Introduction
Fibroproliferative diseases, diseases characterized
by extensive tissue fibrosis, are substantial contributors to
mortality in the developed world (1). Tissue fibrosis refers to a process
by which a damaged organ undergoes a wound repair program, but
instead of regenerating native tissue, it creates a collagen-rich
scar that fails to recapitulate the mechanical environment and
functional capacity of the original tissue. Various diverse organs
undergo fibrosis, including the liver, heart, kidney and skin
(2). However, despite there being
numerous crucial differences among fibrotic pathologies of various
organs, the ubiquity of the fibrotic response across diverse organ
systems demonstrates that there are commonalities amongst these
tissues to the fibrotic program as well (2). One of these commonalities is the
paradigm of fibroblast activation, during which a fibroblast
differentiates and undergoes a phenotypic switch to become a
contractile myofibroblast with a distinct secretory profile of type
I collagen and fibronectin, as well as the expression of smooth
muscle markers, including, smooth muscle α actin (α-SMA) (3).
As myofibroblast contractile activity and excessive
deposition of fibrotic extracellular matrix (ECM) can frequently be
detrimental to health, and is regulated by the cellular
environment, fibroblasts must only become activated into
myofibroblasts during scenarios in which tissue repair is necessary
(3). A class of pathways
responsible for transducing cellular signals from the extracellular
environment is known as the mitogen activated protein kinases
(MAPKs) (4). MAPK pathways have
been implicated in the development or prevention of fibrotic
phenotypes at the cellular level and in various tissues, partially
through their ability to propagate signaling initiated by growth
factors in the extracellular milieu, including the ubiquitous
pro-fibrotic transforming growth factor beta (TGF-β) pathway
(5-15). Thus, an understanding of the
interplay of various stimulating factors in the wound
micro-environment is paramount to the understanding of healthy and
pathological wound healing. Despite this, the manner in which
complex environmental signals are integrated into a coherent wound
healing response, characterized by defined actions of particular
effectors cells, is not well-understood. Thus, the aim of the
present study was to improve the understanding of the effects of
MAPK pathways through the use of chemical inhibitors, in
conjunction with each other and with TGF-β signaling mediators, in
order to improve the understanding of a number of signaling
processes resulting in fibroblast activation.
Materials and methods
Reagents
The primary antibodies used were the following:
α-SMA (cat. no. sc-32251; 1:1,000 for western blotting, 1:100 for
fluorescence microscopy), collagen I (cat. no. sc-8783; 1:100),
ED-A fibronectin (ED-A Fn; cat. no. sc-59826; 1:100), Histone H3
(cat. no. sc-8654-R; 1:1,000) (all from Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), Calponin (clone: CALP; 1:1,000; Dako;
Agilent Technologies, Inc., Santa Clara, CA, USA), SM22α (cat. no.
VPA00048KT; 1:1,000; Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and p-ERK1/2 (cat. no. 4370; 1:1,000; Cell Signaling Technology,
Inc., Danvers, MA, USA). The secondary antibodies used were
AlexaFluor® 488-conjugated donkey anti-goat IgG (cat.
no. A-11055) or AlexaFluor488-conjugated goat anti-mouse IgG (cat.
no. A-11001) (Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) for immunofluorescence and horseradish peroxidase
(HRP)-conjugated for western blotting (Bio-Rad Laboratories, Inc.).
The MAPK inhibitors used were U0126, SP600125 and SB202190 (all
from Santa Cruz Biotechnology, Inc.). The TGF-βR1 inhibitor used
was RepSox (Tocris Biosceince, Bristol, UK). The recombinant human
TGF-β1 used was purchased from Peprotech, Inc. (Rocky Hill, NJ,
USA). The recombinant human fibroblast growth factor 2 used was
purchased from Cell Signaling Technology, Inc. AlexaFluor
568-conjugated Phalloidin (cat. no. A12380) was obtained from
Invitrogen (Thermo Fisher Scientific, Inc.) and used to stain
F-actin.
Cell culture
CRL-2097 and CRL-2352 human dermal fibroblasts were
obtained from American Type Culture Collection (Manassas, VA, USA),
and CT-1005 human dermal fibroblasts were obtained from the
University of Massachusetts Medical School tissue distribution
program (Worcester, MA, USA), derived from the dermis of a human
female patient undergoing a panniculectomy. Fibroblasts were
cultured in 1:1 Dulbecco’s modified Eagle’s medium:Ham’s F12
supplemented with 4 mM L-glutamine (Corning Incorporated, Corning,
NY, USA) and 10% Fetal Clone III (Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA) on Nunclon Delta tissue culture plastic
(Thermo Fisher Scientific, Inc.). Cultures were incubated at 37°C
in an atmosphere containing 5% CO2 and high humidity,
and in the presence or absence of MAPK inhibitors, recombinant FGF2
or TGF-β1, or RepSox. Cells were processed on day 4 in culture,
unless otherwise indicated, for analysis, by trypsinizing cells to
collect them, washing once with PBS, pelleting by centrifugation
for 5 min at 200 × g at room temperature, and snap freezing in
liquid nitrogen for storage at −80°C.
Indirect flow cytometry
CRL-2097 fibroblasts were harvested with 0.05%
trypsin and washed twice in Dulbecco’s PBS (DPBS; Corning
Incorporated). Cells were fixed at 4°C for 20 min in cold methanol
and washed in 1X DPBS. Blocking was performed for 30 min at room
temperature in 5% bovine serum albumin (BSA; VWR International,
Leicestershire, UK) in PBS with 0.05% Tween-20 (PBS-T) followed by
a 30 min incubation in a primary antibody solution (1:200 dilution
of α-SMA antibody) in PBS-T at room temperature. Cells were then
washed again once in 1X DPBS and resuspended in a 1:500 dilution of
secondary antibody solution in PBS-T (AlexaFluor488-conjugated goat
anti-mouse IgG). Cells were counterstained with 500 ng/ml propidium
iodide for 15 min at room temperature in the dark and analyzed on
an Accuri C6 flow cytometer using Accuri-C6 analysis software
(version 1.0.264.21; BD Biosciences, Franklin Lakes, NJ, USA). A
total of 20,000 events were collected per sample.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
RNA was isolated from snap-frozen CRL-2097 cell
pellets with an E.Z.N.A. DNA/RNA Isolation kit (Omega Bio-Tek,
Inc., Norcross, GA, USA), according to manufacturer’s protocols.
Concentration of RNA was determined using a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc.). Complementary
DNA (cDNA) was synthesized using a qScript cDNA Supermix
(Quantabio, Beverly, MA, USA), according to manufacturer’s
protocols, and stored at −20°C. PCR reactions were conducted
according to the manufacturer’s protocols using an AB 7500 (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with PowerUp
SYBR® Master mix (Thermo Fisher Scientific, Inc.), using
5 ng cDNA/reaction and 500 nM concentration/primer. Primer
sequences are detailed in Table
I. Relative expression fold changes were calculated using the
ΔΔCq method (16).
 | Table IPrimer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene name | Protein
encoded | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Amplicon size
(bp) |
|---|
| ACTA2 | α-SMA |
ACTGCCTTGGTGTGTGACAA |
CACCATCACCCCCTGATGTC | 120 |
| CNN1 | Calponin |
AGGTTAAGAACAAGCTGGCCC |
GAGGCCGTCCATGAAGTTGT | 113 |
| TAGLN | SM22α |
CACAAGGTGTGTGTAAGGGTG |
GGCTCATGCCATAGGAAGGAC | 132 |
| CCN2 | CCN2 |
GTGCCTGCCATTACAACTGTC |
TCTCACTCTCTGGCTTCATGC | 98 |
| ED-A Fn | ED-A Fn |
CAGTGGAGTATGTGGTTAGTGTC |
GTGACCTGAGTGAACTTCAGG | 119 |
| Col1A1 | Col1A1 |
GTCAGGCTGGTGTGATGGG |
GCCTTGTTCACCTCTCTCGC | 182 |
| Col1A2 | Col1A2 |
CTGGAGAGGCTGGTACTGCT |
AGCACCAAGAAGACCCTGAG | 62 |
| Col3A1 | Col3A1 |
GGACACAGAGGCTTCGATGG |
CTCGAGCACCGTCATTACCC | 190 |
| MMP1 | MMP1 |
GCATATCGATGCTGCTCTTTC |
GATAACCTGGATCCATAGATCGTT | 110 |
| LOX | LOX |
CTCTTGCTGTCCTCCGCTC |
ATCTTGGTCGGCTGGGTAAG | 155 |
| MYOCD | MYOCD |
AGAGGCCATAAAAGGTAACCAGG |
GGGGGTCTTCACTTCGAGTC | 116 |
| TGFB1 | TGF-β1 |
CATTGGTGATGAAATCCTGGT |
TGACACTCACCACATTGTTTTTC | 110 |
| TGFB2 | TGF-β2 |
GAGCGACGAAGAGTACTACG |
TTGTAACAACTGGGCAGACA | 89 |
| TGFBR1 | TGF-βR1 |
GCAGACTTAGGACTGGCAGTAAG |
AGAACTTCAGGGGCCATGT | 104 |
| TGFBR2 | TGF-βR2 |
TGGACCCTACTCTGTCTGTG |
CTGGAGCCATGTATCTTGCAG | 72 |
| TGFBR3 | Betaglycan |
CTGGTGTGGCATCTGAAGAC |
GGACCACAGAACCCTCAGAC | 79 |
| GAPDH | GAPDH |
GAGTCCACTGGCGTCTTCAC |
TTCACACCCATGACGAACAT | 119 |
SDS-PAGE and western blotting
Protein was isolated from CRL-2097, CRL-2352 or
CT-1005 fibroblasts using cold 2X Laemmli sample buffer
supplemented with protease and phosphatase inhibitor cocktail
(Thermo Fisher Scientific, Inc.). Protein concentration was
quantified with a Bradford protein assay (Bio-Rad Laboratories,
Inc.). Cells were lysed on ice and DNA was sheared on ice using a
Missonix CL-2000 ultrasonic cell disruptor. Samples were boiled for
10 min on a heat block, and then 4 µg total protein/sample
was separated on 12% SDS-PAGE. For western blot analysis, protein
from the gel was transferred to a polyvinylidene fluoride membrane
(EMD Millipore, Billerica, MA, USA) using a mini-PROTEAN Tetra Cell
(Bio-Rad Laboratories, Inc.). The membrane was blocked in 5%
fat-free dry milk in buffer containing 1X TBS and 0.1% Tween-20
(TBS-T; pH=8.0) for 1 h at room temperature. The membrane was
incubated and rotated overnight at 4°C in the primary antibody
solution (α-SMA, Histone H3, Calponin, SM22α or p-ERK1/2) at a
pre-determined, antibody-specific dilution in 1% fat-free dry milk
in TBS-T. The membrane was washed four times for 10 min each in
TBS-T and incubated in a species-specific, HRP-conjugated secondary
antibody (dilution, 1:5,000) in 1% fat-free dry milk in TBS-T and
rotated at room temperature for 2 h. Subsequently, the membrane was
washed four times for 10 min each in TBS-T, and the signal was
visualized with a ChemiDoc XRS system (Bio-Rad Laboratories, Inc.)
