Introduction
Alzheimer's disease (AD), due to neuron loss and
synaptic degeneration, is characterized by a significant decline in
cognitive function and poor prognosis, which accounts for 60-70% of
all types of dementia (1,2). Although the aggregated amyloid β
(Aβ) titanium and hyper-phosphorylated (p-) Tau may exhibit
neurotoxicity in the development of AD, the exact therapeutic
targets have not been successfully identified due to the complex
mechanism associated with the pathogenesis of this disease
(3). Among the various
hypotheses, the occurrence of oxidative stress and neuronal
apoptosis in AD has been accepted. Oxidative stress, represented by
the over-accumulation of reactive oxygen species (ROS), is
responsible for the damage to the mitochondria, membrane lipids,
and nucleic acids (4). Oxidative
stress promotes Aβ aggregation, which exhibits a direct toxic
effect to its surrounding neurons, leading to the susceptibility of
neurons to free radicals, particularly ROS (5,6).
As a major cellular source of ROS, mitochondria
serve an important role in the pathophysiology of
neurode-generative diseases (7).
The disruption of mitochondrial homeostasis caused by the high
levels of ROS further leads to the over-production of ROS and other
cytokines, such as cytochrome c, which induces the cell
apoptosis program (8,9). The extremely high levels of
glutamate (Glu) associated with ROS accumulation, a central nervous
neurotransmitter, damages neurons (10); however, during the onset of
oxidative stress, the transcription factor NF-E2p45-related factor
2 (Nrf2) helps maintain cellular redox homeostasis, and supports
the structure and functional integrity of the mitochondria
(11). Furthermore, the deletion
of Nrf2 can increase the intracellular levels of Aβ in mice with AD
(12).
The occurrence of AD is rapidly increasing
worldwide, and according to statistics, there are >40 million
patients globally (13). As the
prevention and treatment of AD has been a serious challenge,
potential agents applied in clinics have failed to effectively
treat patients with AD. In addition, the adverse effects of these
agents have been noted, including gastrointestinal discomfort,
difficulty sleeping and muscle spasms (14). Herbal compounds have been reported
as a large and underappreciated source of potential agents to treat
or prevent AD (15,16). Evodiamine treatment resulted in
the return of AD-associated symptoms via modulating oxidative
stress-mediated apoptosis in L-glutamate (L-Glu)-damaged HT22
cells, and a mouse model with AlCl3- and D-galactose
(D-gal)-induced AD (17).
Polysaccharides extracted from Sparassis crispa (18) and
Armillaria mellea (19)
have been successfully confirmed to exert neuroprotective effects
against AD. Isoastilbin (IAB), a dihydroflavonol glycoside compound
(Fig. 1A), is widely distributed
in Rhizoma Smilacis glabrae and Astragalus
membranaceus. The majority studies of IAB have focused on its
extraction and purification from herbs (20,21). A study reported the antioxidative
effects of IAB (22); however,
astilbin, the isomer of IAB, was reported to improve the cognitive
abilities of a transgenic mouse with AD via regulating the protein
kinase B/glycogen synthase kinase-3β signaling pathway (23). Based on the pharmacological
activity of astilbin and the antioxidative properties of IAB, we
hypothesized that IAB may exhibit neuroprotective effects against
AD.
In the present study, the protective effects of IAB
on restoring L-Glu-induced PC12 cell apoptosis, and reducing
AlCl3- and D-gal-induced AD-associated symptoms in
Balb/c male mice were investigated. To the best of our knowledge,
the present study is the first to report of the potential
therapeutic properties of IAB against AD in an apoptotic cell model
and in mice with AD-associated symptoms. The results may provide
insight into the application of IAB in the adjuvant therapy of
AD.
Materials and methods
Cell culture
PC12 cells (CRL-1721; American Type Culture
Collection, Manassas, VA, USA) were cultured in Dulbecco's Modified
Eagle's medium (DMEM; Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) containing 5% horse serum (HS; Invitrogen; Thermo
Fisher Scientific, Inc.), 10% fetal bovine serum (FBS; Invitrogen
Thermo Fisher Scientific, Inc.), penicillin (100 U/ml) and
streptomycin (100 µg/ml) (Invitrogen; Thermo Fisher
Scientific, Inc.), under a humidified atmosphere containing 5%/95%
CO2/air at 37°C. PC12 cells underwent differentiation
for 48 h at 37°C with 50 ng/ml nerve growth factor (Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) dissolved in DMEM containing 1%
FBS, 1% HS and 100 U/ml of penicillin/streptomycin.
Cell viability and caspase activity
analysis
PC12 cells were seeded into 96-well plates (8,000
cells/well/100 µl), pre-treated with 10 or 30 µM of
IAB (purity, ≥95%; Shanghai Yuanye Biotechnology Co., Ltd.,
Shanghai, China) for 3 h at 37°C, and then co-incubated with 25 mM
of L-Glu for another 24 h at 37°C. Cell viability was detected by a
3-(4,5-dimethyl-2-thiazolyl)-2,5-di-phenyl-2-H-tetrazolium bromide
(MTT; cat. no. R20228, Shanghai Yuanye Biotechnology Co., Ltd.,
Shanghai, China) assay (19), it
was used according to the manufacturer's protocols. A total of 100
µl dimethyl sulfoxide was used to solubilize purple formazan
crystals, then analyzing the absorbance using a microplate reader
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) at a wavelength of
490 nm. The activity of caspase-3 was analyzed using a commercial
kit, it was used according to the manufacturer's protocols (cat.
no. G007; Nanjing Jiancheng Bioengineering Institute, Nanjing,
China). The experiments were repeated eight times.
Cell apoptosis assay
PC12 cells were seeded into 6-well plates at
5×105 cells/well/ml, and pre-treated with 10 or 30
µM of IAB for 3 h at 37°C, and then co-incubated with 25 mM
of L-Glu for another 24 h at 37°C. The rate of cell apoptosis was
determined via the early apoptosis and late apoptosis of cells,
which was analyzed by propidium iodide/Annexin V staining (EMD
Millipore, Billerica, MA, USA) using a Muse™ Cell Analyzer flow
cytometer (EMD Millipore, Billerica, MA, USA); the data was
analyzed by Muse 1.4 Analysis. The experiments were repeated eight
times.
ROS levels and the dissipation of
mitochondrial membrane potential (MMP)
PC12 cells were seeded into 6-well plates at
5×105 cells/well/ml, and pre-treated with 10 and 30
µM of IAB for 3 h, and then co-incubated with 25 mM of L-Glu
for another 12 h at 37°C. The ROS levels were analyzed by staining
with 2,7-dichlorofluorescein diacetate (DCFH-DA; Sigma-Aldrich;
Merck KGaA) according to a previous study (24). The alterations in MMP were
analyzed with
5,5′,6,6′-tetra-chloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine
iodide (Sigma-Aldrich; Merck KGaA) staining as described (25). Alterations in fluorescent
intensity were analyzed using a fluorescent microscope
(magnification, ×200; CCD camera, Axio Observer Z1; Zeiss AG,
Oberkochen, Germany). The experiments were repeated eight times.
Quantification of data was conducted with Image J software version
1.46 (National Institutes of Health, Bethesda, MD, USA) and
expressed as the green fluorescence intensity for intracellular ROS
levels, and the ratio of red to green fluorescence intensity for
MMP detection.
Experimental protocol on the mouse model
with AD
The present study was approved by the Animal Ethics
Committee of Jilin University (no. 20170206). A total of 36 Balb/c
male mice (10-weeks old, 23-35 g) were obtained from Bethune
Medical College, Jilin University (Changchun, China) were housed in
cages under a temperature of 23±1°C and humidity of 40-60% with
sufficient water and food, and under a 12 h light/dark cycle.
A total of 24 mice were subcutaneously injected with
120 mg/kg of D-gal and intragastrically administered 20 mg/kg of
AlCl3 once per day for 56 days. At the 29th day, the
mice were randomly divided into two groups and intra-gastrically
administered normal saline solution (n=12) or 40 mg/kg of IAB
(n=12) once daily for 28 days. The other 12 mice were treated with
normal saline solution for 56 days and served as control group. On
days 54, 55 and 56, behavioral tests, including a Morris water maze
test (17), an open field test
(17) and the Y maze test
(26), were respectively
performed as previously described without modifications. At the end
of the experiments, blood samples were collected from the caudal
vein under anesthesia with 400 mg/kg (10%) chloral hydrate, and the
whole brains, kidney and spleen were collected following euthanasia
by injection with 150 mg/kg (1.5%) pentobarbital, after standing at
25°C for 30 min, blood samples were centrifuged at 300 × g for 10
min at 25°C twice to obtain serum.
