3.97 |
| F-value | | 11.354 | 1.803 | 80.080 | 7.013 | 0.976 |
| P-value | | <0.001 | 0.169 | <0.001 | 0.001 | 0.418 |
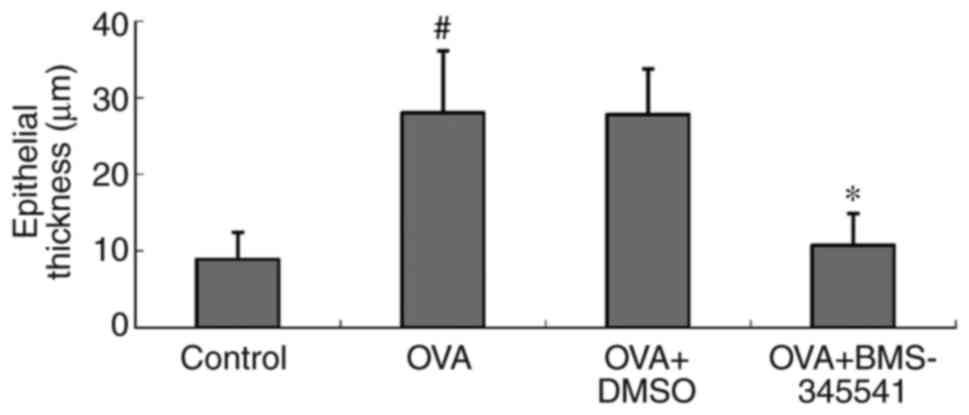
The epithelial thicknesses were examined to assess
the features of airway remodeling. A quantitative assessment of
epithelial thickness demonstrated significant increases in the OVA
group (28.02±8.09 µm) and the OVA + DMSO group (27.80±5.93
µm). There was a significant difference between the OVA and
the control groups (P<0.001). However, the epithelial thickness
was markedly alleviated by the administration of BMS-345541
(10.71±4.18 µm). There was also a significant difference
between the OVA + BMS-345541 and the OVA groups (P<0.001)
(Fig. 5, Table IV).
| Table IVEpithelial thickness. |
Table IV
Epithelial thickness.
| Group | N | Epithelial
thickness (µm) |
|---|
| Control | 8 | 8.85±3.15 |
| OVA | 8 | 28.02±8.09a |
| OVA + DMSO | 8 | 27.81±5.93 |
| OVA +
BMS-345541 | 8 | 10.71±4.18b |
| F-value | | 27.545 |
| P-value | | <0.001 |
Effect of BMS-345541 on the
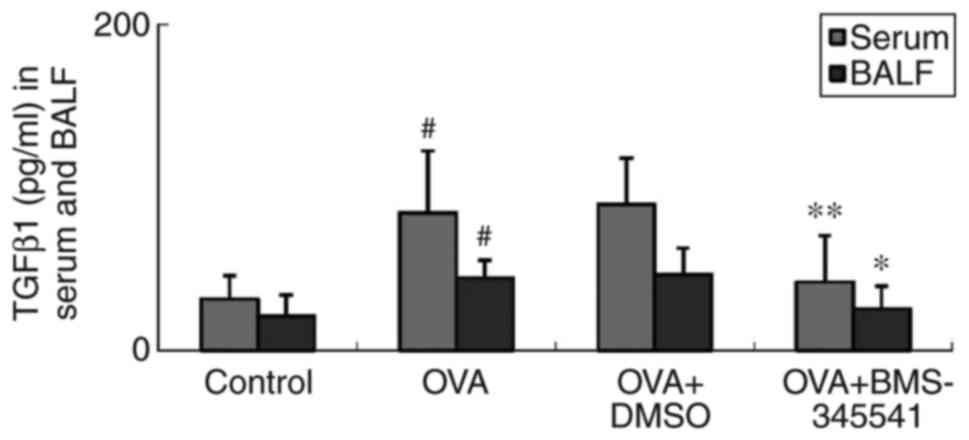
concentrations of TGFβ1 in serum and BALF
TGFβ1 is an important cytokine involved in airway
remodeling, and is important in EMT (25). TGFβ1 mediates tissue inflammation
in asthma (26). Concentrations
of TGFβ1 in the serum and BALF were detected by ELISA, and it was
found that the OVA group had markedly elevated levels of serum and
BALF TGFβ1, compared with the control group. No difference between
the OVA and OVA + DMSO groups was found. However, unlike in the OVA
+ DMSO group, the concentrations of TGFβ1 in the BMS-345541-treated
OVA group were significantly decreased compared with those in the
OVA group (Fig. 6, Table V).
