Introduction
Nucleotides, including extracellular adenine (ATP
and ADP) and uracil (UTP, UDP, UDP-glucose and UDP-galactose)
nucleotides, are signaling molecules involved in a wide range of
physiological processes in response to pathological stimuli and
cellular injury (1,2). These actions are mediated by seven
ionotropic (P2X) and eight metabotropic (P2Y) receptor subtypes. On
the other hand, cysteinyl leukotrienes (CysLTs), including
LTC4, LTD4 and LTE4, are potent
proinflammatory mediators. CysLTs are implicated in diverse
pathologies, such as respiratory diseases, and inflammatory
conditions, including cardiovascular, gastrointestinal, and immune
disorders, as well as neurodegenerative responses, which are
mediated by CysLT receptor 1 and CysLT receptor 2 (3-7).
Both nucleotides and CysLTs are proven inducers of brain
inflammation, and have important roles in ischemic/inflammatory
conditions and neurodegenerative responses (2,7).
Currently, G protein-coupled receptor 17 (GPR17) has
been described as a G (i)-coupled dual receptor linked to P2Y and
CysLT receptors for uracil nucleotides and CysLTs, respectively
(8-11). GPR17 is highly expressed in brain,
heart, and kidney tissues undergoing ischemic injury (9,12).
In the brain, GPR17 is expressed in neurons, oligodendrocyte
precursor cells (OPCs), and ependymal cells, but not in astrocytes,
under physiological conditions (9,13-16). Following ischemic, traumatic, or
demyelinating diseases, GPR17 is transiently and sequentially
upregulated in neurons, microglia/macrophages, and oligodendrocyte
precursors (9,14,17-19). Mounting evidence highlights the
role of GPR17 in modulating oligodendrocyte precursor maturation,
and GPR17 is considered a novel target for innovative therapeutic
approaches to demyelinating diseases, such as multiple sclerosis
(19-23).
Increasing evidence confirms that GPR17 is also
involved in a broad range of pathological processes, such as
cerebral ischemic injury and spinal cord damage (9,14,17,18). In acute ischemic stroke, GPR17
upregulation in neurons is associated with enhanced cell death,
while its knockdown markedly attenuates ischemic damage (9,14,17). Beside its role in neuronal injury,
GPR17 is a key player in brain remodeling and repair. In chronic
ischemic stroke, activated microglia/macrophages induce GPR17
upregulation, and its inhibition prevents the progression of
chronic ischemic injury by reducing brain atrophy while
ameliorating chronic neuronal death and micro-gliosis (14,17). Similar expression and modulation
of GPR17 were demonstrated in traumatic spinal cord lesions
(18). Thus, these findings
indicated that GPR17 might become a new therapeutic target in
neurodegenerative diseases.
However, whether ischemia-like neuronal injury and
microglial activation in vitro are mediated by GPR17
activation remains unclear. A better understanding of the related
mechanism is crucial for developing effective therapeutics for
ischemic stroke. The present study comprehensively determined the
role of GPR17 in neuronal injury and microglial activation in
oxygen-glucose deprivation/recovery (OGD/R)-, LTD4-, or
UDP-induced injury in vitro. The present study aimed to
address the following questions: Determine the spatiotemporal
profiles and localization of GPR17 in different cell cultures in
vitro; assess whether GPR17 is correlated with neuronal injury
and microglial activation following ischemia-like injury in
vitro; and explore the mechanism by which GPR17 may regulate
ischemia-like injury.
Materials and methods
Cell culture and OGD/R
Primary neurons were obtained from cerebral cortices
of neonatal SD rats (Experimental Animal Center, Zhejiang Academy
of Medicine Sciences, Hangzhou, China), as described in previous
studies (5,24). Briefly, cerebral cortices were
dissected and digested with 0.25% EDTA-free trypsin (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) at 37°C for 10 min.
Then, the dissociated cells were immediately seeded onto
poly-L-lysine (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
pre‑coated flasks, and incubated in high glucose DMEM (Gibco;
Thermo Fisher Scientific, Inc.) containing 10% fetal bovine serum
(FBS; Hangzhou Sijiqing Biotechnology Co., Hangzhou, China), 10%
horse serum (Gibco; Thermo Fisher Scientific, Inc.), 2 mM
glutamine, 100 µg/ml streptomycin, and 100 units/ml penicillin
(Sigma-Aldrich; Merck KGaA) for 24 h under normal culture
conditions. After 24 h of culture, the medium was changed to high
glucose DMEM containing 5% horse serum, 2 mM glutamine, 100
units/ml penicillin, 100 µg/ml streptomycin, 0.04% B27, and 0.01%
N2 (Gibco; Thermo Fisher Scientific, Inc.). The day of plating was
recorded as DIV 0 in vitro. On DIV 3, 10 µM cytosine
arabinoside (Sigma-Aldrich; Merck KGaA) was added for 24 h to
inhibit the growth of glial cells in cultured neurons; the medium
was refreshed every 3 days. On DIV 10, >95% of the cultured
cells were neurons, as determined by immunofluorescent staining for
microtubule-associated protein-2 (MAP-2; Millipore, Billerica, MA,
USA).
For the neuron-glial mixed cultures of cortical
cells, similar procedures were used, except for cytosine
arabinoside supplementation, as previously described (25). On DIV 10, ~28% of the cultured
cells were neurons, 7% were microglia and most of the remaining
cells were astrocytes, as assessed by immunofluorescent staining
for anti‑MAP‑2 (1:200; cat. no. AB5622; Millipore), anti-glial
fibrillary acidic protein (GFAP; 1:800; cat. no. MAB3402;
Millipore), and anti-ionized calcium-binding adapter molecule 1
(Iba1; 1:1,000; cat. no. 019-19741; Wako Pure Chemical Industries,
Ltd., Osaka, Japan) antibodies.
Primary astrocytes were prepared from cerebral
cortices of neonatal SD rats, as previously described (4,26).
Briefly, cerebral cortices were dissected and digested with 0.25%
trypsin for 15 min. Then, the dissociated cells were immediately
plated onto poly‑L‑lysine pre‑coated flasks, and incubated in high
glucose DMEM containing 10% FBS, 2 mM glutamine, 100 U/ml
penicillin and 100 µg/ml streptomycin under normal culture
conditions. Thereafter, the medium was refreshed every 3 days. On
DIV 14, confluent cultures were shaken overnight at 250 rpm at 37°C
to reduce microglial contamination. Subsequently, adherent cells
were digested with 0.25% trypsin and replated in the growth medium.
More than 95% of the cultured cells were astrocytes, as assessed by
immunofluorescent staining for GFAP.
Primary microglia were prepared from cerebral
cortices of neonatal SD rats, as described previously (5,27).
Briefly, cerebral cortices were digested with 0.25% trypsin for 10
min; then, the dissociated cells were immediately plated onto
poly‑L‑lysine pre‑coated flasks in antibiotics‑free minimum Eagle's
medium (Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS
at 37°C. On DIV 10, the microglia cells were harvested by shaking
for 30 min at 250 rpm. The cells were centrifuged and reseeded in
the growth medium. More than 95% of the cultured cells were
microglia, as determined by immunofluorescent staining for the
microglial marker Iba-1.
To evaluate the effect of the microglial conditioned
medium on primary neurons, cell supernatants were collected from
the microglia pretreated with or without OGD/R as well as small
interfering (si)RNA (5). The
conditioned media was collected and centrifuged to remove cell
debris. The primary neurons were incubated with the conditioned
medium from the above microglia for 24 h. Neuronal death was
assessed subsequently.
For OGD (4,5,28),
cells were rinsed twice and cultured in Earle's solution without
glucose. Then, the cells were incubated in an anaerobic chamber
containing 95% N2 and 5% CO2 at 37°C for 1 h.
Control cells were incubated in Earle's solution containing 25 mM
glucose under normal culture conditions for 1 h. Following OGD, the
cells were returned to the common incubator and cultured in regular
medium to allow recovery.
The nonselective GPR17 agonists leukotriene D4
(LTD4) and uridine 5′-diphosphate (UDP; both Sigma-Aldrich; Merck
KGaA) were utilized for receptor activation. LTD4 or UDP
was added to the medium at final concentrations of 0.1‑1,000 nM and
0.1-1,000 µM for 24 h, respectively.
All animal experiments were performed in accordance
with the National Institutes of Health Guide for Care and Use of
Laboratory Animals. The experimental protocols were approved by the
Ethics Committee of Laboratory Animal Care and Welfare, School of
Medicine, Zhejiang University (Hangzhou, China).
GPR17 silencing by RNA interference
In the present study, siRNA was used to effectively
downregulate GPR17 protein levels, for lack of a selective GPR17
antagonist. The specifically designed target sequence for rat GPR17
was CCG TAT AGA GAA GCA CCT CAA (GenePharma Co., Ltd., Shanghai,
China) (17,29). To rule out possible non-specific
effects, a non-effective sequence (sequence, CCU ACG CCA CCA AUU
UCG UTT) was used as negative control (NC). Transfection of siRNA
duplexes was performed according to the manufacturer's
instructions. Briefly, 24 h prior to transfection, the medium was
changed to appropriate antibiotic-free medium containing 10% FBS.
Then, GPR17 siRNA or NC siRNA was transiently transfected with
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) at
a final concentration of 100 nM for 6 h; the cells were cultured
for an additional 48 h prior to exposure to OGD/R, UDP or
LTD4.