using SuperSignal West Dura Extended Duration Substrate (Thermo
Fisher Scientific, Inc.). Densitometry was performed in ImageJ
1.8.0 (National Institutes of Health, Bethesda, MD, USA).
Fluorescent microscopy
CRL-2097 fibroblasts were plated on glass coverslips
inside of 24-well tissue culture plates for 4 days in the presence
or absence of 10 µM MAPK inhibitors and/or 10 ng/ml
exogenous recombinant human TGF-β1. At the time of analysis, cells
were fixed in 4% paraformaldehyde at room temperature for 20 min,
and then permeabilized with 0.1% Triton X-100 in 1X PBS. Blocking
was performed in 5% BSA in PBS-T for 30 min at room temperature.
Subsequently, Cells were incubated overnight at 4°C with agitation
in 250 µl/well of primary antibody (α-SMA, collagen I or
ED-A fibronectin) in PBS-T using antibody-specific dilutions. Cells
were then washed three times for 5 min each in PBS-T prior to the
addition of a 1:500 dilution of AlexaFluor 488-conjugated secondary
antibodies or AlexaFluor-conjugated phalloidin (both purchased from
Invitrogen; Thermo Fisher Scientific, Inc.), incubated at room
temperature in the dark for 30 min, and protected from light
thereafter. Cells were then washed three times for 5 min each in
PBS-T prior to being incubated in 500 ng/ml Hoechst 33342 in 1X
PBS-T (Thermo Fisher Scientific, Inc.) for 15 min at room
temperature to stain the nuclei. Cells were washed twice for 5 min
each in PBS and coverslips were mounted onto glass slides using
Prolong Gold (Thermo Fisher Scientific, Inc.) and stored at 4°C
until imaging. Images were collected using an Axiovert 200M (Carl
Zeiss AG, Oberkochen, Germany) at ×20 magnification or using
identical exposure times and settings between treatments, to allow
for direct qualitative comparison of protein expression between
different treatments.
Statistical analysis
All statistics were performed in GraphPad Prism 7
(GraphPad Software, Inc., La Jolla, CA, USA). All statistics were
performed with Student’s t-tests or one-way analysis of variance
with Holm-Sidak post-hoc analysis. P<0.05 was considered to
indicate a statistically significant difference. All data are
presented as the mean ± standard deviation.
Results
Fibroblast activation is promoted by
inhibition of ERK or JNK and antagonized by inhibition of p38
Firstly, the effects of MAPK inhibitors on
activation of human fibroblasts was investigated. Treatment of
CRL-2097 human neonatal foreskin fibroblasts with 10 µM
U0126 (ERK inhibitor) or 10 µM SP600125 (JNK inhibitor) was
sufficient to induce upregulation of three myofibroblast-associated
transcripts, α-SMA (ACTA2), calponin 1 (CNN1), and
transgelin (TAGLN), the genes that encode α-SMA, calponin,
and TAGLN (SM22α), respectively. In contrast, treatment with 10
µM SB202190 (p38 inhibitor) significantly downregulated the
expression of these transcripts, compared with the control
fibroblasts (P<0.05) (Fig.
1A). Western blot analysis coincided with the RT-qPCR data and
demonstrated that treatment with these inhibitors upregulated or
downregulated the expression of the myofibroblast markers encoded
by these genes in a qualitatively similar manner (Fig. 1B). In order to improve the
understanding of the effects of MAPK inhibition on expression of
α-SMA, the major myofibroblast marker, at the cellular level,
indirect flow cytometry was performed. Analysis of the data
revealed that the mean level of α-SMA protein expression/cell was
significantly increased in cells cultured in the presence of ERK or
JNK inhibitors, or in the presence of exogenous TGF-β1, and
significantly decreased in the presence of p38 inhibitor, compared
with fibroblasts cultured under control conditions (P<0.05)
(Fig. 1C). Immunofluorescent
imaging revealed that fibroblasts treated with ERK or JNK
inhibitors exhibited increased levels of filamentous actin,
arranged into stress fibers, and increased fraction of cells
stained positive for α-SMA, compared with control fibroblasts
cultured in the absence of MAPK inhibitors. In contrast,
fibroblasts cultured in the presence of p38 inhibitor exhibited a
notable reduction of filamentous actin stress fiber formation, as
well as a reduction in the expression of α-SMA protein (Fig. 1D). Subsequently, the effects of
MAPK inhibition on fibroblast activation were confirmed to be
generalizable across multiple lines of human dermal fibroblasts,
rather than specific to one cell line. It was demonstrated by
western blot analysis that two primary adult human dermal
fibroblast lines, CRL-2352 and CT-1005, exhibited a qualitatively
similar increase in α-SMA expression when treated with ERK or JNK
inhibitor, and a qualitatively similar decrease in α-SMA expression
when treated with p38 inhibitor, as was demonstrated in CRL-2097
fibroblasts (Fig. 1E).
Collectively, these data demonstrated that culturing fibroblasts
with ERK or JNK inhibitors is sufficient to induce fibroblast
activation, and that culturing fibroblasts with an inhibitor of p38
is sufficient to antagonize fibroblast activation. Additionally, it
was determined that these effects are generalizable across multiple
lines of primary human dermal fibroblasts.
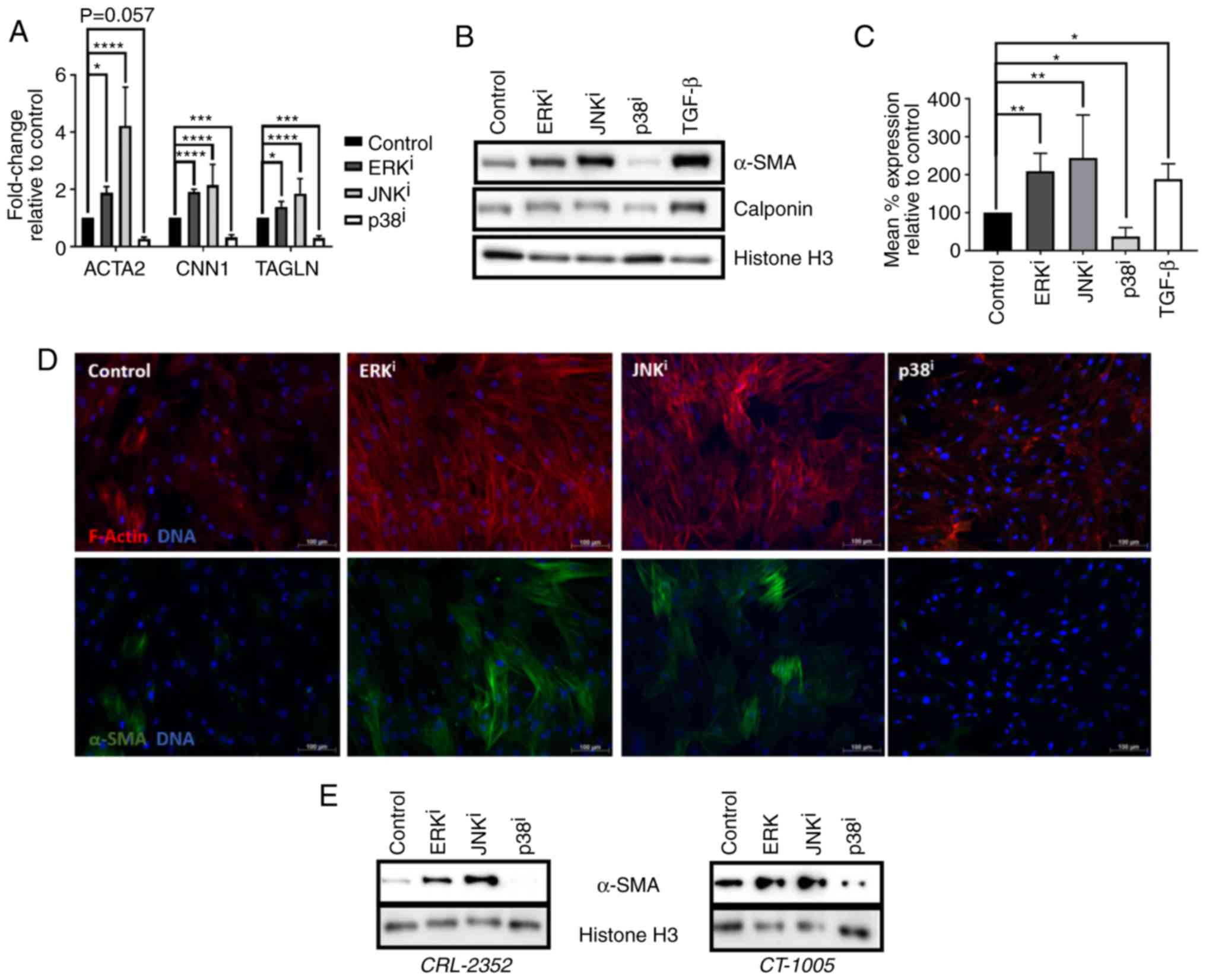 | Figure 1Effects of MAPK inhibitors on
fibroblast activation. (A–D) CRL-2097 human dermal fibroblasts were
cultured under control conditions, or in the presence of 10
µM U0126 (ERKi), 10 µM SP600125
(JNKi), 10 µM SB202190 (p38i) or 10
ng/ml exogenous recombinant human TGF-β1, and harvested at day 4 in
culture. (A) Expression of myofibroblast marker transcripts
ACTA2, CNN1 and TAGLN were determined by
reverse transcription-quantitative polymerase chain reaction,
normalized to the expression of GAPDH, and expressed as fold
change relative to the expression of control samples. There were 4
biological replicates/condition. Error bars represent standard
deviation. Statistics were performed by one-way ANOVA with multiple
comparisons using the Holm-Sidak post-hoc test.