| Table VTGFβ1 concentration in serum and
BALF. |
Table V
TGFβ1 concentration in serum and
BALF.
| Group | N | Serum (pg/ml) | BALF (pg/ml) |
|---|
| Control | 8 | 31.53±14.41 | 20.98±13.29 |
| OVA | 8 | 84.41±38.18a | 44.36±10.98a |
| OVA + DMSO | 8 | 89.63±28.62 | 46.58±16.27 |
| OVA +
BMS-345541 | 8 | 42.04±28.38c | 25.45±14.15b |
| F-value | | 8.405 | 7.109 |
| P-value | | <0.001 | 0.001 |
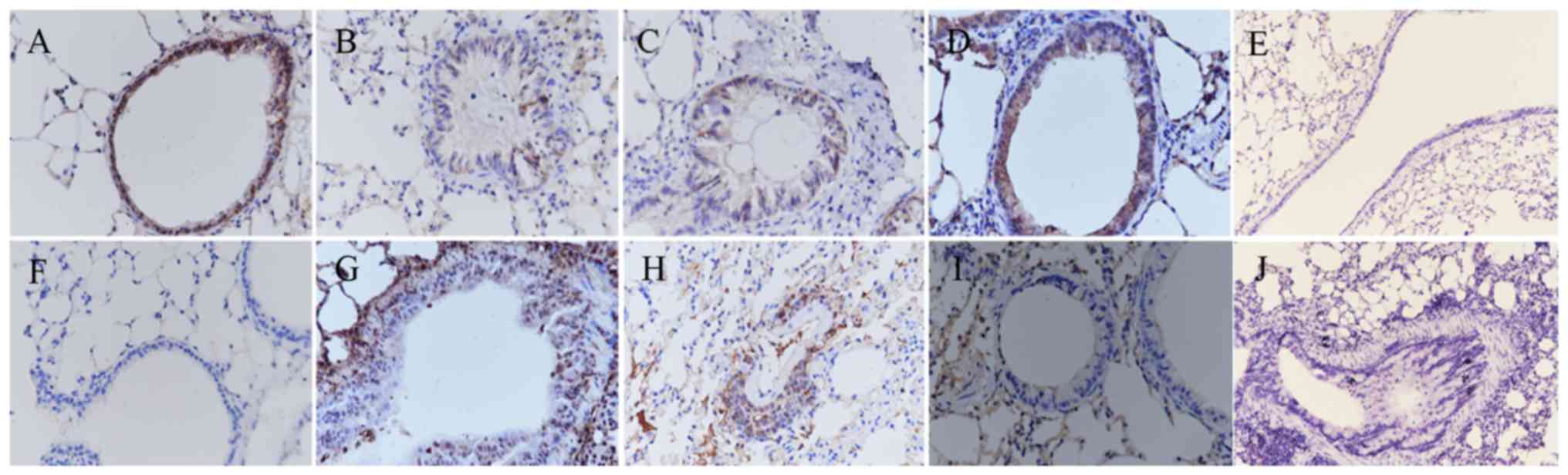
Effect of BMS-345541 on the expression of
E-cadherin and vimentin as assessed by immunohistochemistry,
RT-qPCR and western blot analyses
The epithelial marker E-cadherin and the mesenchymal
marker vimentin are important markers of EMT. The expression levels
of E-cadherin and vimentin were estimated by immunohistochemical
image analysis. The mRNA and protein levels of E-cadherin and
vimentin were determined by RT-qPCR and western blot analyses. As
shown in Fig. 7A–G, the results
of the immunohistochemistry indicated a significant decrease in the
expression of E-cadherin and an increase in the expression of
vimentin in the epithelium of the OVA-induced mice (Fig. 7B and G). Following the daily
administration of BMS-345541, the expression of E-cadherin was
increased, whereas the expression of vimentin was decreased, as
shown in Fig. 7D and I. No
E-cadherin or vimentin was identified in the negative controls
(Fig. 7E and J).