MTT and lactate dehydrogenase (LDH)
release assays
Following treatments, cell viability was evaluated
by the MTT assay. Briefly, MTT (Sigma‑Aldrich; Merck KGaA) was
added to each well at a final concentration of 0.5 mg/ml and
incubated for 4 h at 37°C. After careful removal of the medium, 100
µl DMSO (Sigma-Aldrich; Merck KGaA) was added for 10 min with
shaking. Finally, absorbance was measured at 490 nm on a microplate
reader (Elx800; BioTek Instruments, Inc., Winooski, VT, USA). LDH
released into the medium was quantified with a LDH detection kit
(Jiancheng Co., Ltd., Nanjing, China), according to the
manufacturer's instructions. LDH was detected at 450 nm on a
microplate reader. Data were expressed as % of controls.
Immunofluorescence
Cells on cover slips in 24-well plates were fixed in
cold methanol for 5 min, followed by reaction with 10% normal goat
serum (Zhongshan Biotechnology Co., Beijing, China) for 2 h to
block non‑specific IgG binding. Then, the cells were incubated with
anti-MAP-2 (1:200; cat. no. AB5622; Millipore) anti-NeuN (1:500;
cat. no. MAB377; Millipore), anti-GFAP (1:800; cat. no. MAB3402;
Millipore), anti-Iba1 (1:1,000; cat. no. 019-19741; Wako Pure
Chemical Industries, Ltd.), and anti-GPR17 (1:500; lab made)
primary antibodies at 4°C overnight. Following rinsing with PBS,
the cells were incubated with Cy3- or fluorescein isothiocyanate
(FITC)-conjugated secondary antibody (1:200; cat. nos. AP187C and
AP124F; Millipore) for 2 h at room temperature. After mounting with
anti-fade medium (Invitrogen; Thermo Fisher Scientific, Inc.), the
stained cells were visualized by fluorescence microscopy (BX51;
Olympus Corporation, Tokyo, Japan). Negative control cover slips
were incubated with normal goat serum instead of primary antibody.
The polyclonal rabbit antibody against rat GPR17 used in the
immunofluorescence is specific as reported (9,14,17,30).
Assessment of cell death
To quantify cell death, cells grown on cover slips
in 24-well plates were stained with 10 µg/ml propidium iodide (PI;
Sigma-Aldrich; Merck KGaA) for 10 min. Following rinsing and
fixation in cold methanol for 5 min, immunofluorescent staining was
performed by incubation with antibodies against NeuN (neuron
marker), GFAP (astrocyte marker), and Iba-1 (microglia marker),
respectively. After mounting with anti-fade medium, images were
captured by fluorescent microcopy. The % of cell death was
determined as the number of red PI-positive cells to total cells
(as measured by DAPI staining). An observer blinded to the
experimental treatments counted the cells.
Immunoblotting
Total cellular protein extraction was performed
according to the manufacturer's instructions (Kangcheng Co., Ltd.,
Shanghai, China). Briefly, the cells were washed with PBS and lysed
in Cell and Tissue Protein Extraction buffer on ice for 30 min.
Then, the homogenate was collected, centrifuged at 12,000 x g for
30 min at 4°C, and protein amounts were quantified by Coomassie
brilliant blue staining. Equal amounts of protein (60 µg) for each
sample were separated by 10% SDS-PAGE and transferred onto
nitrocellulose membranes. The membranes were incubated with either
goat polyclonal antibody against GPR17 (1:200; cat. no. sc-74791;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) or mouse
monoclonal antibody against GAPDH, (1:5,000; cat. no. KC-5G4;
Kangcheng Co., Ltd., Shanghai, China) overnight at 4°C.
Subsequently, the membranes were treated with IRDye-800CW donkey
anti-goat IgG (1:3,000; cat. no. 926-32214; LI-COR Biosciences,
Lincoln, NE, USA) or IRDye-800CW goat anti-mouse IgG (1:5,000; cat.
no. 610-132-121; Rockland Immunochemicals, Inc., Gilbertsville, PA,
USA) for 2 h at room temperature. Images were scanned on an Odyssey
fluorescence system (LI‑COR Biosciences). Optical densities of
GPR17 (41 kDa) and GAPDH (36 kDa) were quantitatively assessed by
Quantity One software (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Microglial phagocytosis
To evaluate the phagocytic activity of primary
microglia, 1 mm-diameter fluorescent carboxylate‑modified
microspheres (Millipore) were added to the cells for 1 h.
Subsequently, the cells were rinsed in PBS, immediately digested
and resuspended, and transferred to flow‑cytometry tubes. Finally,
fluorescence was detected in the FL‑3 channel by flow cytometry (BD
Biosciences, Franklin Lakes, NJ, USA).
To evaluate the phagocytosis of microglia in mixed
cortical cells, cover slips were placed in 24-well plates prior to
cell seeding. Fluorescent microspheres were added to the cells for
1 h following treatment. After fixation and rinsing with PBS, the
cells were incubated with 10% normal goat serum for 2 h at room
temperature. To label the microglia, cells were treated with
primary rabbit polyclonal antibody targeting Iba‑1 (1:1,000) at 4°C
overnight. Following two washes, the cells were incubated with
FITC-conjugated secondary antibody for 2 h at room temperature.
This was followed by mounting with anti-fade medium; phagocytosis
and the levels of the microglial marker Iba‑1 were captured by
fluorescence microscopy. Image analysis (31,32) was performed based on a
semi-quantitative method using ImageJ software (version 1.61;
National Institutes of Health, Bethesda, MD, USA).
Inflammatory cytokine release
measurement
Release of inflammatory cytokines, including tumor
necrosis factor‑α (TNF-α) and interleukin-1β (IL-1β), was detected
with TNF-α (cat. no. RTA00; R&D Systems, Inc., Minneapolis, MN,
USA) and IL-1β (cat. no. EK0393; Boster Co., Ltd., Wuhan, China)
ELISA kits, respectively. In brief, cultured cell supernatants were
collected following treatment, and cleared by centrifugation (670 x
g for 10 min). ELISA was performed according to the manufacturers'
instructions. Optical densities were measured at 450 nm within 30
min, and the amounts of released cytokine were derived from
standard curves.
Statistical analysis
Data are mean ± standard error of the mean. Groups
were compared by one-way analysis of variance, followed by
Dunnett's post hoc test, using SPSS 10.0 for Windows (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression patterns of GPR17 in primary
cortical cells
To determine the localization of GPR17 in
vitro, double immunostaining was performed to assess the
colocalization of GPR17 with the specific markers NeuN (neurons),
GFAP (astrocytes), and CD11b (microglia) in neuron-glial mixed
cultures with or without OGD/R. In control cells, most GPR17
expression was observed in NeuN-positive neurons, with low amounts
in CD11b-positive microglia (Fig.
1A). Following OGD/R, GPR17 immunoreactivity was increased and
evenly localized in NeuN-positive neurons and CD11b-positive
microglia (Fig. 1A). By contrast,
no colocalization was observed in GFAP-positive astrocytes, with or
without OGD/R (Fig. 1A).
Consistent with the immunostaining results, western blot analysis
demonstrated that the GPR17 protein expression levels were
significantly increased following exposure to OGD for 1 h and
recovery for 24 or 72 h (Fig.
1B).
 | Figure 1GPR17 distribution and expression in
neuron-glial mixed cultures of cortical cells. (A) Representative
images from microscopy analysis showing immunoreactivity to
anti‑GPR17 antibody (red) and the specific markers NeuN (neurons),
CD11b (microglia) and GFAP (astrocytes) in neuron‑glial mixed
cultures of cortical cells, with or without ischemia-like injury.
Ischemia-like injury was achieved by OGD for 1 h and recovery for
24 h. Scale bar, 50 µm. (B) Western blotting revealed that GPR17
protein levels increased 24 and 72 h following recovery from OGD
for 1 h. (C) Reduced GPR17 expression in neuron‑glial mixed
cultures of cortical cells following treatment with specific GPR17
siRNA, but not a randomly designed NC siRNA. Data are presented as
mean ± standard error of the mean (n=5). **P<0.01
compared with control. GPR17, G protein‑coupled receptor 17; GFAP,
glial fibrillary acidic protein; OGD, oxygen-glucose deprivation;
si, small interfering; NC, negative control; R, recovery. |
Effects of GPR17 knockdown on
OGD/R‑induced injury in neuron‑glial mixed cultures of cortical
cells
To further confirm the role of GPR17 in
vitro, ischemia-like injury was assessed in neuron-glial mixed
cultures, a cellular environment simulating the intact brain.
Western blot analysis indicated that GPR17 siRNA treatment
successfully downregulated GPR17 at the protein level, not only in
neuron-glial mixed cultures (Fig.
1C), but also in primarily cultured neurons and microglia (data
not shown); these findings were also verified by immunostaining
(data not shown), indicating consistent GPR17 knockdown.
The results demonstrated that OGD for 1 h followed
by recovery for 24 h resulted in decreased cell viability and
increased LDH release in neuron-glial mixed cultures; these effects
were attenuated by GPR17 siRNA (Fig.
2C and D). Thus, GPR17 knockdown exhibited remarkable
protective effects on OGD/R-induced ischemic injury.