Levels of neurotransmitters and factors
associated with oxidative stress, and Aβ1-42 detection
In each group, six mice were randomly selected for
the detection of biochemical indexes. The levels of ROS (cat. no.
E-20634), superoxide dismutase (SOD; cat. no. E-20347), and
glutathione peroxidase (GSH-Px; cat. no. E-20584) and Aβ1-42 (cat.
no. E-20118) in the serum and brains, and the levels of
acetylcholine (Ach) (cat. no. E-20535), Ach esterase (AchE; cat.
no. E-2143) and choline acetyltransferase (ChAT; cat. no. E-21422)
in brains were detected by commercialized ELISA kits (Shanghai
Yuanye Biological Technology Co., Ltd.).
Immunohistochemistry
Similar to a previous study, immuno-histochemical
analysis was conducted to analyze the levels of Aβ1-42 and p-Tau
(Ser404) in the brain (27). In
each group, the brain tissues of six other mice were fixed with 4%
formalin solution at 25°C for 1 week, then sliced into 5
µm-thick sections, which prepared for analysis. Briefly,
after antigen retrieval via heating in 0.01 mol/l citrate buffer at
98°C, the slides were blocked in 3% hydrogen peroxide for 10 min,
and 2% goat serum for 2 h at room temperature, respectively. The
slides were then exposed to antibodies of Aβ1-42 (1:200, cat. no.
bs-0877R; BIOSS, Beijing, China) and p-Tau (Ser404, 1:200, cat. no.
bs-2392R; BIOSS) overnight at 4°C, followed by incubation with a
horseradish peroxidase-conjugated secondary antibody (cat. no.
SH-0032; Bejing Dingguo Changsheng Biotechnology Co., Ltd.,
Beijing, China) at a dilution of 1:2,000 for 4 h at 4°C.
3,3′-diaminobenzidine and Mayer's hematoxylin were applied for 5
min at 25°C, the slides were solidified with a neutral resin and
photographed by the Olympus IX73 optical microscope (Olympus
Corporation, Tokyo, Japan).
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick end labeling
(TUNEL) assay
TUNEL was applied to analyze the occurrence of
apoptosis in the brain by observing the number of positive cells in
the field of view (19).
Following deparaffinization, the brain slides from six mice of each
group were washed with PBS and covered with permeabilization
reagent (20 µg/ml Proteinase K). The brain slides were
incubated with TdT reaction mixture in the dark for 1 h at 37°C,
and then analyzed three fields per view under a Nikon Eclipse TE
2000-S fluorescence microscope (magnification, ×200) Images were
collected with a CCD camera.
Histological analysis
The whole brains, kidney and spleen of six mice from
each group were preserved in 10% formaldehyde solution at 25°C for
1 week. Following dehydration using 100, 95, 80, 70 and 50% ethanol
and distilled water in sequence, the samples were embedded in
paraffin wax and sliced into joint cavity sections of 5 µm
thickness. Following H&E staining for 10 min at 25°C, light
microscopy was performed at ×100 (for brain slides) or ×200 (for
kidney and splenic slides) to analyze histopathological alterations
within the tissues, three fields per view were analyzed.
Western blotting
PC12 cells were seeded into 6-well plates at
5×105 cells/well/1 ml, and pre-treated with 10 or 30
µM of IAB for 3 h, and then co-incubated with 25 mM of L-Glu
for another 24 h. Radioimmunoprecipitation assay buffer
(Sigma-Aldrich; Merck KGaA) containing 1% protease inhibitor
cocktail (Sigma-Aldrich; Merck KGaA) was used to lyse the treated
cells and collected brain tissues. The nuclear fractions of cells
and tissue samples to determine Nrf2 expression were obtained using
a nuclear pulp separation kit (cat. no. BB-36021-2; BestBio,
Shanghai, China), which was used according to the manufacturer's
protocols, The protein concentration of the samples was detected by
a Bicinchoninic Acid kit; 40 µg of samples were separated
via 12% SDS-PAGE, electrotrans-ferred onto 0.45 µm
nitrocellulose membranes (Bio Basic, Inc., Toronto, Canada), and
then incubated with primary antibodies including B-cell lymphoma 2
(Bcl-2; sc-70411), B-cell lymphoma-extra large (Bcl-xL; sc-8392),
Bcl-2-associated X (Bax; sc-7480), cleaved caspase-3 (sc-136219),
cleaved caspase-9 (sc-56076), Nrf2 (sc-722), heme oxygenase-1
(HO-1; sc-390991), HO-2 (sc-17786), superoxide dismutase 1 (SOD1;
sc-271014), oxidative enzyme catalase (CAT; sc-271803) and GAPDH
(sc-365062 Santa Cruz Biotechnology, Inc., Dallas, TX, SA) at
dilution of 1:1,000 at 4°C overnight. The membranes were then
exposed to horseradish peroxidase-conjugated secondary antibodies
(sc-516102; Santa Cruz Biotechnology, Inc.) at dilution of 1:2,000
for 4 h at 4°C. The protein blots were detected using enhanced
chemiluminescence detection kits (GE Healthcare, Chicago, IL, USA),
and analyzed with ImageJ software version 1.46 (National Institutes
of Health, Bethesda, MD, USA).
Statistical analysis
Data were expressed as the mean ± standard deviation
for in vitro experiments and the mean ± standard error of
mean for in vivo experiments. Software SPSS 16.0 (SPSS,
Inc., Chicago. IL, USA) was used to analyze the data via one-way
analysis of variance, followed by a Duncan's multiple range test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
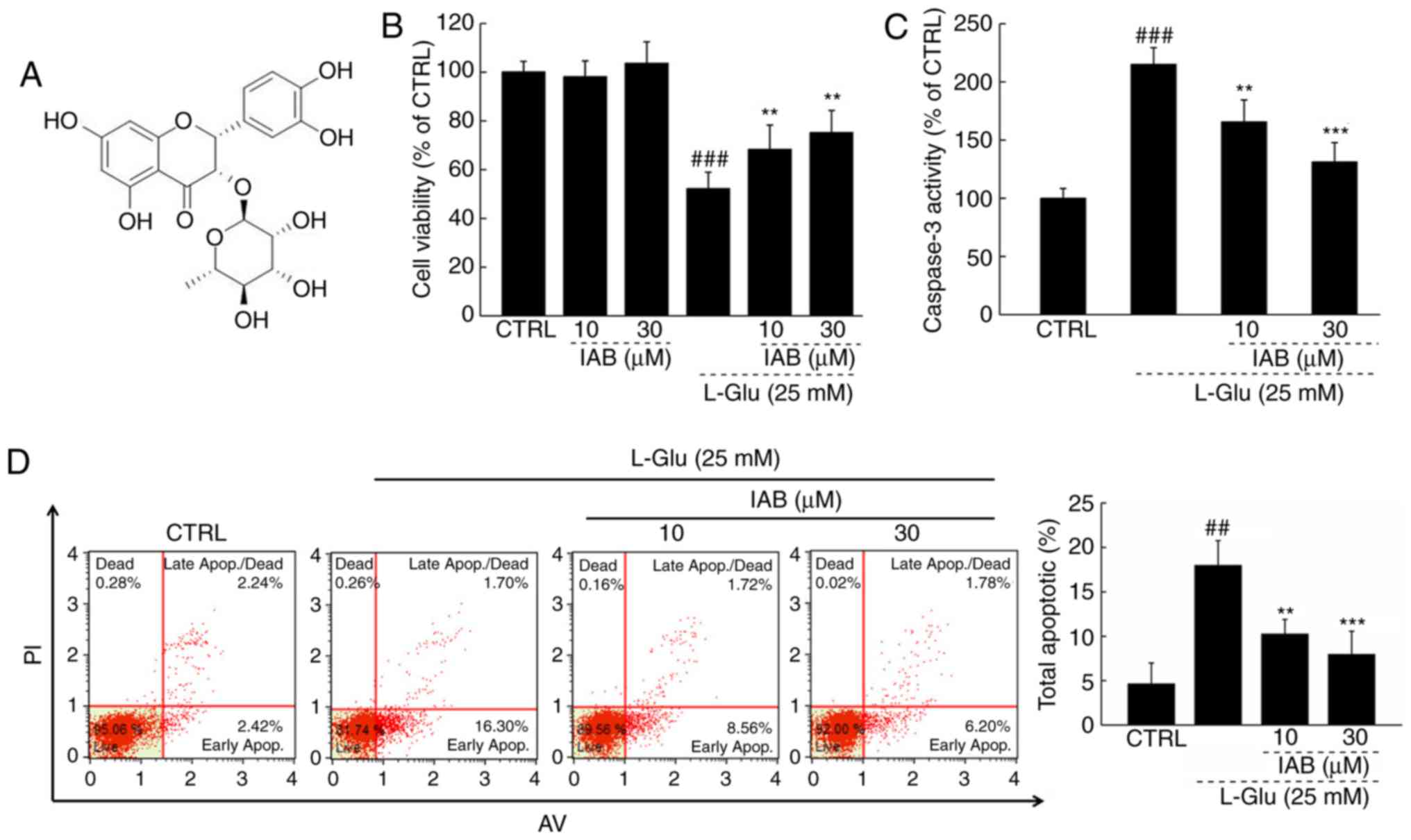
IAB protects PC12 cells against
L-Glu-induced damage
IAB significantly improved the viability of >30%
L-Glu-damaged PC12 cells compared with cells treated with L-Glu
alone (P<0.01; Fig. 1B);
however, IAB alone exhibited no signifi-cant effects on cell
viability in normal PC12 cells (Fig.