*P<0.05, ***P<0.001 and
****P<0.0001. (B) Western blot analysis was performed
on myofibroblast markers α-SMA and calponin on whole cell lysates.
Histone H3 was used as a loading control. (C) Fibroblasts were
fixed and subjected to indirect flow cytometry, and the expression
of α-SMA was compared with the expression in control fibroblasts.
There were 4 biological replicates/condition. Statistics were
performed by one-way ANOVA with multiple comparisons using the
Holm-Sidak post-hoc test. *P<0.05 and
**P<0.01. (D) Fibroblasts were fixed and stained by
immunofluorescence for α-SMA, and F-actin was visualized using
fluorescently-labeled phalloidin. Nuclei were counterstained with
Hoechst. Scale bars, 100 µm. (E) CRL-2352 and CT-1005
fibroblasts were cultured under control conditions or in the
presence of a MAPK inhibitor, and harvested at day 4 in culture.
Western blot analysis was performed for detection of α-SMA. Histone
H3 was used as a loading control. MAPK, mitogen-activated protein
kinase; ERK, extracellular signal-regulated kinase; JNK, c-Jun
N-terminal kinase; ANOVA, analysis of variance; ACTA2, α-SMA;
α-SMA, smooth muscle α actin; CNN1, calponin 1; TAGLN, transgelin;
TGF-β, transforming growth factor-β; i, inhibitor. |
Pro-fibrotic gene expression paradigm is
induced by inhibition of ERK or JNK and antagonized by inhibition
of p38
Since it was demonstrated that MAPK inhibition
variably affected fibroblast activation as determined by the
expression of canonical myofibroblast markers, it was subsequently
investigated whether this inhibition also affected the expression
of other genes associated with ECM deposition and myofibroblast
activation. Analysis by RT-qPCR demonstrated that inhibition of ERK
or JNK resulted in the significant upregulation (P<0.05,
compared with control fibroblasts) of collagen type 1 α 1
(Col1A1) and Col1A2, the genes encoding the chains of
type I collagen, and the major ECM component of fibrotic scar
tissue, as well as Col3A1, the gene encoding type III
collagen. Concordantly, inhibition of ERK or JNK also resulted in
significant downregulation of transcription of matrix
metallopeptidase 1 (MMP1), the gene encoding the
interstitial collagenase MMP1 (P<0.05, compared with control
fibroblasts), further indicating an activated fibroblast phenotype
associated with excessive deposition of pathologic ECM (Fig. 2A). Inhibition of ERK or JNK also
resulted in significantly increased expression of ED-A fibronectin
(ED-A Fn), the transcript encoding the
myofibroblast-specific fibronectin isoform known as ED-A Fn, as
well as myocardin (MYOCD) and connective tissue growth
factor (CCN2), the genes encoding the myogenic transcription
factor MYOCD and the pro-fibrotic matricellular protein CNN2,
respectively (P<0.05, compared with control fibroblasts)
(Fig. 2B). In contrast,
inhibition of p38 resulted in significantly decreased expression of
these myofibroblast-associated genes, consistent with its apparent
ability to antagonize fibroblast activation (P<0.05, compared
with control fibroblasts). Inhibition of p38 also resulted in
significant downregulation of lysyl oxidase (LOX)
(P<0.05, compared with control fibroblasts), the transcript
encoding LOX, a copper-dependent protein that is responsible for
the crosslinking of immature collagen precursors into mature
collagen fibrils (17) (Fig. 2A). Additionally, immunofluorescent
analysis demonstrated that inhibition of ERK or JNK resulted in
increased levels of type I collagen and ED-A Fn deposition, and
that inhibition of p38 resulted in diminished levels of type I
collagen and ED-A Fn deposition (Fig.
2C). Collectively, these data indicate that inhibition of ERK
or JNK results in increased deposition of fibrotic ECM through
upregulation of transcription of fibrosis-associated and
collagen-encoding genes, as well as through downregulation of the
expression of the metalloproteinase-encoding gene MMP1.
Additionally, these data demonstrated that inhibition of p38
results in decreased deposition of fibrotic ECM through
downregulation of fibrosis-associated and collagen-encoding genes,
as well as through downregulation of the gene encoding the collagen
maturation enzyme LOX.
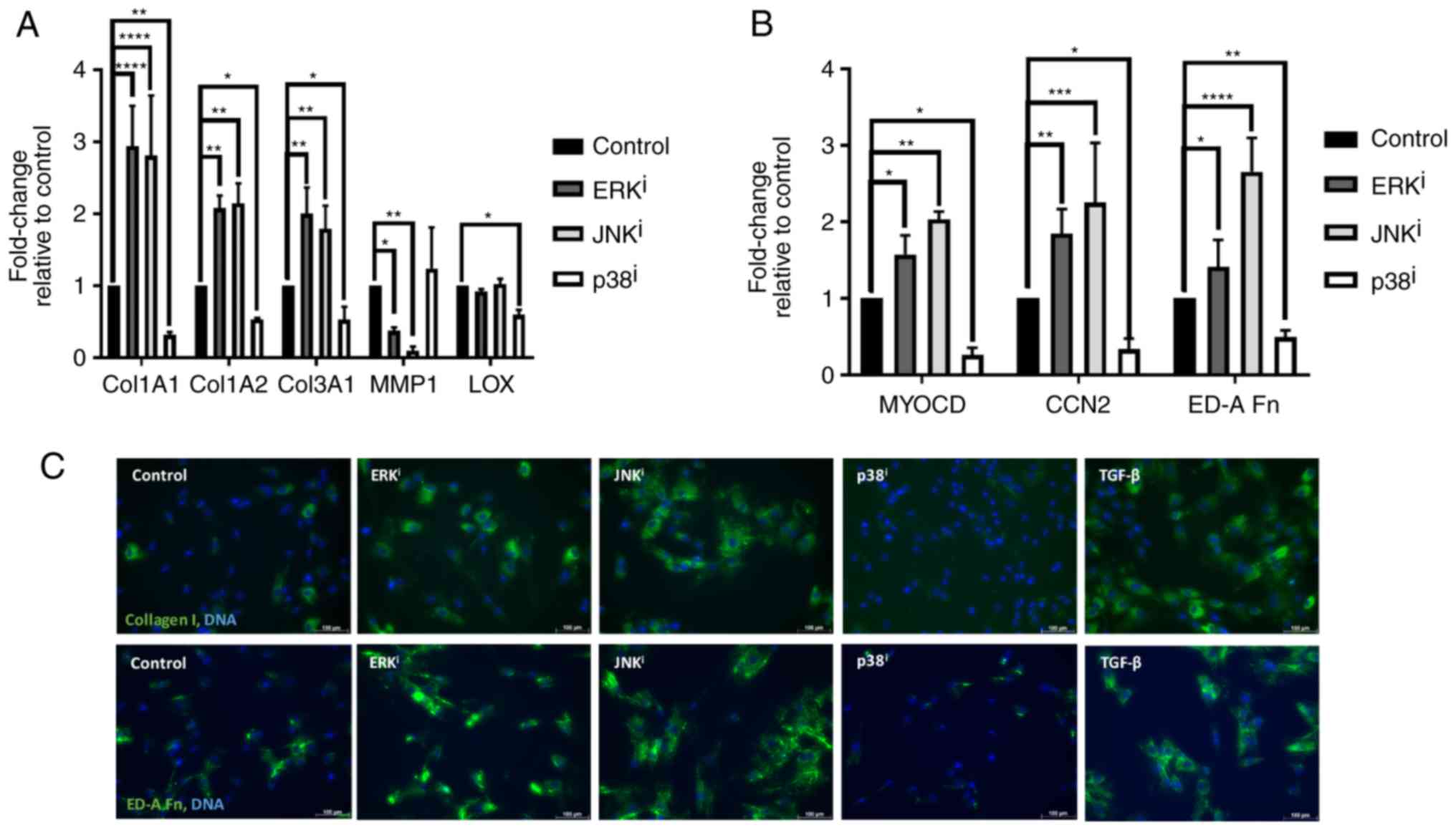 | Figure 2Effects of mitogen-activated protein
kinase inhibitors on the ECM. CRL-2097 human dermal fibroblasts
were cultured in the presence or absence of 10 µM U0126
(ERKi), 10 µM SP600125 (JNKi) or 10
µM SB202190 (p38i), and harvested at day 4.