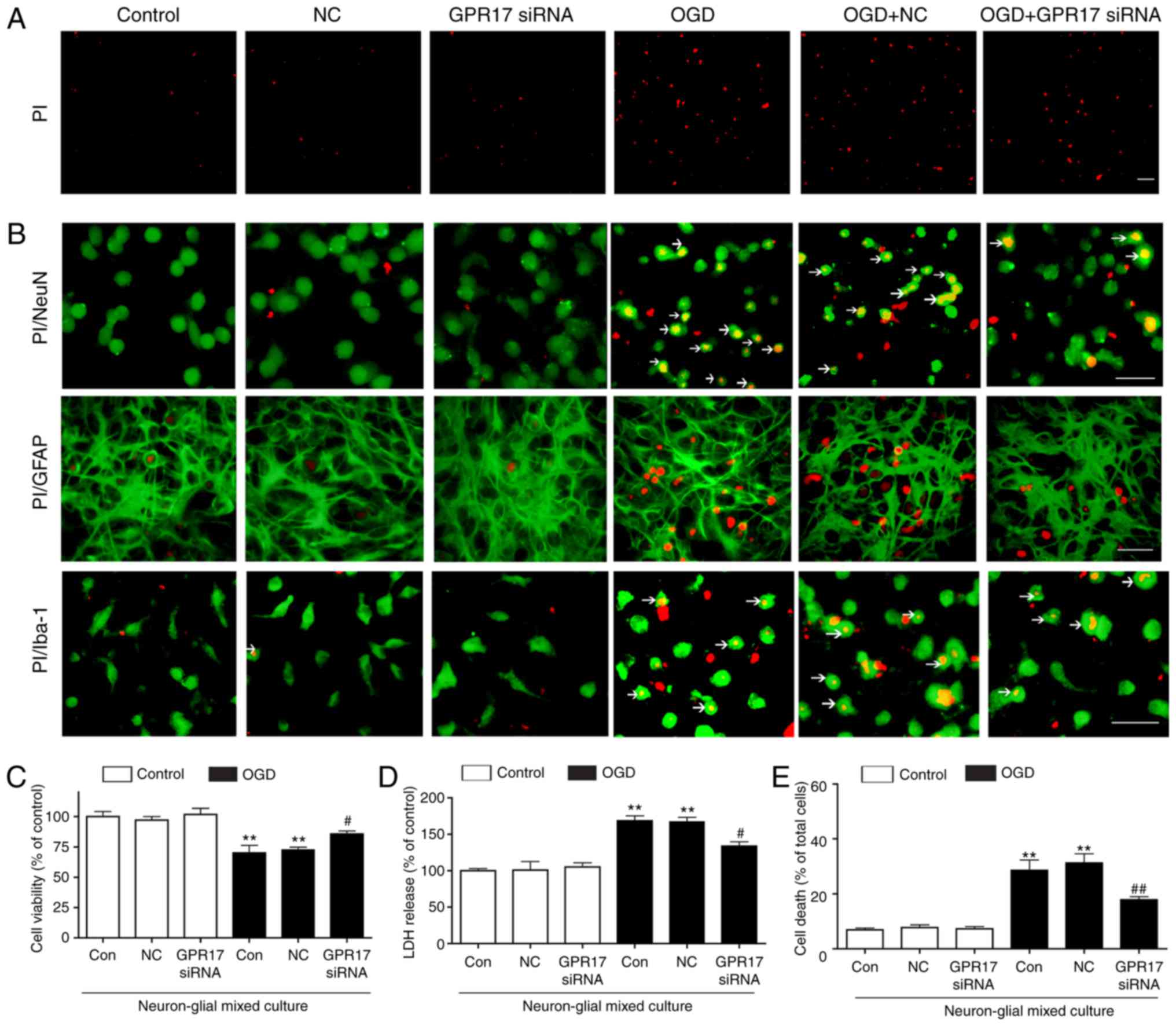 | Figure 2GPR17 mediates OGD/R-induced cell
injury in neuron-glial mixed cultures of cortical cells. (A) Cell
death in the neuron-glial mixed cultures was determined by PI
staining (red). (B) Representative images from microscopy analysis
showing co‑staining of PI (red) with the specific markers NeuN,
GFAP, or Iba‑1 (green), with or without OGD/R. Arrows indicated the
co-staining of PI with the specifc marker (NeuN, GFAP, or Iba-1).
(C) Cell viability was measured by MTT assay. (D) LDH release. (E)
Quantification of PI staining as a measure of cell death. Data are
presented as mean ± standard error of the mean (n=6‑8).
**P<0.01 compared with control; #P<0.05
and ##P<0.01 compared with the OGD alone. Scale bar,
50 µm. GPR17, G protein-coupled receptor 17; OGD/R, oxygen-glucose
deprivation/recovery; PI, propidium iodide; GFAP, glial fibrillary
acidic protein; LDH, lactate dehydrogenase; si, small interfering;
NC, negative control. |
PI/Hoechst 33342 staining demonstrated that OGD for
1 h followed by recovery for 24 h (OGD/R) induced obvious cell
death (such as necrosis and pyroptosis) in neuron-glial mixed
cultures, but apoptosis in very low levels (Fig. 2A). Next, cell death was assessed
by double immunostaining with PI to char-acterize cell injury. In
neuron-glial mixed cultures, OGD/R induced remarkable cell death in
neurons and lower levels in Iba-1-positive microglia, with nearly
no PI/GFAP-double positive astrocytes observed (Fig. 2B). Application of GPR17 siRNA
significantly ameliorated OGD/R‑induced increase of cell death
(Fig. 2A and E); the negative
control siRNA had no such effects. Therefore, GPR17 knockdown
exerted obvious protective effects on OGD/R-induced ischemic cell
damage in neuron-glial mixed cultures. These results indicated that
GPR17 might mediate OGD/R-induced ischemic injury in neuron-glial
mixed cultures.
Effects of GPR17 knockdown on
agonist‑induced cell injury in neuron‑glial mixed cultures of
cortical cells
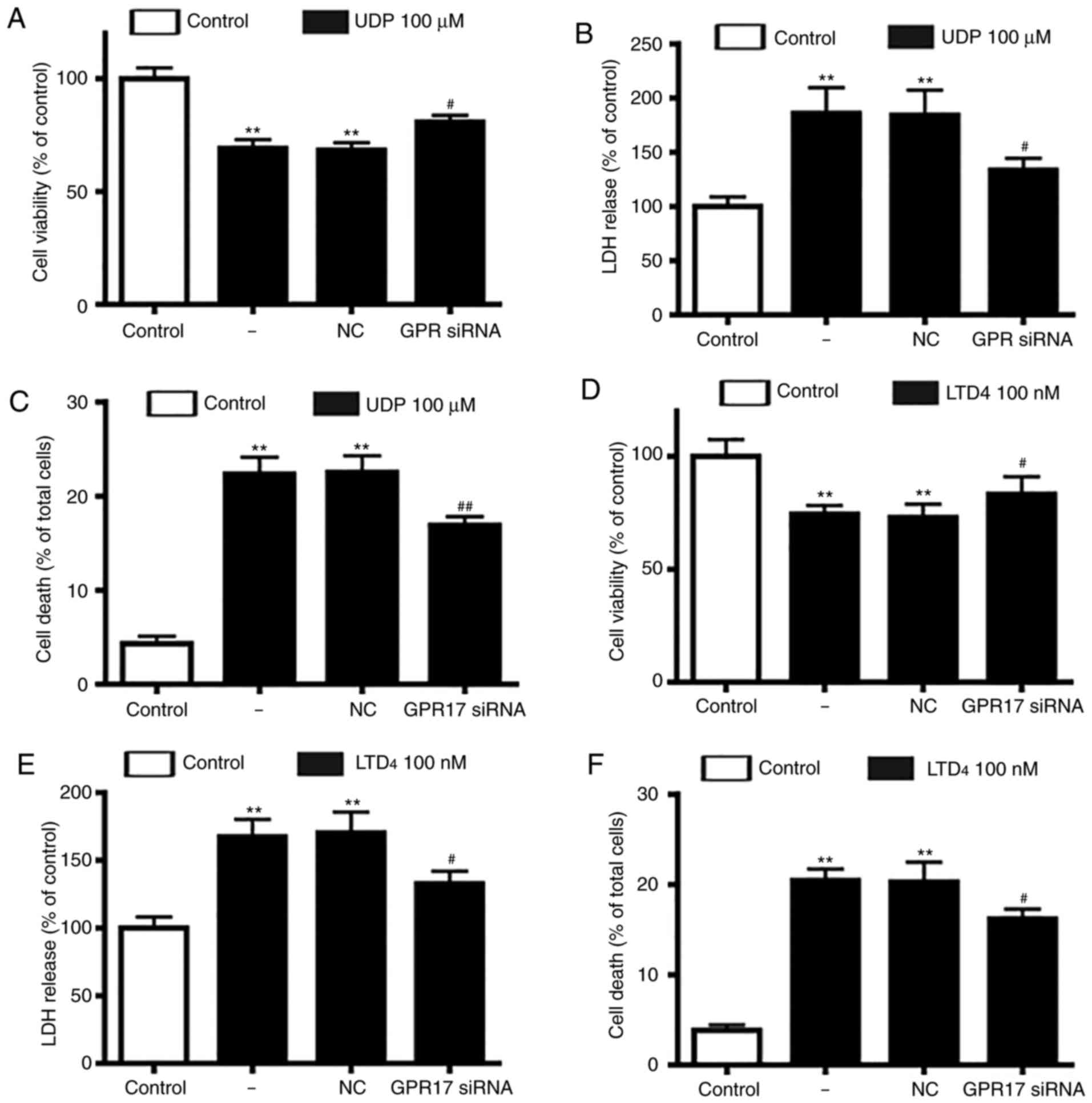
Next, the present study assessed whether GPR17
agonists had a direct injury effect in neuron-glial mixed cultures,
similar to OGD/R-induced ischemic injury. Following treatment with
UDP (10-1,000 µM) or LTD4 (10-1,000 nM), it was observed
that GPR17 agonists decreased cell viability and increased LDH
release in a dose and time-dependent manner (data not shown).