1B). Compared with L-Glu-treated PC12 cells, IAB, particularly
at 30 µM, significantly reduced caspase-3 activity by 39.1%
(P<0.001; Fig. 1C). PC12 cells
exposed to 25 mM L-Glu demonstrated an apoptosis rate of 18%, which
decreased to 7.98% in 30 µM IAB pre-treated PC12 cells
(P<0.05; Fig. 1D).
IAB restores ROS-mediated MMP dissipation
in L-Glu- damaged PC12 cells
A significant accumulation of intracellular ROS
(P<0.001; Fig. 2A) and
dissipation of MMP (P<0.001; Fig.
2B) were observed in PC12 cells exposed to 25 mM L-Glu for 12 h
compared with the control. Pre-incubation with IAB significantly
suppressed the over-accumulation of ROS, as indicated by the
reduced green fluorescence (P<0.05; Fig. 2A), and restored the dissipation of
MMP, as indicated by the enhanced red/green fluorescence
(P<0.01; Fig. 2B).
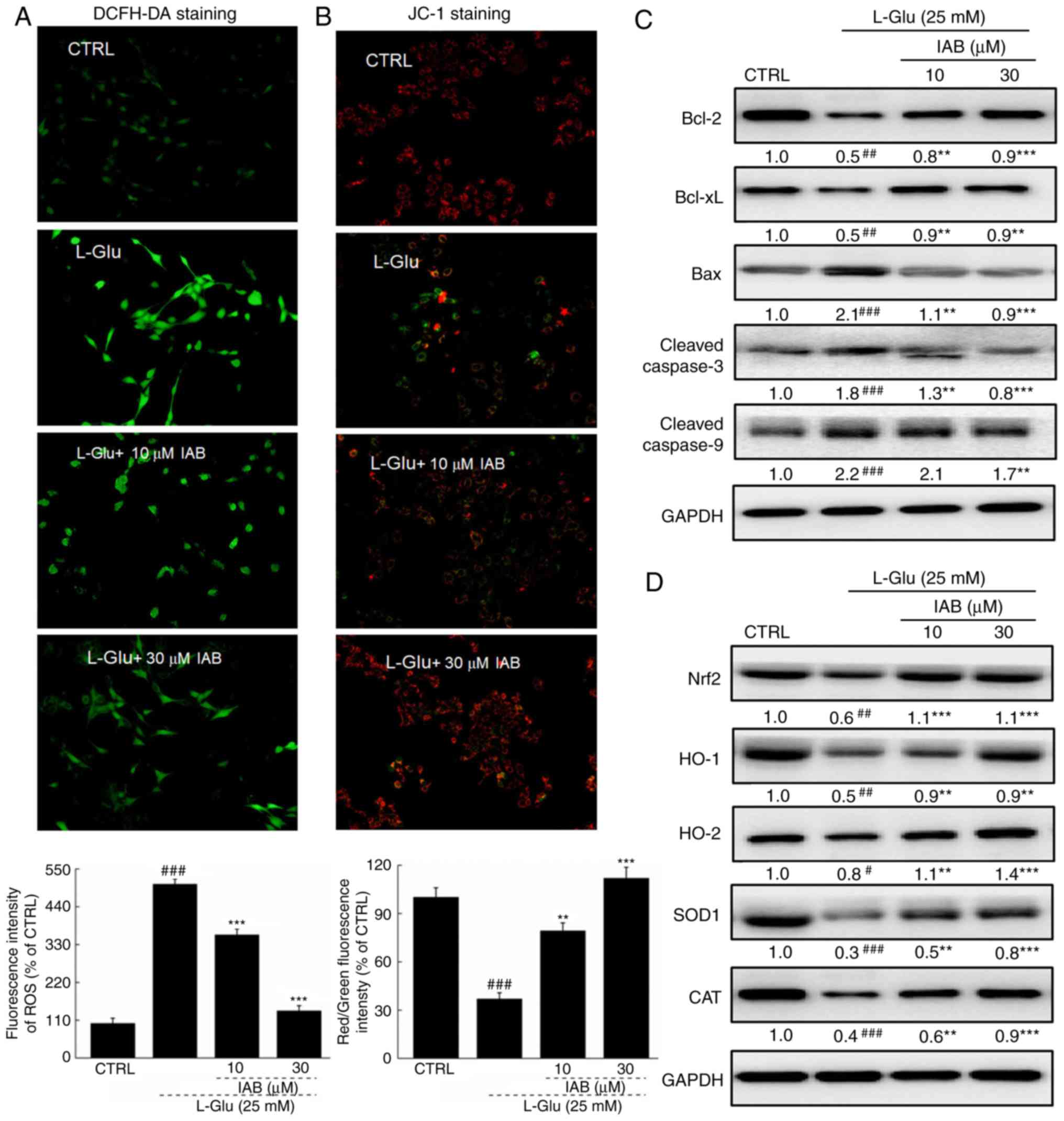 | Figure 2IAB ameliorates L-Glu-induced
mitochondrial apoptosis in PC12 cells via regulation the Nrf2
pathway. (A) IAB suppressed the over-accumulation of ROS in PC12
cells exposed to L-Glu for 12 h (n=9). (B) IAB restored
L-Glu-induced mitochondrial membrane potential dissipation after
12-h co-incubation (n=9). Data are expressed as mean ± standard
deviation. (n=9). (C) IAB enhanced the expression levels of Bcl-2
and Bcl-xL, and reduced the expression levels of Bax, cleaved
caspases-3 and -9 in PC12 cells exposed to L-Glu for 24 h (n=6).
(D) IAB strongly enhanced the expression levels of Nrf2, HO-1,
HO-2, SOD1 and CAT in PC12 cells exposed to L-Glu for 24 h (n=6).
Quantification data was normalized by GAPDH. The mean fold of band
intensity compared with CTRL group was presented respectively.
#P<0.05, ##P<0.01 and
###P<0.001 vs. CTRL. **P<0.01 and
***P<0.001 vs. L-Glu-treated cells. Bcl-2, B-cell
lymphoma-2; Bax, Bcl-2-associated X; Bcl-xL, Bcl-extra large; CAT
catalase; CTRL, control; HO, heme oxygenase; IAB, isoastilbin;
L-Glu, L-glutamic acid; Nrf2, NF-E2p45-related factor 2; ROS,
reactive oxygen species; SOD, superoxide dismutase. |
Significantly low expression levels of Bcl-2
(P<0.01) and Bcl-xL (P<0.01), and high expression levels of
Bax (P<0.001) and cleaved casapase-3 (P<0.001) and -9
(P<0.001) in PC12 cells were noted after 24-h exposure to L-Glu
compared with the control (Fig.
2C). Conversely, 3-h pre-incubation with IAB significantly
reversed these alterations induced by L-Glu (P<0.01, Fig. 2C).
L-Glu exposure resulted in significant reductions in
the expression of Nrf2 (P<0.01), HO-1 (P<0.01), HO-2
(P<0.05), SOD1 (P<0.001) and CAT (P<0.001) in PC12 cells
compared with the control, but were strongly enhanced by IAB
treatment (10 and 30 µM) after 24-h co-incubation
(P<0.01; Fig. 2D).
IAB alleviates the behavioral symptoms
and reduced the apoptotic rate, Aβ1-42 levels, and P-Tau
aggregations in brains of AD mice
The experimental protocol conducted with mice was
presented in Fig. 3A. In
addition, IAB did not exhibit adverse effects on organs, including
the brain (Fig. 3B), kidneys
(Fig. 3C), and spleen (Fig. 3D) of AD mice as analyzed by
H&E staining.