Expression levels of ECM-associated transcripts (A) Col1A1,
Col1A2, Col3A1, MMP1 and LOX, and (B)
MYOCD, CCN2 and ED-A Fn were determined
relative to fibroblasts cultured under control conditions by
reverse transcription-quantitative polymerase chain reaction.
GAPDH expression was used as an internal control. Expression
levels per transcript were compared using a one-way analysis of
variance and post-hoc Holm-Sidak analysis. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. There were 2≤n≤6 biological
replicates/condition. Error bars represent standard deviation. (C)
Fibroblasts were fixed and stained for immunofluorescence for
collagen I or ED-A Fn. Nuclei were counterstained with Hoechst.
Scale bars, 100 µm. ECM, extracellular matrix; Col1A1,
collagen type 1 α 1; MMP1, matrix metallopeptidase 1; LOX, lysyl
oxidase; MYOCD, myocardin; CCN2, connective tissue growth factor;
ED-A Fn, ED-A fibronectin; i, inhibitor. |
ERK or JNK inhibition cooperatively
enhances TGF-β- mediated fibroblast activation
As TGF-β1 is a fibrogenic cytokine and canonical
myofibroblast activator, and specific paradigms of non-canonical
TGF-β signaling have been demonstrated to proceed via activation of
MAPKs, including ERK and JNK (18), the present study determined
whether ERK and JNK inhibition would antagonize or enhance
TGF-β1-mediated fibroblast activation. Western blot analysis
demonstrated that treatment with exogenous TGF-β1, as well as
treatment with ERK or JNK inhibitors individually, was sufficient
to induce the expression of myofibroblast maker proteins α-SMA,
calponin and SM22α above the baseline expression levels of control
fibroblasts. Additionally, treatment of fibroblasts with TGF-β1 and
one of the fibrogenic MAPK inhibitors (ERK inhibitor U0126 or JNK
inhibitor SP600125) enhanced myofibroblast activation, as indicated
by a notable increase in the expression of the identical
myofibroblast marker proteins, compared with the expression levels
observed in cells treated with exogenous TGF-β1 or pro-fibrotic
MAPK inhibitor alone (Fig. 3A).
These observations were confirmed by immunofluorescence, in which
co-treatment of TGF-β1 with ERK of JNK inhibitors increased the
number and intensity of α-SMA+ cells, compared with
treatment with TGF-β1 alone (Fig.
3B). Collectively, these data indicated that inhibition of ERK
or JNK cooperatively enhances the effects of exogenous TGF-β1 on
fibroblast activation.
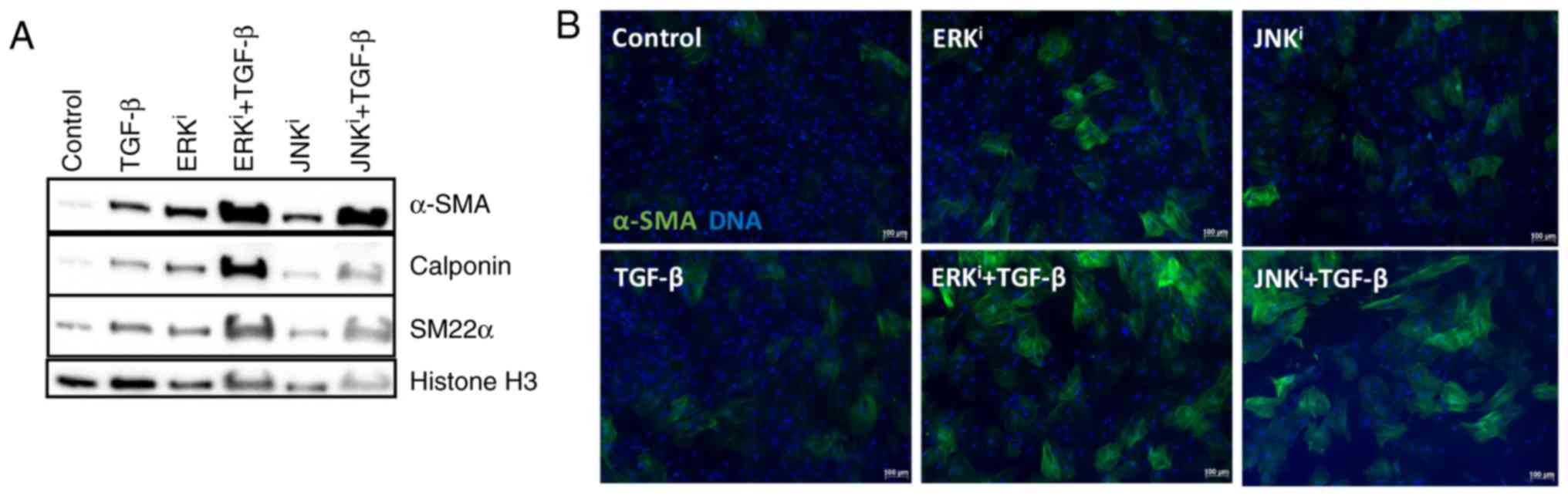 | Figure 3Cooperative effects of
mitogen-activated protein kinase inhibition and exogenous TGF-β on
fibroblast activation. CRL-2097 human dermal fibroblasts were
cultured under control conditions or in the presence of 10
µM U0126 (ERKi), 10 µM SP600125
(JNKi), and/or 10 ng/ml exogenous recombinant human
TGF-β1, and analyzed at day 4 in culture. (A) Protein lysates were
examined for expression of myofibroblast-associated protein markers
α-SMA, calponin, and SM22α by western blot analysis. Histone H3 was
used as a loading control. (B) Fibroblasts were fixed and stained
for the myofibroblast marker protein α-SMA and nuclei were
counterstained with Hoechst. Scale bar, 100 µm. TGF-β,
transforming growth factor-β; ERK, extracellular signal-regulated
kinase; JNK, c-Jun N-terminal kinase; α-SMA, smooth muscle α actin;
i, inhibitor. |
Inhibition of p38 antagonizes fibroblast
activation induced by ERK or JNK inhibition
Previous studies implicated MAPK pathways in
fibroblast activation, and demonstrated the ability of MAPKs to act
as signal transduction mediators of growth factors, demonstrating
fibrotic and anti-fibrotic activities (5-15).
Since it had been previously demonstrated that different MAPK
inhibitors have varied effects on fibroblast activation, and it had
been previously demonstrated that inhibition of p38 was sufficient
to attenuate fibroblast activation induced by exogenous TGF-β1
(15), the present study sought
to determine whether these MAPK inhibitors had the ability to
antagonize the activity of each other. Measurement of
myofibroblast-associated transcripts ACTA2, CNN1 and
TAGLN demonstrated that treatment with the p38 inhibitor
antagonized fibroblast activation induced by treatment with ERK or
JNK inhibitors (P<0.05 for fibroblasts treated with
ERKi or JNKi and p38i vs.
fibroblasts treated with ERKi or JNKi alone)
(Fig. 4A). Similarly, western
blot analysis demonstrated that treatment with the p38 inhibitor
antagonized the expression of α-SMA, calponin, and SM22α protein
induced by treatment with ERK or JNK inhibitors, by comparing
levels of these proteins in fibroblasts treated with ERK or JNK and
p38 inhibitors vs. fibroblasts treated with ERK or JNK inhibitor
alone (Fig. 4B).
Immunofluorescent analysis corroborated the Western blot analysis
data, demonstrating that treatment with p38 inhibitor decreased the
number and intensity of α-SMA+ cells, by comparing
fluorescence intensity in fibroblasts treated with ERK or JNK and
p38 inhibitors vs. fibroblasts treated with ERK or JNK inhibitor
alone (Fig. 4C). Collectively,
these data indicate that inhibition of p38 is sufficient to
antagonize fibroblast activation induced by inhibition of ERK or
JNK, as assessed by transcript and protein levels of canonical
myofibroblast markers.