Moderate amounts of LTD4 (100 nM) or UDP (100 µM) for 48
h were selected as the optimal conditions in subsequent
experiments. Notable, GPR17 siRNA silencing, and not negative
control siRNA, significantly alleviated the reduced cell viability
and enhanced LDH release induced by UDP (Fig. 3A and B) or LTD4
(Fig. 3D and E).
Subsequently, the present study evaluated whether
GPR17 was involved in LTD4 or UDP-induced cell death.
The results demonstrated that LTD4 or UDP induced
significant cell death in neuron-glial mixed cultures, and
application of GPR17 siRNA significantly ameliorated the
LTD4 or UDP-induced cell death (Fig. 3C and F); by contrast, negative
control siRNA did not affect cell death. These findings indicated
that GPR17 knockdown could alleviate LTD4 or UDP-induced
ischemia-like injury in neuron-glial mixed cultures.
Effects of GPR17 RNA knockdown on
OGD/R‑induced microg‑ lial activation in neuron‑glial mixed
cultures of cortical cells
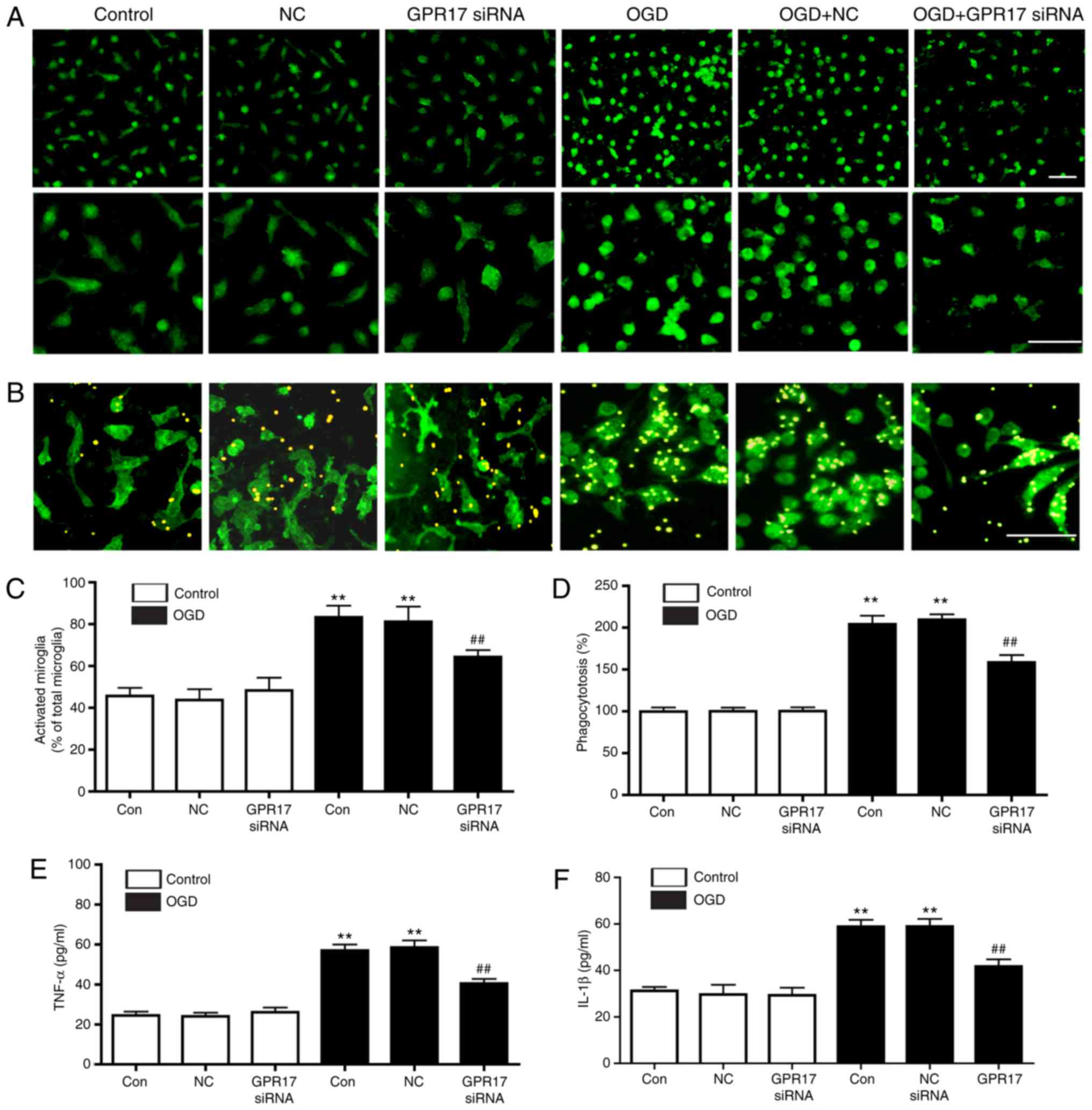
To clarify the regulation of microglial activation
by GPR17, changes of microglial morphology, phagocytosis, and
release of proinflammatory cytokines, as indicators of microglial
activation in neuron-glial mixed cultures, were assessed. In
control conditions, ramified Iba-1-positive microglia persisted at
a relatively high ratio, whereas microglia with activated
appearance (rounded or amoeboid macrophage-like) progressively
increased following exposure to OGD/R. GPR17 siRNA treatment
ameliorated the ratio of activated microglia, whereas the negative
control siRNA had no effect (Fig. 4A
and C). In addition, OGD/R significantly enhanced microglial
phagocytosis, an effect significantly inhibited by GPR17 siRNA as
assessed microscopically (Fig. 4B and
D). Furthermore, OGD/R promoted the release of inflammatory
cytokines TNF-α (Fig. 4E) and
IL-1β (Fig. 4F), and this effect
was significantly inhibited by GPR17 siRNA (Fig. 4E and F). These findings indicated
that GPR17 mediated microglial activation, including morphological
changes, phagocytosis and inflammatory cytokine release in
neuron‑glial mixed cultures.
Effects of GPR17 knockdown on
OGD/R‑induced cell injury in primary neurons and astrocytes
To explore the detailed mechanisms involved in the
protective effects of GPR17 knockdown against OGD/R-induced
ischemia-like injury, primary neurons and astrocytes were assessed,
respectively.
In primary astrocyte cultures, GPR17 was not even
expressed following exposure to OGD/R (data not shown). Consistent
with previous reports (26), OGD
for 1 h and recovery for 24 h (simulating moderate injury) did not
induce significant changes in cell viability and LDH release in
primary astrocyte cultures, and GPR17 siRNA did not affect
astrocyte viability, LDH release and cell damage following exposure
to OGD/R (Fig. 5A and C–E). GPR17
or OGD/R did not induce detectable necrosis in primary astrocyte
cultures (Fig. 5E). In primary
neuron cultures, OGD for 1 h and recovery for 24 h resulted in
decreased neuronal viability, and increased LDH release; however,
in contrast to the findings from neuron‑glial mixed cultures, the
OGD/R-induced injury was not attenuated by GPR17 siRNA (Fig. 5B, F and G). In addition, OGD/R
induced remarkable neuronal death in primary neuron cultures,
however, increased neuronal injury was not inhibited by GPR17 siRNA
(Fig. 5B and H).