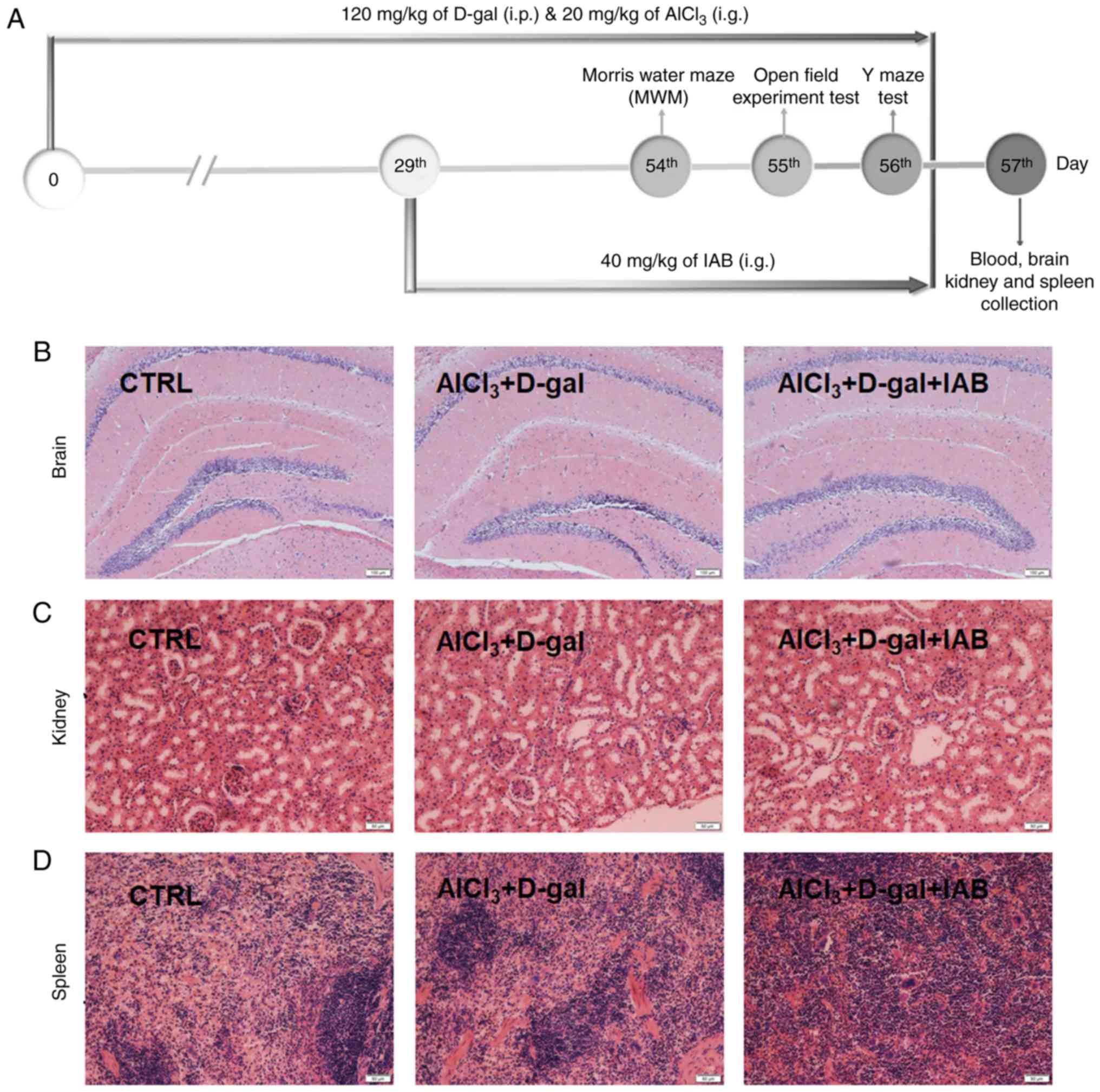 | Figure 3(A) Schematic of animal experiments.
No notable pathological alterations were noted in the (B) brain
(magnification, ×100; scale bar, 100 µm), (C) kidney
(magnification, ×200; scale bar, 50 µm) and (D) spleen
(magnification, ×200; scale bar, 50 µm) among all
experimental mice (n=6). CTRL, control; D-gal, D-galactose; IAB,
isoastilbin. |
The learning and memory abilities of mice with mice
were analyzed by behavioral tests. In an open field test, a
significant time spent in the central area with chaotic movement
without purpose was observed in AD mice; however, this duration was
significantly reduced in IAB-treated mice (P<0.01; Fig. 4A). In the Morris water maze test,
compared with heathy mice, AD mice spent more time finding the
platform hidden in the water and exhibited more chaotic movement.
Conversely, IAB administration led to a 32.8% reduction in escape
latency time (P<0.01; Fig.
4B). In the Y maze test, it took AD mice nearly twice as long
to find hidden food compared with the heathy mice (P<0.001;
Fig. 4C). IAB administration led
to a 30.9% reduction in the seeking time for hidden food
(P<0.01; Fig. 4C).
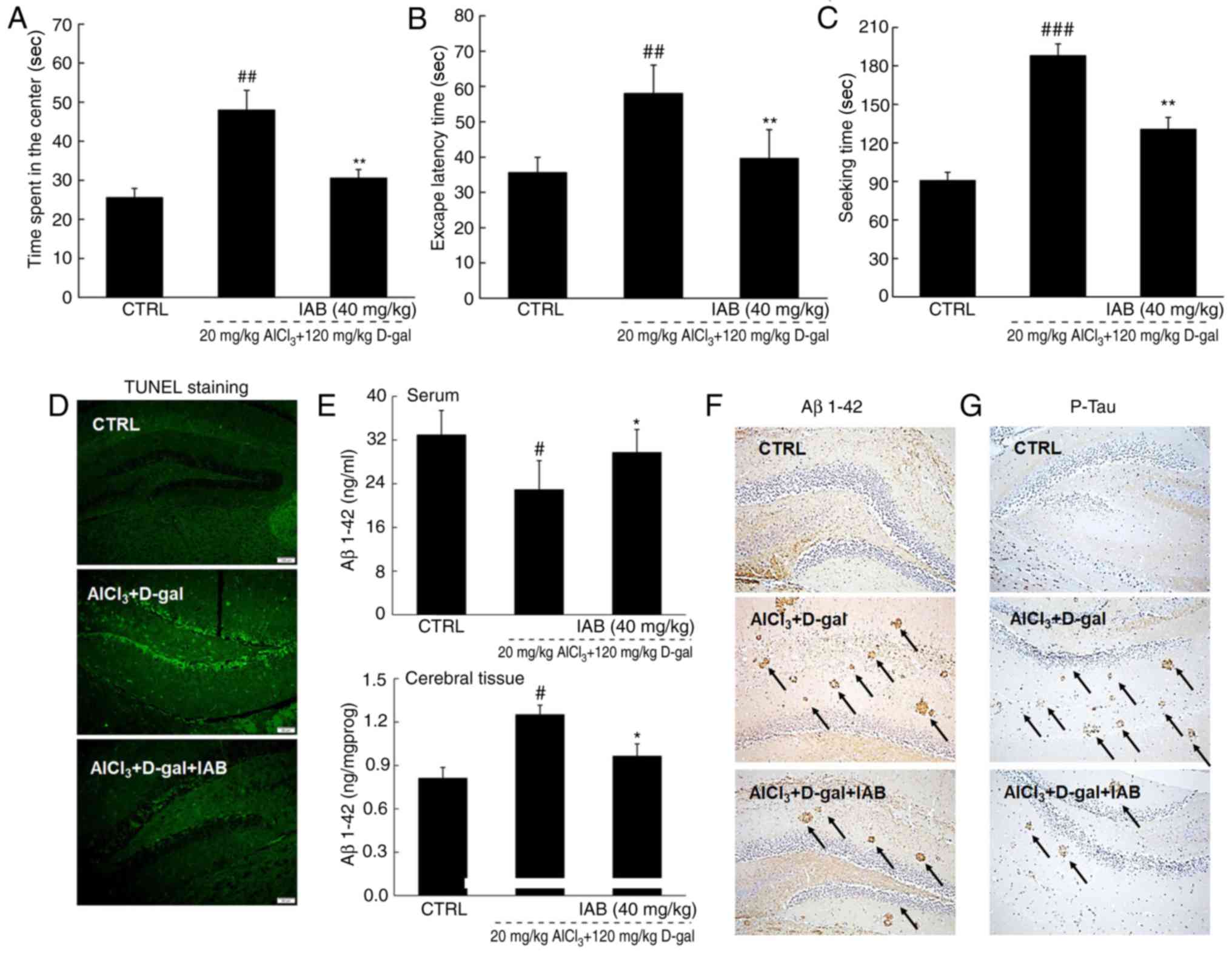 | Figure 4IAB improves AD-like behaviors, and
inhibits Aβ1-42 deposition and p-Tau accumulation in mice with
D-gal and AlCl3-induced AD. Compared with
vehicle-treated AD mice, 28-day administration of IAB reduced (A)
the escape latency time in the Morris water maze test, (B) the time
spent in the central area in open field test and (C) the time to
seek hidden food in the Y maze test. Data are expressed as mean ±
standard error of the mean. (n=12). ##P<0.01 and
###P<0.001 vs. CTRL, **P<0.01 vs.
vehicle-treated AD mice. (D) IAB suppressed the apoptosis in the
brain of AD mice (n=12). (E) IAB enhanced the serum levels of
Aβ1-42, and reduced the cerebral levels of Aβ1-42 in AD mice as
detected via ELISA. Data are expressed as mean ± standard error of
the mean. (n=6). #P<0.05 vs. CTRL,
*P<0.05 vs. vehicle-treated AD mice. (F) Aβ deposits
in the brain of AD mice were significantly decreased by IAB
detecting by immunohistochemistry. Magnification, ×200 (n=6).