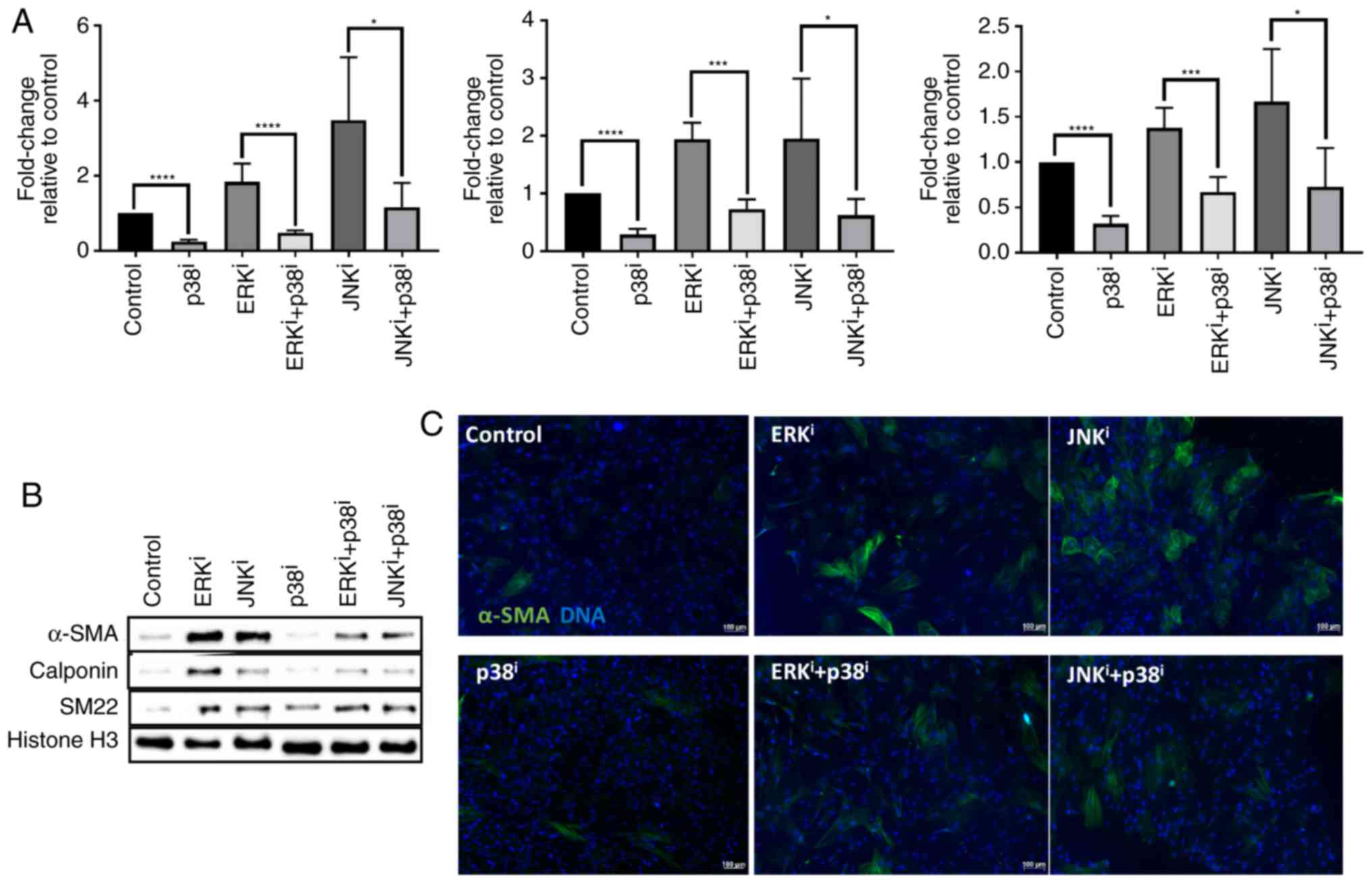 | Figure 4Antagonism of p38 inhibitor towards
ERK and JNK inhibitor-mediated fibroblast activation. CRL-2097
human dermal fibroblasts were cultured under control conditions or
in the presence of 10 µM U0126 (ERKi), 10
µM SP600125 (JNKi), and/or 10 µM SB202190
(p38i), and analyzed at day 4 in culture. (A) Expression
levels of myofibroblast-associated transcripts ACTA2,
CNN1 and TAGLN were determined relative to
fibroblasts cultured under control conditions by reverse
transcription-quantitative polymerase chain reaction. GAPDH
expression was used as an internal control. Student’s t-tests were
performed in order to compare the expression between each culture
condition and its p38i-treated counterpart.
*P<0.05, ***P<0.001 and
****P<0.0001. There were 4 biological
replicates/condition. Error bars depict standard deviation. (B)
Protein lysates were examined for expression of
myofibroblast-associated marker proteins α-SMA, calponin and SM22α
by western blot analysis. Histone H3 was used as a loading control.
(C) Fibroblasts were fixed and stained for the myofibroblast marker
protein α-SMA and nuclei were counterstained with Hoechst. Scale
bars, 100 µm. ACTA2, α-SMA; TAGLN, transgelin; CNN1,
calponin 1; ERK, extracellular signal-regulated kinase; JNK, c-Jun
N-terminal kinase; α-SMA, smooth muscle α actin; i,
inhibitor. |
Similar effects of ERK and JNK inhibitors
on fibroblast activation are not a function of non-specific
inhibition of p-ERK by JNK inhibitor
Due to the observation that ERK and JNK
inhibitor-mediated fibroblast activation were associated with
qualitatively similar expression changes for each gene analyzed,
and in consideration of previously reported activity of SP600125 as
a modest inhibitor of MAPK kinase 1 (MKK1) (19), the present study sought to confirm
that the reason for these similarities was not due to nonspecific
inhibitory activity of ERK signaling by the JNK inhibitor. Western
blot analysis demonstrated that, while ERK inhibitor treatment
decreased ERK phosphorylation at 15-60 min post-treatment as
expected, treatment with JNK inhibitor failed to inhibit ERK
phosphorylation at these time-points post-treatment (Fig. 5A).
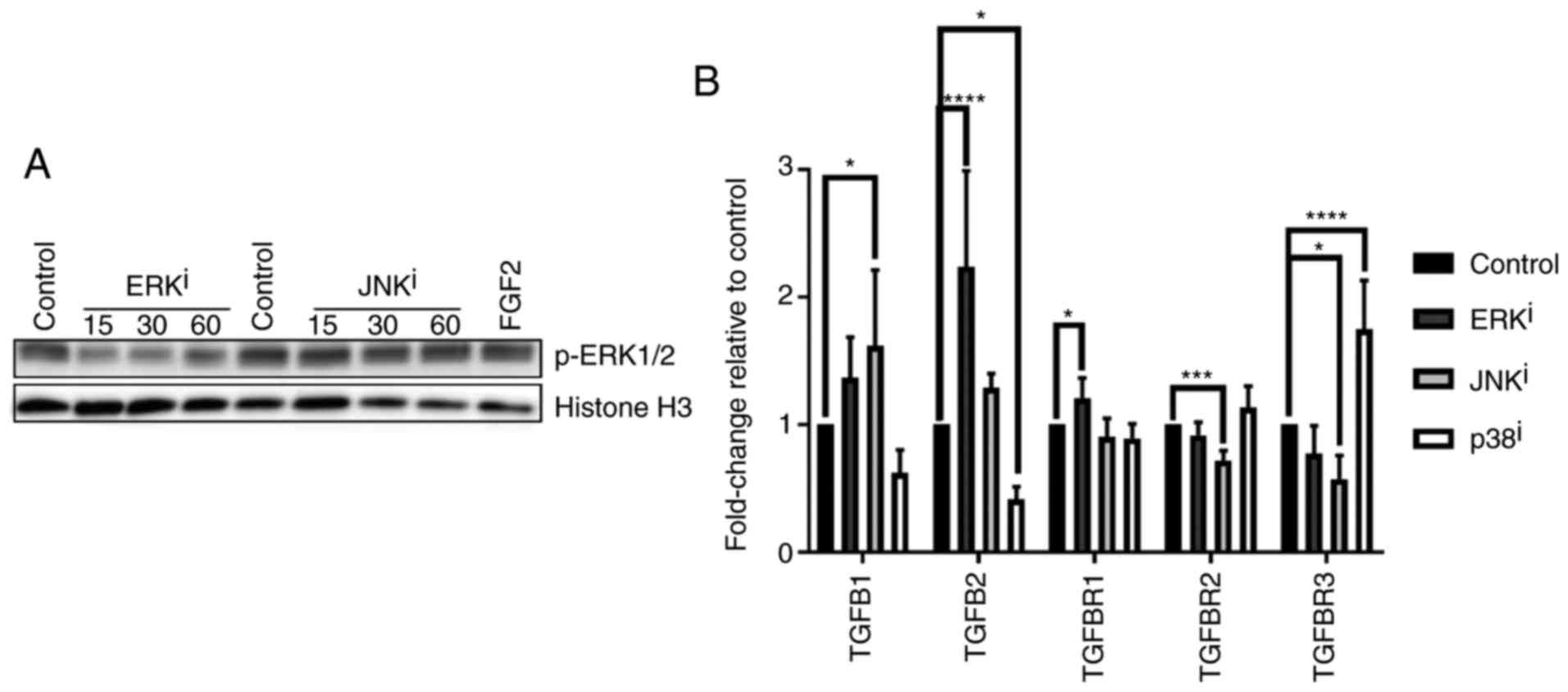 | Figure 5TGF-β-associated gene expression
profiles induced by treatment with mitogen-activated protein kinase
inhibitors. (A) CRL-2097 human dermal fibroblasts were cultured for
15-60 min in the presence or absence of 10 µM U0126
(ERKi) or 10 µM SP600125 (JNKi) and
subjected to western blot analysis for p-ERK1/2. Histone H3 was
used as a loading control. Fibroblasts incubated with recombinant
human FGF2 for 30 min were used as a positive control for p-ERK1/2
expression. (B) CRL-2097 human dermal fibroblasts were cultured in
the presence or absence of 10 µM U0126 (ERKi), 10
µM SP600125 (JNKi) or 10 µM SB202190
(p38i) until day 4. Expression levels of
TGF-β-associated transcripts TGFB1, TGFB2,
TGFBR1, TGFBR2 and TGFBR3 were determined
relative to fibroblasts cultured under control conditions by
reverse transcription-quantitative polymerase chain reaction.