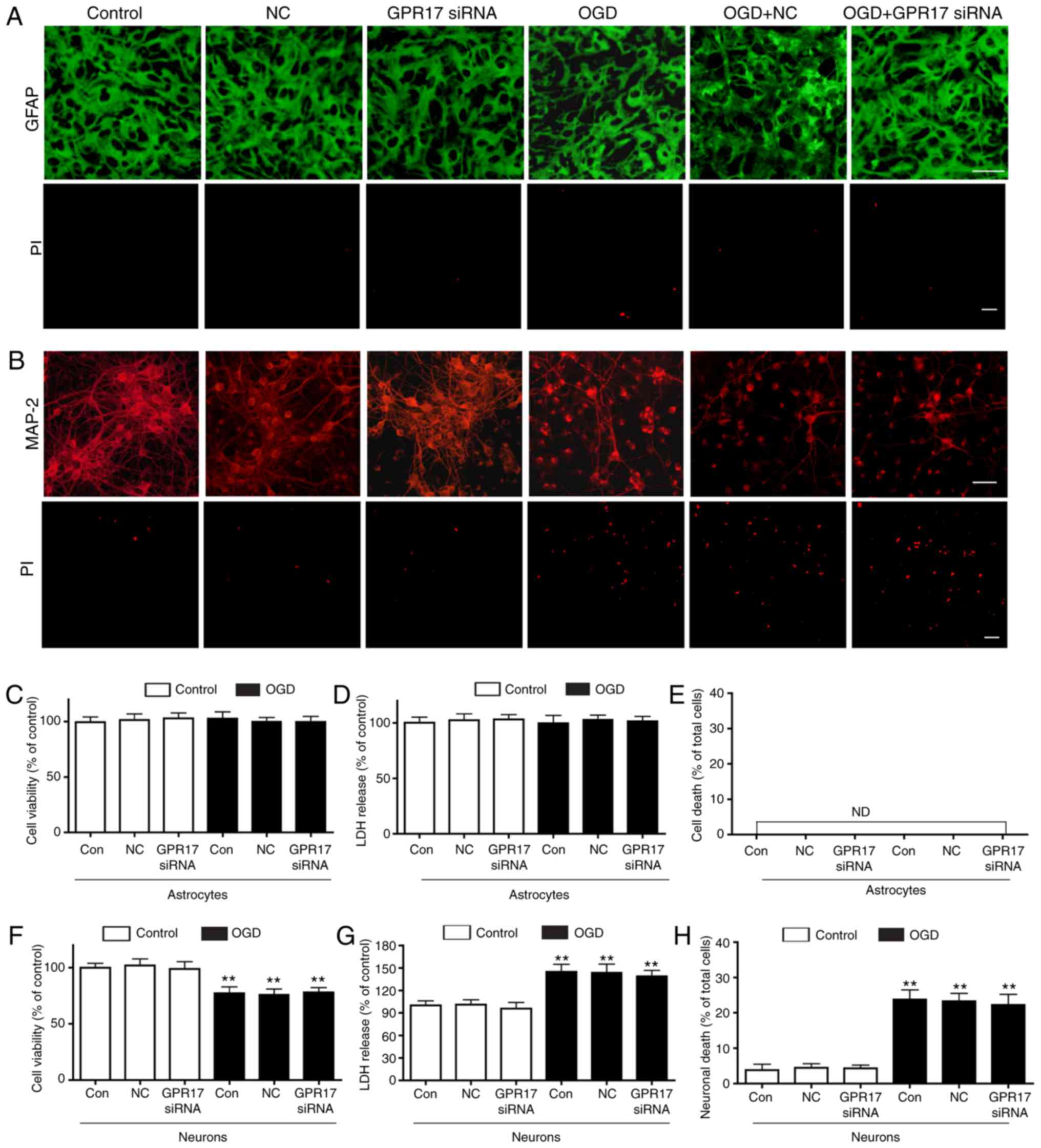 | Figure 5GPR17 is not involved in
OGD/R-induced injury in primary neurons or astrocytes.
Representative images from microscopy analysis showing the
morphology of (A) astrocytes and (B) neurons immunostained with
GFAP or MAP-2, respectively. Cell death was evaluated by PI
staining. Scale bar, 50 µm. (C) Cell viability was detected by the
MTT assay in astrocytes. (D) LDH release in astrocytes. (E)
Quantification of cell death by PI staining in astrocytes. (F) Cell
viability was detected by the MTT assay in neurons. (G) LDH release
in neurons. (H) Quantification of cell death by PI staining in
neurons. Data are presented as mean ± standard error of the mean
(n=6-8). **P<0.01 compared with control. GPR17, G
protein-coupled receptor 17; OGD/R, oxygen-glucose
deprivation/recovery; GFAP, glial fibrillary acidic protein; MAP‑2,
microtubule‑associated protein‑2; PI, propidium iodide; LDH,
lactate dehydrogenase; si, small interfering; NC, negative control;
ND, not detectable. |
These findings indicated that the protective effects
of GPR17 knockdown against OGD/R-induced ischemic neuronal injury
were not directly on neurons, at least following moderate OGD/R
treatment, which cannot explain the in vitro findings for
neuron‑glial mixed cultures, as well as in previous in vivo
data (9,14,17). Therefore, it is possible that
GPR17 might regulate ischemic neuronal injury via interactions
between neurons and surrounding cells, such as astrocytes and
microglia. However, the results demonstrated that astrocytes were
not involved in the protective effects of GPR17 knckdown. Thus, it
can be speculated that other cells in the mixed cortical cell
cultures used in the present study, perhaps microglia, may be
associated with the protective effects of GPR17 knockdown, which
might eventually cause neuronal injury.
Effects of GPR17 knockdown on
OGD/R‑induced microglial activation in primary microglia
cultures
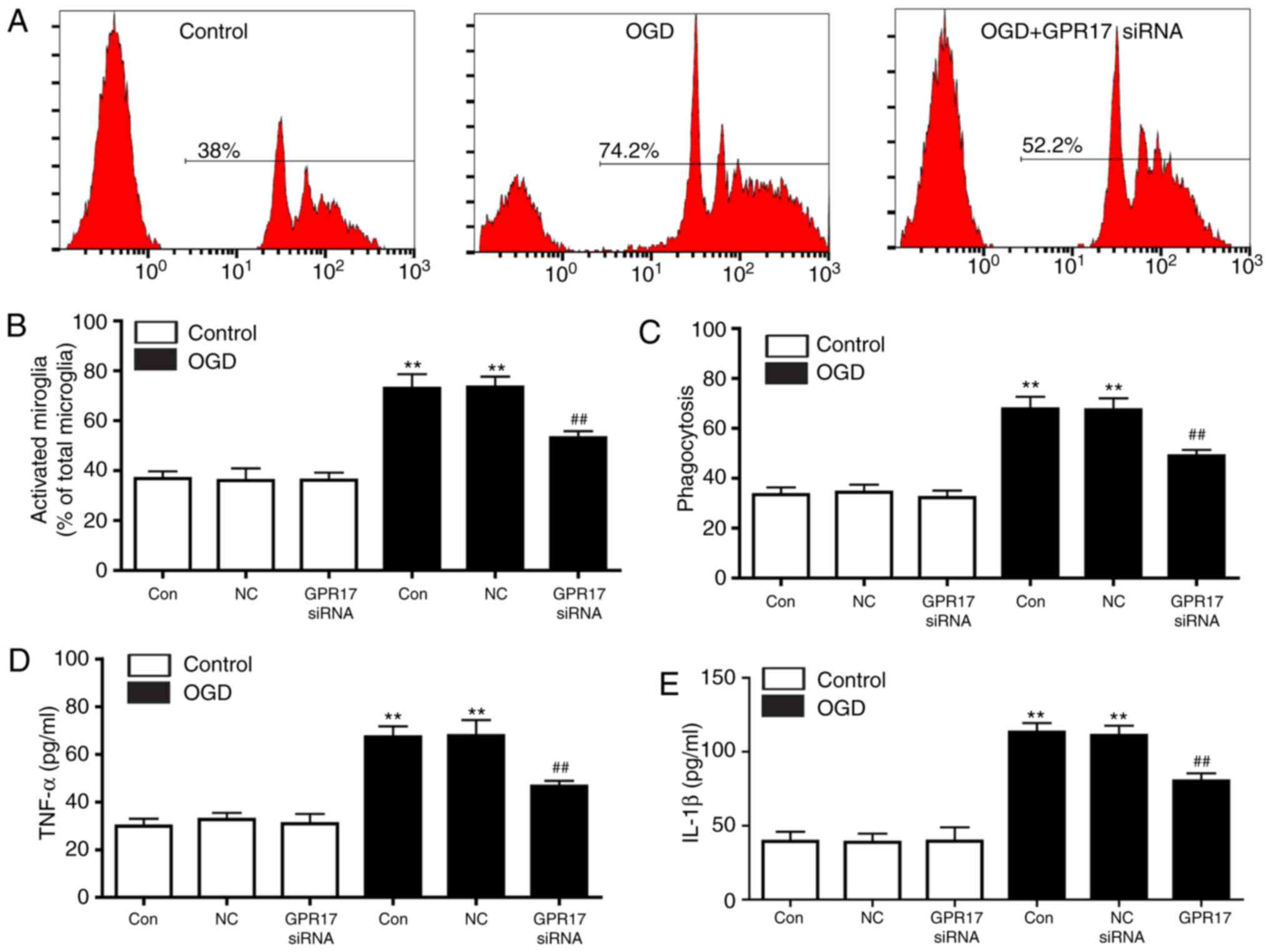
To explore whether GPR17 directly regulates
microglial activation, microglial phagocytosis and proinflammatory
cytokine release were assessed in primary microglia cultures. Flow
cytometry analysis demonstrated that OGD/R significantly enhanced
microglial phagocytosis, and this effect was significantly
suppressed by GPR17 siRNA (Fig. 6A
and C). In addition, in control conditions, microglia with
activated appearance (rounded or amoeboid macrophage-like)
persisted at a relatively low ratio, whereas activated microglia
progressively increased following exposure to OGD/R (Fig. 6B). GPR17 siRNA treatment
ameliorated the ratio of activated microglial, whereas the negative
control siRNA had no effect (Fig.
6B). Finally, OGD/R significantly increased the release of the
proinflammatory cytokines TNF‑α and IL-1β, however, this effect was
significantly inhibited by GPR17 siRNA (Fig. 6D and E). These findings indicated
that GPR17 knockdown directly inhibited OGD/R-induced microglial
activation in primary microglia cultures.