Arrows show Aβ deposit. (G) Increased expression of p-Tau in the
brain of AD mice was significantly suppressed by IAB as detected by
immunohistochemistry. Magnification, ×200 (n=6). Arrows show
neurofibrillary tangle induced by P-Tau. Aβ, amyloid β; AD,
Alzheimer's disease; CTRL, control; D-gal, D-galactose; IAB,
isoastilbin. |
The apoptotic status of neurons was detected by
TUNEL staining, and a number of TUNEL-positive cells were observed
in the brains of the vehicle-treated AD mice, as indicated by the
enhanced green fluorescence intensity (Fig. 4D). The number of TUNEL-positive
cells was notably reduced following 4 weeks of treatment with IAB
(Fig. 4D).
Central to pathogenesis of AD, the levels of Aβ, the
principal constituent of neuritic plaques (28), were detected in the present study.
Compared with healthy mice, significantly low serum levels of
Aβ1-42 and high cerebral levels of Aβ1-42 were noted in the
vehicle-treated AD mice compared with in healthy mice (P<0.05;
Fig. 4E); however, IAB
administration for 4 weeks resulted in a 29.8% increase in the
serum levels of Aβ1-42 (P<0.05) and a 22.9% reduction in the
cerebral levels of Aβ1-42 (P<0.05; Fig. 4E). Immunohistochemical analysis
further confirmed that the suppressive activities of IAB on Aβ1-42
in the mouse brains resulted in fewer neuritic plaques in
IAB-treated mice than in AD mice (Fig. 4F). Furthermore, the expression
levels of p-Tau were notably enhanced in the brains of
vehicle-treated AD mice, but were significantly reduced upon
treatment with IAB (Fig. 4G).
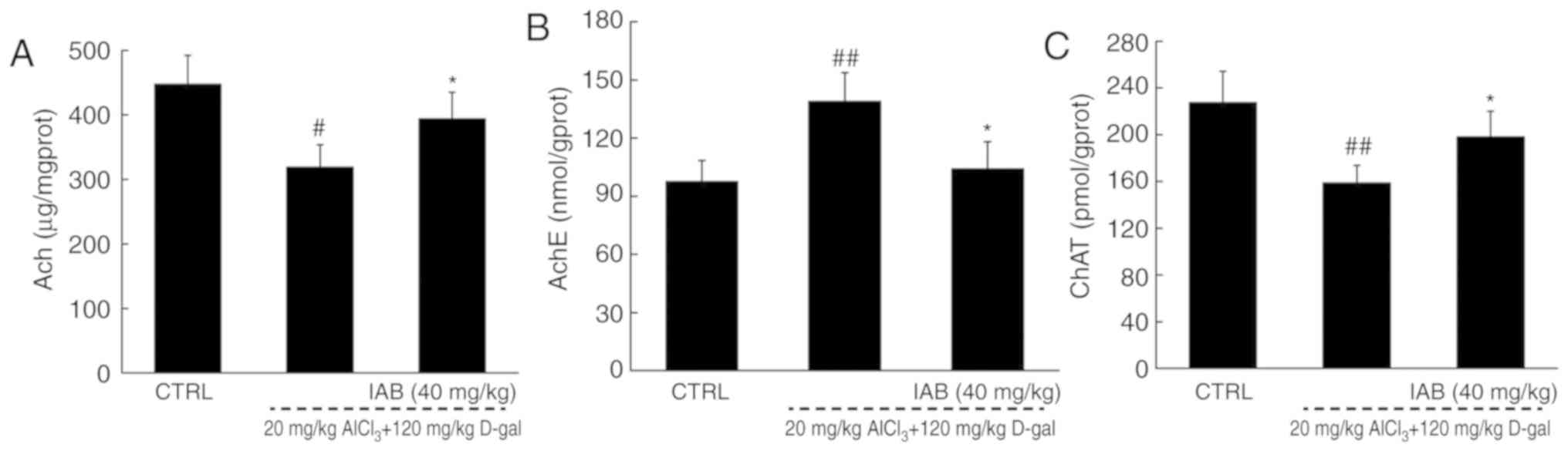
IAB regulates the levels of cholinergic
neurotransmitters in mice with AD
The dysregulated levels of cholinergic
neurotransmitters is responsible for the characteristic memory
impairment associated with AD (29). Significant reductions in the
levels of Ach (P<0.05) and ChAT (P<0.01), and increased
levels of AchE (P<0.01) were observed in the brains of the
vehicle-treated AD mice compared with the control (Fig. 5). After 4 weeks of treatment with
IAB, a 24.9 and 25.3% increase in the cerebral levels of Ach
(P<0.05; Fig. 5A) and ChAT
(P<0.05; Fig. 5C) were
observed, respectively, and a 30.6% reduction in the cerebral
levels of AchE were detected (P<0.05; Fig. 5B). These findings suggests that
may IAB improve cholinergic dysfunction.
IAB regulates Nrf2 signaling to modulate
the levels of pro- and antioxidative factors in mice with AD
The brain is sensitive to oxidative stress;
consequently, the over-accumulation of ROS may affect the
expression of β-amyloid precursor protein (APP) and mitochondrial
DNA, which further leads to neuronal apoptosis (30). Compared with in healthy mice,
significantly higher levels of ROS, and notably lower levels of SOD
and GSH-Px were detected in the serum and brains of vehicle-treated
AD mice (P<0.05; Fig. 6A-F).
In the serum of AD mice, IAB significantly reduced ROS levels by
17.9% (P<0.05; Fig. 6A), and
enhanced the levels of SOD by 21.4% (P<0.05; Fig. 6B) and GSH-Px by 71.6% (P<0.01;
Fig. 6C). In the brains of mice
with AD, IAB significantly reduced ROS levels by 29.7% (P<0.01;
Fig. 6D), and enhanced the levels
of SOD by 94.2% (P<0.001; Fig.
6E) and GSH-Px by 80.7% (P<0.001; Fig. 6F).