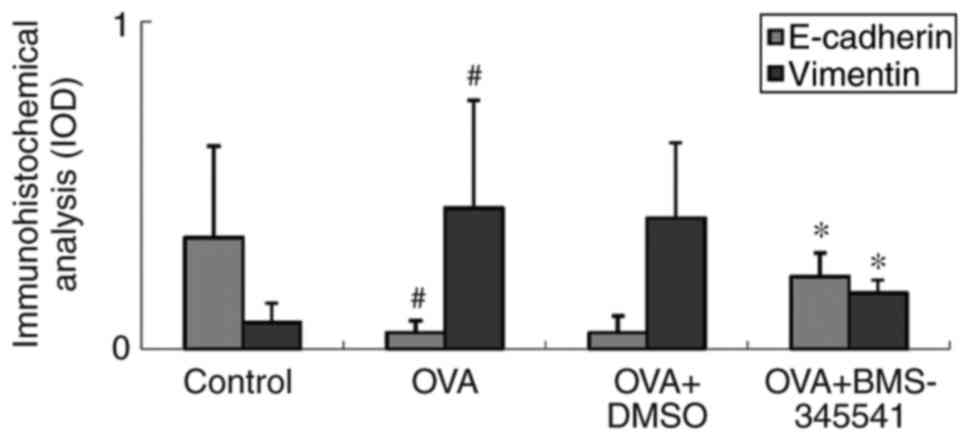
As shown in Fig. 8
and Table VI, the expression of
EMT markers was further investigated by IOD measurement. It was
found that the IOD value of vimentin was higher in the OVA group
and the OVA + DMSO group, whereas the IOD value in the OVA +
BMS-345541 group decreased significantly. By contrast, it was found
that the E-cadherin IOD values were decreased in the OVA and OVA +
DMSO groups, and increased in the OVA + BMS-345541 group following
BMS-345541 administration. There were significant differences
between the OVA and the control groups (P<0.01), and the OVA and
OVA + BMS-345541 groups (P<0.05). No significant difference was
observed between the OVA and the OVA + DMSO groups.
| Table VIIOD of E-cadherin and vimentin
expression. |
Table VI
IOD of E-cadherin and vimentin
expression.
| Group | N | E-cadherin | Vimentin |
|---|
| Control | 8 | 0.34±0.28 | 0.08±0.06 |
| OVA | 8 | 0.05±0.04a | 0.43±0.33a |
| OVA + DMSO | 8 | 0.05±0.05 | 0.40±0.23 |
| OVA +
BMS-345541 | 8 | 0.22±0.07b | 0.17±0.04b |
| F-value | | 7.164 | 5.716 |
| P-value | | 0.001 | 0.003 |
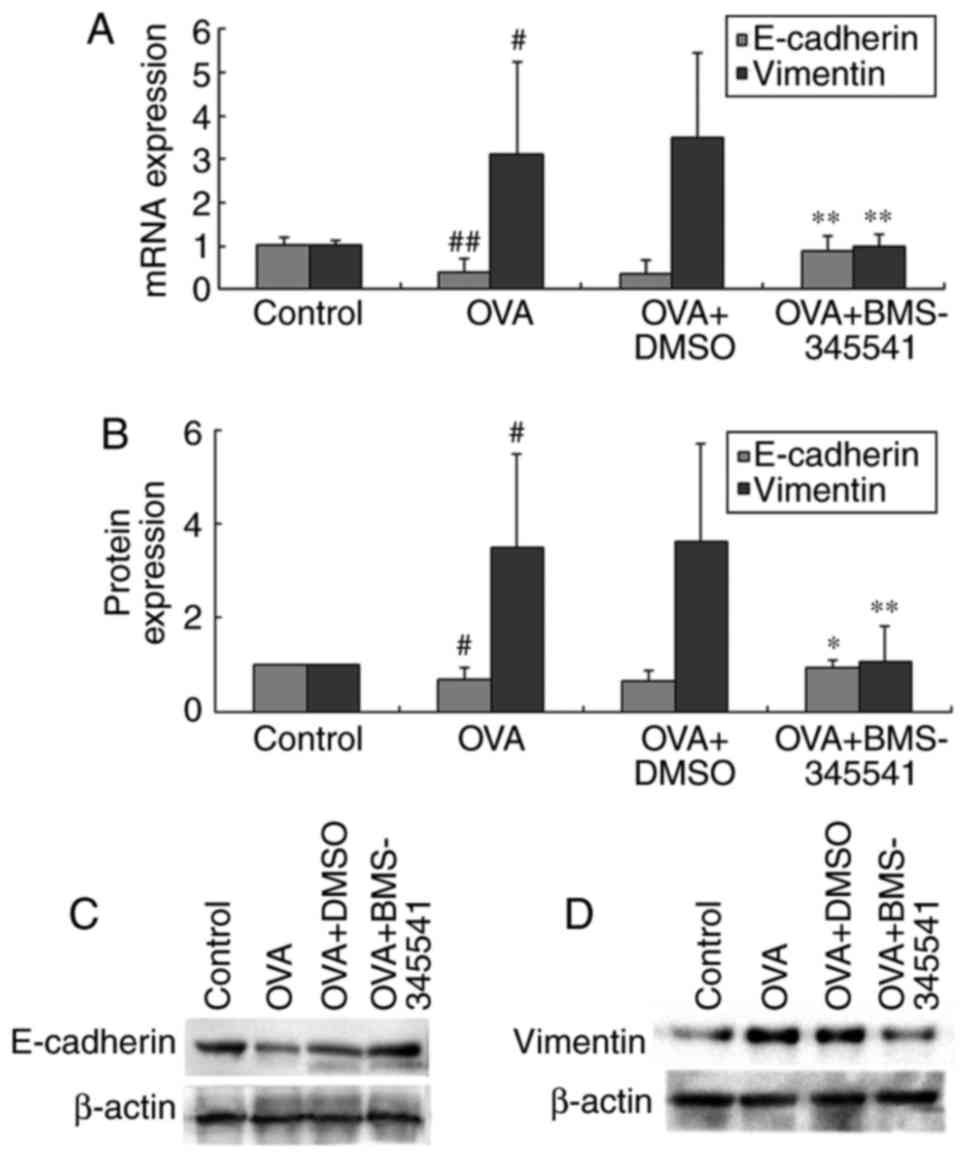
The mRNA and protein levels showed the same results,
which were consistent with the immunohistochemical results. As
shown in Fig. 9A–D and Table VII, the same results were
observed for the expression of E-cadherin, which was
down-regulated, and expression of vimentin, which was upregulated,
in the OVA group mice. BMS-345541 supressed the expression of
vimentin and promoted the expression of E-cadherin. These results
indicated that BMS-345541 regulated the EMT process in mice with
asthma, whereas DMSO did not have a similar effect.
| Table VIImRNA and protein expression levels of
E-cadherin and vimentin. |
Table VII
mRNA and protein expression levels of
E-cadherin and vimentin.
| Group | N | E-cadherin
| Vimentin
|
|---|
| mRNA | Protein | mRNA | Protein |
|---|
| Control | 8 | 1.03±0.16 | 1.00±0.00 | 1.02±0.10 | 1.00±0.00 |
| OVA | 8 | 0.38±0.33b | 0.69±0.24a | 3.10±2.14a | 3.48±2.02a |
| OVA + DMSO | 8 | 0.36±0.33 | 0.66±0.22 | 3.49±1.96 | 3.62±2.09 |
| OVA +
BMS-345541 | 8 | 0.89±0.35d | 0.94±0.16c | 1.00±0.28d | 1.05±0.78d |
| F-value | | 10.370 | 7.805 | 6.670 | 7.501 |
| P-value | | <0.001 | 0.001 | 0.002 | 0.001 |