Effects of GPR17 knockdown on
microglial‑conditioned medium‑induced neuronal injury
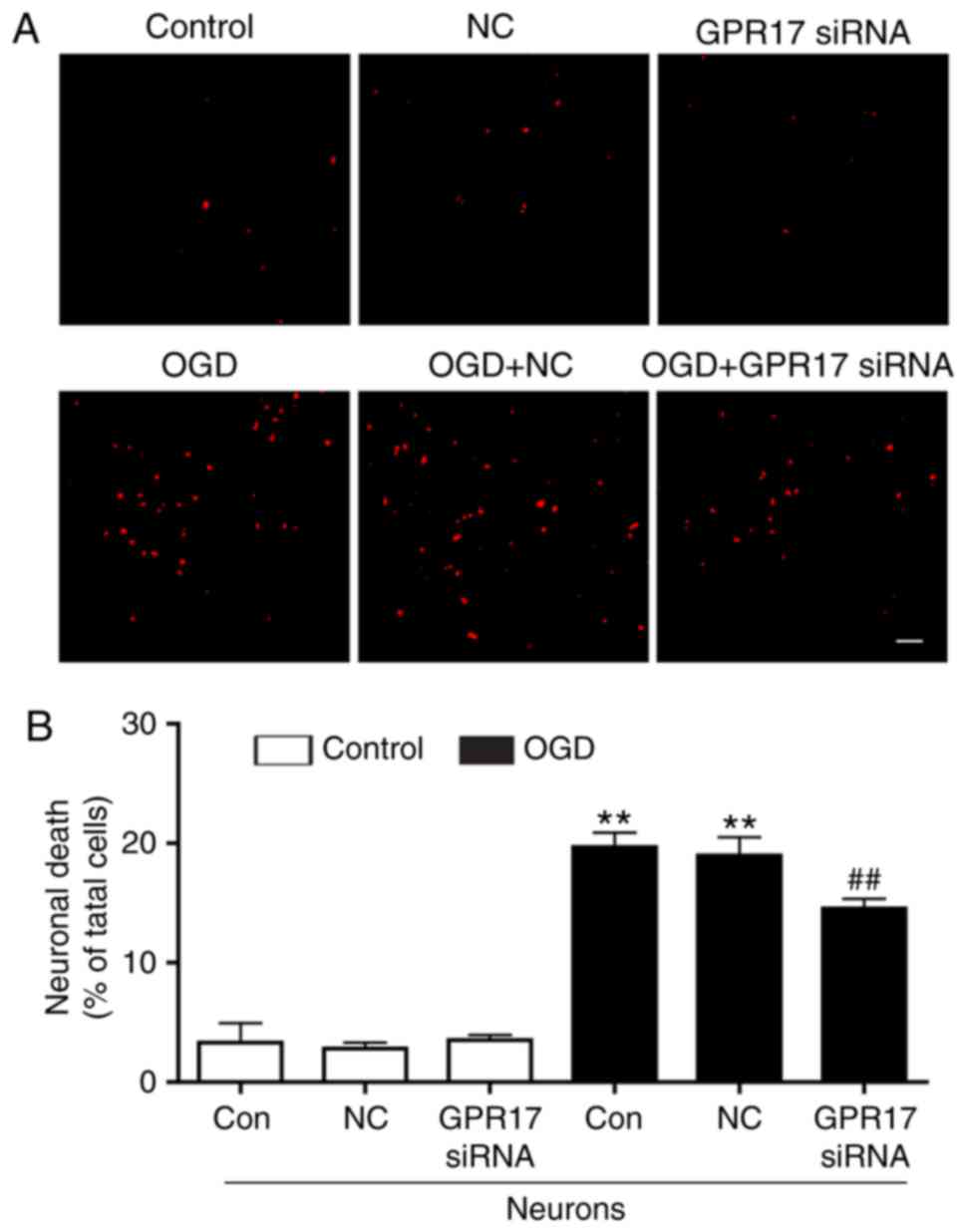
Finally, neuronal death was investigated in primary
neurons induced by the conditioned medium from microglia,
pretreated with or without OGD/R, as well as siRNA. As illustrated
in Fig. 7, the conditioned medium
pretreated with OGD/R was able to induce neuronal death, and this
effect was significantly inhibited by GPR17 siRNA pretreatment,
while control siRNA had no effect (Fig. 7A and B).
Taken together, these findings suggested that GPR17
might directly mediate microglial activation, and this effect might
be the primary way for regulating ischemic neuronal injury in mixed
cultures of cortical cells. However, whether GPR17 regulates
neuronal injury mediated by oligodendrocytes remains to be
investigated.
Discussion
The present study confirmed and extended previous
studies which reported that GPR17 upregulation is spatiotemporally
correlated with neuronal injury and microglial activation in
vivo. In addition, the present study provided further evidence
that GPR17 knockdown attenuated ischemia-like neuronal injury only
in neuron-glial mixed cultures of cortical cells, and not in
primary neuron cultures, indicating the participation of glia cells
in the process. In addition, GPR17 knockdown was demonstrated to
suppress OGD/R-induced microglial activation, inducing phagocytosis
and inflammatory cyto-kine release, in both neuron-glial mixed
cortical cells and primary microglia. An important finding of the
present study was that the conditioned medium from microglia
pretreated with OGD/R induced neuronal death, and the injury was
significantly inhibited by GPR17 siRNA pretreatment. To the best of
our knowledge, this is the first description of functional effects
of GPR17 in a native in vitro system mimicking the intact
brain, as well as its role in ischemia-like microglial activation
and neuronal injury in vitro.
The current results also confirmed that GPR17,
activated by the agonists UDP and LTD4, enhanced LDH
release and neuronal death, while reducing cell viability; these
effects were attenuated by GPR17 knockdown in neuron-glial mixed
cultures of cortical cells. These findings corroborated previous
reports demonstrating that GPR17 is phylogenetically linked to P2Y
receptors for nucleotides and cysteinyl leukotriene receptors for
CysLTs (8-10). However, with regard to the
activation of GPR17 by nucleotides and cysteinyl leukotrienes,
results from different laboratories remain controversial and
require further study (33-38).
Currently, GPR17 has mainly been assessed for its
roles in regulating OPC differentiation and myelination (19-22). Mounting evidence indicates that
GPR17 represents one of the potential G protein-coupled receptor
(GPCR) drug targets for demyelinating diseases, such as multiple
sclerosis (20,39,40). Previous studies reported that
GPR17 also acts as a damage sensor in cerebral ischemic injury and
spinal cord damage, and might serve important roles in the
modulation of both ischemic neuronal injury and late
remodeling/repair responses (9,14,17,18). Following cerebral ischemia, GPR17
upregulation in neurons was reported to be associated with enhanced
cell damage, especially within 24 h of reperfusion (microglia not
markedly activated); GPR17 knockdown attenuated acute ischemic
injury, strongly indicating its role in neuronal death in
vivo (9,14,17). While in the late phase (14-28
days) of focal cerebral ischemia, GPR17 level increase is mainly
localized in microglia/microphages in the penumbra and ischemic
core, its knockdown results in attenuated brain atrophy and
post-ischemic microgliosis, indicating GPR17 involvement in the
modulation of microglial activation (14,17). However, whether ischemia-like
neuronal injury and microglial activation in vitro are
mediated by GPR17 is poorly understood. A better understanding of
the underlying mechanism is crucial for the development of
effective therapeutics to ischemic stroke.
In the present study, neuron-glial mixed cultures
were used to simulate the brain environment. Consistent with
previous reports, ~28% of cultured cells were neurons, 7% were
microglia cells, and most of the remaining cells were astrocytes in
the neuron-glial mixed cultures of cortical cells (25). Following moderate ischemia-like
injury (OGD for 1 h and recovery for 24 h), neuronal death and
microglial activation were significantly increased, whereas
astrocytes were not injured (nearly no necrosis or morphological
changes). GPR17 immunoreactivity was observed in neurons and
microglia, but not in astrocytes even after OGD/R. As demonstrated
in the current study, GPR17 siRNA attenuated ischemic neuronal
injury only in neuron-glial mixed cultured cells, and not in
primary neuron cultures. Combined with in vivo findings
(9,14,17), it can be speculated that GPR17 on
glial cells might directly mediate responses to uracil nucleotides
and CysLTs, and secondarily induce ischemia-like neuronal injury.
However, GPR17 siRNA did not affect astrocyte responses in both
neuron-glial mixed cultured cells and primary astrocyte cultures.
In addition, oligodendrocyte linkage persisted at very low levels
in the neuron-glial mixed cultures of cortical cells (5,25).
Based on these findings, the present study was designed to further
investigate the effect of microglial activation. Nevertheless, the
present experiments did not remove all of the oligodendrocyte
linkage in neuron-glial mixed cultured cells, and thus the
possibility that oligodendrocyte linkage may have a regulatory role
in the present study cannot be excluded.
Microglia constitute the first immune defense in the
central nervous system, and have dual roles in physiological and
pathological conditions (41,42). Activated microglia can protect
neurons against damage by phagocytosis of cellular debris, release
of neurotrophic and anti-inflammatory mediators (41,43). Nevertheless, uncontrolled or
overactivated microglia can trigger neurotoxicity by releasing
harmful substances, such as inflammatory cytokines, reactive oxygen
species, and proteinases (42,44). Thus, microglial activation and the
subsequent inflammation-mediated neurotoxicity have crucial roles
in the pathogenesis of neurodegenerative diseases, including
cerebral ischemia.