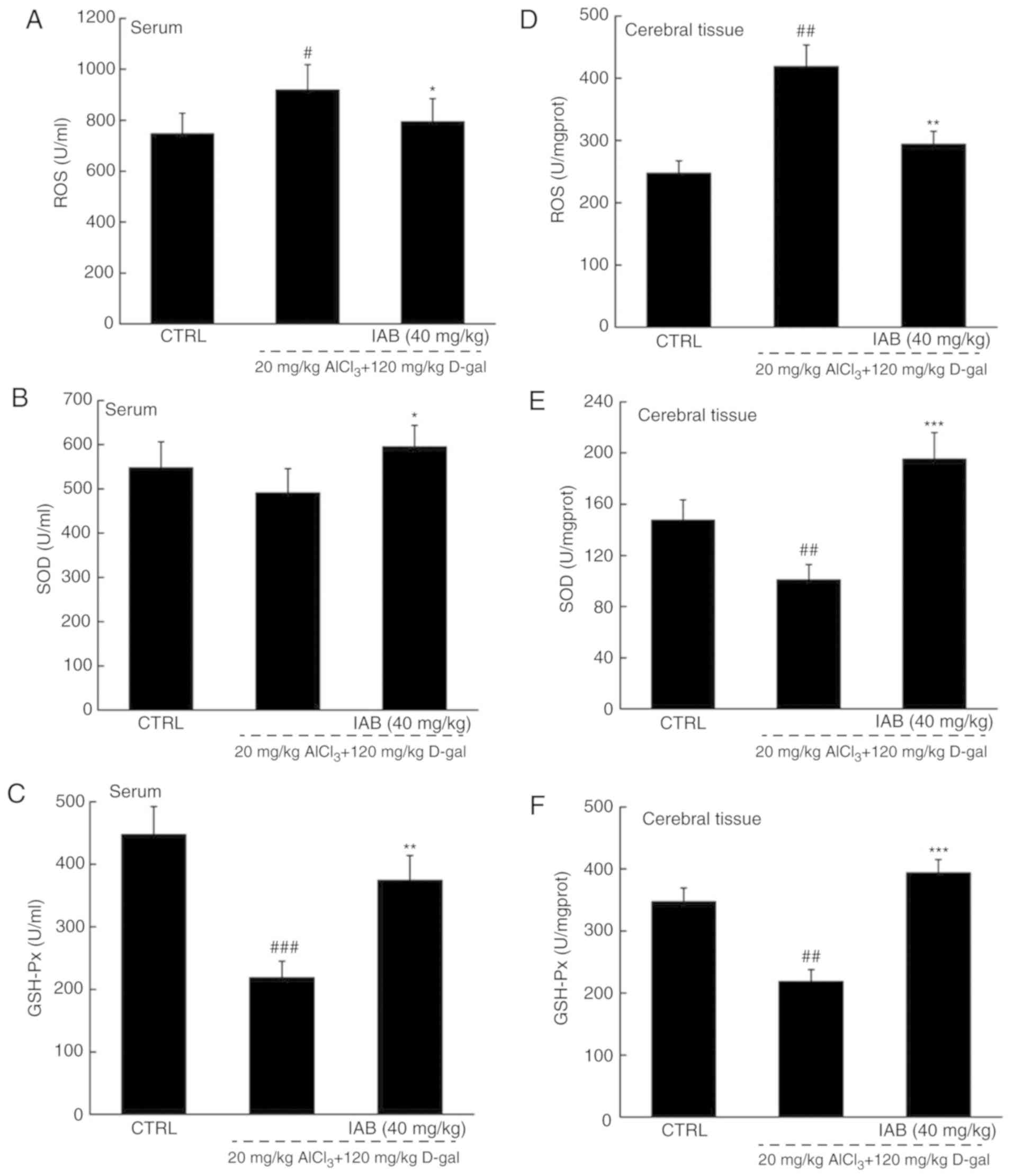 | Figure 6IAB relieves oxidative stress in mice
with AD via the regulation of the Nrf2 pathway. Compared with
vehicle-treated AD mice, 28-day administration of IAB reduced the
levels of (A and D) ROS, and enhanced the levels of (B and E) SOD
and (C and F) GSH-Px in the serum and whole brains of mice with AD.
Data are expressed as mean ± standard error of the mean (n=6). (G)
IAB regulated the expression of proteins associated apoptosis and
oxidative stress in the brains of AD mice (n=6). Quantification
data was normalized by GAPDH. The mean fold of band intensity
compared with CTRL group was presented respectively.
#P<0.05, ##P<0.01 and
###P<0.001 vs. CTRL mice. *P<0.05,
**P<0.01 and ***P<0.001 vs.
vehicle-treated AD mice. AD, Alzheimer's disease; Bcl-2, B-cell
lymphoma-2; Bax, Bcl-2-associated X; Bcl-xL, Bcl-extra large; CAT
catalase; CTRL, control; D-gal, D-galactose; GSH-Px, glutathione
peroxidase; HO, heme oxygenase; IAB, isoastilbin; L-Glu, L-glutamic
acid; Nrf2, NF-E2p45-related factor 2; ROS, reactive oxygen
species; SOD, superoxide dismutase. |
In the brains of AD mice, notably increased
expression levels of pro-apoptotic proteins, including Bax
(P<0.001; Fig. 6G) and cleaved
caspases-3 (P<0.01; Fig. 6G)
and -9 (P<0.001; Fig. 6G), and
the low levels of anti-apoptotic proteins, such as Bcl-2
(P<0.01) and Bcl-xL (P<0.01), were observed (Fig. 6G). The expression levels were
similar to the control after 4 weeks of treatment with IAB
(P<0.01; Fig. 6G), all were
strongly improved to a healthy standard after four-week IAB
administration (Fig. 6G).
Additionally, the notably reduced levels of proteins associated
with antioxidation, including Nrf2 (P<0.001), HO-1 (P<0.01),
HO-2 (P<0.01), SOD1 (P<0.001) and CAT (P<0.001), were
detected in the brains of mice with AD (Fig. 6G). On the contrary, the suppressed
expression of these proteins induced by the occurrence of oxidative
stress within the AD process markedly recovered following IAB
treatment, which resulted in >50% upregulation (P<0.01;
Fig. 6G).
Discussion
Apoptotic neurons have been detected in patients
with AD (31). Based on our
preliminary experiments, the neuroprotective effects of numerous
natural compounds including IAB, astilbin and curculigoside were
screened in L-Glu-damaged PC12 cells, and the results revealed that
IAB exhibited the best protective effects than the other compounds.
In the present study, IAB significantly reduced L-Glu-induced
apoptosis of PC12 cells and apoptosis in the brains of mice with
AD, which was induced with AlCl3 and D-gal. The notably
high levels of Glu in brains is responsible for the occurrence of
oxidative stress, as indicated by the over-accumulation of ROS,
which opens mitochondrial permeability transition pores, resulting
in the further dissipation of MMP (32). As of the feedback loop between the
increased production of ROS and mitochondrial dysfunction,
pro-apoptotic cytokines, such as cytochrome c are released
from the mitochondria and bind to apoptotic protease activating
factor 1 (33,34). This complex recruits caspase-9 to
activate caspase-3 via proteolytic cleavage (35,36). Consequently, caspase-3 is the
executor of the apoptotic program (37). In L-Glu-damaged PC12 cells and the
brains of mice with AD, IAB significantly reduced the expression of
caspase-9 and -3, and regulated the expression of Bcl-2 family
members, which suggests the inhibition of mitochondria-mediated
apoptosis. Bcl-2 family members, which are located in the membrane
of mitochondria, affect mitochondrial apoptosis (38). The ratio of pro- and
anti-apoptotic members can directly reflect the function of
mitochondria (39,40). Upregulated Bax levels accelerate
apoptosis by permeabilizing mitochondria (41). Conversely, the enhanced expression
of Bcl-2 and Bcl-xL may help improve MMP (39).
The transcription factor Nrf2 supports the
structural and functional integrity of the mitochondria (42). In response to apoptosis, Nrf-2
becomes activated and released, and binds to corresponding proteins
in the nucleus to form dimers (43). This results in the activation and
transactivation of HO-1 and HO-2, which consequently leads to the
enhanced activities of SOD and CAT (44,45). As an antioxidant enzyme, SOD1
helps suppress the toxicity of superoxide radicals (46). HO-1 and -2 can be activated by
Nrf2 under conditions of oxidative stress, which provides powerful
protection against oxidative injury (47). These cascade activations may be
associated with the etiology of AD and provide the possibility for
screening therapeutic targets; we speculate that IAB enhanced
cascade activation within the Nrf2 pathway, which may be involved
in the neuroprotection of IAB against AD.
During the development of AD in mice, long-term
D-gal and AlCl3 administration leads to the accumulation
of ROS and damage to polyunsaturated fatty acids contained in the
brain, which is responsible for the appearance of AD-like symptoms,
including cognitive disorders and dysmnesia (48,49). During this process, the
aggregation of Aβ can be observed, which induces cascade reactions,
such as mitochondrial dysfunction (40,50), and triggers the pathological Tau,
which in turn contributes to the formation of neurofibrillary
tangles (27). However, Aβ plaque
deposition in the brain disturbs the anti- and pro-oxidation
equilibrium and enhances ROS production in particular (51). Additionally, IAB not only
suppressed the abnormal accumulation of Aβ1-42 and p-Tau in the
mouse brain, but also regulated the redox system via modulating the
expression levels including Bcl-2, Bcl-xL, Bax, cleaved caspase-9
and -3 and the activation of the Nrf2 pathway in mice with AD.
Combined with the occurrence of oxidative stress, oxidized
biomolecules and ROS accumulate in cells, which consequently
promotes amyloidogenic APP processing and Aβ overproduction
(52). Under these conditions,
Nrf2 activates a series of kinases, including SOD1, CAT, and HO-1
and -2, which in eliminate the accumulated ROS (53). SOD and GSH-Px, which are
representative endogenous antioxidants, have been recognized as the
first-line drugs to defend against oxidative damage (54). Combined with the suppressed ROS
generation, the cerebral deposition of Aβ can be removed from
brains, as evidenced by enhanced peripheral levels (55). As of the activated Nrf2, the
inhibited deposition of Aβ leads to the fragmentation of p-Tau
aggregation (56,57). These data suggest that the ability
of IAB to improve the cognitive performance of mice with AD may be
associated with Nrf2-mediated oxidative stress.
The loss of cholinergic neurotransmission in
cerebral areas is responsible for the cognitive deterioration in
patients with AD (58).