Discussion
Asthma is a chronic inflammatory disorder, and the
predisposing factors and pathogenesis of asthma are complicated. In
the present study, mice were challenged with OVA to produce an
allergy model of asthma. Normal control and DMSO therapeutic
control groups were set up in this experiment. On the basis of this
mouse asthma model, EMT characteristics, airway inflammation,
airway remodeling, and airway reactivity were investigated in the
asthmatic mice. Additionally, the treatment effects of BMS-345541
on OVA-induced asthmatic mice were observed, and the molecular
regulation and intervention mechanisms of this treatment in the
airway remodeling of asthma were examined.
In the experiment, mice in the OVA group exhibited a
variety of asthma-related symptoms. AHR is an important
characteristic feature of asthma. Several factors contribute to its
development. The administration of Ach at progressively increasing
doses led to a marked increase in RL in the OVA asthma group. These
results showed that the OVA-induced asthmatic mice exhibited
AHR.
Airway inflammation is central to asthma
pathophysiology and considered to be the key trigger of AHR. The
development of airway inflammation involves a large number of
inflammatory cells, particularly eosinophils, infiltrating the lung
tissue, gathering around the bronchus, and periodically
infiltrating into the BALF. Eosinophils are required for the
occurrence of airway remodeling. The hypersecretion of airway
mucus, produced and secreted from goblet cells, is an important
cause of airway obstruction, asthma deterioration and fatal asthma
(27). In the present study, the
OVA group exhibited significant inflammatory cell infiltration
around the blood vessels and bronchi, submucosal edema, increased
numbers of total cells, and an increased percentage of eosinophils
in the BALF, compared with the control group. The lung tissue from
mice in the OVA group exhibited airway remodeling, including
enlargement of airway smooth muscle, basement membrane thickening,
epithelial damage, subepithelial fibrosis, goblet cell hyperplasia,
and mucus hypersecretion. All these observations suggested that the
asthmatic signs of airway inflammation and airway remodeling were
established successfully in the OVA-induced mouse model.