In neuron-glial mixed cultures, GPR17 knockdown
inhibited OGD/R-induced microglial activation, inducing
phagocytosis and the release of inflammatory cytokines TNF‑α and
IL-1β. Thereafter, primary microglia cultures were used to explore
whether GPR17 could directly regulate microglial activation. The
current findings revealed that GPR17 knockdown inhibited
OGD/R-induced microglial activation by decreasing microglial
phagocytosis, and inhibiting the release of TNF-α and IL-1β. In
line with these findings, colleagues have demonstrated that GPR17
RNA interference inhibited phagocytosis, suppressed the expression
of iNOS and IL-1β, following OGD/R in BV2 microglial cells
(unpublished data). In addition, the microglial conditioned medium
pretreated with OGD/R was able to induce neuronal death, which was
significantly inhibited by GPR17 siRNA pretreatment. Combined with
the present results, it can be speculated that GPR17 might directly
mediate microglial activation and, in turn, cause neuronal injury
in the mixed culture of cortical cells, although further direct
evidence is required for confirmation.
The regulatory role of GPR17 in microglial
activation in ischemic/inflammatory diseases might be a response to
the changes of extracellular nucleotides and CysLTs. The above
changes were supported by the following evidences: Neurons can
release both adenine (such as ATP) and uracil (such as UTP and UDP)
nucleotides into the extracellular space in response to
ischemic/inflammatory damage (45,46); 1321N1 human astrocytoma cells
release uracil nucleotides (UTP) and nucleotide sugar (UDP-glucose)
in response to various stimuli (47,48); excitatory kainic acid can increase
extracellular UTP in vivo and in vitro (45). In addition, CysLT levels are
significantly elevated at 3 h, and persisted for 24 h in the
ischemic brain (49,50); OGD/R can increase CysLT release in
cortical cells (4,26). Under these conditions, GPR17 might
operate as a sensor molecule regulating microglial activation.
The present study cannot exclude the possibility
that GPR17 in oligodendrocyte linkage has a role in modulating
neuronal injury and microglial activation, although
oligodendrocytes existed in minute amounts in the present
neuron-glial mixed cultures. Additionally, there is evidence of
probable associations of GPR17 with other G protein coupled
receptors that serve important roles in allergic pulmonary
inflammation and oligodendrocyte myelination processes (8,51,52). CysLT1 and
CysLT2 receptors exhibit expression and localization
patterns similar to those of GPR17 in the brain, and are remarkably
upregulated and localized in microglia during the late phases of
ischemic brain (53,54). The aforementioned evidence
suggests that other regulatory mechanisms may be present in the
role of GPR17 in OGD/R-induced microglial activation and neuronal
injury, and this requires further investigation.
In summary, the present study confirmed that GPR17
upregulation was associated with microglial activation and neuronal
injury in vitro. GPR17 knockdown attenuated ischemia-like
neuronal injury only in neuron-glial mixed cultures, and not
directly in neurons or astrocytes. Additionally, GPR17 knockdown
attenuated OGD/R-induced microglial activation, inducing
phagocytosis and inflammatory cytokine release, in both
neuron-glial mixed cultures and primary microglia cultures.
Finally, the conditioned medium of microglia pretreated with OGD/R
induced neuronal death, and the neuronal death was significantly
inhibited by GPR17 siRNA treatment. These findings suggest that
GPR17 may mediate ischemia-like neuronal injury by regulating
microglial activation in vitro.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81401566 and 31301933) and
the Zhejiang Provincial Natural Science Foundation (grant nos.
LQ15H090005 and Y16H150006).
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
BZ, EQW and QJS designed the research. BZ, QJS, HW,
CXL, SWS, SHF performed experiments and data collection. BZ, HW and
QJS analyzed the data and wrote the manuscript.
Ethics approval and consent to
participate
Experimental protocols involving animals were
approved by the Ethics Committee of Laboratory Animal Care and
Welfare, School of Medicine, Zhejiang University (Hangzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Cekic C and Linden J: Purinergic
regulation of the immune system. Nat Rev Immunol. 16:177–192. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Idzko M, Ferrari D and Eltzschig HK:
Nucleotide signalling during inflammation. Nature. 509:3103172014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ni NC, Yan D, Ballantyne LL,
Barajas-Espinosa A, St Amand T, Pratt DA and Funk CD: A selective
cysteinyl leukotriene receptor 2 antagonist blocks myocardial
ischemia/reperfusion injury and vascular permeability in mice. J
Pharmacol Exp Ther. 339:768–778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang XQ, Zhang XY, Wang XR, Yu SY, Fang
SH, Lu YB, Zhang WP and Wei EQ: Transforming growth factor
β1-induced astrocyte migration is mediated in part by activating
5-lipoxygenase and cysteinyl leukotriene receptor 1. J
Neuroinflammation. 9:1452012. View Article : Google Scholar
|
|
5
|
Zhang XY, Wang XR, Xu DM, Yu SY, Shi QJ,
Zhang LH, Chen L, Fang SH, Lu YB, Zhang WP and Wei EQ: HAMI 3379, a
CysLT2 receptor antagonist, attenuates ischemia-like neuronal
injury by inhibiting microglial activation. J Pharmacol Exp Ther.
346:328–341. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shi QJ, Wang H, Liu ZX, Fang SH, Song XM,
Lu YB, Zhang WP, Sa XY, Ying HZ and Wei EQ: HAMI 3379, a CysLT2R
antagonist, dose- and time-dependently attenuates brain injury and
inhibits microglial inflammation after focal cerebral ischemia in
rats. Neuroscience. 291:53–69. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ghosh A, Chen F, Thakur A and Hong H:
Cysteinyl leukotrienes and their Receptors: Emerging therapeutic
targets in central nervous system disorders. CNS Neurosci Ther.
22:943–951. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Daniele S, Trincavelli ML, Gabelloni P,
Lecca D, Rosa P, Abbracchio MP and Martini C: Agonist-induced
desensitization/resensitization of human G protein-coupled receptor
17: A functional cross-talk between purinergic and
cysteinyl-leukotriene ligands. J Pharmacol Exp Ther. 338:559–567.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ciana P, Fumagalli M, Trincavelli ML,
Verderio C, Rosa P, Lecca D, Ferrario S, Parravicini C, Capra V,
Gelosa P, et al: The orphan receptor GPR17 identified as a new dual
uracil nucleotides/ cysteinyl-leukotrienes receptor. EMBO J.
25:4615–4627. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Temporini C, Ceruti S, Calleri E, Ferrario
S, Moaddel R, Abbracchio MP and Massolini G: Development of an
immobilized GPR17 receptor stationary phase for binding
determination using frontal affinity chromatography coupled to mass
spectrometry. Anal Biochem. 384:123–129. 2009. View Article : Google Scholar
|
|
11
|
Marucci G, Dal Ben D, Lambertucci C,
Santinelli C, Spinaci A, Thomas A, Volpini R and Buccioni M: The G
protein-coupled receptor GPR17: Overview and update. ChemMedChem.
11:2567–2574. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cosentino S, Castiglioni L, Colazzo F,
Nobili E, Tremoli E, Rosa P, Abbracchio MP, Sironi L and Pesce M:
Expression of dual nucleotides/cysteinyl-leukotrienes receptor
GPR17 in early trafficking of cardiac stromal cells after
myocardial infarction. J Cell Mol Med. 18:1785–1796. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Franke H, Parravicini C, Lecca D, Zanier
ER, Heine C, Bremicker K, Fumagalli M, Rosa P, Longhi L, Stocchetti
N, et al: Changes of the GPR17 receptor, a new target for
neurorepair, in neurons and glial cells in patients with traumatic
brain injury. Purinergic Signal. 9:451–462. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lecca D, Trincavelli ML, Gelosa P, Sironi
L, Ciana P, Fumagalli M, Villa G, Verderio C, Grumelli C, Guerrini
U, et al: The recently identified P2Y‑like receptor GPR17 is a
sensor of brain damage and a new target for brain repair. PLoS One.
3:e35792008. View Article : Google Scholar
|
|
15
|
Boda E, Viganò F, Rosa P, Fumagalli M,
Labat-Gest V, Tempia F, Abbracchio MP, Dimou L and Buffo A: The
GPR17 receptor in NG2 expressing cells: Focus on in vivo cell
maturation and participation in acute trauma and chronic damage.
Glia. 59:1958–1973. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ren H, Cook JR, Kon N and Accili D: Gpr17
in AgRP neurons regulates feeding and sensitivity to insulin and
leptin. Diabetes. 64:3670–3679. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao B, Zhao CZ, Zhang XY, Huang XQ, Shi
WZ, Fang SH, Lu YB, Zhang WP and Wei EQ: The new P2Y-like receptor
G protein-coupled receptor 17 mediates acute neuronal injury and
late microgliosis after focal cerebral ischemia in rats.
Neuroscience. 202:42–57. 2012. View Article : Google Scholar
|
|
18
|
Ceruti S, Villa G, Genovese T, Mazzon E,
Longhi R, Rosa P, Bramanti P, Cuzzocrea S and Abbracchio MP: The
P2Y-like receptor GPR17 as a sensor of damage and a new potential
target in spinal cord injury. Brain. 132:2206–2218. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fumagalli M, Lecca D and Abbracchio MP:
CNS remyelination as a novel reparative approach to
neurodegenerative diseases: The roles of purinergic signaling and
the P2Y-like receptor GPR17. Neuropharmacology. 104:82–93. 2016.
