Introduction
Heat shock protein (HSP) is a highly conserved
protein synthesized by organisms in response to stress. According
to the molecular size, it can be divided into HSP100, HSP90, HSP70,
HSP60 and small heat shock protein. HSP has been shown to serve an
important role in the development and progression of
atherosclerosis (AS). HSP27 exerts its anti-atherosclerotic effect
by restraining the antioxidant stress reaction, reducing the
inflammatory response and inhibiting the proliferation, migration
of vascular smooth muscle (1).
HSP60 causes inflammation in AS by increasing the endothelial
dysfunction via induction of the anti-HSP60 adaptive immune
reaction (2).
HSP22, a type of small molecular weight HSP
(3), was first found in HeLa and
melanoma cells. The HSP can be activated by different proteases and
has molecular chaperone and autokinase bioactivities. HSP22
protects the cells by regulating proliferation and migration, and
inhibiting apoptosis (4).
Marunouchi et al (5)
reported that the expression of HSP22 in cardiomyocytes was
increased on the compensation stage of heart failure following
myocardial infarction, and that HSP22 can protect the mitochondrial
function. A decrease in the phosphorylation of HSP22 was positively
correlated with mitochondrial hypofunction, which resulted in heart
failure, suggesting a protective role of HSP22 in cardiomyocytes
(6). However, the association
between HSP22 and AS remains unclear.
Statins have often been used in AS treatment due to
their pleiotropic effects on inflammation (7). Atorvastatin (ATV) exerts its
anti-atherosclerotic effects by targeting the receptor for advanced
glycation end products in human umbilical vein endothelial cells
(HUVECs) and in Goto Kakisaki rats (8), or by downregulating HSP22 expression
induced by oxidized low-density lipoprotein (ox-LDL) in HUVECs
(9). In addition, statins have a
number of other effects, including anti-inflammation,
anti-oxidative stress and improving endothelial function (10). However, the specific mechanism of
ATV on AS and the effect of ATV on HSP22 expression remain
unknown.
The present study aimed to investigate whether ATV
exerts part of its inhibitory role on the progression of AS by
targeting HSP22. Specifically, the expression of HSP22 and its
downstream p38 signaling, and endothelial nitric oxide synthase
(eNOS) activity in apolipoprotein E-deficient (ApoE−/−)
mice and HUVECs. Moreover, cell proliferation and the cell cycle
were also measured in HUVECs with HSP22 knockdown by shRNA
transfection.
Materials and methods
Animals and diets
A total of 36 male ApoE−/− mice (8 weeks
old, 18-22 g), provided by the Scientific Research Institute
(Shanghai, China), were housed using a 12 h light/dark cycle at a
constant temperature of 25°C, with a relative humidity of 60-70%.
The mice were randomly divided into three groups: The normal diet
group (ND; 12 mice), the high-fat diet group (HFD; 12 mice) (diets
both from Mediscience Ltd., Jiangsu, China) and the HFD plus ATV
group (HFD + ATV; 12 mice; ATV from Pfizer, Inc., Suzhou, China).
Mice in the ATV-treated group were treated with 10 mg/kg/day ATV
via intragastric administration. All the mice were fed for 13 weeks
with an HFD, and otherwise were treated with ATV for 9 weeks
subsequent to being fed for 4 weeks with an HFD. Subsequent to
being fed with HFD or ATV, mice were further fed with a normal diet
for 1 week and anesthetized with 3% sodium pentobarbital (40 mg/kg;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) via intra-peritoneal
injection prior to cervical dislocation. All animal care and
experimental procedures in the current study complied with the
protocol approved by the Second Affiliated Hospital of Nanchang
University (Nanchang, China).
Metabolic profile analysis
Serum collected from the blood samples of the three
different groups was used to measure levels of plasma lipids,
including total cholesterol (TC), triglycerides (TG), low-density
lipoprotein cholesterol (LDL) and high-density lipoprotein
cholesterol (HDL), using a cholesterol kit (cat. no. 000060094),
triglycerides kit (cat. no. 000060104), HDL-cholesterol kit (cat.
no. 000000190) and LDL-cholesterol kit (cat. no. 000000210) (all
BioSino Biotechnology Co., Ltd., Shanghai, China),
respectively.
Immunohistochemistry
Aortas of the three different groups were collected
and quickly frozen in liquid nitrogen, as previously described
(8), and then stored at −80°C for
hematoxylin and eosin (H&E) and immunohistochemical studies, as
previously described (11,12).
Paraffin-embedded aorta sections (4 to 7-μm thick) were fixed in
10% formalin overnight at room temperature, and separately
dehydrated by 50, 70, 85, 95 and 100% ethanol for 2 h, then
deparaffinized by dimethylbenzene for 15 min and separately
hydrated by 100, 95, 85 and 75% ethanol for 5 min at room
temperature. Following blocking with 2% bovine serum albumin (Sigma
Aldrich; Merck KGaA) in PBS for 1 h at room temperature, the tissue
sections were incubated with anti-HSP22 (1:100 dilution; cat. no.
ab151552; Abcam, Cambridge, MA, USA) and anti-phosphorylated p38
(anti-p-p38) (1:1,600; cat. no. 9212; Cell Signaling Technology,
Inc., Danvers, MA, USA) primary antibodies at 4°C overnight, and
then incubated with biotinylated secondary antibodies, horseradish
peroxidase goat anti-rabbit IgG (1:100 dilution; cat. no.
111-035-008) and goat anti-mouse IgG (H+L) (1:100 dilution; cat.
no. 111-035-003) (noth Jackson ImmunoResearch, West Grove, PA, USA)
for 1 h at room temperature. Five fields from each slide were
examined using microscopy (×200 magnification) and images were
captured using a light microscope equipped with a camera (Olympus
BX-50; Olympus Corporation, Tokyo, Japan).
Enzyme-linked immunosorbent assay
(ELISA)
Secretion of HSP22 from serum samples of the three
different groups was determined by ELISA using the Heat Shock 22kDa
Protein 8 (HSPB8) ELISA kit (cat. no. ABIN425260) according to the
manufacturer’s protocols (Beijing 4A Biotech Co., Ltd., Beijing,
China).
Cell culture
HUVECs were purchased from Eidia Ltd.; Sekisui
Chemical Co., Ltd. (Tokyo, Japan) and cultured in The Second
Affiliated Hospital of Nanchang University. HUVECs at passage 3-4
were used for experiments. The cells were cultured with serum-free
RPMI-1640 medium for 24 h prior to treatment. The HUVECs were
randomly assigned to the indicated groups as follows: The control
group, the ox-LDL (160 µg/ml) group, the ATV (40 µM)
group and the ox-LDL (160 µg/ml) plus ATV (40 µM)
group. HUVECs were treated with ox-LDL or ATV alone for 24 h. For
the combined group, HUVECs were treated with ox-LDL for 24 h,
followed by ATV for 24 h. Cells were cultured at 37°C for 24 h with
5% CO2.