TGFβ has three isomers (TGFβ1, TGFβ2, and TGFβ3),
which show a high degree of homology. Among them, TGFβ1 is
important in biological functions. It remains controversial whether
TGFβ1 has different roles in the differentiation, expression of
contractile proteins, and mediation of the proliferation of airway
cells (28). The mechanisms
underlying the effects of TGFβ1 on asthma remain to be fully
elucidated. There are various hypotheses regarding the role of
TGFβ1 in asthma. The majority of studies have shown that TGFβ1 can
trigger EMT (29) and that it is
important in the onset of asthma and airway remodelling (25). TGFβ1 is a remodeling related
mediator; additionally, the increased secretion of TGFβ1 induced by
asthmatic chronic inflammation may result in EMT, which is one of
the most important mechanisms of airway inflammation and airway
remodeling in asthma (7). The
present study detected TGFβ1 concentrations in mouse serum and BALF
samples by ELISA. The mice in the OVA group exhibited increased
expression of TGFβ1 in serum and BALF. These findings indicated
that TGFβ1 was associated with the inflammation and airway
hyperactivity in asthma.
The E-cadherin and vimentin proteins are considered
to be critical biomarkers of EMT (30). The loss of E-cadherin, a cell-cell
adhesion protein, disrupts epithelial barrier function and causes
the bronchial epithelium to lose its structural stability and
polarity, which is necessary for bronchial epithelial cell
migration. The increased expression of vimentin, an intermediate
filament protein, alters the cytoskeletal protein composition and
thus drives the transformation of epithelial-derived cubic-shaped
cells into spindle-shaped fibre-like cells, which subsequently gain
the ability to migrate (31). A
previous study found that the expression of E-cadherin decreased
and that of vimentin increased in airway epithelial cells from
asthmatic mice exposed to dust mites (32). Furthermore, lower expression of
E-cadherin in patients with asthma has been associated with more
severe loss of airway barrier function, which promotes the
development of airway remodelling (33). In the present study, the
expression of E-cadherin was downregulated and the expression of
vimentin was upregulated in mice in the OVA group. These results
further confirmed the role of TGFβ1-induced EMT on airway
remodeling, and suggested that it promotes airway inflammation and
is involved in the pathogenesis of asthma.
The NF-κB signaling pathway is central in the
pathogenesis of airway inflammation in asthma (34). NF-κB is already under
consideration as a promising novel target for the treatment of
asthma. Inhibition of the NF-κB pathway by inhibitors has been
shown to attenuate allergic airway inflammation in mice (35). IKKβ, the target of BMS-345541, is
a key positive regulator of NF-κB. The inhibition of IKKβ inhibits
the activation of NF-κB. BMS-345541 is a highly selective inhibitor
of IκB kinase, which binds at an allosteric site of the enzyme and
inhibits NF-κB-dependent transcription in mice (36). BMS-345541 does not bind to the ATP
binding site, and oral BMS-345541 is well tolerated by mice. A
previous study found that mice administered with a prophylactic
dose of BMS-345541 exhibited no toxicological changes, even at the
efficacious dose of 100 mg/kg (15). Therefore, BMS-345541 is considered
safe and particularly well suited to the investigation of the use
of IKK inhibitors in murine models of inflammation. BMS-345541
remains at micromolar serum drug levels for several hours following
an oral dose and is biochemically active in mice (37). Treatment with varying doses of
BMS-345541 (10–100 mg/kg, p.o.) can inhibit inflammation in mice to
different degrees, with a high dosage (100 mg/kg) of BMS-345541
showing therapeutic effects comparable with the effects of
glucocorticoid treatments (15).
In the preliminary experiments of the present study,
normal female BALB/c mice (aged 6–8 weeks, weighing 25±3 g) were
fed with BMS-345541 or DMSO for 7 days. None of the mice showed
abnormal behavior or death during this process. In addition, lung
samples from the mice were stained with H&E to observe their
pathological features, and no changes were found from the
observations in the control mice. Therefore, BMS-345541 and DMSO
were considered safe and non-toxic. This finding was similar to the
results of previous studies (15,38).