Abnormally low levels of Ach and ChAT, and upregulated levels of
AchE were noted in the brains of mice with AD in the present study,
all of which were notably restored by IAB. Ach, controlled by the
terminating enzyme AchE and the synthesizing enzyme ChAT (59), stimulates cholinergic function,
and contributes to the storage and recovery of long-term memory
(60); due to increased AchE and
decreased ChAT, cognitive function gradually decreases (29). Under oxidative stress, increased
levels of AchE promote the formation and deposition of Aβ plaques
(61), which severely damages the
cholinergic system (62). Based
on the findings of the present study, the protective role of IAB in
the cholinergic system against AD were proposed; however, the
association between the cholinergic transmitter and Nrf2-mediated
oxidative stress was not clearly elaborated. This will be
investigated in our ongoing research.
In conclusion, L-Glu-induced apoptosis of PC12
cells, and AlCl3- and D-gal-induced AD mice models were
generated in the present study. To the best of our knowledge, the
present study is the first to report of the neuroprotection of IAB,
and this property, may be, at least partly, associated with the
regulation of IAB on Nrf2-mediated oxidative stress. The results
provide insight for further study into the protective effects of
IAB and its possibility of clinical application in AD in the
future.
Funding
The present study was supported by the Special
Projects of Cooperation among Jilin University and Jilin (China;
grant no. SXGJSF2017-1) and the Science Foundation in Jilin of
China (China; grant no. 20180101098JC).
Availability of data and materials
All data generated and analyzed during the present
study are included in this published article.
Authors' contributions
HB made substantial contributions to the design of
the present study. HY, BY, QC and CW performed the experiments and
analyzed the data; HB, HY and BY wrote the paper, and HB revised
the paper.
Ethics approval and consent to
participate
The experimental animal protocol was approved by the
Animal Ethics Committee of Jilin University (no. 20170206).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Ach
|
acetylcholine
|
|
AchE
|
acetylcholinesterase
|
|
AlC13
|
aluminum trichloride
|
|
Aβ
|
amyloid β
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
Bcl-xL
|
B-cell lymphoma-extra large
|
|
CAT
|
catalase
|
|
ChAT
|
choline acetyltransferase
|
|
D-gal
|
D-galactose
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
FBS
|
fetal bovine serum
|
|
GSH-Px
|
glutathione peroxidase
|
|
HO-1
|
heme oxygenase-1
|
|
HO-2
|
heme oxygenase-2
|
|
HS
|
horse serum
|
|
L-Glu
|
L-glutamate
|
|
MMP
|
mitochondrial transmembrane
potential
|
|
MTT
|
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium
bromide
|
|
Nrf2
|
NF-E2-related factor 2
|
Acknowledgments
Not applicable.
References
|
1
|
Cheignon C, Tomas M, Bonnefont-Rousselot
D, Faller P, Hureau C and Collin F: Oxidative stress and the
amyloid beta peptide in Alzheimer's disease. Redox Biol.
14:450–464. 2018. View Article : Google Scholar
|
|
2
|
Coimbra JRM, Marques DFF, Baptista SJ,
Pereira CMF, Moreira PI, Dinis TCP, Santos AE and Salvador JAR:
Highlights in BACE1 Inhibitors for Alzheimer's disease treatment.
Front Chem. 6:1782018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosello A, Warnes G and Meier UC: Cell
death pathways and autophagy in the central nervous system and its
involvement in neurodegeneration, immunity and central nervous
system infection: To die or not to die-that is the question. Clin
Exp Immunol. 168:52–57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Angelova PR and Abramov AY: Role of
mitochondrial ROS in the brain: From physiology to
neurodegeneration. FEBS Lett. 592:692–702. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
de Oliveira RB, Gravina FS, Lim R, Brichta
AM, Callister RJ and van Helden DF: Heterogeneous responses to
antioxidants in noradrenergic neurons of the Locus coeruleus
indicate differing susceptibility to free radical content. Oxid Med
Cell Longev. 2012:8202852012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Daulatzai MA: Cerebral hypoperfusion and
glucose hypometabolism: Key pathophysiological modulators promote
neurodegeneration, cognitive impairment, and Alzheimer's disease. J
Neurosci Res. 95:943–972. 2017. View Article : Google Scholar
|
|
7
|
Nesi G, Sestito S, Digiacomo M and
Rapposelli S: Oxidative stress, mitochondrial abnormalities and
proteins deposition: Multitarget approaches in Alzheimer's disease.
Curr Top Med Chemy. 17:3062–3079. 2017.
|
|
8
|
Tyagi N, Ovechkin AV, Lominadze D, Moshal
KS and Tyagi SC: Mitochondrial mechanism of microvascular
endothelial cells apoptosis in hyperhomocysteinemia. J Cell
Biochem. 98:1150–1162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu X, Wang J, Lu C, Zhu C, Qian B, Li Z,
Liu C, Shao J and Yan J: The role of lysosomes in BDE 47-mediated
activation of mitochondrial apoptotic pathway in HepG2 cells.
Chemosphere. 124:10–21. 2015. View Article : Google Scholar
|
|
10
|
Luo P, Fei F, Zhang L, Qu Y and Fei Z: The
role of glutamate receptors in traumatic brain injury: Implications
for postsynaptic density in pathophysiology. Brain Res Bull.
85:313–320. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dinkova-Kostova AT and Abramov AY: The
emerging role of Nrf2 in mitochondrial function. Free Radic Biol
Med. 88:179–188. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Joshi G, Gan KA, Johnson DA and Johnson
JA: Increased Alzheimer's disease-like pathology in the APP/ PS1ΔE9
mouse model lacking Nrf2 through modulation of autophagy. Neurobiol
Aging. 36:664–679. 2015. View Article : Google Scholar
|
|
13
|
McDade E and Bateman RJ: Stop alzheimer's
before it starts. Nature. 547:153–155. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Querfurth HW and LaFerla FM: Alzheimer's
Disease. N Engl J Med. 362:329–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jesky R and Hailong C: Are herbal
compounds the next frontier for alleviating learning and memory
impairments? An integrative look at memory, dementia and the
promising therapeutics of traditional Chinese medicines. Phytother
Res. 25:1105–1118. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Man SC, Chan KW, Lu JH, Durairajan SS, Liu
LF and Li M: Systematic review on the efficacy and safety of herbal
medicines for vascular dementia. Evid Based Complement Alternat
Med. 2012:4262152012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Wang J, Wang C, Li Z, Liu X,
Zhang J, Lu J and Wang D: Pharmacological basis for the use of
evodiamine in alzheimer's disease: Antioxidation and antiapoptosis.
Int J Mol Sci. 19:pii: E1527. 2018.
|
|
18
|
Hu S, Wang D, Zhang J, Du M, Cheng Y, Liu
Y, Zhang N, Wang D and Wu Y: Mitochondria related pathway is
essential for polysaccharides purified from Sparassis crispa
mediated neuro-protection against glutamate-induced toxicity in
differentiated PC12 cells. Int J Mol Sci. 17:pii: E133. 2016.
View Article : Google Scholar
|
|
19
|
An S, Lu W, Zhang Y, Yuan Q and Wang D:
Pharmacological basis for use of Armillaria mellea polysaccharides
in Alzheimer's disease: Antiapoptosis and antioxidation. Oxid Med
Cell Longev. 2017:41845622017. View Article : Google Scholar :
|
|
20
|
Du Q, Li L and Jerz G: Purification of
astilbin and isoastilbin in the extract of Smilax glabra rhizome by
high-speed counter-current chromatography. J Chromatogr A.
1077:98–101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou X, Xu Q, Li JX and Chen T: Structural
revision of two flavanonol glycosides from Smilax glabra. Planta
Med. 75:654–655. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu CL, Zhu W, Wang M, Xu XJ and Lu CJ:
Antioxidant and anti-inflammatory activities of phenolic-enriched
extracts of Smilax glabra. Evid Based Complement Alternat Med.
2014:9104382014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang D, Li S, Chen J, Liu L and Zhu X: The
effects of astilbin on cognitive impairments in a transgenic mouse
model of Alzheimer's disease. Cell Mol Neurobiol. 37:695–706. 2017.
View Article : Google Scholar
|
|
24
|
Zhang X, Chen Y, Cai G, Li X and Wang D:
Carnosic acid induces apoptosis of hepatocellular carcinoma cells
via ROS-mediated mitochondrial pathway. Chem Biol Interact.
277:91–100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li Z, Chen X, Lu W, Zhang S, Guan X, Li Z
and Wang D: Anti-oxidative stress activity is essential for Amanita
caesarea mediated neuroprotection on glutamate-induced apoptotic
HT22 cells and an Alzheimer's disease mouse model. Int J Mol Sci.