Short hairpin RNA (shRNA)
transfection
HSP22 shRNA-1, 5′-GCTGGGAGCCTGTCAGTTTAT-3′; HSP22
shRNA-2, 5′-GGATCCTGTGACAGTATTTGC-3′; and HSP22 shRNA-3,
5′-GCAGTTTCAACAACGAGCTTC-3′, designed for targeting human SLC44A5
mRNA, were cloned into a lentiviral vector (pLKO.1-EGFP; Addgene,
Cambridge, MA, USA). The cells were transfected with
pLKO.1-EGFP-HSP22-shRNA (40 nM) using Lipofectamine 2000 (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) according the
manufacturer’s protocol. A non-specific scramble shRNA sequence was
used as negative control (NC; 5′-TTCTCCGAACGTGTCACGT-3′). Cell
proliferation and the cell cycle were analyzed at 48 h
post-transfection.
MTT assay
HUVECs were treated with ox-LDL (20, 40, 80, 160 and
320 µg/ml) or ATV (20, 40, 80, 160 and 320 µM),
respectively, while HUVECs with HSP22 shRNA transfection were
treated with ox-LDL (160 µg/ml), ATV (40 µM) or
ox-LDL (160 µg/ml) plus ATV (40 µM), and incubated
for 24, 48 and 72 h. Next, the HUVECs were cultured with 20
µl MTT (5 mg/ml) for 4 h. The intracellular formazan
crystals formed were solubilized with acidic isopropanol
(Sigma-Aldrich; Merck KGaA) and the absorbance was read on a
microplate reader (Utrao Medical Instrument, Shanghai, China) at
570 nm.
Cell cycle assay
HUVECs with HSP22 shRNA transfection
(5×105 cells/well) were treated with ox-LDL (160
µg/ml), ATV (40 µM) or ox-LDL (160 µg/ml) plus
ATV (40 µM), and subsequently incubated with propidium
iodide (PI) and 0.5 µg/µl RNase A for 30 min.
Thereafter, the cells were analyzed on a flow cytometer (BD
Biosciences, San Diego, CA, USA).
mRNA quantization by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA isolated from aortas or HUVECs using
TRIzol and purified with an RNAeasy kit (both Thermo Fisher
Scientific, Inc.) was reversed transcribed to cDNA using the
Prime-Script RT reagent kit (Takara Bio, Inc., Otsu, Japan).
RT-qPCR was performed with the ABI 7500 (Applied Biosystems, Foster
City, CA, USA) using SYBR Premix Ex Taq (Takara Bio). Primers were
as follows: HSP22 sense, 5′-CAGGTCCCTCCTTACTCA-3′ and antisense,
5′-CCCGCACCCTCTAACA T-3′; and β-actin sense,
5′-AGGGGCCGGACTCGTCATACT-3′ and antisense,
5′-GGCGGCACCACCATGTACCCT-3′. The HSP22 mRNA level was normalized by
internal β-actin mRNA. The following thermo-cycling conditions were
used for the PCR: 95°C for 10 min, followed by 40 cycles of 95°C
for 15 sec and 60°C for 45 sec, and a final extension step of 95°C
for 15 sec, 60°C for 1 min, 95°C for 15 sec and 60°C for 15 sec.
The relative quantification values for the gene expression were
calculated using the 2−ΔΔCq method (13).
Western blot analysis
Total protein was extracted from aortas or HUVECs
using radioimmunoprecipitation buffer (JRDUN Biotechnology Co.,
Ltd. Shanghai, China). The total protein concentration in each
sample was measured using a Lowry protein assay kit (Bio Rad
Laboratories, Inc., Hercules, CA, USA). Equal amounts of extracted
protein (50 μg) were separated by SDS-PAGE on a 10% gel and
transferred onto polyvinylidene difluoride membranes (Roche
Diagnostics GmbH, Mannheim, Germany), followed by blocking in 5%
skimmed milk overnight at 4°C. The protein abundance was detected
with antibodies against HSP22 (1:1,000 dilution; cat. no. ab151552;
Abcam), p-eNOS [1:500 dilution; cat. no. YS-(kt)-0446; YS
Biotechnology Co., Ltd., Shanghai, China], eNOS (1:1,000 dilution;
cat. no. ab76198; Abcam), p-p38 (1:1,000 dilution; cat. no. 4511),
p38 (1:1,000 dilution; cat. no. 9212), anti-GAPDH (1:1,500; cat.
no. 5174) and anti-β-actin (1:1,000; cat. no. 4970) (all Cell
Signaling Technology, Inc.) for 2 h at room temperature. Next, the
membranes were incubated with the aforementioned fluorescence
secondary antibodies for 1 h at 37°C. Chemiluminescence detection
was conducted using Western Lightning Chemiluminescence Reagent
Plus (PerkinElmer, Inc., Waltham, MA, USA). and signal intensity
was determined using ImageJ software version 1.46 (National
Institutes of Health, Bethesda, MD, USA).
Statistical analysis
The data are presented as the mean ± standard
deviation of at least two independent studies each performed in
triplicate. Statistical analyses were performed using GraphPad
Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Comparisons
of means between groups were analyzed using one way analysis of
variance followed by Tukey’s post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
ATV reduces atherosclerotic lesion
formation in ApoE−/− mice
Fig. 1A shows the
data on the TC, TG, LDL and HDL levels of the ApoE−/−
mice. Statistical analysis showed that HFD treatment for 12 weeks
significantly increased the levels of TC, TG and LDL compared with
the ND group, with HDL as an exception. However, the data indicated
that ATV can markedly reduce the effects of HF on the levels of TC,
TG, LDL and HDL in ApoE−/− mice. Moreover, the H&E
staining results of aortic root showed the presence of more
atherosclerotic plaques in the ApoE−/− mice with HFD
treatment compared with that in the ND group (Fig. 1B). Whereas, in ApoE−/−
mice, the ATV treatment showed fewer atherosclerotic plaques than
in HFD group. These data show that ATV can inhibit the formation of
atherosclerotic areas in the aortic roots of ApoE−/−
mice.
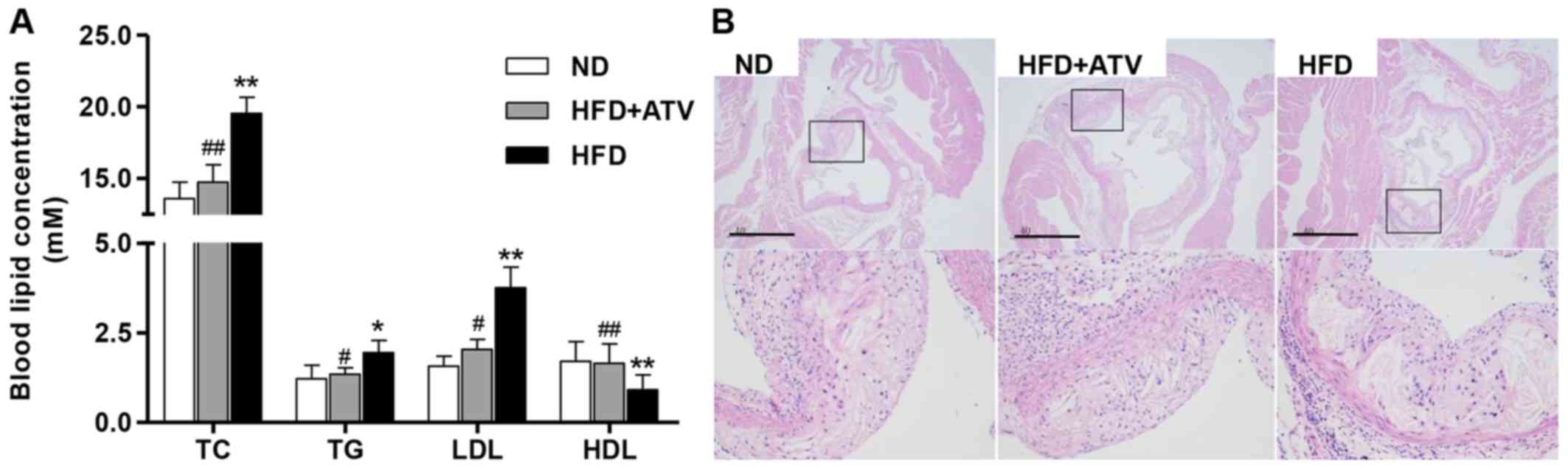 | Figure 1Atherosclerotic lesion formation in
aortic sections from ApoE−/− mice. (A) There were
significantly increased TC, TG and LDL levels, and decreased HDL
levels in the mice in the HFD group. (B) Atherosclerotic lesions in
the aortic root, as measured by hematoxylin and eosin staining.