GAPDH expression was used as an internal control. Expression
levels per transcript were compared using an one-way analysis of
variance and post-hoc Holm-Sidak analysis. *P<0.05,
***P<0.001 and ****P<0.0001. There were
6 biological replicates/condition. Error bars represent the
standard deviation. TGFB1, transforming growth factor-β1; TGFBR1,
TGF-β receptor 1; p-ERK, phosphoextracellular signal-regulated
kinase; JNK, c-Jun N-terminal kinase; FGF2, fibroblast growth
factor 2; i, inhibitor. |
MAPK inhibitors variably affect the
transcription of genes encoding TGF-β ligands and receptors
Given the importance of TGF-β signaling as a
ubiquitous central modulator of myofibroblast activation, the
present study sought to determine whether the observed effects of
MAPK inhibitors on fibroblast activation were accompanied by
changes in the expression of TGF-β-associated genes. Analysis of
transcript expression (Fig. 5B)
demonstrated that treatment with the ERK inhibitor resulted in the
significant upregulation of TGF-β2 (TGFB2) and TGF-βR1
(TGFBR1), the genes encoding TGF-β2 and TGF-βR1 respectively,
while treatment with the JNK inhibitor resulted in the significant
upregulation of TGFB1 and significant downregulation of
TGFBR3, the genes encoding TGF-β1 and betaglycan,
respectively, as well as an unexpected significant downregulation
of TGFBR2, the gene encoding TGF-βR2 (P<0.05, compared
with control fibroblasts). These data also demonstrated that
treatment with the p38 inhibitor decreased the expression of
TGFB2 and induced the expression of TGFBR3, in
accordance with expectations that p38 antagonizes the expression of
fibrotic TGF-β signaling mediators, as indicated by the
aforementioned results. Collectively, these data indicated that the
effects of the MAPK inhibitor treatments on fibroblast activation
may be partially due to their effects on the expression of TGF-β
signaling-associated genes.
Fibroblast activation induced by ERK or
JNK inhibition is antagonized by co-inhibition of TGF-βR1
Since ERK and JNK inhibition resulted in fibroblast
activation and were associated with modulation of the expression of
genes encoding TGF-β ligands and receptors, the present study
sought to determine whether ERK or JNK inhibitor-mediated
fibroblast activation could be antagonized by inhibition of TGF-β
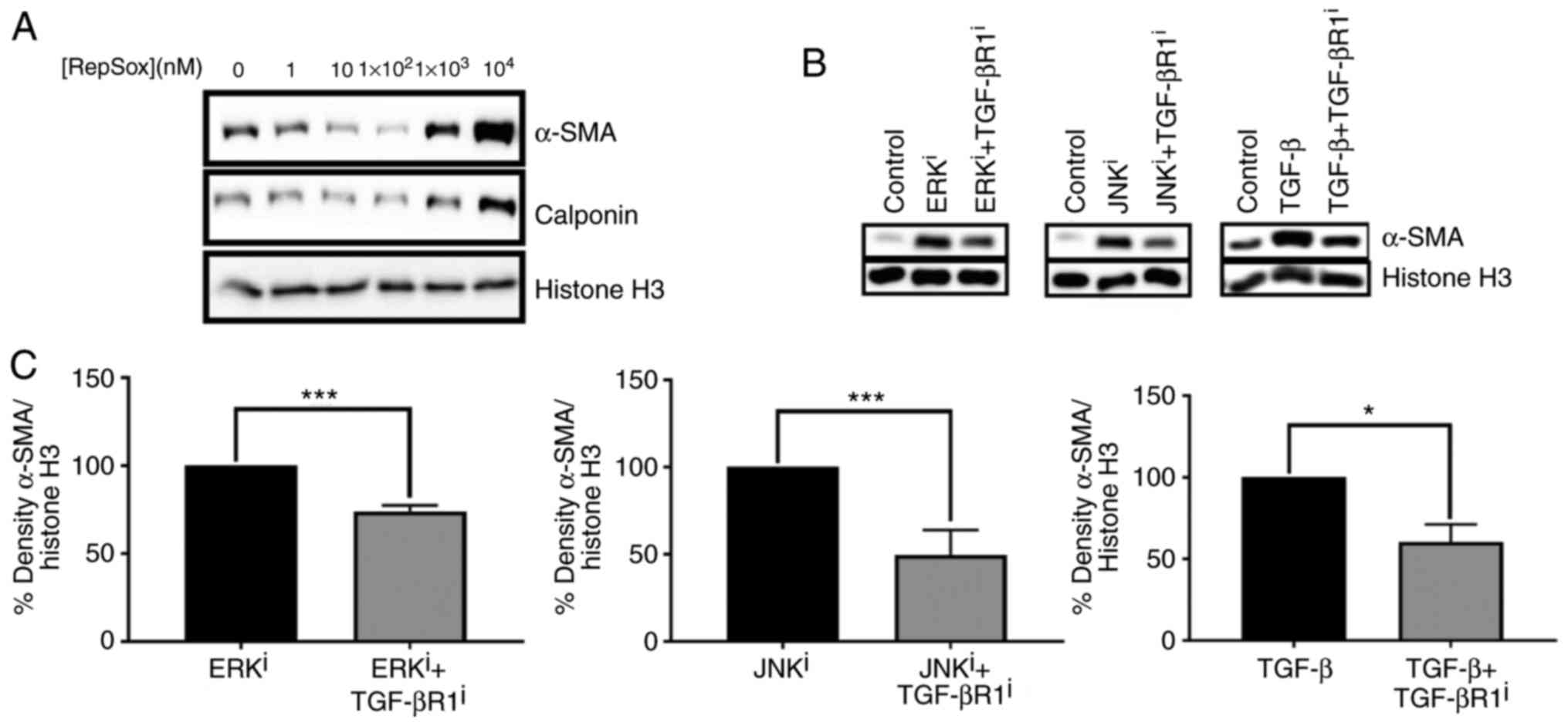
signaling. A dose-response curve demonstrated that treatment of
fibroblasts with 100 nM Repsox, a TGF-βR1 inhibitor, was sufficient
to decrease basal expression of myofibroblast markers α-SMA and
calponin (Fig. 6A). Thus, 100 nM
Repsox was selected for use in subsequent experiments. Notably,
increasing concentrations of Repsox up to 10 µM were
determined to substantially induce fibroblast activation, as
demonstrated by notable upregulation of myofibroblast-associated
proteins α-SMA and calponin, in contrast to intended effects as an
antagonist of TGF-β signaling.
Western blot analysis demonstrated that treatment
with 100 nM TGF-β R1 inhibitor partially antagonized ERK and JNK
inhibitor-mediated fibroblast activation, as indicated by the
expression levels of α-SMA protein (Fig. 6B). Densitometry and subsequent
statistical analysis confirmed that the antagonistic effects of
TGF-βR1 inhibitor on ERK inhibitor-mediated, JNK inhibitor-mediated
and exogenous TGF-β1-mediated fibroblast activation were
statistically significant (P<0.05 for fibroblasts treated with
ERKi+TGFBR1i, compared with ERKi,
JNKi+TGFBR1i, compared with JNKi,
and TGF-β+TGF-βR1i, compared with TGF-β; Fig. 6C). Collectively, these data
demonstrate that fibroblast activation induced by small
molecule-mediated inhibition of ERK or JNK is partially dependent
on signaling via TGF-βR1.
Discussion
One of the dominant mechanisms by which a cell
receives signals from its extracellular environment and converts
these signals to actionable responses is through signaling via
growth factors (20-23). Growth factors are one vehicle used
to relay signals containing information regarding the extracellular
environment in order to induce a functional response in cells.
Growth factor signaling pathways are defined by the ligands that
initiate the cellular signal, the growth factor receptor that
transduces this signal from the extracellular environment across
the plasma membrane, and the intracellular effector proteins that
work to initiate and propagate a functional response in the target
cell (20-23). The mechanisms by which growth
factor/growth factor receptor complexes transduce a signal and
result in cellular activity vary among different pathways, but one
of the most common paradigms is transduction via MAPK signaling
(24,25). As myofibroblasts are one of many
important effector cells in the mammalian wound healing response,
and as fibroblasts are activated to myofibro-blasts by TGF-β
signaling (3), the present study
investigated the effects of MAPK inhibitors on TGF-β signaling and
fibroblast activation.
The present data demonstrates that treatment of
proliferative fibroblasts with ERK or JNK inhibitors is sufficient
to induce fibroblast activation and upregulate the expression of
myofibroblast marker proteins, and that treatment with a p38
inhibitor is sufficient to antagonize fibroblast activation
(Fig. 1), in accordance with our
previously published data (15).
These fibroblast activation dynamics also predicted the changes
observed in ECM-associated gene and protein expression (Fig. 2), where it was determined that
treatment of fibroblasts with ERK or JNK inhibitors induced a
relative gene expression profile associated with fibroblast
activation and conducive to fibrotic ECM deposition. Additionally,
it was indicated that treatment of fibroblasts with a p38 inhibitor
induced a relative gene expression profile contrary to fibroblast
activation and averse to pro-fibrotic ECM deposition.
There is precedent for the role of p38 in
fibroproliferative diseases. Constitutive activation of p38 was
demonstrated in fibroblasts derived from patients with scleroderma,
and inhibition of p38 activation via small molecules or transient
transfection with a p38 dominant negative mutant was sufficient to
reduce the high levels of type I collagen and fibronectin
associated with scleroderma fibroblasts (26). Cardiac fibroblast-specific
deletion of MAPK14, the gene encoding p38α, resulted in a
reduction of fibrotic damage induced at a later time in a murine
ischemic model, and deletion of MAPK14 in existing
myofibroblasts resulted in increased tolerance to myocardial
infarct injury. In concordance with these data, cardiac
fibroblast-specific overexpression of MKK6, which is responsible
for activation of p38 signaling, resulted in an enhanced fibrotic
response (11). In a mouse model
of nephro-pathic fibrosis, small molecule-mediated inhibition,
alone and cooperatively alongside administration of a TGF-βR1
inhibitor, of p38 reduced intestinal fibrosis (9). This indicates that the pro-fibrotic
effects of p38 signaling are partially distinct from canonical
TGF-β/TGF-βR/SMAD signaling. This is plausible considering the
understanding of the ability of TGF-βR1 and TGF-βR2 to directly
contribute to MAPK phosphorylation (27,28), as well as in light of the numerous
examples of crosstalk between TGF-β and MAPK signaling (29-35). Collectively, these data alongside
other reports demonstrating the anti-fibrotic effects of p38
inhibition (36-40), indicate that p38 activation is
necessary for the differentiation and maintenance of
myofibroblasts, such that inhibition of p38 or its downstream
effectors may be a viable therapeutic option for specific
pathological fibroproliferative states.