View Article : Google Scholar
|
|
20
|
Chen Y, Wu H, Wang S, Koito H, Li J, Ye F,
Hoang J, Escobar SS, Gow A, Arnett HA, et al: The
oligodendrocyte-specific G protein-coupled receptor GPR17 is a
cell-intrinsic timer of myelination. Nat Neurosci. 12:1398–1406.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun Z, Ou Y, Lin L, You N, Liu X, Li H, Ma
Y, Cao L, Han Y, Liu M, et al: Olig2-targeted G-protein-coupled
receptor Gpr17 regulates oligodendrocyte survival in response to
lysolecithin-induced demyelination. J Neurosci. 36:10560–10573.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Simon K, Hennen S, Merten N, Blättermann
S, Gillard M, Kostenis E and Gomeza J: The orphan G protein-coupled
receptor GPR17 negatively regulates oligodendrocyte differentiation
via Gαi/o and its downstream effector molecules. J Biol Chem.
291:705–718. 2016. View Article : Google Scholar
|
|
23
|
Coppi E, Maraula G, Fumagalli M, Failli P,
Cellai L, Bonfanti E, Mazzoni L, Coppini R, Abbracchio MP, Pedata F
and Pugliese AM: UDP-glucose enhances outward K(+) currents
necessary for cell differentiation and stimulates cell migration by
activating the GPR17 receptor in oligodendrocyte precursors. Glia.
61:1155–1171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meloni BP, Majda BT and Knuckey NW:
Establishment of neuronal in vitro models of ischemia in 96-well
microtiter strip-plates that result in acute, progressive and
delayed neuronal death. Neuroscience. 108:17–26. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao B, Zhang M, Han X, Zhang XY, Xing Q,
Dong X, Shi QJ, Huang P, Lu YB, Wei EQ, et al: Cerebral ischemia is
exacerbated by extracellular nicotinamide phosphoribosyltransferase
via a non-enzymatic mechanism. PLoS One. 8:e854032013. View Article : Google Scholar
|
|
26
|
Huang XJ, Zhang WP, Li CT, Shi WZ, Fang
SH, Lu YB, Chen Z and Wei EQ: Activation of CysLT receptors induces
astrocyte proliferation and death after oxygen-glucose deprivation.
Glia. 56:27–37. 2008. View Article : Google Scholar
|
|
27
|
Ni M and Aschner M: Neonatal rat primary
microglia: Isolation, culturing, and selected applications. Curr
Protoc Toxicol Chapter 12: Unit. 12:172010.
|
|
28
|
Goldberg MP and Choi DW: Combined oxygen
and glucose deprivation in cortical cell culture: Calcium-dependent
and calcium-independent mechanisms of neuronal injury. J Neurosci.
13:3510–3524. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Daniele S, Lecca D, Trincavelli ML, Ciampi
O, Abbracchio MP and Martini C: Regulation of PC12 cell survival
and differentiation by the new P2Y-like receptor GPR17. Cell
Signal. 22:697–706. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qi LL, Lu YB, Shi WZ, Zhao CZ, Zhang YM,
Chen LP, Zhang LH, Fang SH, Bao JF, Shen JG and Wei EQ: Preparation
and identification of a polyclonal antibody against novel cysteinyl
leukotriene receptor GPR17. Zhejiang Da Xue Xue Bao Yi Xue Ban.
38:357–361. 2009.In Chinese. PubMed/NCBI
|
|
31
|
Zhang Z, Luo J, Huang J, Liu Z, Fang S,
Zhang WP, Wei E and Lu Y: Leukotriene D4 activates BV2 microglia in
vitro. Zhejiang Da Xue Xue Bao Yi Xue Ban. 42:253–260. 2013.In
Chinese. PubMed/NCBI
|
|
32
|
Neher JJ, Neniskyte U, Hornik T and Brown
GC: Inhibition of UDP/P2Y6 purinergic signaling prevents
phagocytosis of viable neurons by activated microglia in vitro and
in vivo. Glia. 62:1463–1475. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qi AD, Harden TK and Nicholas RA: Is GPR17
a P2Y/leukotriene receptor? examination of uracil nucleotides,
nucleotide sugars, and cysteinyl leukotrienes as agonists of GPR17.
J Pharmacol Exp Ther. 347:38–46. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Norregaard K, Benned-Jensen T and
Rosenkilde MM: EBI2, GPR18 and GPR17-three structurally related,
but biologically distinct 7TM receptors. Curr Top Med Chem.
11:618–628. 2011. View Article : Google Scholar
|
|
35
|
Benned-Jensen T and Rosenkilde MM:
Distinct expression and ligand‑binding profiles of two
constitutively active GPR17 splice variants. Br J Pharmacol.
159:1092–1105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hennen S, Wang H, Peters L, Merten N,
Simon K, Spinrath A, Blättermann S, Akkari R, Schrage R, Schröder
R, et al: Decoding signaling and function of the orphan G
protein-coupled receptor GPR17 with a small-molecule agonist. Sci
Signal. 6:ra932013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Simon K, Merten N, Schröder R, Hennen S,
Preis P, Schmitt NK, Peters L, Schrage R, Vermeiren C, Gillard M,
et al: The orphan receptor Gpr17 Is unresponsive to
uracil-nucleotides and cysteinyl-leukotrienes. Mol Pharmacol.
91:518–532. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Boccazzi M, Lecca D, Marangon D, Guagnini
F, Abbracchio MP and Ceruti S: A new role for the P2Y-like GPR17
receptor in the modulation of multipotency of oligodendrocyte
precursor cells in vitro. Purinergic Signal. 12:661–672. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Harden TK: Enigmatic GPCR finds a
stimulating drug. Sci Signal. 6:pe342013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fumagalli M, Bonfanti E, Daniele S,
Zappelli E, Lecca D, Martini C, Trincavelli ML and Abbracchio MP:
The ubiquitin ligase Mdm2 controls oligodendrocyte maturation by
intertwining mTOR with G protein-coupled receptor kinase 2 in the
regulation of GPR17 receptor desensitization. Glia. 63:2327–2339.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim JY, Kim N and Yenari MA: Mechanisms
and potential therapeutic applications of microglial activation
after brain injury. CNS Neurosci Ther. 21:309–319. 2015. View Article : Google Scholar :
|
|
42
|
Ma Y, Wang J, Wang YA and Yang GY: The
biphasic function of microglia in ischemic stroke. Prog Neurobiol.
157:247–272. 2017. View Article : Google Scholar
|
|
43
|
Yang X, Lou Y, Liu G, Wang X, Qian Y, Ding
J, Chen S and Xiao Q: Microglia P2Y6 receptor is related to
Parkinson's disease through neuroinflammatory process. J
Neuroinflammation. 14:382017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Block ML, Zecca L and Hong JS:
Microglia-mediated neurotoxicity: Uncovering the molecular
mechanisms. Nat Rev Neurosci. 8:57–69. 2007. View Article : Google Scholar
|
|
45
|
Koizumi S, Shigemoto-Mogami Y, Nasu-Tada
K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S
and Inoue K: UDP acting at P2Y6 receptors is a mediator of
microglial phagocytosis. Nature. 446:1091–1095. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Günther A, Manaenko A, Franke H, Dickel T,
Berrouschot J, Wagner A, Illes P and Reinhardt R: Early biochemical
and histological changes during hyperbaric or normobaric
reoxygenation after in vitro ischaemia in primary corticoencephalic
cell cultures of rats. Brain Res. 946:130–138. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lazarowski ER, Shea DA, Boucher RC and
Harden TK: Release of cellular UDP-glucose as a potential
extracellular signaling molecule. Mol Pharmacol. 63:1190–1197.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kreda SM, Seminario-Vidal L, Heusden C and
Lazarowski ER: Thrombin-promoted release of UDP-glucose from human
astrocytoma cells. Br J Pharmacol. 153:1528–1537. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou Y, Wei EQ, Fang SH, Chu LS, Wang ML,
Zhang WP, Yu GL, Ye YL, Lin SC and Chen Z: Spatio-temporal
properties of 5-lipoxygenase expression and activation in the brain
after focal cerebral ischemia in rats. Life Sci. 79:1645–1656.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ciceri P, Rabuffetti M, Monopoli A and
Nicosia S: Production of leukotrienes in a model of focal cerebral
ischaemia in the rat. Br J Pharmacol. 133:1323–1329. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Maekawa A, Balestrieri B, Austen KF and
Kanaoka Y: GPR17 is a negative regulator of the cysteinyl
leukotriene 1 receptor response to leukotriene D4. Proc Natl Acad
Sci USA. 106:11685–11690. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Maekawa A, Xing W, Austen KF and Kanaoka
Y: GPR17 regulates immune pulmonary inflammation induced by house
dust mites. J Immunol. 185:1846–1854. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fang SH, Wei EQ, Zhou Y, Wang ML, Zhang
WP, Yu GL, Chu LS and Chen Z: Increased expression of cysteinyl
leukotriene receptor-1 in the brain mediates neuronal damage and
astrogliosis after focal cerebral ischemia in rats. Neuroscience.
140:969–979. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhao CZ, Zhao B, Zhang XY, Huang XQ, Shi
WZ, Liu HL, Fang SH, Lu YB, Zhang WP and Wei EQ: Cysteinyl
leukotriene receptor 2 is spatiotemporally involved in neuron
injury, astrocytosis and microgliosis after focal cerebral ischemia
in rats. Neuroscience. 189:1–11. 2011. View Article : Google Scholar : PubMed/NCBI
|