Data are presented as the mean ± standard deviation (n=6).
*P<0.05 and **P<0.01 compared with the
ND group; #P<0.05 and ##P<0.01 compared
with the HFD group. Scale bars, 40 µm. ND, normal diet; HFD,
high-fat diet; ApoE−/−, apolipoprotein E-deficient; TC,
total cholesterol; TG, triglycerides; LDL, low-density lipoprotein
cholesterol; HDL, high-density lipoprotein cholesterol; ATV,
atorvastatin. |
ATV alters the content of HSP22, p-eNOS
and p-p38 in atherosclerotic lesions
Aorta cross-section immunochemistry results showed
that there was significantly less HSP22 in the aortic plaques
following intervention with ATV in ApoE−/− mice when
compared with HFD treatment alone (Fig. 2A). The similar effects of HFD and
ATV treatment on the content of HSP22 were also found at the serum
(Fig. 2B) and aortic tissue
(Fig. 2C and D) levels. In
addition, compared with the ND group, the HFD group exhibited
decreased p-eNOS expression in the aortic tissue (Fig. 2C and E), while the p-p38 increased
(Fig. 2C, E and F). However, ATV
did significantly augment p-eNOS and reduce p-p38 in the aortic
tissue of the HFD group. These data show that ATV can activate the
eNOS signaling pathway and inhibit the p38 mitogen-activated
protein kinase (MAPK) signaling pathway, which have already been
shown to serve a key role in anti-atherosclerotic effects (14).
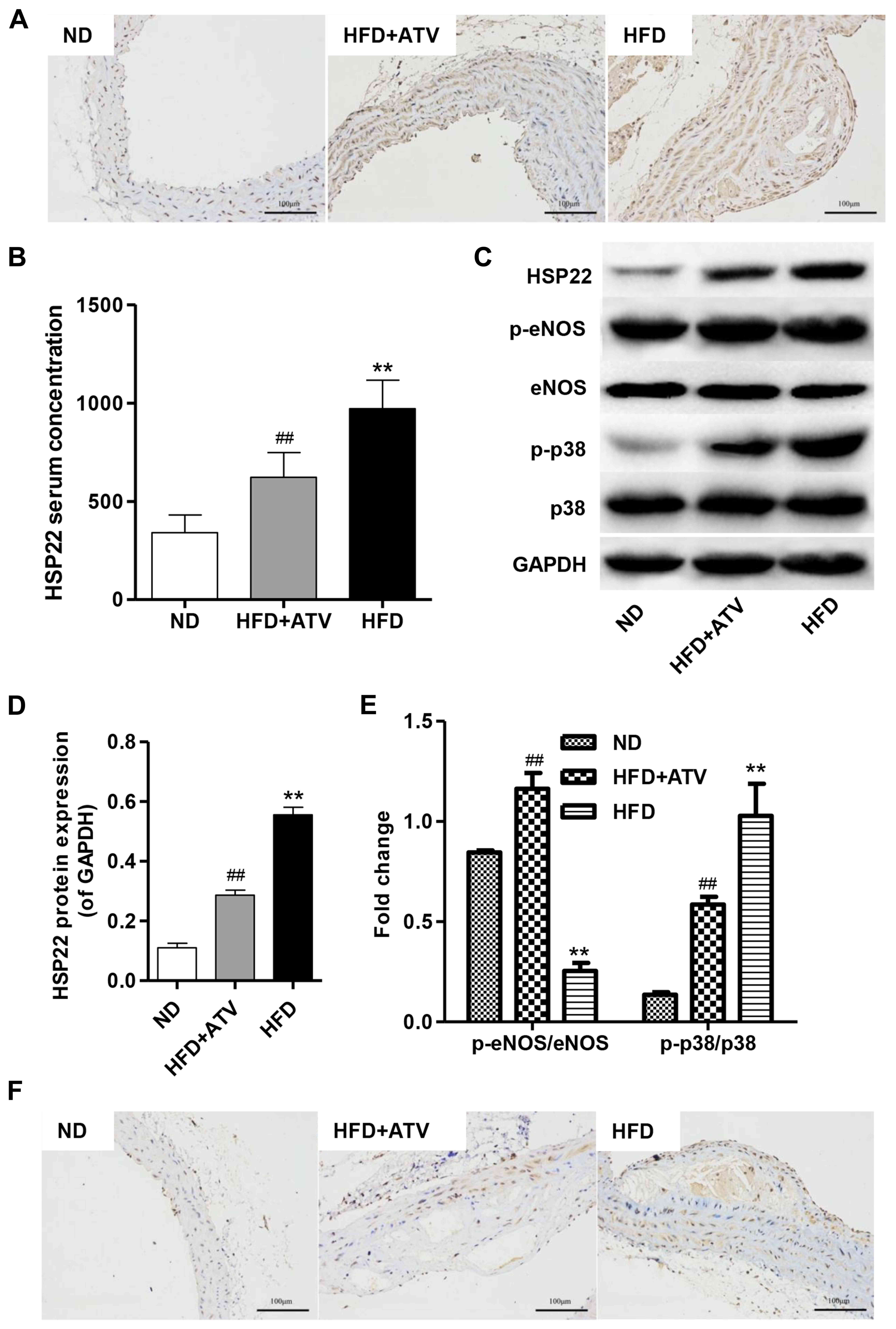 | Figure 2Effect of HFD on HSP22, eNOS and p38
MAPK in atherosclerotic lesions. The expression of HSP22 in
ApoE−/− mice with an HFD and/or ATV treatment was
measured by (A) immunohistochemistry, (B) enzyme-linked
immunosorbent assay and (C and D) western blot assay, respectively.
(C and E) The expression level of p-eNOS was examined by western
blot assay in ApoE−/− mice with an HFD and/or ATV
treatment. The expression level of p-p38 MAPK was examined by (C
and E) western blot assay and (F) immunohistochemistry in
ApoE−/− mice with an HFD and/or ATV treatment. Data are
presented as the mean ± standard deviation (n=6).