It was also demonstrated that treatment with ERK or
JNK inhibitors cooperatively enhanced fibroblast activation induced
by treatment with exogenous TGF-β1 (Fig. 3), and that treatment with a p38
inhibitor partially inhibited fibroblast activation induced by
treatment with ERK or JNK inhibitors (Fig. 4), indicating crosstalk between the
MAPK and TGF-β pathways, as well as between individual MAPK
pathways. Additionally, it was determined that fibroblasts treated
with MAPK inhibitors demonstrated distinct expression profiles of
TGF-β-associated genes, compared with control fibroblasts (Fig. 5). Notably, these changes in gene
expression spanned TGF-β ligands and receptors, including the
non-signal transducing receptor TGF-βR3, also known as betaglycan,
which has the ability to sequester pro-fibrotic TGF-β ligands
(41,42). Treatment with a JNK inhibitor
downregulated betaglycan transcript expression, and treatment with
a p38 inhibitor upregulated betaglycan transcript expression, both
of which were expected considering the observation that JNK
inhibition induces and p38 inhibition antagonizes fibroblast
activation. Accordingly, a synthetic peptide derived from the
ligand-binding domain of betaglycan has been demonstrated to
attenuate fibrosis in in vivo models of myocardial (43), hepatic (44) and dermal fibrosis (45), and stable overexpression of
betaglycan attenuates TGF-β signaling (SMAD-dependent and
SMAD-independent) in lung fibroblasts (46). It was also determined that the
activation of fibroblasts mediated by treatment with ERK or JNK
inhibitors was partially antagonized by co-treatment with a small
molecule inhibitor of TGF-βR1 (Fig.
6), indicating that fibroblast activation induced by these
inhibitors is partially due to their effects on TGF-β/TGF-βR
activity. The present data demonstrating that treatment with JNK
inhibitor induced fibroblast activation and a pro-fibrotic TGF-β
gene expression paradigm are also consistent with previous reports
indicating the ability of the JNK inhibitor SP600125 to induce SMAD
phosphorylation and cooperate with exogenous TGF-β to enhance SMAD
signaling (47,48).
The mechanisms through which treatment with ERK and
JNK inhibitors induces fibroblast activation downstream of their
effects on MAPK signaling, even in the absence of exogenous
stimulation with a growth factor, are not completely understood,
but a number of possibilities can be considered. It is possible
that ERK or JNK inhibitors are inhibiting basal levels of MAPK
signaling resulting from serum stimulation or from autocrine or
paracrine signaling of secreted growth factors, including
fibroblast growth factors (49).
Additionally, the regulatory linker region of SMAD proteins has
been demonstrated to undergo phosphorylation by ERK (50,51) and JNK (52,53), among numerous other kinases
(18). As SMAD signaling is
critical to fibroblast activation and fibrosis induced by TGF-β,
modulation of SMAD signaling via MAPK inhibition may be partially
responsible for the effects of MAPK inhibitors on fibroblast
activation, perhaps at least in part through further modulation of
TGF-β signaling at the transcriptional level (Fig. 5).
The present data shed light on the effects of MAPK
inhibition on human fibroblasts, in the context of fibroblast
activation and subsequent pro-fibrotic, myofibroblast-associated
gene expression. Developing an understanding of MAPK inhibitor
effects on fibroblast phenotype is important, particularly
considering the interest in tyrosine kinase inhibitors as
therapeutics for various diseases, including fibrotic diseases
(54-57). As further research yields an
improved understanding of the effects of MAPK inhibitors on
fibroblasts, and the mechanisms through which these activities are
induced, the potential benefits and pitfalls of modulating these
highly pleiotropic pathways for treatment or prevention of
fibroproliferative diseases can be assessed in an improved
manner.
Acknowledgments
The authors would like to thank Ms Victoria Huntress
(Worcester Polytechnic Institute, Worcester, MA, USA), of the WPI
microscopy core, for her assistance with fluorescent imaging. The
abstract was presented at Experimental Biology 2018 on April 21-25
2018 in San Diego, CA and published as abstract no. 660.6 in The
FASEB Journal (Supp 1): 2018.
Funding
The present study was supported by a National
Institutes of Health award to Tanja Dominko (grant no. R01GM85456)
and the National Science Foundation Integrative Graduate Education
and Research Traineeship (grant no. DGE 1144804) awarded to David
Dolivo.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
DD, SL and TD conceived the experiments. DD and SL
performed the experiments. DD drafted the manuscript. DD, SL and TD
edited and revised manuscript. DD, SL and TD approved the final
draft of the manuscript prior to submission.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wynn TA: Common and unique mechanisms
regulate fibrosis in various fibroproliferative diseases. J Clin
Invest. 117:524–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zeisberg M and Kalluri R: Cellular
mechanisms of tissue fibrosis. 1. Common and organ-specific
mechanisms associated with tissue fibrosis. Am J Physiol Cell
Physiol. 304:C216–C225. 2013. View Article : Google Scholar :
|
|
3
|
Hinz B: Myofibroblasts. Exp Eye Res.
142:56–70. 2016. View Article : Google Scholar
|
|
4
|
Cargnello M and Roux PP: Activation and
function of the MAPKs and their substrates, the MAPK-activated
protein kinases. Microbiol Mol Biol Rev. 75:50–83. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mulsow JJ, Watson RW, Fitzpatrick JM and
O’connell PR: Transforming growth factor-beta promotes pro-fibrotic
behavior by serosal fibroblasts via PKC and ERK1/2 mitogen
activated protein kinase cell signaling. Ann Surg. 242:880–889.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stambe C, Atkins RC, Tesch GH, Masaki T,
Schreiner GF and Nikolic-Paterson DJ: The role of p38α
mitogen-activated protein kinase activation in renal fibrosis. J Am
Soc Nephrol. 15:370–379. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grella A, Kole D, Holmes W and Dominko T:
FGF2 overrides TGFβ1-driven integrin ITGA11 expression in human
dermal fibroblasts. J Cell Biochem. 117:1000–1008. 2016. View Article : Google Scholar
|
|
8
|
Wu W, Muchir A, Shan J, Bonne G and Worman
HJ: Mitogen-activated protein kinase inhibitors improve heart
function and prevent fibrosis in cardiomyopathy caused by mutation
in lamin A/C gene. Circulation. 123:53–61. 2011. View Article : Google Scholar
|
|
9
|
Li J, Campanale NV, Liang RJ, Deane JA,
Bertram JF and Ricardo SD: Inhibition of p38 mitogen-activated
protein kinase and transforming growth factor-β1/Smad signaling
pathways modulates the development of fibrosis in
adriamycin-induced nephropathy. Am J Pathol. 169:1527–1540. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
See F, Thomas W, Way K, Tzanidis A, Kompa
A, Lewis D, Itescu S and Krum H: p38 mitogen-activated protein
kinase inhibition improves cardiac function and attenuates left
ventricular remodeling following myocardial infarction in the rat.
J Am Coll Cardiol. 44:1679–1689. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Molkentin JD, Bugg D, Ghearing N, Dorn LE,
Kim P, Sargent MA, Gunaje J, Otsu K and Davis J:
Fibroblast-specific genetic manipulation of p38 Mitogen-activated
protein kinase in vivo reveals its central regulatory role in
fibrosis. Circulation. 136:549–561. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Furukawa F, Matsuzaki K, Mori S, Tahashi
Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki
Y, et al: p38 MAPK mediates fibrogenic signal through Smad3
phosphorylation in rat myofibroblasts. Hepatology. 38:879–889.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meyer-Ter-Vehn T, Gebhardt S, Sebald W,
Buttmann M, Grehn F, Schlunck G and Knaus P: p38 inhibitors prevent
TGF-beta-induced myofibroblast transdifferentiation in human Tenon
fibroblasts. Invest Ophthalmol Vis Sci. 47:1500–1509. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sousa AM, Liu T, Guevara O, Stevens J,
Fanburg BL, Gaestel M, Toksoz D and Kayyali US: Smooth muscle
alpha-actin expression and myofibroblast differentiation by TGFbeta
are dependent upon MK2. J Cell Biochem. 100:1581–1592. 2007.
View Article : Google Scholar
|
|
15
|
Dolivo DM, Larson SA and Dominko T:
FGF2-mediated attenuation of myofibroblast activation is modulated
by distinct MAPK signaling pathways in human dermal fibroblasts. J
Dermatol Sci. 88:339–348. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative CT method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar
|
|
17
|
Kagan HM and Li W: Lysyl oxidase:
Properties, specificity, and biological roles inside and outside of
the cell. J Cell Biochem. 88:660–672. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Burch ML, Zheng W and Little PJ: Smad
linker region phosphorylation in the regulation of extracellular
matrix synthesis. Cell Mol Life Sci. 68:97–107. 2011. View Article : Google Scholar
|
|
19
|
Bain J, Plater L, Elliott M, Shpiro N,
Hastie CJ, Mclauchlan H, Klevernic I, Arthur JS, Alessi DR and
Cohen P: The selectivity of protein kinase inhibitors: A further
update. Biochem J. 408:297–315. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maddaluno L, Urwyler C and Werner S:
Fibroblast growth factors: Key players in regeneration and tissue
repair. Development. 144:4047–4060. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Johnson KE and Wilgus TA: Vascular
endothelial growth factor and angiogenesis in the regulation of
cutaneous wound repair. Adv Wound Care (New Rochelle). 3:647–661.