Subsequent experiments performed in the present
study aimed to examine the therapeutic effect of BMS-345541, in
which 50 mg/kg BMS-345541 was used to treat OVA-induced asthma in
mice, with DMSO used as the therapeutic control vehicle
simultaneously. It was found that the asthma symptoms were relieved
by BMS-345541 treatment, with RL values significantly decreased,
compared with the values in the OVA and OVA + DMSO groups. In
addition, following BMS-345541 treatment, there were a series of
other changes, whereas DMSO had no similar effects. In the OVA +
BMS-345541 group, histopathological changes of lung lesions,
including epithelial thickening, bronchospasm, inflammatory cell
infiltration, and mucus secretion, were significantly reduced. The
BMS-345541 treatment group exhibited less inflammatory cell
infiltration, and the cell count and eosinophil percentage were
markedly decreased. The levels of TGFβ1 in the BALF and serum were
also decreased. BMS-345541 treatment was accompanied by the
upregulated expression of the epithelial marker E-cadherin and
downregulated expression of the mesenchymal marker vimentin, which
was verified by immunohistochemistry, RT-qPCR and western blot
analyses. These changes in EMT marker protein expression were
inhibited by BMS-345541 treatment. Therefore, these results further
confirmed that BMS-345541 inhibited the production of TGFβ1 and
ameliorated airway hyperresponsiveness via the inhibition of IKKβ.
BMS-345541 treatment protected from OVA-induced allergic airway
inflammation and attenuated EMT in airway remodeling in asthmatic
mice.
The present study had a number of limitations,
including the fact that there was no investigation of the effect of
BMS-345541 on lung immune cells, including CD3, CD4, CD8,
regulatory T, T helper 17, and B cells. There was also no
investigation of changes resulting from BMS-345541 to major
secretory mucins, including MUC5AC and MUC5B, nor were changes to
major extracellular matrix components investigated. There was also
no investigation of fundamental inflammatory mediators in the
pathogenesis of bronchial asthma, including IgE, interleukin
(IL)-4, IL-5, IL-5 receptor, and IL-13. In the future, these
limitations are to be addressed. Similar to previous studies, as
bronchial specimens are not easily acquired in mice, the lungs of
the mice were selected and harvested for histopathology to obtain
supernatants following lung homogenization and to obtain nuclear
and protein extracts to detect the mRNA and protein levels of
E-cadherin and vimentin. Future investigations aim to obtain
bronchial specimens for confirmation of the results. In addition,
only the preliminary experiments included BMS-345541 alone and DMSO
alone control groups, which were not included in later experiments
and presents another limitation of the study. Further improvements
are required in the future.
In conclusion, an allergic asthmatic mouse model can
be established successfully by OVA sensitization and challenge.
Asthmatic mice in this model have the typical airway inflammation,
airway hyperresponsiveness and airway remodeling observed in
asthma. Airway remodeling is observed in OVA-treated asthmatic
mice, in addition to evidence of EMT. The increased secretion of
TGFβ1 by asthmatic inflammation may result in EMT. TGFβ1, NF-κB and
EMT are important for asthma. The IKKβ inhibitor, BMS-345541, had a
therapeutic effect on asthma airway inflammation and airway
remodeling. The therapeutic mechanism may be achieved through
decreasing the expression of TGFβ1, inhibiting the activation of
NF-κB and reducing EMT. The results of the present study provide an
experimental basis for the potential use of IKKβ inhibitors as
therapeutics in the clinical treatment of asthma. IKKβ inhibitors
may be promising candidates for pharmacologic agents to use in
future human asthma therapy.
Acknowledgments
The authors would like to acknowledge the facilities
supported by the Clinical Laboratory and the Central Laboratory of
Jiangxi Children's Hospital, and the Central Laboratory of Jiangxi
People's Hospital.
Funding
This study was supported by the Technology and
Science Foundation of Jiangxi Province (no. 20151BBB70267).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XZ performed all the experiments and statistical
analyses. QL, GH and JW were involved in the design of the study.
QH, ZL, GW and YZ performed part of the experiments. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the Medical
Ethics Committee of Jiangxi People's Hospital (2015084).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Killeen K and Skora E: Pathophysiology,
diagnosis, and clinical assessment of asthma in the adult. Nurs
Clin North Am. 48:11–23. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Holgate ST: Pathogenesis of asthma. Clin
Exp Allergy. 38:872–897. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bergmann KC: Bronchial asthma-many types,
different therapies. Dtsch Med Wochenschr. 141:687–692. 2016.In
German. PubMed/NCBI
|
|
4
|
Hirota N and Martin JG: Mechanisms of
airway remodeling. Chest. 144:1026–1032. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gras D, Bourdin A, Chanez P and Vachier I:
Airway remodeling in asthma: Clinical and functional correlates.