**P<0.01 compared with the ND group;
##P<0.01 compared with the HFD group. Scale bars, 100
µm. ND, normal diet; HFD, high-fat diet; HSP22, heat shock
protein 22; eNOS, endothelial nitric oxide synthase; MAPK,
mitogen-activated protein kinase; ApoE−/−,
apolipoprotein E-deficient; ATV, atorvastatin;
p-phosphorylated. |
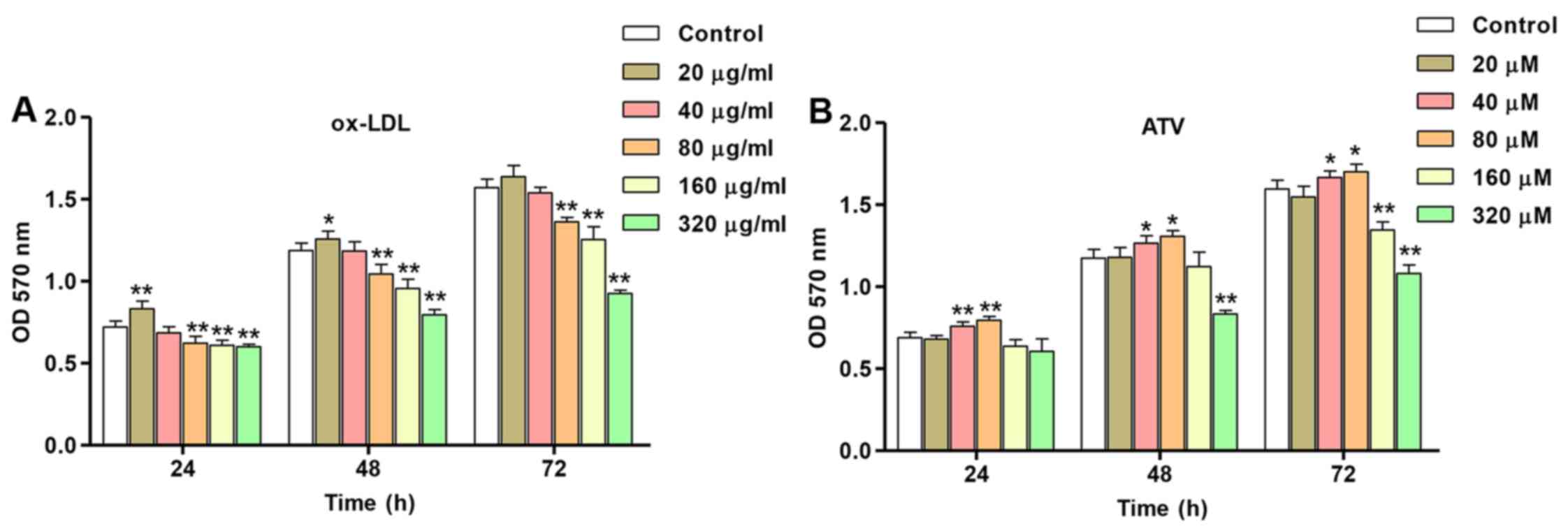
Effects of ox-LDL and ATV on HUVEC
proliferation
The concentration-dependent effect of ox-LDL on
HUVEC proliferation is shown in Fig.
3A. As shown in Fig. 3A,
compared with the ND group, HUVEC proliferation was increased with
ox-LDL stimulation at lower concentration (20 µg/ml) at 24
and 48 h; however, incubation with different concentrations of
ox-LDL (80, 160 and 320 µg/ml) led to a significant
decrease. The initial significant decrease in proliferation
compared with the control was observed following incubation of
HUVECs with 80 µg/ml ox-LDL at 24, 48 and 72 h. To
investigate the effects of ATV on the cytotoxicity of HUVECs, the
proliferation of HUVECs treated with ATV at different
concentrations and for time periods was also measured. Stimulation
of HUVECs with 40 and 80 µM ATV led to a significant
increase in HUVEC proliferation that was highest at 72 h post-ATV
application, while decreased proliferation was noted when using
higher concentrations of ATV treatment (160 and 320 µM) in
HUVECs at 72 h (Fig. 3B). These
results indicate that ox-LDL and ATV induce the proliferation of
HUVECs in a concentration- and time-dependent manner. Therefore,
160 µg/ml ox-LDL and 40 µM ATV were used for
subsequent experiments.
ATV inhibits the effects of ox-LDL on the
expression of HSP22, p-eNOS and p-p38 in HUVECs
Fig. 4A and B show
that 160 µg/ml ox-LDL treatment for 24 h significantly
increased the mRNA and protein expression of HSP22 in HUVECs
compared with the control group, but that 40 µM ATV
treatment for 24 h significantly decreased HSP22 expression. Prior
to the ATV treatment, pretreatment with ox-LDL at a concentration
of 160 µg/ml for 24 h significantly decreased the mRNA and
protein expression of HUSP22 in HUVECs compared with the group with
ox-LDL treatment alone. Fig. 4C and
D show that stimulation of ox-LDL caused decreased p-eNOS and
increased p-p38 levels in HUVECs compared with the control group,