2014. View Article : Google Scholar :
|
|
22
|
Barrientos S, Stojadinovic O, Golinko MS,
Brem H and Tomic-Canic M: Growth factors and cytokines in wound
healing. Wound Repair Regen. 16:585–601. 2008. View Article : Google Scholar
|
|
23
|
Werner S and Grose R: Regulation of wound
healing by growth factors and cytokines. Physiol Rev. 83:835–870.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Krishna M and Narang H: The complexity of
mitogen-activated protein kinases (MAPKs) made simple. Cell Mol
Life Sci. 65:3525–3544. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Plotnikov A, Zehorai E, Procaccia S and
Seger R: The MAPK cascades: Signaling components, nuclear roles and
mechanisms of nuclear translocation. Biochim Biophys Acta.
1813.1619–1633. 2011.
|
|
26
|
Ihn H, Yamane K and Tamaki K: Increased
phosphorylation and activation of mitogen-activated protein kinase
p38 in scleroderma fibroblasts. J Invest Dermatol. 20:247–255.
2005. View Article : Google Scholar
|
|
27
|
Galliher AJ and Schiemann WP: Src
phosphorylates Tyr284 in TGF-beta type II receptor and regulates
TGF-beta stimulation of p38 MAPK during breast cancer cell
proliferation and invasion. Cancer Res. 67:3752–3758. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee MK, Pardoux C, Hall MC, Lee PS,
Warburton D, Qing J, Smith SM and Derynck R: TGF-beta activates Erk
MAP kinase signalling through direct phosphorylation of ShcA. EMBO
J. 26:3957–3967. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chapnick DA, Warner L, Bernet J, Rao T and
Liu X: Partners in crime: The TGFβ and MAPK pathways in cancer
progression. Cell Biosci. 1:422011. View Article : Google Scholar
|
|
30
|
Xie L, Law BK, Chytil AM, Brown KA, Aakre
ME and Moses HL: Activation of the Erk pathway is required for
TGF-beta1-induced EMT in vitro. Neoplasia. 6:603–610. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gui T, Sun Y, Shimokado A and Muragaki Y:
The roles of mitogen-activated protein kinase pathways in
TGF-β-induced epithelial-mesenchymal transition. J Signal
Transduct. 2012.289243:2012.
|
|
32
|
Selvamurugan N, Kwok S, Alliston T, Reiss
M and Partridge NC: Transforming growth factor-beta1 regulation of
collagenase-3 expression in osteoblastic cells by cross-talk
between the Smad and MAPK signaling pathways and their components,
Smad2 and Runx2. J Biol Chem. 279:19327–19334. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Terai K, Call MK, Liu H, Saika S, Liu CY,
Hayashi Y, Chikama T, Zhang J, Terai N, Kao CW and Kao WW:
Crosstalk between TGF-beta and MAPK signaling during corneal wound
healing. Invest Ophthalmol Vis Sci. 52:8208–8215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xiao YQ, Malcolm K, Worthen GS, Gardai S,
Schiemann WP, Fadok VA, Bratton DL and Henson PM: Cross-talk
between ERK and p38 MAPK mediates selective suppression of
pro-inflammatory cytokines by transforming growth factor-β. J Biol
Chem. 277:14884–14893. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kolosova I, Nethery D and Kern JA: Role of
Smad2/3 and p38 MAP kinase in TGF-β1-induced epithelial-mesenchymal
transition of pulmonary epithelial cells. J Cell Physiol.
226:1248–1254. 2011. View Article : Google Scholar :
|
|
36
|
Nishida M, Okumura Y, Sato H and Hamaoka
K: Delayed inhibition of p38 mitogen-activated protein kinase
ameliorates renal fibrosis in obstructive nephropathy. Nephrol Dial
Transplant. 23:2520–2524. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Raia V, Maiuri L, Ciacci C, Ricciardelli
I, Vacca L, Auricchio S, Cimmino M, Cavaliere M, Nardone M, Cesaro
A, et al: Inhibition of p38 mitogen activated protein kinase
controls airway inflammation in cystic fibrosis. Thorax.
60:773–780. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gao F, Wang Y, Li S, Wang Z, Liu C and Sun
D: Inhibition of p38 mitogen-activated protein kinases attenuates
renal interstitial fibrosis in a murine unilateral ureteral
occlusion model. Life Sci. 167:78–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sugiyama N, Kohno M and Yokoyama T:
Inhibition of the p38 MAPK pathway ameliorates renal fibrosis in an
NPHP2 mouse model. Nephrol Dial Transplant. 27:1351–1358. 2012.
View Article : Google Scholar
|
|
40
|
Wilde JM, Gumucio JP, Grekin JA, Sarver
DC, Noah AC, Ruehlmann DG, Davis ME, Bedi A and Mendias CL:
Inhibition of p38 mitogen-activated protein kinase signaling
reduces fibrosis and lipid accumulation after rotator cuff repair.
J Shoulder Elbow Surg. 25:1501–1518. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
López-Casillas F, Payne HM, Andres JL and
Massagué J: Betaglycan can act as a dual modulator of TGF-beta
access to signaling receptors: Mapping of ligand binding and GAG
attachment sites. J Cell Biol. 124:557–568. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Eickelberg O, Centrella M, Reiss M,
Kashgarian M and Wells RG: Betaglycan Inhibits TGF-beta signaling
by preventing type I-type II receptor complex formation.
Glycosaminoglycan modifications alter betaglycan function. J Biol
Chem. 277:823–829. 2002. View Article : Google Scholar
|
|
43
|
Hermida N, López B, González A, Dotor J,
Lasarte JJ, Sarobe P, Borrás-Cuesta F and Díez J: A synthetic
peptide from transforming growth factor-beta1 type III receptor
prevents myocardial fibrosis in spontaneously hypertensive rats.
Cardiovasc Res. 81:601–609. 2009. View Article : Google Scholar
|
|
44
|
Ezquerro IJ, Lasarte JJ, Dotor J,
Castilla-Cortazar I, Bustos M, Peñuelas I, Blanco G, Rodríguez C,
Lechuga Mdel C, Greenwel P, et al: A synthetic peptide from
transforming growth factor beta type III receptor inhibits liver
fibrogenesis in rats with carbon tetrachloride liver injury.
Cytokine. 22:12–20. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Santiago B, Gutierrez-Cañas I, Dotor J,
Palao G, Lasarte JJ, Ruiz J, Prieto J, Borrás-Cuesta F and Pablos
JL: Topical application of a peptide inhibitor of transforming
growth factor-beta1 ameliorates bleomycin-induced skin fibrosis. J
Invest Dermatol. 125:450–455. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ahn JY, Park S, Yun YS and Song JY:
Inhibition of type III TGF-β receptor aggravates lung fibrotic
process. Biomed Pharmacother. 64:472–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lin Y, Zhang B, Liang H, Lu Y, Ai X, Zhang
B and Chen X: JNK inhibitor SP600125 enhances TGF-β-induced
apoptosis of RBE human cholangiocarcinoma cells in a Smad-dependent
manner. Mol Med Rep. 8:1623–1629. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wu S, Kasisomayajula K, Peng J and
Bancalari E: Inhibition of JNK enhances TGF-beta1-activated Smad2
signaling in mouse embryonic lung. Pediatr Res. 65:381–386. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hough C, Radu M and Doré JJ: Tgf-beta
induced Erk phosphorylation of smad linker region regulates smad
signaling. PLoS One. 7:e425132012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hayashida T, Decaestecker M and Schnaper
HW: Cross-talk between ERK MAP kinase and Smad signaling pathways
enhances TGF-beta-dependent responses in human mesangial cells.
FASEB J. 17:1576–1578. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Engel ME, McDonnell MA, Law BK and Moses
HL: Interdependent SMAD and JNK signaling in transforming growth
factor-beta-mediated transcription. J Biol Chem. 274:37413–37420.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Mori S, Matsuzaki K, Yoshida K, Furukawa
F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa
M, et al: TGF-beta and HGF transmit the signals through
JNK-dependent Smad2/3 phosphorylation at the linker regions.
Oncogene. 23:7416–7429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Qu K, Huang Z, Lin T, Liu S, Chang H, Yan
Z, Zhang H and Liu C: New Insight into the anti-liver fibrosis
effect of multi-targeted tyrosine kinase inhibitors: From molecular
target to clinical trials. Front Pharmacol. 6:3002016. View Article : Google Scholar
|
|
55
|
Kompa AR: Do p38 mitogen-activated protein
kinase inhibitors have a future for the treatment of cardiovascular
disease. J Thorac Dis. 8:E1068–E1071. 2016. View Article : Google Scholar
|
|
56
|
Raghu G and Selman M: Nintedanib and
pirfenidone. New antifibrotic treatments indicated for idiopathic
pulmonary fibrosis offer hopes and raises questions. Am Thoracic
Soc. 191:252–254. 2015.
|
|
57
|
Hartmann JT, Haap M, Kopp HG and Lipp HP:
Tyrosine kinase inhibitors-a review on pharmacology, metabolism and
side effects. Curr Drug Metab. 10:470–481. 2009. View Article : Google Scholar : PubMed/NCBI
|