Med Sci (Paris). 27:959–965. 2011.In French. View Article : Google Scholar
|
|
6
|
Fischer KD and Agrawal DK: Vitamin D
regulating TGF-β induced epithelial-mesenchymal transition. Respir
Res. 15:1462014. View Article : Google Scholar
|
|
7
|
Tian X, Tian X, Huo R, Chang Q, Zheng G,
Du Y, Chen Y and Niu B: Bacillus Calmette-Guerin alleviates airway
inflammation and remodeling by preventing TGF-β1 induced
epithelial-mesenchymal transition. Hum Vaccin Immunother.
13:1758–1764. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kalita M, Tian B, Gao B, Choudhary S, Wood
TG, Carmical JR, Boldogh I, Mitra S, Minna JD and Brasier AR:
Systems approaches to modeling chronic mucosal inflammation. Biomed
Res Int. 2013:5058642013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ijaz T, Pazdrak K, Kalita M, Konig R,
Choudhary S, Tian B, Boldogh I and Brasier AR: Systems biology
approaches to understanding Epithelial Mesenchymal transition (EMT)
in mucosal remodeling and signaling in asthma. World Allergy Organ
J. 7:132014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Loffredo LF, Abdala-Valencia H, Anekalla
KR, Cuervo-Pardo L, Gottardi CJ and Berdnikovs S: Beyond
epithelial-to-mesenchymal transition: Common suppression of
differentiation programs underlies epithelial barrier dysfunction
in mild, moderate, and severe asthma. Allergy. 72:1988–2004. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berair R and Brightling CE: Asthma therapy
and its effect on airway remodelling. Drugs. 74:1345–1369. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao Y, Tian B, Sadygov RG, Zhang Y and
Brasier AR: Integrative proteomic analysis reveals reprograming
tumor necrosis factor signaling in epithelial mesenchymal
transition. J Proteom. 148:126–138. 2016. View Article : Google Scholar
|
|
13
|
Gagliardo R, Chanez P, Profita M, Bonanno
A, Albano GD, Montalbano AM, Pompeo F, Gagliardo C, Merendino AM
and Gjomarkaj M: IκB kinase-driven nuclear factor-κB activation in
patients with asthma and chronic obstructive pulmonary disease. J
Allergy Clin Immunol. 128:635–645. e1–e2. 2011. View Article : Google Scholar
|
|
14
|
Tian F, Zhou P, Kang W, Luo L, Fan X, Yan
J and Liang H: The small-molecule inhibitor selectivity between
IKKα and IKKβ kinases in NF-κB signaling pathway. J Recept Signal
Transduct Res. 35:307–318. 2015. View Article : Google Scholar
|
|
15
|
McIntyre KW, Shuster DJ, Gillooly KM,
Dambach DM, Pattoli MA, Lu P, Zhou XD, Qiu Y, Zusi FC and Burke JR:
A highly selective inhibitor of I kappa B kinase, BMS-345541,
blocks both joint inflammation and destruction in collagen-induced
arthritis in mice. Arthritis Rheum. 48:2652–2659. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Caramori G, Adcock IM and Ito K:
Anti-inflammatory inhibitors of IkappaB kinase in asthma and COPD.
Curr Opin Investig Drugs. 5:1141–1147. 2004.PubMed/NCBI
|
|
17
|
Pattoli MA, MacMaster JF, Gregor KR and
Burke JR: Collagen and aggrecan degradation is blocked in
interleukin-1-treated cartilage explants by an inhibitor of IkappaB
kinase through suppression of metalloproteinase expression. J
Pharmacol Exp Ther. 315:382–388. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ping H, Yang F, Wang M, Niu Y and Xing N:
IKK inhibitor suppresses epithelial-mesenchymal transition and
induces cell death in prostate cancer. Oncol Rep. 36:1658–1664.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li H, Han W, Polosukhin V, Yull FE, Segal
BH, Xie CM and Blackwell TS: NF-κB inhibition after cecal ligation
and puncture reduces sepsis-associated lung injury without altering
bacterial host defense. Mediators Inflamm. 2013:5032132013.