but that ATV had an inverse effect. Prior to the ATV treatment,
pretreatment with ox-LDL significantly increased p-eNOS and
decreased p-p38 levels in HUVECs compared with using the ox-LDL
treatment alone.
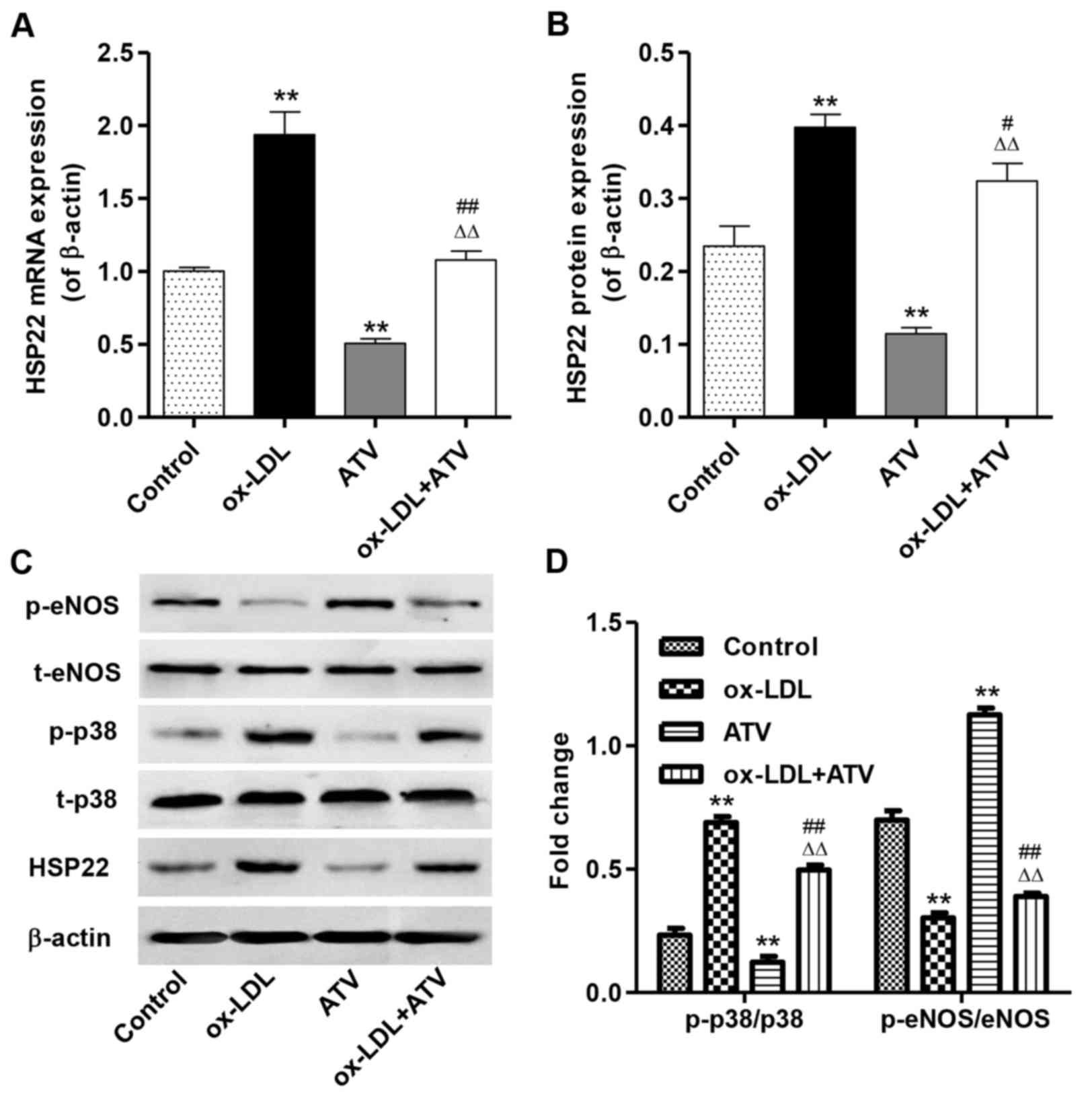 | Figure 4Effect of ATV on HSP22, eNOS and p38
MAPK in HUVECs. The expression of HSP22 in HUVECs with an HFD
and/or ATV treatment was measured by (A) reverse
transcription-quantitative polymerase chain reaction and (B)
western blot assay, respectively. (C and D) The expression levels
of p-eNOS and p-p38 MAPK were examined by western blot assay in
HUVECs with an HFD and/or ATV treatment. **P<0.01
compared with the control group; #P<0.05 and
##P<0.01 compared with the ox-LDL group;
△△P<0.01 compared with the ATV group. ox-LDL,
oxidized low-density lipoprotein; ATV, atorvastatin; HUVECs, human
umbilical vein endothelial cells; HSP22, heat shock protein 22;
eNOS, endothelial nitric oxide synthase; MAPK, mitogen-activated
kinase; HFD, high-fat diet; p-, phosphorylated; t-, total. |
HSP22 shRNA alters the expression of
p-eNOS and p-p38 in HUVECs induced by ox-LDL
To elucidate whether the cytoprotection of ATV was
associated with its down-regulation of HSP22 in ox-LDL-stimulated
HUVECs, three shRNAs targeting HSP22 were cloned into a lentiviral
vector for HUSP22-knockdown. shRNA-2 showed a minimal HSP22 protein
level compared with the other two shRNAs in the HUVECs, but
exhibited no effect in NC-transfected HUVECs (Fig. 5A). Moreover, shRNA transfection in
HUVECs significantly decreased HSP22 expression in ox-LDL, ATV and
ox-LDL plus ATV treatment groups compared with corresponding NC
groups (Fig. 5B). Notably,
increased p-eNOS levels were found only in HUVECs with shRNA or
shRNA plus ox-LDL treatment groups compared with the corresponding
NC groups (Fig. 5C and D).
Similarly, HSP22 shRNA markedly decreased the p-p38 levels in
ox-LDL, ATV and ox-LDL plus ATV treatment groups compared with that
in the corresponding NC groups (Fig.
5C and D). These findings suggest that the inhibition of the
cytotoxicity of ATV is associated with its downregulation of
HSP22.
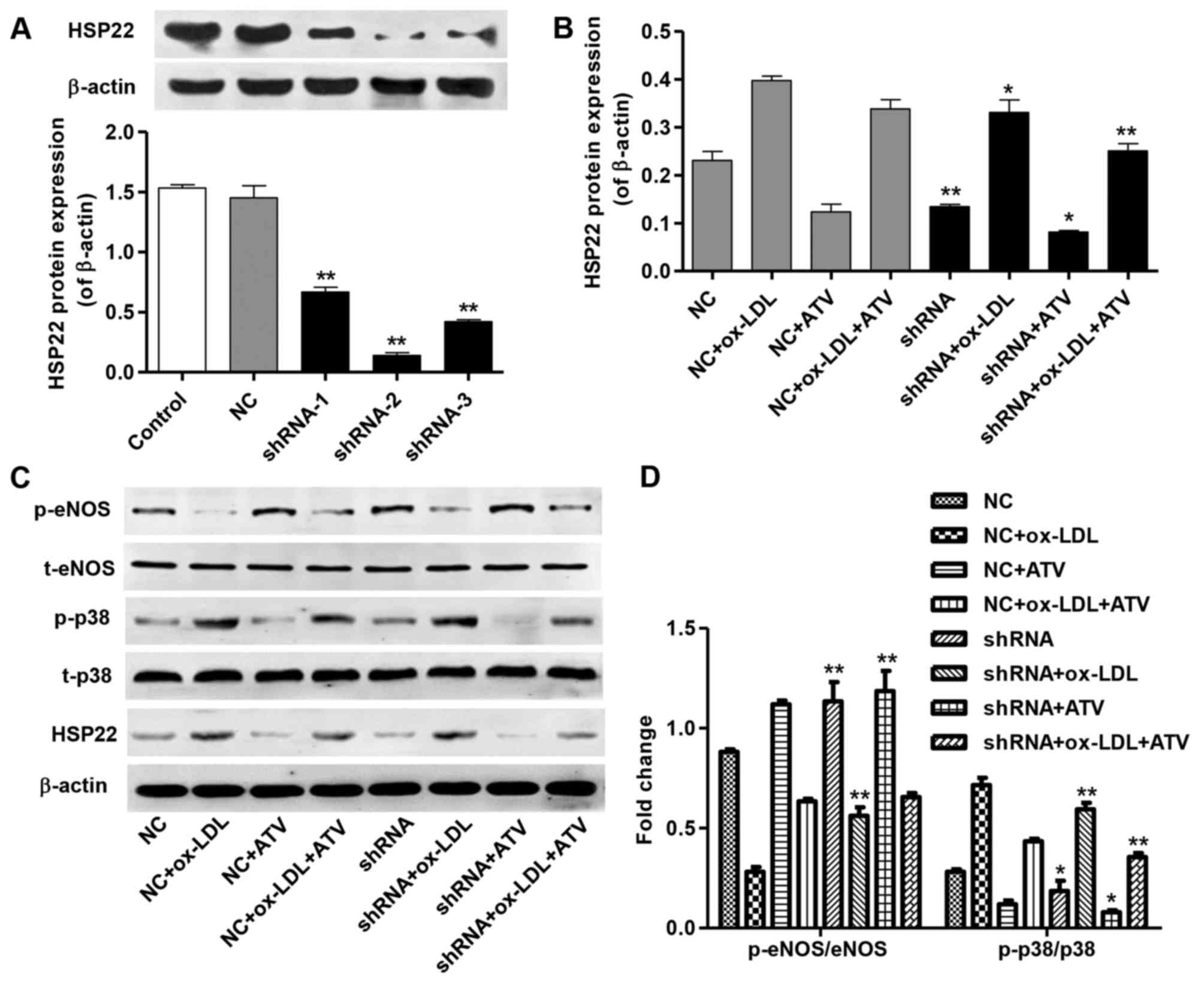 | Figure 5HSP22-knockdown inhibits
ox-LDL-induced p-eNOS decrease and p-p38 increase in HUVECs. (A)