18:pii: E1623. 2017. View Article : Google Scholar
|
|
26
|
Parikh A, Kathawala K, Li J, Chen C, Shan
Z, Cao X, Wang YJ, Garg S and Zhou XF: Self-nanomicellizing solid
dispersion of edaravone: Part II: In vivo assessment of efficacy
against behavior deficits and safety in Alzheimer's disease model.
Drug Des Devel Ther. 12:2111–2128. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao JM, Li L, Chen L, Shi Y, Li YW, Shang
HX, Wu LY, Weng ZJ, Bao CH and Wu HG: Comparison of the analgesic
effects between electro-acupuncture and moxibustion with visceral
hypersensitivity rats in irritable bowel syndrome. World J
Gastroenterol. 23:2928–2939. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chun KA: Beta-amyloid imaging in dementia.
Yeungnam Univ J Med. 35:1–6. 2018. View Article : Google Scholar
|
|
29
|
Yamini P, Ray RS and Chopra K: Vitamin
D3 attenuates cognitive deficits and neuroinflammatory
responses in ICV-STZ induced sporadic Alzheimer's disease.
Inflammopharmacology. 26:39–55. 2018. View Article : Google Scholar
|
|
30
|
Tönnies E and Trushina E: Oxidative
stress, synaptic dysfunction, and Alzheimer's disease. J Alzheimers
Dis. 57:1105–1121. 2017. View Article : Google Scholar
|
|
31
|
Koh CH, Whiteman M, Li QX, Halliwell B,
Jenner AM, Wong BS, Laughton KM, Wenk M, Masters CL, Beart PM, et
al: Chronic exposure to U18666A is associated with oxidative stress
in cultured murine cortical neurons. J Neurochem. 98:1278–1289.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Resseguie EA, Staversky RJ, Brookes PS and
O'Reilly MA: Hyperoxia activates ATM independent from mitochondrial
ROS and dysfunction. Redox Biol. 5:176–185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Skulachev VP: Cytochrome c in the
apoptotic and antioxidant cascades. FEBS Lett. 423:275–280. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Reubold TF and Eschenburg S: A molecular
view on signal transduction by the apoptosome. Cell Signal.
24:1420–1425. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Allan LA and Clarke PR: Apoptosis and
autophagy: Regulation of caspase-9 by phosphorylation. FEBS J.
276:6063–6073. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Espin R, Roca FJ, Candel S, Sepulcre MP,
González-Rosa JM, Alcaraz-Pérez F, Meseguer J, Cayuela ML, Mercader
N and Mulero V: TNF receptors regulate vascular homeostasis in
zebrafish through a caspase-8, caspase-2 and P53 apoptotic program
that bypasses caspase-3. Dis Model Mech. 6:383–396. 2013.
View Article : Google Scholar :
|
|
38
|
Gross A and Katz SG: Non-apoptotic
functions of BCL-2 family proteins. Cell Death Differ.
24:1348–1358. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang X, Wu J, Yu C, Tang Y, Liu J, Chen H,
Jin B, Mei Q, Cao S and Qin D: Lychee seed saponins improve
cognitive function and prevent neuronal injury via inhibiting
neuronal apoptosis in a rat model of Alzheimer's disease.
Nutrients. 9:pii: E105. 2017. View Article : Google Scholar
|
|
40
|
Tong Y, Bai L, Gong R, Chuan J, Duan X and
Zhu Y: Shikonin protects PC12 cells against beta-amyloid
peptide-induced cell injury through antioxidant and antiapoptotic
activities. Sci Rep. 8:262018. View Article : Google Scholar
|
|
41
|
Lai YC, Li CC, Sung TC, Chang CW, Lan YJ
and Chiang YW: The role of cardiolipin in promoting the membrane
pore-forming activity of BAX oligomers. Biochim Biophys Acta
Biomembr. 1861:268–280. 2019. View Article : Google Scholar
|
|
42
|
O'Connell MA and Hayes JD: The Keap1/Nrf2
pathway in health and disease: From the bench to the clinic.
Biochem Soc Trans. 43:687–689. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chu H, Yu H, Ren D, Zhu K and Huang H:
Plumbagin exerts protective effects in nucleus pulposus cells by
attenuating hydrogen peroxide-induced oxidative stress,
inflammation and apoptosis through NF-κB and Nrf-2. Int J Mol Med.
37:1669–1676. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu L, Liu Z, Feng Z, Hao J, Shen W, Li X,
Sun L, Sharman E, Wang Y, Wertz K, et al: Hydroxytyrosol protects
against oxidative damage by simultaneous activation of
mitochondrial biogenesis and phase II detoxifying enzyme systems in
retinal pigment epithelial cells. J Nutr Biochem. 21:1089–1098.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Khalaj L, Nejad SC, Mohammadi M, Zadeh SS,
Pour MH, Ahmadiani A, Khodagholi F, Ashabi G, Alamdary SZ and
Samami E: Gemfibrozil pretreatment proved protection against acute
restraint stress-induced changes in the male rats' hippocampus.
Brain Res. 1527:117–130. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kaur SJ, McKeown SR and Rashid S: Mutant
SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene.
577:109–118. 2016. View Article : Google Scholar
|
|
47
|
Kansanen E, Kuosmanen SM, Leinonen H and
Levonen AL: The Keap1-Nrf2 pathway: Mechanisms of activation and
dysregulation in cancer. Redox Biol. 1:45–49. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Qu Z, Zhang J, Yang H, Huo L, Gao J, Chen
H and Gao W: Protective effect of tetrahydropalmatine against
d-galactose induced memory impairment in rat. Physiol Behav.
154:114–125. 2016. View Article : Google Scholar
|
|
49
|
Jiang T, Sun Q and Chen S: Oxidative
stress: A major pathogenesis and potential therapeutic target of
antioxidative agents in Parkinson's disease and Alzheimer's
disease. Prog Neurobiol. 147:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xu T, Niu C, Zhang X and Dong M:
β-Ecdysterone protects SH-SY5Y cells against β-amyloid-induced
apoptosis via c-Jun N-terminal kinase- and Akt-associated
complementary pathways. Lab Invest. 98:489–499. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim DI, Lee KH, Gabr AA, Choi GE, Kim JS,
Ko SH and Han HJ: Aβ-Induced Drp1 phosphorylation through Akt
activation promotes excessive mitochondrial fission leading to
neuronal apoptosis. Biochim Biophys Acta. 1863:2820–2834. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Leuner K, Schütt T, Kurz C, Eckert SH,
Schiller C, Occhipinti A, Mai S, Jendrach M, Eckert GP, Kruse SE,
et al: Mitochondrion-derived reactive oxygen species lead to
enhanced amyloid beta formation. Antioxid Redox Signal.
16:1421–1433. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fujita K, Yamafuji M, Nakabeppu Y and Noda
M: Therapeutic approach to neurodegenerative diseases by medical
gases: Focusing on redox signaling and related antioxidant enzymes.
Oxid Med Cell Longev. 2012:3242562012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Islam MT: Oxidative stress and
mitochondrial dysfunction-linked neurodegenerative disorders.
Neurol Res. 39:73–82. 2017. View Article : Google Scholar
|
|
55
|
Fei M, Jianghua W, Rujuan M, Wei Z and
Qian W: The relationship of plasma Aβ levels to dementia in aging
individuals with mild cognitive impairment. J Neurol Sci.
305:92–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Park SY and Ferreira A: The generation of
a 17 kDa neurotoxic fragment: An alternative mechanism by which tau
mediates beta-amyloid-induced neurodegeneration. J Neurosci.
25:5365–5375. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Reifert J, Hartung-Cranston D and
Feinstein SC: Amyloid beta-mediated cell death of cultured
hippocampal neurons reveals extensive Tau fragmentation without
increased full-length tau phosphorylation. J Biol Chem.
286:20797–20811. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bartus RT: On neurodegenerative diseases,
models, and treatment strategies: Lessons learned and lessons
forgotten a generation following the cholinergic hypothesis. Exp
Neurol. 163:495–529. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ferreira-Vieira TH, Guimaraes IM, Silva FR
and Ribeiro FM: Alzheimer's disease: Targeting the cholinergic
system. Curr Neuropharmacol. 14:101–115. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Deiana S, Platt B and Riedel G: The
cholinergic system and spatial learning. Behav Brain Res.
221:389–411. 2011. View Article : Google Scholar
|
|
61
|
Lushchekina S, Kots E, Novichkova D,
Petrov K and Masson P: Role of Acetylcholinesterase in β-amyloid
aggregation studied by accelerated molecular dynamics.
Bionanoscience. 7:396–402. 2017. View Article : Google Scholar
|
|
62
|
Nitta A, Itoh A, Hasegawa T and Nabeshima
T: beta-Amyloid protein-induced Alzheimer's disease animal model.
Neurosci Lett. 170:63–66. 1994. View Article : Google Scholar : PubMed/NCBI
|