View Article : Google Scholar
|
|
20
|
Wang ZW, Li RK, Ren Y, Liu XF, Cheng XL
and Tuo HY: Establishment and evaluation of a mouse model of
bronchial asthma with Yin deficiency syndrome. Zhongguo Ying Yong
Sheng Li Xue Za Zhi. 31:556–560. 2015.
|
|
21
|
Yao J, Jiang M, Zhang Y, Liu X, Du Q and
Feng G: Chrysin alleviates allergic inflammation and airway
remodeling in a murine model of chronic asthma. Int
Immunopharmacol. 32:24–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yao T, Ying X, Zhao Y, Yuan A, He Q, Tong
H, Ding S, Liu J, Peng X, Gao E, et al: Vitamin D receptor
activation protects against myocardial reperfusion injury through
inhibition of apoptosis and modulation of autophagy. Antioxid Redox
Signal. 22:633–650. 2015. View Article : Google Scholar :
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Ordoñez CL, Khashayar R, Wong HH, Ferrando
R, Wu R, Hyde DM, Hotchkiss JA, Zhang Y, Novikov A, Dolganov G and
Fahy JV: Mild and moderate asthma is associated with airway goblet
cell hyperplasia and abnormalities in mucin gene expression. Am J
Respir Crit Care Med. 163:517–523. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi J, Chen M, Ouyang L, Huang L, Lin X,
Zhang W, Liang R, Lv Z, Liu S and Jiang S: Airway smooth muscle
cells from ovalbumin-sensitized mice show increased proliferative
response to TGFβ1 due to up-regulation of Smad3 and TGFβRII. J
Asthma. 54:467–475. 2017. View Article : Google Scholar
|
|
26
|
Dimitropoulou C, Drakopanagiotakis F,
Chatterjee A, Snead C and Catravas JD: Estrogen replacement therapy
prevents airway dysfunction in a murine model of allergen-induced
asthma. Lung. 187:116–127. 2009. View Article : Google Scholar
|
|
27
|
Rogers DF: Physiology of airway mucus
secretion and pathophysiology of hypersecretion. Respir Care.
52:1134–1149. 2007.PubMed/NCBI
|
|
28
|
Ojiaku CA, Yoo EJ and Panettieri RA Jr:
Transforming growth factor β1 function in airway remodeling and
hyperresponsiveness. The missing link? Am J Respir Cell Mol Biol.
56:432–442. 2017. View Article : Google Scholar :
|
|
29
|
Yang YC, Zhang N, Van Crombruggen K, Hu
GH, Hong SL and Bachert C: Transforming growth factor-beta1 in
inflammatory airway disease: A key for understanding inflammation
and remodeling. Allergy. 67:1193–1202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Scanlon CS, Van Tubergen EA, Inglehart RC
and D'Silva NJ: Biomarkers of epithelial-mesenchymal transition in
squamous cell carcinoma. J Dent Res. 92:114–121. 2013. View Article : Google Scholar :
|
|
31
|
Kokkinos MI, Wafai R, Wong MK, Newgreen
DF, Thompson EW and Waltham M: Vimentin and epithelial-mesenchymal
transition in human breast cancer-observations in vitro and in
vivo. Cells Tissues Organs. 185:191–203. 2007. View Article : Google Scholar
|
|
32
|
Fischer KD, Hall SC and Agrawal DK:
Vitamin D supplementation reduces induction of
epithelial-mesenchymal transition in allergen sensitized and
challenged mice. Plos One. 11:e01491802016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
de Boer WI, Sharma HS, Baelemans SM,
Hoogsteden HC, Lambrecht BN and Braunstahl GJ: Altered expression
of epithelial junctional proteins in atopic asthma: Possible role
in inflammation. Can J Physiol Pharmacol. 86:105–112. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schuliga M: NF-kappaB signaling in chronic
inflammatory airway disease. Biomolecules. 5:1266–1283. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gu X, Zhang Q, Du Q, Shen H and Zhu Z:
Pinocembrin attenuates allergic airway inflammation via inhibition
of NF-κB pathway in mice. Int Immunopharmacol. 53:90–95. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Burke JR, Pattoli MA, Gregor KR, Brassil
PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad
J, et al: BMS-345541 is a highly selective inhibitor of I kappa B
kinase that binds at an allosteric site of the enzyme and blocks
NF-kappa B-dependent transcription in mice. J Biol Chem.
278:1450–1456. 2003. View Article : Google Scholar
|
|
37
|
MacMaster JF, Dambach DM, Lee DB, Berry
KK, Qiu Y, Zusi FC and Burke JR: An inhibitor of IkappaB kinase,
BMS-345541, blocks endothelial cell adhesion molecule expression
and reduces the severity of dextran sulfate sodium-induced colitis
in mice. Inflamm Res. 52:508–511. 2003. View Article : Google Scholar
|
|
38
|
Carneiro PJ, Clevelario AL, Padilha GA,
Silva JD, Kitoko JZ, Olsen PC, Capelozzi VL, Rocco PR and Cruz FF:
Bosutinib therapy ameliorates lung inflammation and fibrosis in
experimental silicosis. Front Physiol. 8:1592017. View Article : Google Scholar : PubMed/NCBI
|