The transfection efficiency of HSP22 shRNAs in HUVECs was measured
by western blot assay. (B) The expression of HSP22 in HUVECs with
an HFD and/or ATV treatment was measured by western blot assay. (C
and D) The expression of p-eNOS and p-p38 in HUVECs with an HFD
and/or ATV treatment in the absence or presence of HSP22 shRNA was
measured by western blot assay. *P<0.05 and
**P<0.01 compared with the corresponding NC group.
ox-LDL, oxidized low-density lipoprotein; ATV, atorvastatin;
HUVECs, human umbilical vein endothelial cells; HSP22, heat shock
protein 22; eNOS, endothelial nitric oxide synthase; MAPK,
mitogen-activated kinase; HFD, high-fat diet; p-, phosphorylated;
t-, total; NC, negative control; shRNA, short hairpin RNA. |
HSP22 shRNA reduces proliferation
inhibition and cell cycle arrest of HUVECs induced by ox-LDL
The effects of HSP22-knockdown on cell proliferation
and the cell cycle progression of HUVECs were measured by MTT and
flow cytometry analysis, respectively. ATV treatment markedly
reversed the inhibition of proliferation of HUVECs with NC or HSP22
shRNA transfection caused by ox-LDL treatment at 24, 48 and 72 h
(Fig. 6A-C). More importantly,
HSP22 shRNA had a similar effect to ATV, which showed increased
proliferation of HUVECs compared with the corresponding NC groups.
Furthermore, ATV or HSP22 shRNA treatment reduced the
ox-LDL-induced G2/M cell cycle arrest of HUVECs (Fig. 6D and E). However, treatment with
ATV alone had no effect on the cell cycle of the HUVECs.
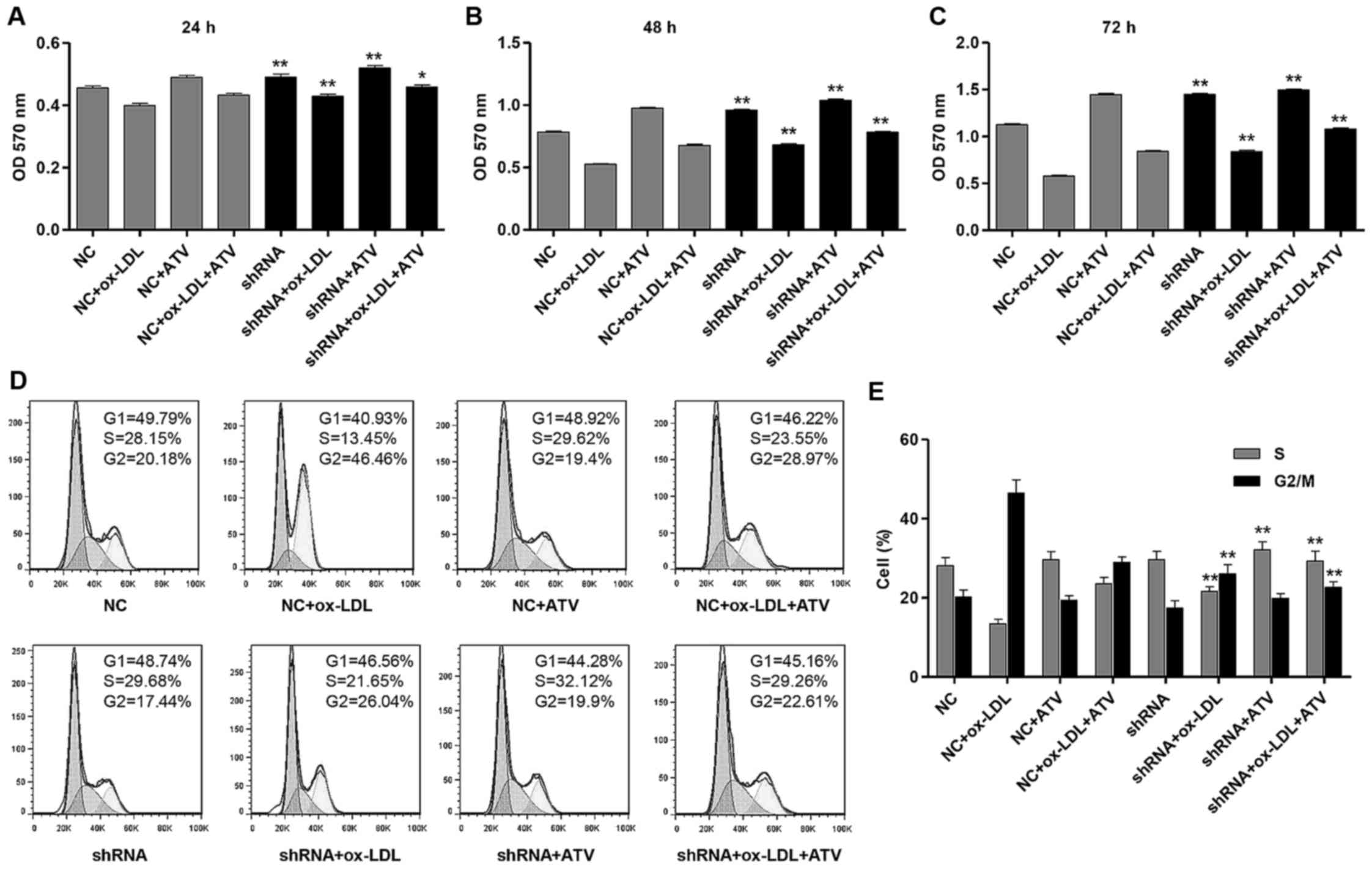 | Figure 6HSP22-knockdown reduces
ox-LDL-induced cell proliferation inhibition and cell cycle arrest
in HUVECs. Cell proliferation was measured by MTT assay in HUVECs
with an HFD and/or ATV treatment in the absence or presence of
HSP22 shRNA for (A) 24 h, (B) 48 h and (C) 72 h. (D and E) The cell
cycle was measured by flow cytometry assay in HUVECs with an HFD
and/or ATV treatment in the absence or presence of HSP22 shRNA.
*P<0.05 and **P<0.01 compared with the
corresponding NC group. ox-LDL, oxidized low-density lipoprotein;
ATV, atorvastatin; HUVECs, human umbilical vein endothelial cells;
HSP22, heat shock protein 22; eNOS, endothelial nitric oxide
synthase; MAPK, mitogen-activated kinase; HFD, high-fat diet; p-,
phosphorylated; t-, total; NC, negative control; shRNA, short
hairpin RNA; OD, optical density. |
Discussion
Coronary artery AS is the single largest killer of
men and women in the world; it is the principal cause of coronary
artery disease, in which atherosclerotic changes are present within
the walls of the coronary arteries (15). The accumulation of lipoproteins
not only damages the endothelial cells, but also modulates the
expression levels of adhesive molecules and inflammatory factors,
resulting in the progression of AS (16). Depending on the model used,
animals fed an HFD usually develop hyperlipidemia. ATV therapy is
effective in lowering the serum lipid levels in these animals,
including the cholesterol and TG levels (17,18). In the present study, the lipid
metabolism of ApoE−/− mice was abnormal following HFD
intervention, showing increased serum levels of TC, TG and LDL, and
a decreased HDL serum level. Atherosclerotic plaques were also
observed in HFD-treated ApoE−/− mice, which indicated
that an AS mouse model had been successfully established.
HSP22 is widely distributed in a number of tissues,
particularly in skeletal muscle, smooth muscle, the myocardium and
the brain (19). In the present
study, the expression of HSP22 was measured by western blot
analysis and immunochemistry assay. It was found that the HFD model
exhibited a higher level of HSP22 expression compared with the ND
group in ApoE−/− mice. LDL, particularly ox-LDL, is the
main cause for AS in hyperlipidemia mice. In the present study, it
was also found that ox-LDL can stimulate HUVECs and then increase
the expression of HSP22. HSPs can be detected in the serum of
various diseases; for example, HSP70, is considered to be the
predictor for acute coronary syndrome and the risk factor for
cardiovascular disease (20). The
present results found that the content of HSP22 in the serum of
ApoE−/− mice was significantly increased following HFD
treatment.
Endothelial dysfunction, a critical and initial
factor for AS, is dependent on the expression of eNOS (21). Under anoxic conditions, the
increased expression of HSP22 in the myocardial cells and
mitochondria can upregulate the expression of mitochondrial NOS. NO
generation by eNOS possesses a protective effect on cardiomyocytes
through decreasing the levels of oxidative phosphorylation,
reactive oxygen and the opening of mitochondrial permeability
transition pores (22). In the
present study, it was found that HFD or ox-LDL stimulation
decreased the levels of p-eNOS/eNOS, in contrast to the expression
of HSP22 in ApoE−/− mice and HUVECs, which was a similar
result to a previous study (23).
Activation of AMP-activated protein kinase has been
reported to possess anti-atherosclerotic effects by upregulating
the protein kinase B/eNOS/NO signaling pathway, leading to the
suppression of p38-mediated nuclear factor-κB activation and
consequent suppression of downstream inflammatory responses
(24,25). The suppression of MAPK has also
been reported to have beneficial effects on AS through inhibition
of adhesion molecules and anti-inflammatory effects, as well as
increase in the stability of carotid plaques (26,27). In the present study, the
expression of p-p38 MAPK was markedly increased in the
ApoE−/− mice and the HUVECs. In the HFD-induced AS
model, inhibition of p38 MAPK promotes vasculogenic cells survival,
activates the downstream mitogen- and stress-activated protein
kinase and the cyclic adenosine monophosphate response element
binding protein, and reduces endothelial dysfunction and AS
progression (28).
ATV, as the hydroxy-3-methylglutaryl-CoA reductase
inhibitor, has been reported to serve a protective role in
cardiovascular disease, including AS. However, the mechanisms
through which ATV attenuates the progress of AS is complex and not
completely understood. Notably, in the present study, ATV decreased
HSP22 and p-p38 MAPK expression, and increased p-eNOS/eNOS
expression in the ApoE−/− mice and the HUVECs. A
previous study showed that ATV reduced endothelial apoptosis
through suppressing advanced glycation end product-induced injury
and increasing the expression of HSP70 (29), while the upregulation of HSP22 was
able to promote the inflammation of rheumatoid arthritis (30). According to these findings, we
propose that ATV protects against AS by directly inhibiting the
expression of HSP22. In thre present study, HSP22 downregulation
increased p-eNOS/eNOS and decreased p-p38 MAPK expression in the
HUVECs, suggesting that ATV reduced AS through suppressing
HSP22-dependent inactivation of eNOS signaling and activation of
p38 MAPK, which is in line with another previous study (31). Ye (32) reported that the inhibition of
HSP22 aggravated the cell apoptosis induced by ox-LDL, indicating a
protective effect of HSP22 on cells through improving
ox-LDL-induced lesions. However, the present results observed that
inhibition of HSP22 by shRNA transfection in HUVECs reduced cell
proliferation inhibition and cell cycle G2/M arrest induced by
ox-LDL.
A limitation of the present study is that it could
not provide direct evidence of HSP22 activating the p38 MAPK
signaling pathway. The definitive mechanisms of action for ATV
require further investigation in vitro by culturing primary
aortic endothelial cells. In the present study, ATV was shown to
suppress ox-LDL-induced HSP22 expression, which suggests that the
powerful anti-atherosclerotic effects of ATV may activate eNOS
signaling and inhibit p38 MAPK signaling by suppressing HFD- or
ox-LDL-induced HSP22 expression. Investigation of the HSP22
expression induced by HFD- or ox-LDL may provide a clue to
understanding the pathophysiology of AS, and may lead to a novel
and promising therapeutic strategy.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 30800467).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors’ contributions
QC and JX conceived and designed the experiments.
RG, HF, CX and YW performed the experiments. CX and HZ analyzed the
data. HZ and YW contributed with regards to the
reagents/materials/analysis tools. QC, JX and YW wrote the paper.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal care and experimental procedures in the
current study complied with the protocol approved by the Second
Affiliated Hospital of Nanchang University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Cuerrier CM, Chen YX, Tremblay D, Rayner
K, McNulty M, Zhao X, Kennedy CR, de BelleRoche J, Pelling AE and
O’Brien ER: Chronic overexpression of heat shock protein 27
attenuates atherogenesis and enhances plaque remodeling: A combined
histological and mechanical assessment of aortic lesions. PLoS One.
8:e558672013. View Article : Google Scholar
|
|
2
|
Almanzar G, Öllinger R, Leuenberger J,
Onestingel E, Rantner B, Zehm S, Cardini B, van der Zee R,
Grundtman C and Wick G: Autoreactive HSP60 epitope-specific T-cells
in early human atherosclerotic lesions. J Autoimmun. 39:441–450.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Smith CC, Yu YX, Kulka M and Aurelian L: A
novel human gene similar to the protein kinase (PK) coding domain
of the large subunit of herpes simplex virus type 2 ribonucleotide
reductase (ICP10) codes for a serine-threonine PK and is expressed
in melanoma cells. J Biol Chem. 275:25690–25699. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li XS, Xu Q, Fu XY and Luo WS: Heat shock
protein 22 over-expression is associated with the progression and
prognosis in gastric cancer. J Cancer Res Clin Oncol.
140:1305–1313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marunouchi T, Abe Y, Murata M, Inomata S,
Sanbe A, Takagi N and Tanonaka K: Changes in small heat shock
proteins HSPB1, HSPB5 and HSPB8 in mitochondria of the failing
heart following myocardial infarction in rats. Biol Pharm Bull.
36:529–539. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marunouchi T, Inomata S, Sanbe A, Takagi N
and Tanonaka K: Protective effect of geranylgeranylacetone via
enhanced induction of HSPB1 and HSPB8 in mitochondria of the
failing heart following myocardial infarction in rats. Eur J
Pharmacol. 730:140–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pantan R, Tocharus J, Suksamrarn A and
Tocharus C: Synergistic effect of atorvastatin and
Cyanidin-3-glucoside on angiotensin II-induced inflammation in
vascular smooth muscle cells. Exp Cell Res. 342:104–112. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Feng B, Xu L, Wang H, Yan X, Xue J, Liu F
and Hu JF: Atorvastatin exerts its anti-atherosclerotic effects by
targeting the receptor for advanced glycation end products. Biochim
Biophys Acta. 1812:1130–1137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang M, Zhou SH, Li XP, Shen XQ, Fang ZF,
Liu QM, Qiu SF and Zhao SP: Atorvastatin downregulates BMP-2
expression induced by oxidized low-density lipoprotein in human
umbilical vein endothelial cells. Circ J. 72:807–812. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin CP, Chen YH, Lin WT, Leu HB, Liu TZ,
Huang SL and Chen JW: Direct effect of statins on
homocysteine-induced endo-thelial adhesiveness: Potential impact to
human atherosclerosis. Eur J Clin Invest. 38:106–116. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zeng Y, Li C, Guan M, Zheng Z, Li J, Xu W,
Wang L, He F and Xue Y: The DPP-4 inhibitor sitagliptin attenuates
the progress of atherosclerosis in apolipoprotein-E-knockout mice
via AMPK- and MAPK-dependent mechanisms. Cardiovasc Diabetol.
13:322014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang XF, Zhu J, Geng WY, Zhao SJ, Jiang
CW, Cai SR, Cheng M, Zhou CY and Liu ZB: Electroacupuncture at
Feishu (BL13) and Zusanli (ST36) down-regulates the expression of
orexins and their receptors in rats with chronic obstructive
pulmonary disease. J Integr Med. 12:417–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real time quantitative PCR and
the 2(Delta Delta C(T)) method. Methods. 25(402): 4082001.
View Article : Google Scholar
|
|
14
|
Cuerrier CM, Chen YX, Tremblay D, Rayner
K, McNulty M, Zhao X, Kennedy CR, de BelleRoche J, Pelling AE and
O’Brien ER: Chronic over-expression of heat shock protein 27
attenuates atherogenesis and enhances plaque remodeling: A combined
histological and mechanical assessment of aortic lesions. PLoS One.
8:e558672013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hodis HN, Mack WJ, Azen SP, Lobo RA,
Shoupe D, Mahrer PR, Faxon DP, Cashin-Hemphill L, Sanmarco ME,
French WJ, et al: Women’s Estrogen-Progestin Lipid-Lowering Hormone
Atherosclerosis Regression Trial Research Group: Hormone therapy
and the progression of coronary-artery atherosclerosis in
postmenopausal women. N Engl J Med. 349:535–545. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hermida N and Balligand JL: Low-density
lipoprotein-cholesterol-induced endothelial dysfunction and
oxidative stress: The role of statins. Antioxid Redox Signal.
20:1216–1237. 2014. View Article : Google Scholar
|
|
17
|
Ji G, Zhao X, Leng L, Liu P and Jiang Z:
Comparison of dietary control and atorvastatin on high fat diet
induced hepatic steatosis and hyperlipidemia in rats. Lipids Health
Dis. 10:232011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Paraskevas KI, Pantopoulou A, Vlachos IS,
Agrogiannis G, Iliopoulos DG, Karatzas G, Tzivras D, Mikhailidis DP
and Perrea DN: Comparison of fibrate, ezetimibe, low- and high-dose
statin therapy for the dyslipidemia of the metabolic syndrome in a
mouse model. Angiology. 62:144–154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Acunzo J, Katsogiannou M and Rocchi P:
Small heat shock proteins HSP27 (HspB1), αB-crystallin (HspB5) and
HSP22 (HspB8) as regulators of cell death. Int J Biochem Cell Biol.
44:1622–1631. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang X, Xu Z, Zhou L, Chen Y, He M, Cheng
L, Hu FB, Tanguay RM and Wu T: Plasma levels of Hsp70 and
anti-Hsp70 antibody predict risk of acute coronary syndrome. Cell
Stress Chaperones. 15:675–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vergnani L, Hatrik S, Ricci F, Passaro A,
Manzoli N, Zuliani G, Brovkovych V, Fellin R and Malinski T: Effect
of native and oxidized low-density lipoprotein on endothelial
nitric oxide and superoxide production: Key role of L-arginine
availability. Circulation. 101:1261–1266. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Laure L, Long R, Lizano P, Zini R,
Berdeaux A, Depre C and Morin D: Cardiac H11 kinase/Hsp22
stimulates oxidative phosphorylation and modulates mitochondrial
reactive oxygen species production: Involvement of a nitric
oxide-dependent mechanism. Free Radic Biol Med. 52:2168–2176. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Korkmaz Y, Bloch W, Addicks K, Schneider
K, Baumann MA and Raab WH: The Basal phosphorylation sites of
endothelial nitric oxide synthase at serine (Ser)1177, Ser116, and
threonine (Thr)495 in rat molar epithelial rests of Malassez. J
Periodontol. 76:1513–1519. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y, Qiu J, Wang X, Zhang Y and Xia M:
AMP-activated protein kinase suppresses endothelial cell
inflammation through phosphorylation of transcriptional coactivator
p300. Arterioscler Thromb Vasc Biol. 31:2897–2908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ou HC, Hsieh YL, Yang NC, Tsai KL, Chen
KL, Tsai CS, Chen IJ, Wu BT and Lee SD: Ginkgo biloba extract
attenuates oxLDL-induced endothelial dysfunction via an
AMPK-dependent mechanism. J Appl Physiol 1985. 114:274–285. 2013.
View Article : Google Scholar
|
|
26
|
Zhang K, Meng X, Kong J, Liu FF, Yang JM,
Gao F, Zhang Y and Zhang C: Simvastatin increases
Prolyl-4-Hydroxylase α1 expression in atherosclerotic plaque and
ox-LDL-stimulated human aortic smooth muscle cells via p38 MAPK and
ERK1/2 signaling. J Mol Cell Cardiol. 65:43–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park SH, Sung YY, Nho KJ and Kim HK:
Anti-atherosclerotic effects of Polygonum aviculare L. ethanol
extract in ApoE knock-out mice fed a Western diet mediated via the
MAPK pathway. J Ethnopharmacol. 151:1109–1115. 2014. View Article : Google Scholar
|
|
28
|
Seeger FH, Sedding D, Langheinrich AC,
Haendeler J, Zeiher AM and Dimmeler S: Inhibition of the p38 MAP
kinase in vivo improves number and functional activity of
vasculogenic cells and reduces atherosclerotic disease progression.
Basic Res Cardiol. 105:389–397. 2010. View Article : Google Scholar
|
|
29
|
Li Y, Li J, Cui L, Lai Y, Yao Y, Zhang Y,
Pang X, Wang J and Liu X: Inhibitory effect of atorvastatin on
AGE-induced HCAEC apoptosis by upregulating HSF-1 protein. Int J
Biol Macromol. 57:259–264. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Roelofs MF, Boelens WC, Joosten LA,
Abdollahi-Roodsaz S, Geurts J, Wunderink LU, Schreurs BW, van den
Berg WB and Radstake TR: Identification of small heat shock protein
B8 (HSP22) as a novel TLR4 ligand and potential involvement in the
pathogenesis of rheumatoid arthritis. J Immunol. 176:7021–7027.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qiu H, Lizano P, Laure L, Sui X, Rashed E,
Park JY, Hong C, Gao S, Holle E, Morin D, et al: H11 kinase/heat
shock protein 22 deletion impairs both nuclear and mitochondrial
functions of STAT3 and accelerates the transition into heart
failure on cardiac overload. Circulation. 124:406–415. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ye YP: The effect of HSP22 and
Atorvastatin in the injury of HUVECs induced by ox-LDL. Nanchang
University. 2014.
|